
Desorption Kinetics of Legacy Soil Phosphorus: Implications for Non-Point Transport and Plant Uptake


Abstract
:1. Introduction
1.1. Background
1.2. Soil P Desorption Theory
2. Materials and Methods
2.1. Soil Collection and Characterization
2.2. Batch and Flow-Through Experiments to Determine P Desorption Kinetics
2.2.1. Differences between Batch Tests and Flow-Through: Time, Volume, Concentration
2.2.2. Traditional Batch Test Using Constant Volume and Variable Time
2.2.3. Standard Reaction Order
2.2.4. Flow-Through Experiment
2.2.5. Batch Simulated Flow-Through (Variable Time and Volume)
2.2.6. Batch Hybrid (Constant Time, Variable Volume)
2.3. Data Analysis
3. Results and Discussion
3.1. Soil Characterization
3.2. Batch Desorption Tests
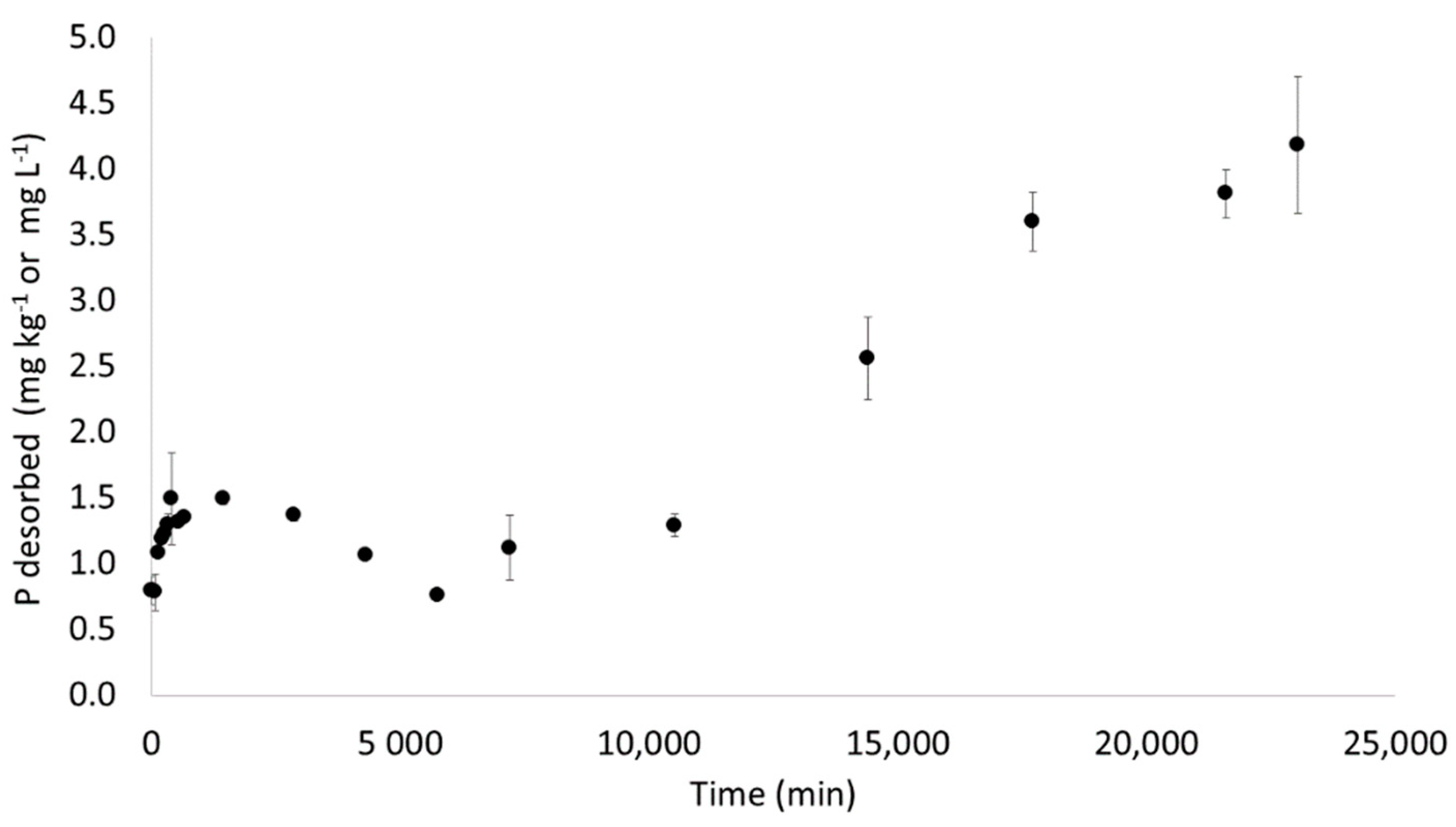
3.2.1. Traditional Batch Test
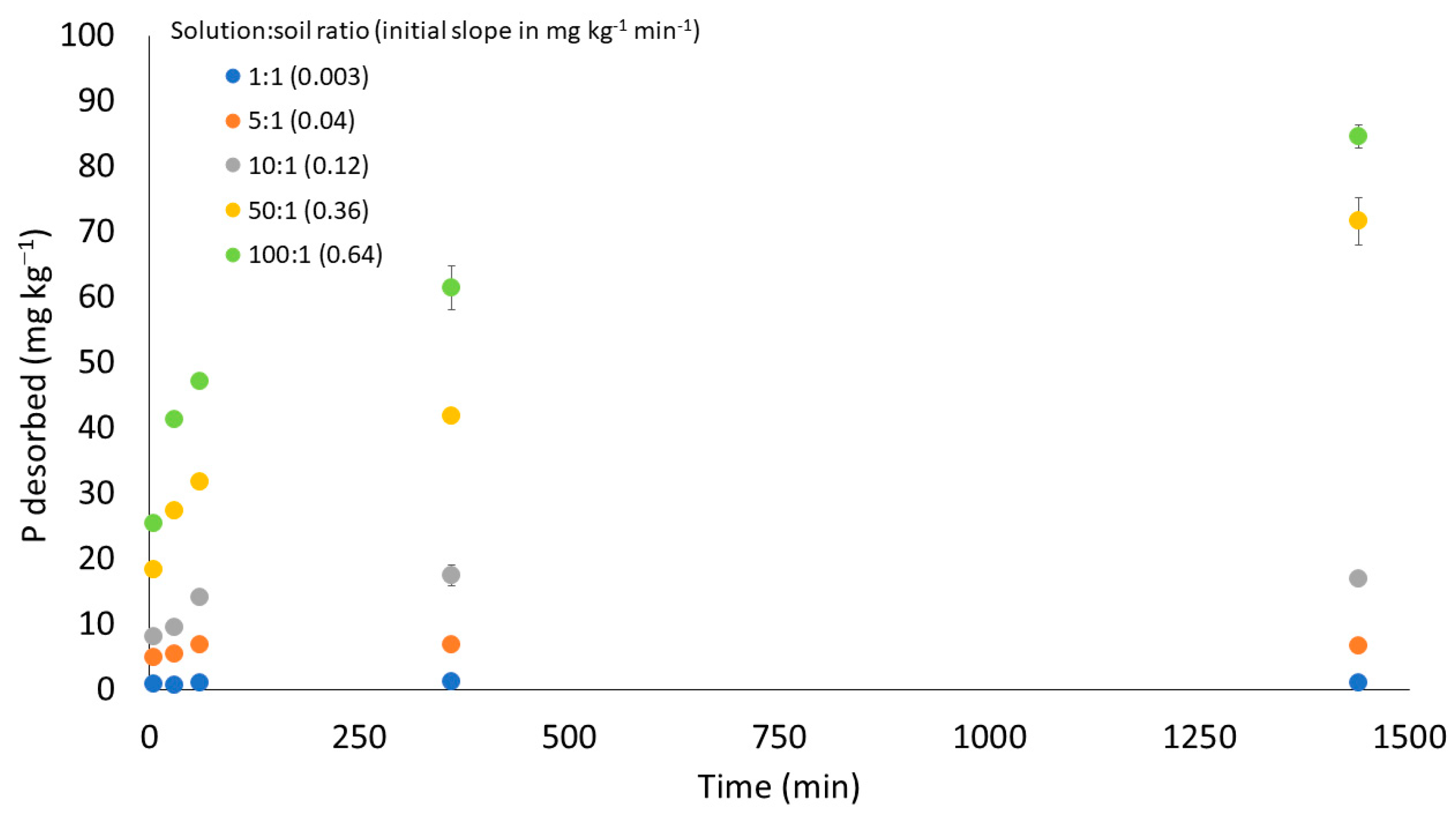
3.2.2. Batch Hybrid: Constant Time and Variable Volume
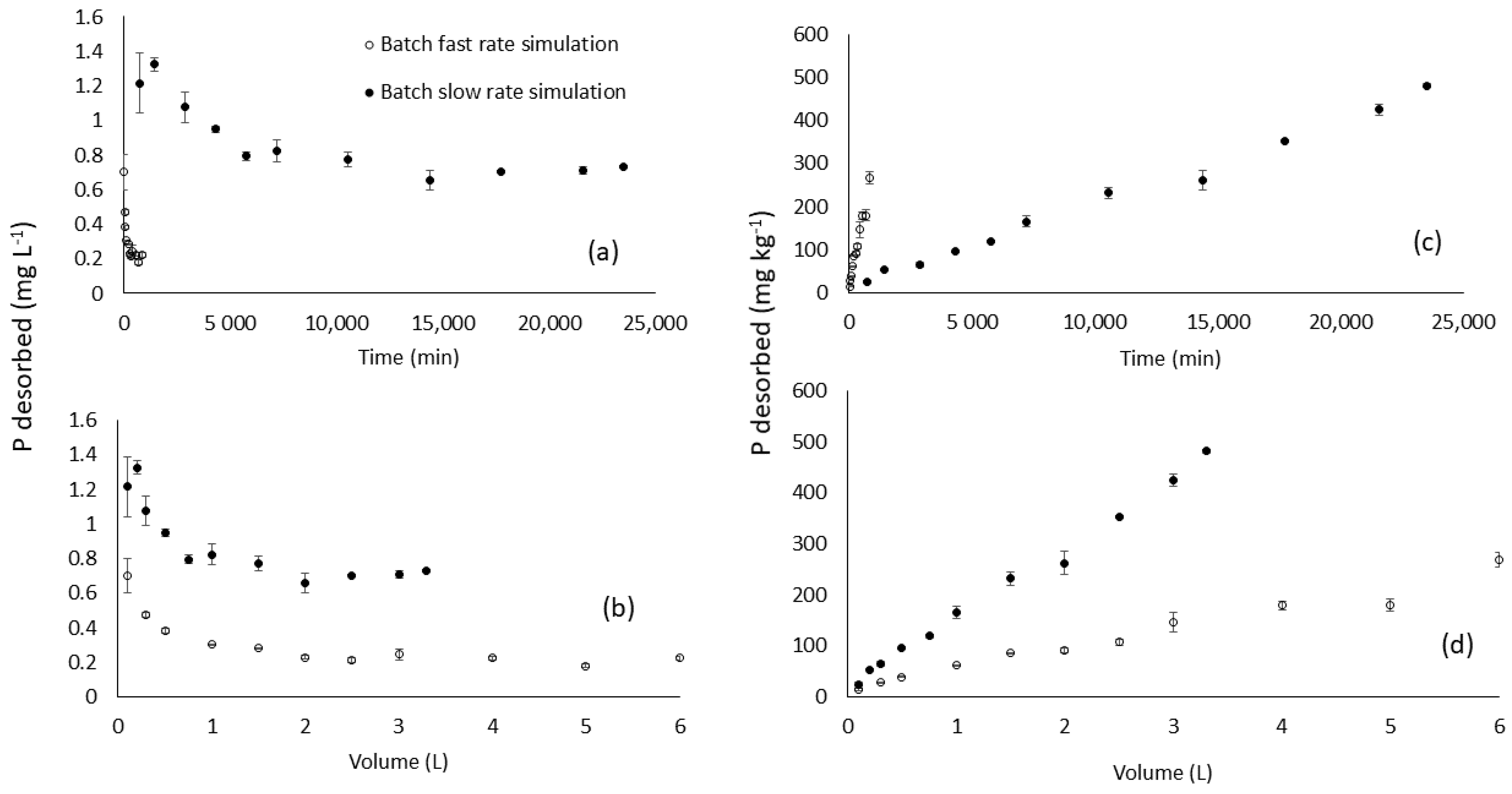
3.2.3. Batch Simulated Flow-Through Test: Variable Time and Variable Volume
3.2.4. Reaction Order Experiment
3.3. Flow-Through Experiment
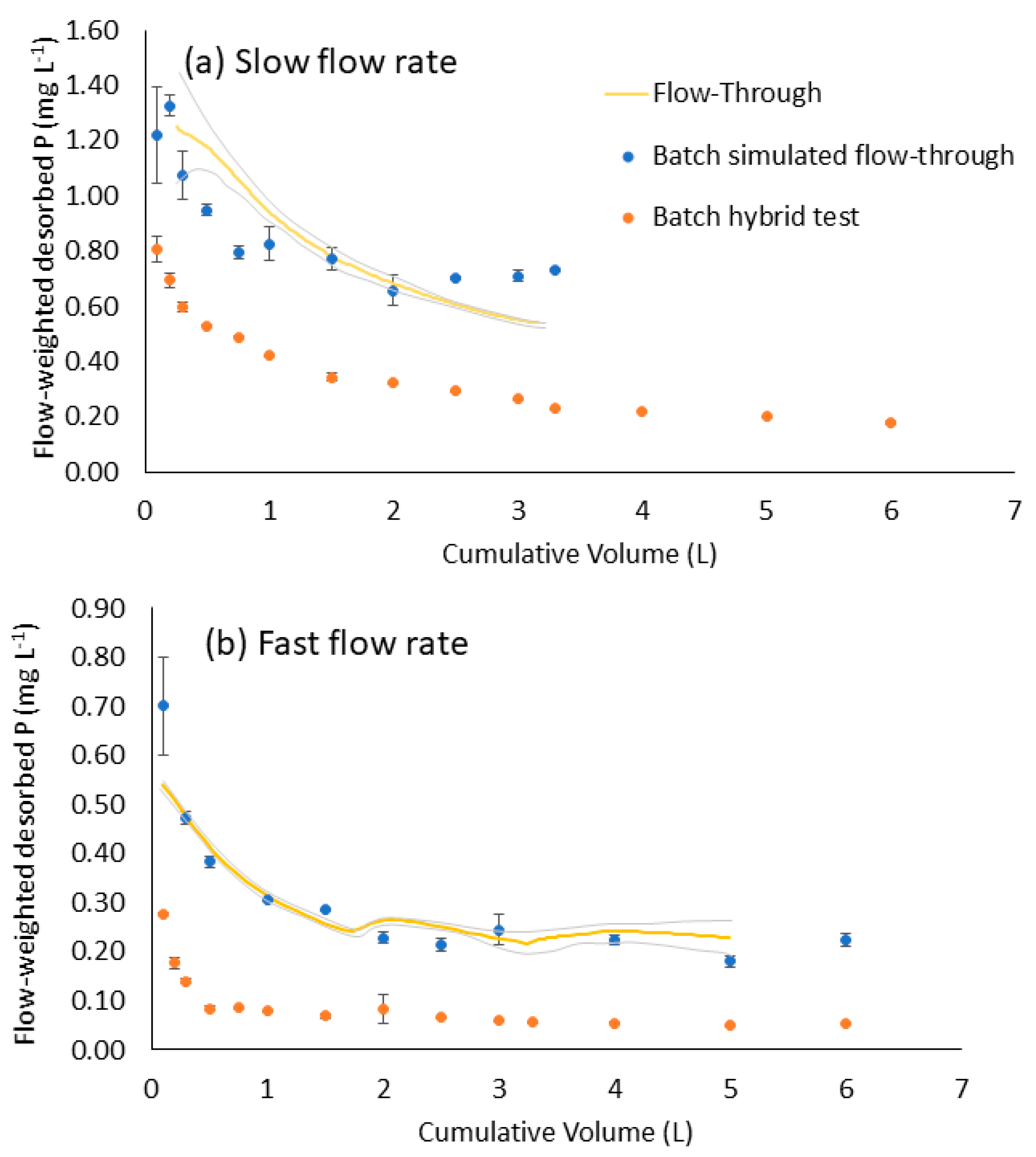
3.4. Comparison of Flow-Through Experiment and Batch Test Desorption
3.5. Differences in P Desorption between Fast and Slow Flow Rates
3.6. A Closer Look at the Physio-Chemical Process of P Buffering and Desorption Kinetics
3.7. Implications for Water Quality and Fertility Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haygarth, P.M.; Jarvie, H.P.; Powers, S.M.; Sharpley, A.N.; Elser, J.J.; Shen, J.; Peterson, H.M.; Chan, N.-I.; Howden, N.J.K.; Burt, T.; et al. Sustainable phosphorus management and the need for a long-term perspective: The legacy hypothesis. Environ. Sci. Technol. 2014, 48, 8417–8419. [Google Scholar] [CrossRef] [PubMed]
- Wurtsbaugh, W.A.; Paerl, H.W.; Dodds, W.K. Nutrients, eutrophication and harmful algal blooms along the freshwater to marine continuum. WIREs Water 2019, 6, e1373. [Google Scholar] [CrossRef]
- Van Der Zee, S.; Van Riemsdijk, W. Sorption kinetics and transport of phosphate in sandy soil. Geoderma 1986, 38, 293–309. [Google Scholar] [CrossRef]
- Freese, D.; Van Riemsdijk, W.; Van Der Zee, S. Modelling phosphate-sorption kinetics in acid soils. Eur. J. Soil Sci. 1995, 46, 239–245. [Google Scholar] [CrossRef]
- Wilson, G.V.; Rhoton, F.E.; Selim, H.M. Modeling the impact of ferrihydrite on adsorption-desorption of soil phosphorus. Soil Sci. 2004, 169, 271–281. [Google Scholar] [CrossRef]
- Sims, J.T.; Maguire, R.O.; Leytem, A.B.; Gartley, K.L.; Pautler, M.C. Evaluation of Mehlich 3 as an agri-environmental soil phosphorus test for the Mid-Atlantic United States of America. Soil Sci. Soc. Am. J. 2002, 66, 2016–2032. [Google Scholar] [CrossRef]
- Penn, C.J.; Mullins, G.L.; Zelazny, L.W.; Sharpley, A.N. Estimating dissolved phosphorus concentrations in runoff from three physiographic regions of Virginia. Soil Sci. Soc. Am. J. 2006, 70, 1967–1974. [Google Scholar] [CrossRef]
- McDowell, R.; Sharpley, A. Approximating phosphorus release from soils to surface runoff and subsurface drainage. J. Environ. Qual. 2001, 30, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.W.; Peaslee, D.E. Testing soils for phosphorus. In Soil Testing and Plant Analysis; Walsh, L.M., Beaton, J.D., Eds.; Soil Science Society of America, Inc.: Madison, WI, USA, 1973; pp. 115–132. [Google Scholar]
- Cox, F.R.; Lins, I.D.G. A phosphorus soil test interpretation for corn grown on acid soils varying in crystalline clay content 1. Commun. Soil Sci. Plant. Anal. 1984, 15, 1481–1491. [Google Scholar] [CrossRef]
- Davis, R.L.; Zhang, H.; Schroder, J.L.; Wang, J.J.; Payton, M.E.; Zazulak, A. Soil Characteristics and Phosphorus Level Effect on Phosphorus Loss in Runoff. J. Environ. Qual. 2005, 34, 1640–1650. [Google Scholar] [CrossRef]
- Amer, F.; Bouldin, D.R.; Black, C.A.; Duke, F.R. Characterization of soil phosphorus by anion exchange resin adsorption and P32-equilibration. Plant. Soil 1955, 6, 391–408. [Google Scholar] [CrossRef]
- Bissani, C.A.; Tedesco, M.J.; Camargo, F.; Miola, G.L.; Gianello, C. Anion-exchange resins and iron oxide-impregnated filter paper as plant available phosphorus indicators in soils. Commun. Soil Sci. Plant. Anal. 2002, 33, 1119–1130. [Google Scholar] [CrossRef]
- Qian, P.; Schoenau, J.J. Practical applications of ion exchange resins in agricultural and environmental soil research. Can. J. Soil Sci. 2002, 82, 9–21. [Google Scholar] [CrossRef]
- Yang, J.E.; Skogley, E.O.; Georgitis, S.J.; Schaff, B.E.; Ferguson, A.H. Phytoavailability Soil Test: Development and Verification of Theory. Soil Sci. Soc. Am. J. 1991, 55, 1358–1365. [Google Scholar] [CrossRef]
- Daverede, I.C.; Kravchenko, A.N.; Hoeft, R.G.; Nafziger, E.D.; Bullock, D.G.; Warren, J.J.; Gonzini, L.C. Phosphorus runoff: Effect of tillage and soil phosphorus levels. J. Environ. Qual. 2003, 32, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Sharpley, A.N. An innovative approach to estimate bioavailable phosphorus in agricultural runoff using iron oxide-impregnated paper. J. Environ. Qual. 1993, 22, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Gaston, L.A.; Drapcho, C.M.; Tapadar, S.; Kovar, J.L. Phosphorus Runoff Relationships for Louisiana Coastal Plain Soils Amended with Poultry Litter. J. Environ. Qual. 2003, 32, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Vadas, P.A.; Kleinman, P.J.A.; Sharpley, A.N.; Turner, B.L. Relating Soil Phosphorus to Dissolved Phosphorus in Runoff: A Single Extraction Coefficient for Water Quality Modeling. J. Environ. Qual. 2005, 34, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrman, J.K.; Zhang, H.; Schroder, J.L.; Davis, R.L.; Payton, M.E. Water-soluble phosphorus as affected by soil to extractant ratios, extraction times, and electrolyte. Commun. Soil Sci. Plant Anal. 2005, 36, 925–935. [Google Scholar] [CrossRef]
- Roswall, T.; Lucas, E.; Yang, Y.-Y.; Burgis, C.; Scott, I.S.P.C.; Toor, G.S. Hotspots of legacy phosphorus in agricultural landscapes: Revisiting water-extractable phosphorus pools in soils. Water 2021, 13, 1006. [Google Scholar] [CrossRef]
- Adams, F.; Burmester, C.; Hue, N.V.; Long, F.L. A Comparison of Column-Displacement and Centrifuge Methods for Obtaining Soil Solutions. Soil Sci. Soc. Am. J. 1980, 44, 733–735. [Google Scholar] [CrossRef]
- Sharpley, A.N.; Ahuja, L.R.; Menzel, R.G. The Release of Soil Phosphorus to Runoff in Relation to the Kinetics of Desorption. J. Environ. Qual. 1981, 10, 386–391. [Google Scholar] [CrossRef]
- van der Paauw, F. An effective water extraction method for the determination of plant-available soil phosphorus. Plant. Soil 1971, 34, 467–481. [Google Scholar] [CrossRef]
- Wuenscher, R.; Unterfrauner, H.; Peticzka, R.; Zehetner, F. A comparison of 14 soil phosphorus extraction methods applied to 50 agricultural soils from Central Europe. Plant. Soil Environ. 2015, 61, 86–96. [Google Scholar] [CrossRef]
- Self-Davis, M.L.; Moore, P.A., Jr.; Joern, B.C. Determination of water-and/or dilute salt-extractable phosphorus. In Methods of Phosphorus Analysis for Soils, Sediments, Residuals, and Waters; Southern Cooperative Series Bulletin: Fayetteville, AR, USA, 2000; Volume 396, pp. 24–26. [Google Scholar]
- Osterholz, W.R.; Hanrahan, B.R.; King, K.W. Legacy phosphorus concentration–discharge relationships in surface runoff and tile drainage from Ohio crop fields. J. Environ. Qual. 2020, 49, 675–687. [Google Scholar] [CrossRef]
- Daniel, T.C.; Edwards, D.R.; Sharpley, A.N. Effect of Extractable Soil Surface Phosphorus on Runoff Water Quality. Trans. ASAE 1993, 36, 1079–1085. [Google Scholar] [CrossRef] [Green Version]
- Raven, K.P.; Hossner, L.R. Soil phosphorus desorption kinetics and its relationship with plant growth. Soil Sci. Soc. Am. J. 1994, 58, 416–423. [Google Scholar] [CrossRef]
- Raven, K.P.; Hossner, L.R. Phosphorus Desorption Quantity-Intensity Relationships in Soils. Soil Sci. Soc. Am. J. 1993, 57, 1501–1508. [Google Scholar] [CrossRef]
- Probert, M.; Moody, P.W. Relating phosphorus quantity, intensity, and buffer capacity to phosphorus uptake. Soil Res. 1998, 36, 389–394. [Google Scholar] [CrossRef]
- Koopmans, G.F.; Chardon, W.; Ehlert, P.A.I.; Dolfing, J.; Suurs, R.A.A.; Oenema, O.; Van Riemsdijk, W.H. Phosphorus availability for plant uptake in a phosphorus-enriched noncalcareous sandy soil. J. Environ. Qual. 2004, 33, 965–975. [Google Scholar] [CrossRef]
- Beauchemin, S.; Simard, R.R.; Cluis, D. Phosphorus sorption-desorption kinetics of soil under contrasting land uses. J. Environ. Qual. 1996, 25, 1317–1325. [Google Scholar] [CrossRef]
- Sparks, D.L.; Zelazny, L.W.; Martens, D.C. Kinetics of Potassium Desorption in Soil using Miscible Displacement. Soil Sci. Soc. Am. J. 1980, 44, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Mansell, R.S.; Selim, H.M.; Kanchanasut, P.; Davidson, J.M.; Fiskell, J.G.A. Experimental and simulated transport of phosphorus through sandy soils. Water Resour. Res. 1977, 13, 189–194. [Google Scholar] [CrossRef]
- Amacher, M.C. Methods of Obtaining and Analyzing Kinetic Data. Rates Soil Chem. Process. 1991, 19–59. [Google Scholar] [CrossRef]
- Sparks, D.L. Kinetics of Soil Chemical Processes; Academic Press: San Diego, CA, USA, 1989. [Google Scholar]
- Sparks, D.L.; Rechcigl, J.E. Comparison of Batch and Miscible Displacement Techniques to Describe Potassium Adsorption Kinetics in Delaware Soils. Soil Sci. Soc. Am. J. 1982, 46, 875–877. [Google Scholar] [CrossRef] [Green Version]
- Penn, C.; Camberato, J. A Critical Review on Soil Chemical Processes that Control How Soil pH Affects Phosphorus Availability to Plants. Agriculture 2019, 9, 120. [Google Scholar] [CrossRef] [Green Version]
- Olsen, S.R.; Watanabe, F.S. A method to determine a phosphorus adsorption maximum of soils as measured by the langmuir isotherm1. Soil Sci. Soc. Am. J. 1957, 21, 144–149. [Google Scholar] [CrossRef]
- Frossard, E.; Demaria, P.; Sinaj, S.; Schärer, M. A flow-through reactor to assess potential phosphate release from agricultural soils. Geoderma 2014, 219–220, 125–135. [Google Scholar] [CrossRef]
- Guedes, R.S.; Melo, L.; Vergutz, L.; Rodríguez-Vila, A.; Covelo, E.F.; Fernandes, A.R. Adsorption and desorption kinetics and phosphorus hysteresis in highly weathered soil by stirred flow chamber experiments. Soil Tillage Res. 2016, 162, 46–54. [Google Scholar] [CrossRef]
- Lookman, R.; Freese, D.; Merckx, R.; Vlassak, K.; Van Riemsdijk, W.H. Long-Term Kinetics of Phosphate Release from Soil. Environ. Sci. Technol. 1995, 29, 1569–1575. [Google Scholar] [CrossRef]
- Lasaga, A.C.; Kirkpatrick, R.J. Kinetics of Geochemical Processes; BookCrafters, Inc.: Chelsea, MI, USA, 1981. [Google Scholar]
- Williams, M.R.; King, K.W.; Ford, W.; Buda, A.R.; Kennedy, C.D. Effect of tillage on macropore flow and phosphorus transport to tile drains. Water Resour. Res. 2016, 52, 2868–2882. [Google Scholar] [CrossRef] [Green Version]
- Shedekar, V.S.; Penn, C.J.; Pease, L.; King, K.W.; Kalcic, M.M.; Livingston, S.J. Performance of a ditch-style phosphorus re-moval structure for treating agricultural drainage water with aluminum-treated steel slag. Water 2020, 12, 2149. [Google Scholar] [CrossRef]
- Nelson, D.W.; Sommers, L.E. Total carbon, organic carbon and organic matter. In Methods of Soil Analysis; Part 3. Chemical Methods; Sparks, D.L., Ed.; Soil Science Society of America, Inc.: Madison, WI, USA, 1996; pp. 961–1010. [Google Scholar]
- Mehlich, A. Mehlich 3 soil test extractant: A modification of Mehlich 2 extractant. Commun. Soil Sci. Plant. Anal. 1984, 15, 1409–1416. [Google Scholar] [CrossRef]
- McKeague, J.A.; Day, J.H. Dithionite-and oxalate-extractable Fe and Al as aids in differentiating various classes of soils. Can. J. Soil Sci. 1966, 46, 13–22. [Google Scholar] [CrossRef]
- Murphy, J.; Riley, J.P. A modified single solution method for the determination of phosphate in natural waters. Anal. Chim. Acta 1962, 27, 31–36. [Google Scholar] [CrossRef]
- Martin, H.W.; Sparks, D.L. Kinetics of Nonexchangeable Potassium Release from Two Coastal Plain Soils1. Soil Sci. Soc. Am. J. 1983, 47, 883–887. [Google Scholar] [CrossRef] [Green Version]
- Jardine, P.M.; Sparks, D.L. Potassium-Calcium Exchange in a Multireactive Soil System: I. Kinetics. Soil Sci. Soc. Am. J. 1984, 48, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Bolan, N.S.; Syers, J.K.; Sumner, M.E. Dissolution of various sources of gypsum in aqueous solutions and in soil. J. Sci. Food Agric. 1991, 57, 527–541. [Google Scholar] [CrossRef]
- Williams, M.R.; Livingston, S.J.; Penn, C.J.; Smith, D.R.; King, K.W.; Huang, C.-H. Controls of event-based nutrient transport within nested headwater agricultural watersheds of the western Lake Erie basin. J. Hydrol. 2018, 559, 749–761. [Google Scholar] [CrossRef]
- King, K.W.; Williams, M.R.; Macrae, M.L.; Fausey, N.R.; Frankenberger, J.; Smith, D.R.; Kleinman, P.J.A.; Brown, L.C. Phos-phorus transport in agricultural subsurface drainage: A review. J. Environ. Qual. 2015, 44, 467–485. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Johnson, L.T.; LaBarge, G.A.; Penn, C.J.; Stumpf, R.P.; Baker, D.B.; Shao, G. Less agricultural phosphorus applied in 2019 led to less dissolved phosphorus transported to Lake Erie. Environ. Sci. Technol. 2020, 55, 283–291. [Google Scholar] [CrossRef]
- Scott, I.S.; Penn, C.J. Estimating the variability of steel slag properties and their influence in phosphorus removal ability. Chemosphere 2021, 276, 130205. [Google Scholar] [CrossRef]
- Penn, C.J.; Bowen, J.M. Design and Construction of Phosphorus Removal Structures for Improving Water Quality; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Williams, M.R.; Coronel, O.; McAfee, S.J.; Sanders, L.L. Preferential flow of surface-applied solutes: Effect of lysimeter design and initial soil water content. Vadose Zone J. 2020, 19. [Google Scholar] [CrossRef]
- Rawls, W.J.; Gimenez, D.; Grossman, R. Use of soil texture, bulk density, and slope of the water retention curve to predict sat-urated hydraulic conductivity. Am. Soc. Agric. Biol. Eng. 1998, 41, 983–988. [Google Scholar] [CrossRef]
- SAS Institute Inc. SAS/STAT 9.1. User’s Guide; SAS Institute Inc.: Cary, NC, USA, 2003. [Google Scholar]
- Culman, S.; Fulford, A.; Camberato, J.; Steinke, K.; Lindsey, L.; LaBarge, G.; Warncke, D. Tri-State Fertilizer Recommendations for Corn, Soybeans, Wheat and Alfalfa. Available online: https://agcrops.osu.edu/FertilityResources/tri-state_info (accessed on 21 November 2021).
- Maguire, R.O.; Sims, J.T. Soil Testing to Predict Phosphorus Leaching. J. Environ. Qual. 2002, 31, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Barrow, N.J.; Shaw, T.C. Effects of solution: Soil ratio and vigour of shaking on the rate of phosphate adsorption by soil. Eur. J. Soil Sci. 1979, 30, 67–76. [Google Scholar] [CrossRef]
- do Carmo Horta, M.; Torrent, J. Phosphorus desorption kinetics in relation to phosphorus forms and sorption properties of portuguese acid soils. Soil Sci. 2007, 172, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Aslyng, H.C. The Lime and Phosphate Potentials of Soils; the Solubility and Availability of Phosphates; LWW: Philadelphia, PA, USA, 1954; Volume 78. [Google Scholar]
- Kressman, T.R.E.; Kitchener, J.A. Cation exchange with a synthetic phenolsulphonate resin. Part, V. Kinetics. Discuss. Faraday Soc. 1949, 7, 90–104. [Google Scholar] [CrossRef]
- Ogwada, R.A.; Sparks, D.L. Kinetics of ion exchange on clay minerals and soil: I. evaluation of methods. Soil Sci. Soc. Am. J. 1986, 50, 1158–1162. [Google Scholar] [CrossRef]
- Miller, D.M.; Sumner, M.E.; Miller, W.P. A Comparison of batch- and flow-generated anion adsorption isotherms. Soil Sci. Soc. Am. J. 1989, 53, 373–380. [Google Scholar] [CrossRef]
- Ogwada, R.A.; Sparks, D.L. Kinetics of Ion Exchange on Clay Minerals and Soil: II. Elucidation of Rate-limiting Steps. Soil Sci. Soc. Am. J. 1986, 50, 1162–1166. [Google Scholar] [CrossRef]
- Ogwada, R.A.; Sparks, D.L. A Critical Evaluation on the Use of Kinetics for Determining Thermodynamics of Ion Exchange in Soils. Soil Sci. Soc. Am. J. 1986, 50, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Sparks, D.L. Kinetics of ionic reactions in clay minerals and soils. Adv. Agron. 1986, 38, 231–266. [Google Scholar] [CrossRef]
- McDowell, R.; Sharpley, A. Phosphorus solubility and release kinetics as a function of soil test P concentration. Geoderma 2003, 112, 143–154. [Google Scholar] [CrossRef]
- Vadas, P.A.; Krogstad, T.; Sharpley, A.N. Modeling phosphorus transfer between labile and nonlabile soil pools: Updating the EPIC model. Soil Sci. Soc. Am. J. 2006, 70, 736–743. [Google Scholar] [CrossRef]
- Akhtar, M.S.; Richards, B.K.; Medrano, P.A.; DeGroot, M.; Steenhuis, T.S. Dissolved phosphorus from undisturbed soil cores: Related to adsorption strength, flow rate, or soil structure? Soil Sci. Soc. Am. J. 2003, 67, 458–470. [Google Scholar] [CrossRef]
- Sharpley, A.N. The effect of storm interval on the transport of soluble phosphorus in runoff. J. Environ. Qual. 1980, 9, 575–578. [Google Scholar] [CrossRef]
- Essington, M.E. Soil and Water Chemistry: An Integrative Approach, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Curtin, D.; Beare, M.H.; Hernandez-Ramirez, G. Temperature and moisture effects on microbial biomass and soil organic matter mineralization. Soil Sci. Soc. Am. J. 2012, 76, 2055–2067. [Google Scholar] [CrossRef]
- Schott, J.; Pokrovsky, O.; Oelkers, E. The link between mineral dissolution/precipitation kinetics and solution chemistry. Rev. Miner. Geochem. 2009, 70, 207–258. [Google Scholar] [CrossRef]
- Barrow, N.J. On the reversibility of phosphate sorption by soils. Eur. J. Soil Sci. 1983, 34, 751–758. [Google Scholar] [CrossRef]
- Penn, C.J.; Rutter, E.B.; Arnall, D.B.; Camberato, J.; Williams, M.; Watkins, P. A Discussion on Mehlich-3 Phosphorus Extraction from the Perspective of Governing Chemical Reactions and Phases: Impact of Soil pH. Agriculture 2018, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Sharpley, A.N. Effect of Soil Properties on the Kinetics of Phosphorus Desorption. Soil Sci. Soc. Am. J. 1983, 47, 462–467. [Google Scholar] [CrossRef]
- Sharpley, A.N. Depth of Surface Soil-runoff Interaction as Affected by Rainfall, Soil Slope, and Management. Soil Sci. Soc. Am. J. 1985, 49, 1010–1015. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, L.R.; Sharpley, A.N.; Yamamoto, M.; Menzel, R.G. The depth of rainfall-runoff-soil interaction as determined by 32 P. Water Resour. Res. 1981, 17, 969–974. [Google Scholar] [CrossRef]
- Ahuja, L.R.; Sharpley, A.N.; Lehman, O.R. Effect of Soil Slope and Rainfall Characteristics on Phosphorus in Runoff. J. Environ. Qual. 1982, 11, 9–13. [Google Scholar] [CrossRef]
- Vadas, P.A.; Sims, J.T.; Leytem, A.B.; Penn, C.J. Modifying FHANTM 2.0 to Estimate phosphorus concentrations in runoff from mid-atlantic coastal plain soils. Soil Sci. Soc. Am. J. 2002, 66, 1974–1980. [Google Scholar] [CrossRef]
- Williams, M.R.; McAfee, S.J. Water storage, mixing, and fluxes in tile-drained agricultural fields inferred from stable water isotopes. J. Hydrol. 2021, 599, 126347. [Google Scholar] [CrossRef]
- King, K.W.; Williams, M.R.; Johnson, L.T.; Smith, D.R.; LaBarge, G.A.; Fausey, N.R. Phosphorus availability in western lake erie basin drainage waters: Legacy evidence across spatial scales. J. Environ. Qual. 2017, 46, 466–469. [Google Scholar] [CrossRef]
- Penn, C.; Livingston, S.; Shedekar, V.; King, K.; Williams, M. Performance of field-scale phosphorus removal structures uti-lizing steel slag for treatment of surface and subsurface drainage. Water 2020, 12, 443. [Google Scholar] [CrossRef] [Green Version]
- Kane, D.D.; Conroy, J.D.; Richards, R.P.; Baker, D.B.; Culver, D.A. Re-eutrophication of Lake Erie: Correlations between trib-utary nutrient loads and phytoplankton biomass. J. Great Lakes Res. 2014, 40, 496–501. [Google Scholar] [CrossRef]
- Barber, S.A. Soil Nutrient Bioavailability: A Mechanistic Approach; John Wiley & Sons: New York, NY, USA, 1995. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Units | Value |
|---|---|---|
| Bulk density | g cm−3 | 0.95 |
| Total carbon | g kg−1 | 19.6 |
| Total nitrogen | g kg−1 | 2.1 |
| Sand | % | 24.9 |
| Silt | % | 64.6 |
| Clay | % | 9.5 |
| Electrical conductivity | µS cm−1 | 358 |
| pH | 6.13 | |
| Water extractable P | mg kg−1, mg L−1 | 10.2, 1.02 |
| Mehlich-3 P | mg kg−1 | 342 |
| Mehlich-3 Ca | mg kg−1 | 3140 |
| Mehlich-3 Al | mg kg−1 | 693 |
| Mehlich-3 Fe | mg kg−1 | 319 |
| DPSM3 | % | 35.1 |
| POx | mg kg−1 | 1803 |
| AlOx | mg kg−1 | 1347 |
| FeOx | mg kg−1 | 5860 |
| DPSOx | % | 37.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penn, C.J.; Williams, M.R.; Camberato, J.; Wenos, N.; Wason, H. Desorption Kinetics of Legacy Soil Phosphorus: Implications for Non-Point Transport and Plant Uptake. Soil Syst. 2022, 6, 6. https://doi.org/10.3390/soilsystems6010006
Penn CJ, Williams MR, Camberato J, Wenos N, Wason H. Desorption Kinetics of Legacy Soil Phosphorus: Implications for Non-Point Transport and Plant Uptake. Soil Systems. 2022; 6(1):6. https://doi.org/10.3390/soilsystems6010006
Chicago/Turabian StylePenn, Chad J., Mark R. Williams, James Camberato, Nicholas Wenos, and Hope Wason. 2022. "Desorption Kinetics of Legacy Soil Phosphorus: Implications for Non-Point Transport and Plant Uptake" Soil Systems 6, no. 1: 6. https://doi.org/10.3390/soilsystems6010006