Abstract
Geogenic chromium (Cr) and vanadium (V) contamination of groundwater in the Piedmont region of North Carolina poses threats to public health. These contaminants are naturally derived from saprolite and aquifer materials, but geochemical variability in these materials makes it difficult to predict specific risks of Cr and V in well water. The objectives of this study were to (1) determine host phases of Cr and V in representative subsurface materials; (2) characterize contaminant binding parameters of chemically variable saprolites; and (3) examine the influence of saprolite chemistry on contaminant sorption, speciation, and phase associations. Isotherm experiments revealed that saprolite samples sorbed roughly an order of magnitude more V than Cr. Chemical extractions and synchrotron-based X-ray fluorescence showed that substantial Cr and V were bound with metal oxide/oxyhydroxides in native and Cr-and V-sorbed saprolites; however, electrostatically bound fractions were also present, representing potentially important sources of groundwater contamination. X-ray absorption-near-edge-structure spectroscopy indicated that sorbed Cr was found as reduced Cr(III), whereas sorbed V was dominated by the oxidized V(V) and intermediate V(IV) species. Results from this study could be used to help parameterize mechanistic models and improve prediction of the Cr and V contamination potential of shallow aquifers.
Keywords:
chromium; vanadium; groundwater contamination; sorption; chemical extraction; spectroscopy 1. Introduction
Contamination of groundwater by the redox-active, oxyanion-forming elements chromium (Cr) and vanadium (V) is of environmental and health significance due to their carcinogenic properties and ability to disrupt insulin signaling in humans [1,2]. Although reduced Cr(III) has low solubility and toxicity, Cr(VI) exists as highly mobile and toxic HCrO4− and CrO42− under a wide pH range and oxidizing conditions [3]. Likewise, V(V) exists as the relatively soluble and toxic H2VO4− and HVO42− oxyanion species under oxidizing conditions, whereas under more reducing conditions, sparingly soluble V(IV) and low-soluble V(III) govern V speciation [3]. Chromium and V contamination have been widely linked to anthropogenic activities such as leather tanning, electroplating, and petroleum refining [4,5,6,7]. However, evidence in the last few decades has shown that rock–water interaction in areas underlain by mafic, ultramafic, andesitic, and felsic rocks may serve as an important geogenic source of these redox-active contaminants to groundwater, occurring over a larger geographic extent than industrial contamination of these elements [8,9]. Once available, the ultimate mobility of these contaminants is largely governed by the strength and rates of sorption to soil and aquifer mineral phases.
Roughly four million people consume groundwater sourced from regolith and aquifer materials rich in geogenic Cr and V across the Piedmont physiographic region of the southeastern United States [8,9,10,11]. In North Carolina (NC), the Piedmont is a geologically diverse grouping of seven tectonically and genetically related terranes, which are belts consisting of metamorphosed intrusive, volcanic, and sedimentary rocks that host Cr and V-bearing primary minerals to various degrees [12] (Figure 1). In the subsurface, where elemental abundances are spatially heterogeneous, Cr and V are thought to share similar geologic sources as each other. Therefore, this distribution and the commonalities in redox chemistry suggest that similar biogeochemical processes may control Cr and V distribution, toxicity, and phase partitioning in groundwater and subsurface environments [8,11,13].
In the subsurface, heterogeneity of natural oxidants and sorbents of Cr and V, such as ubiquitous reactive Mn, Fe, and Al (oxyhydr)oxides [14] and clay minerals, complicates understanding the fate of these redox-sensitive contaminants. Manganese oxide minerals can rapidly oxidize Cr from Cr(III) to Cr(VI) and V(IV) to V(V), thereby enhancing the mobility of Cr and V in subsurface [15,16,17,18,19]. The fate of these species is further influenced by chemical interactions with mineral surfaces, which ultimately determine their mobility and species transformation [20]. The importance of crystalline and poorly ordered Fe and Al oxyhydroxides on the sorption of Cr and V in model synthetic systems has been extensively studied [18,19,20,21,22,23]. However, limited information exists on the sorption, retention, and speciation of these contaminants in natural geologic materials with environmentally relevant abundances of Fe- and Mn-bearing secondary minerals. In recent times, the relationship between speciation of redox-sensitive metals and their host phases in natural materials has been successfully deconvolved using spectroscopic and sequential extraction techniques [24,25,26].
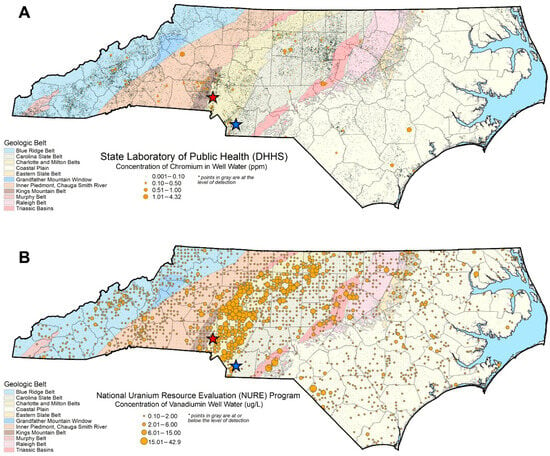
Figure 1.
Well water chromium (A) and vanadium (B) concentrations across NC. Blue and red stars indicate the Union County and Redlair observatory field sites, respectively. The Piedmont physiographic region sits in the center of the state, with the Coastal Plain to the east and the Blue Ridge Belt to the west. Chromium concentration data are from the NCWELL database [27,28]. Vanadium concentration data are from the National Uranium Resource Evaluation (NURE) Hydrogeochemical and Stream Sediment Reconnaissance (HSSR) Program [29].
The overall goal of this study was to quantify controls on Cr and V mobility in representative subsurface materials of the NC Piedmont. Using saprolites weathered from metasedimentary and felsic metavolcanic rocks of the NC Piedmont, we specifically aimed to (1) determine the host phases of Cr and V in representative geogenic subsurface materials, (2) quantify contaminant sorption parameters of chemically variable saprolites, and (3) examine the influence of saprolite heterogeneity on contaminant distribution and speciation. Findings from this study will improve our understanding of the behavior of Cr and V in the natural environment and help parametrize mechanistic models to predict contaminant fate and transport.
2. Material and Methods
2.1. Site Geology Description
In the North Carolina Piedmont region, geological control (that is, the levels of Cr and V in the source rock) is the dominant regional factor that predicts the occurrence of Cr and V in groundwater (Figure 1). To examine this variability, two chemically contrasting saprolite samples were collected for this study from contrasting geological terranes (belts) having contrasting expected Cr/V behavior. We chose saprolite samples because they represent the beginning of the recharge flow path to groundwater, and prior studies have indicated that the actively weathered saprolite zone of the Piedmont can supply high levels of Mn—another period 4 transition metal—to groundwater [11].
A Union County (UC) saprolite sample was collected in a park (34.966° N 80.477° W) within the Carolina Terrane, which contains a relatively high proportion of metamorphosed felsic volcanic and metamorphosed sedimentary rocks, some having volcanic source material. This terrane was inferred to contain less Cr and V due to its less mafic character, and this is reflected in lower groundwater Cr and V occurrence compared to in the adjacent Charlotte Terrane (Figure 1). The UC site was chosen for its central location in the Cid Formation, which consists of an upper, laminated siltstone–mudstone unit having volcanic source components [10]. We inferred that sampling from this area would yield a saprolite sample with relatively low affinity for Cr and V.
A second saprolite sample was collected at the Redlair Observatory (RL) (35.297° N 81.090° W) in Gaston County, North Carolina (Figure 1). The RL sample was collected from the Charlotte Terrane, which consists of meta-intrusive, meta-volcanic, and meta-sedimentary components and which has a relatively high occurrence of felsic metavolcanic rock components [30]. While this reflects a regional pattern rather than a site-specific prediction, we expected that the RL sample would contain some mafic-source mineralogy with the potential for elevated Cr and V, given that these elements preferentially occur in mafic material. Consistent with this more mafic mineralogy, Cr and V are naturally elevated in groundwater of the Charlotte Terrane, relative to other belts (Figure 1, [10]).
2.2. Materials, Sample Collection, and Characterization
All chemicals were reagent grade or higher and provided by Fisher Scientific (Hampton, NH, USA) unless otherwise noted. For Union County saprolite sample collection, a hammer and shovel were used to obtain samples from the surface of the creek wall. Samples were placed in Ziplock bags. Redlair saprolite was collected from the bed of a 2–3 m incised streambed. Saprolite was collected using two adjacent auger holes. Each auger hole was ~2 inches in diameter and augured diagonally downward into the streambank. Before use, saprolite samples were air-dried, passed through a 2 mm sieve, and stored in 50 mL centrifuge tubes.
The bulk mineralogy of the saprolites was characterized by X-ray diffraction (XRD) using a Rigaku benchtop X-ray diffractometer (Tokyo, Japan) equipped with a Cu Kα X-ray radiation source. The saprolite materials were disaggregated using a mortar and pestle, sieved through a 500 µm mesh sieve, and mounted on an aluminum sample holder. Data were collected between 3° and 90° 2θ, with a scan rate of 0.02° 2θ/min. The XRD pattern processing and phase identification for both saprolites were performed using the JADE software (Figures S1 and S2).
Other physicochemical properties of the saprolites were also measured. The pH of the saprolites was measured in 0.01M CaCl2 with a soil solution ratio of 1:2 (w/w) using an Orion Star A111 pH meter (Thermo Fisher Scientific, Hampton, NH, USA). The specific surface areas of each saprolite were analyzed from an 8-point N2-BET isotherm (p/p0 = 0.01–0.23; Autosorb Quantachrome) after degassing the samples at 150 °C. Total concentrations of Cr, V, Fe, and Mn were determined by X-ray fluorescence (XRF) by using a Bruker Tracer 5i (Billerica, MA, USA). Samples were prepared by using a ball mill to grind saprolite materials into a fine powder, adding a binder, and pressing into a pellet with a smooth, flat surface. The handheld XRF was moved over the flat surface of the samples multiple times, and the data were averaged to obtain elemental compositions.
2.3. Chemical Extractions
Parallel chemical extractions were performed on untreated (native) and Cr- and V-loaded samples to quantify the solid-phase fractions of Cr, V, Fe, and Mn. Extractions were modified from existing sequential extraction protocols and targeted ionically bound species (1M magnesium chloride (MgCl2), pH 8), amorphous-oxide-bound phases (0.2 M ammonium oxalate), crystalline-oxide-bound phases (citrate-bicarbonate-dithionite (CBD)), and “environmentally available” phases (strong-acid digestible), utilizing standard protocols [31,32]. Specific procedures are outlined in Table S1. Concentrations of Cr, V, Fe, and Mn in extracts were determined by inductively coupled plasma-mass spectrometry (ICP-MS), as described in Section 2.5.
2.4. Kinetic and Sorption Experiments
Kinetic experiments were conducted to establish rates of Cr and V sorption to the saprolite samples. To establish kinetics, 59 µM Cr(VI) and 60 µM V(V) were prepared from K2Cr2O7 and Na3VO4 in 0.02 M NaCl and set to pH = 6.5 by the addition of 12 M HCl or NaOH in order to achieve the circumneutral conditions common to NC Piedmont aquifers [11]. These concentrations were chosen following pilots that indicated ~50% of the sorption maxima (see isotherm description below). In duplicate, solutions were introduced to the saprolites and monitored over time, with 1 mL aliquots of sample taken at 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, 12, 24, 48, and 72 h for analysis. At every sampling interval, pH was adjusted to 6.5 ± 0.1 by addition of HCl or NaOH. Samples were passed through a 0.22 µm filter, filtrates were acidified with 1 drop of 15.8 M HNO3, and the resulting samples were refrigerated at 4 °C prior to analysis via inductively coupled plasma-mass spectrometry (ICP-MS).
For sorption isotherm experiments, solutions containing seven concentrations of Cr (5 to 100 µM Cr(VI)) and nine concentrations of V (20 to 450 µM V(V)) were prepared by using K2Cr2O7 and Na3VO4 in 0.02 M NaCl. Sorbate controls (i.e., no Cr and V) and soil blanks (no soil) were also included. The sorption experiments were carried out in a batch experiment using 50 mL test tubes with 2 g of saprolite and 40 mL of Cr(VI)- or V(V)-containing solution.
All sorption and control experiments were conducted in triplicate. Slurry pH was adjusted to 6.5 ± 0.2 by the addition of 12 M HCl or NaOH before the start of the experiment and every 24 h thereafter. Vials were covered with aluminum foil and allowed to equilibrate while shaking at 175 rpm on an orbital shaker for 48 h. After 48 h, vials were centrifuged at 4200 rpm for 10 min, pH was adjusted, and solutions were passed through 0.22 µm polyethersulfone (PES) syringe filters. Samples were acidified with 15.8 M HNO3 and stored at 4 °C before analysis by ICP-MS.
The maximum adsorption capacity (q max), Langmuir constant, KL, the Freundlich constants KF, and n were calculated using Langmuir and Freundlich adsorption models [33]. A chi-square test was coupled with the coefficient of determination, R2, to determine the best isotherm model for the resulting data (Table S2). A follow-up experiment to test the impact of the ionic strength of Cr and V adsorption was also conducted using 0.002 and 0.2 M NaCl.
To examine phase associations of sorbed Cr and V, adsorption experiments were performed with 10 mL of 40 µM Cr(VI) or 300 µM V(V), 0.02 M NaCl, and 0.5 g of saprolite. Samples were equilibrated and collected as described above. The resulting solids were characterized by chemical extractions (Section 2.3) and µ-XRF (Section 2.6).
2.5. Aqueous Analysis
Dissolved concentrations of Cr, V, Fe, and Mn from experimental products were measured using a Thermoscientific iCAP-RQ single quadrupole ICP-MS (Hampton, NH, USA) at the Oregon State University Keck Center for Plasma Spectrometry. The instrument detection limits for Cr, V, Fe, and Mn were 1.2, 2.7, 1600, and 10 ng/L, respectively. For follow-up ionic strength experiments, dissolved Cr(VI) was determined via the colorimetric diphenyl carbazide (DPC) method using a Shimadzu 1280 UV-VIS spectrophotometer (Kyoto, Japan). The detection limit for this technique was 5 µg/L.
2.6. Bulk and Micro-Focused X-Ray Absorption Spectroscopy
X-ray absorption spectroscopy (XAS) was conducted at the Stanford Synchrotron Radiation Lightsource (SSRL). Bulk Cr and V K-edge X-ray absorption near edge structure (XANES) spectroscopy data for native saprolite materials, Cr- and V-loaded samples, and standards were collected at beamline 4–3. Solid-phase samples from the second highest and highest loading experiments (60 µM Cr and 450 µM V) were prepared for XAS, and Na3VO4 and K2Cr2O7 reference materials, diluted in BN3 at 1:10, were also analyzed. The samples were loaded onto acrylic sample holders and sealed with Kapton tape (nonadhesive part of Kapton in contact with sample), mounted at 45° to the incident X-ray beam. Absorption spectra were collected in fluorescence mode using a vortex detector positioned 90° to I0, equipped with V and Ti filters and Soller slits for Cr and V data acquisition, respectively. A double-crystal Si (111) LN-cooled monochromator was used for energy selection. Six spectra of each sample were collected.
Data processing for Cr and V speciation and estimates of relative percentages of Cr and V in the samples were made using the linear combination fitting (LCF) function in Athena. For Cr-loaded samples, this procedure was performed using two model compounds, Cr(OH)3, and K2Cr2O7 [34], and spectra from the native saprolite materials (UC sap and RL sap). For Cr analysis, data were plotted in regular energy space and compared with reference materials of known oxidation states (Figure S6), allowing for the determination of proportions of Cr(III) and Cr(VI) in the samples. Vanadium speciation was determined using reference spectra from Wisawapipat and Kretzschmar [25] (Table S3). Fits were based on a minimum of three standards, and the summed contribution was between 97 and 117% for all fits. Fitting ranges were from −20 to 30 eV about the edge energy (E0) of Cr and −20 to 40 eV about the edge energy of V.
Bulk Fe and Mn K-edge XANES spectroscopy data for native saprolite materials were collected at SSRL beamline 11–2. Native saprolite materials were mounted at 45° to the incident beam. The incident beam energy was selected using a Si (220) double crystal monochromator. For each sample, six spectra were collected for Fe in fluorescence mode, using a PIPS detector from −200 to 680 eV about the Fe edge. For Mn XANES, six spectra were collected in fluorescence mode using a 100-element Ge detector from −40 to 90 eV about the Mn edge. Data processing for Mn and Fe and linear combination fitting (LCF) for the determination of Fe (Table S7) speciation were conducted in Athena. Spectra were energy-calibrated by adjusting the maxima of the first derivative of the Fe and Mn foils to 7112 eV and 6539 eV, respectively, and then averaged, deglitched, baseline corrected, and normalized. Fe XANES were fit from −30 to 600 eV using reference spectra from prior studies [35,36,37]. For Mn analysis, because the complexity of phases prevented exact mineralogical determination, data were plotted in derivative space and compared with reference materials [38] of known oxidation states, allowing for the quantitative determination of Mn(II), Mn(III), and Mn(IV) in the samples.
Finally, Cr- and V-loaded saprolite samples for Union County and Redlair Observatory were air-dried and shipped to Spectrum Petrographics Inc. for thin section preparation. Samples were mounted on quartz 4.5 × 2.8 cm quartz glass slides using Epotex-301 resin, cut, and polished before analysis. Spatial distributions of Cr, V, Fe, and Mn in Cr- and V-loaded saprolite samples were determined using µ–X-ray fluorescence (µ-XRF) at SSRL Beamline 2–3. Data were collected in fluorescence mode using harmonic rejection mirrors, a channel-cut Si (111) monochromator, and a 1-channel vortex detector. Beam size was ~5 µm × 5 µm, and µ-XRF images were obtained with incident X-ray energies set to 7200 eV by rastering sample slides in step sizes of 7 µm with 15 ms of dwell time. The areas mapped for UC-1 (60 µM Cr-loaded Union County saprolite), UC-2, (450 µM V-loaded Union County saprolite), and RL-1 (60 µM Cr-loaded Redlair saprolite) were 2 mm × 1.3 mm, 1.9 mm × 1.6 mm, and 2.7 mm × 2.1 mm, respectively.
3. Results
3.1. Saprolite Characterization
Results from XRF analysis revealed that total concentrations of Cr, V, and Mn were higher in the UC saprolite than in the RL sample (Table 1). However, the total concentration of Fe in the RL saprolite was ~2.4 times greater than in the UC saprolite. Chromium was undetectable by XRF in the RL saprolite, but follow-up strong-acid digestion showed that the “environmentally available” Cr was ~0.31 mmol/kg, a value that does not include Cr bound with all silicate minerals.

Table 1.
Physicochemical properties of the saprolites.
Chemical extractions indicated that native Cr and V in both saprolites were associated with crystalline oxides over amorphous oxides and ionically bound species (Table 1). Amorphous and crystalline Fe in the UC saprolite were 1140 and 10,700 mg/kg, respectively, and 530 and ~15,000 mg/kg in the RL saprolite. Amorphous and crystalline Mn oxide in the UC saprolite were ~4 times greater than in the RL saprolite.
X-ray absorption spectroscopy showed that the speciation of Fe, V, and Mn in both the UC and RL saprolites was diverse. In the UC and RL saprolites, the average oxidation states of Fe were 2.6 ± 0.3 and 2.9 ± 0.3, respectively (Figure S3), and the average oxidation states of V were 3.6 ± 0.4 and 3.9 ± 0.7, respectively (Figure S4). Manganese was found in mixed valence (+2/3) forms (Figure S5), and Cr was predominantly found as +3 (Figure S6).
3.2. Kinetics of Chromium and Vanadium Sorption to Saprolite
Chromium and V sorption kinetic data are shown in Figure 2. Kinetic data were fitted with pseudo-first- and pseudo-second-order kinetic models for sorption [39] (Supplementary Materials). The empirical data were best fit with pseudo-second-order kinetic models (Table S8), in which rates were proportional to the square of the dissolved Cr or V concentrations over time, and k2 was the rate constant. Comparing k2 values, sorption of Cr onto UC saprolite was ~2 times faster than that for Cr sorption to RL (Figure 2A,B), and sorption of V on UC was similarly ~2.4 times faster than that for V sorption to the RL saprolite (Figure 2C,D). For both elements and both saprolites, >90% of the observed and maximum sorption occurred by 48 h of reaction in the experiments.
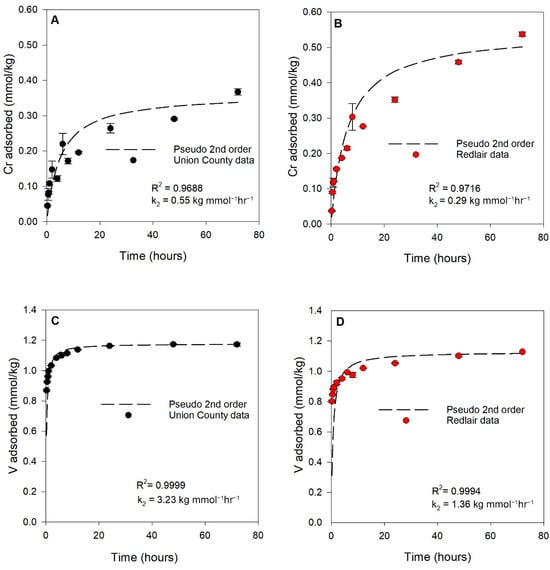
Figure 2.
Sorption kinetics of (A) Cr on Union County saprolite (black data points), (B) Cr on Redlair saprolite (red data points), (C) V on Union County saprolite, and (D) V on Redlair saprolite. Dashed black lines represent pseudo-2nd-order kinetic model fits. Error bars represent the standard error of replicates. Reaction conditions: ionic strength (I) = 0.02 M NaCl; pH = 6.5; initial [Cr(VI)] = 59 µM (3.06 mg/L); initial [V(V)] = 60 µM (3.06 mg/L); solid solution ratio = 2 g/40mL.
3.3. Extent of Sorption of Chromium and Vanadium to Saprolite
Chromium sorption data for both saprolites are shown in Figure 3A. Each isotherm is characterized by a relatively steep slope at small Cr(VI) concentrations with plateaus at greater adsorbate concentrations, characteristic of L-type isotherms (Figure 3A). The sorption of Cr on the UC and RL saprolites was best described by Langmuir models (R2 = 0.977 and 0.994, respectively) as compared to Freundlich models (Figure S7A). The maximum sorption capacities of Cr on both saprolites predicted by the Langmuir models were similar, with values of 0.35 mmol Cr/kg UC saprolite and 0.30 mmol Cr/kg RL saprolite.
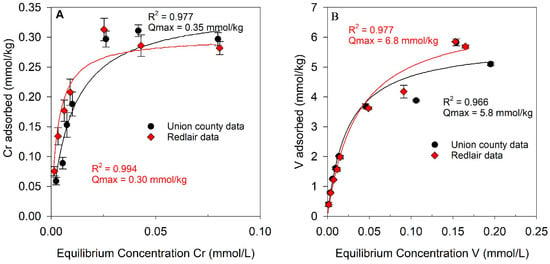
Figure 3.
Sorption of (A) Cr on Union County and Redlair saprolites and (B) V on Union County and Redlair observatory saprolites. For each plot, black and red lines represent Langmuir isotherm models for Union County and Redlair data, respectively. Error bars indicate the standard error of experimental replicates. Reaction conditions: ionic strength (I) = 0.02 M NaCl; pH = 6.5; initial [Cr] = 5–100 µM; initial [V] = 20–450 µM; solid solution ratio = 2 g/40 mL.
Vanadium sorption data for both saprolites are presented in Figure 3B. Compared to Cr sorption isotherms, the sorption of V was greater for both saprolite samples. In comparison, UC and RL saprolites sorbed ~16–23 times more V than Cr (on a molar basis). The V sorption data were well explained by both Langmuir (Figure 3B) and Freundlich models (Figure S7B), but a chi-square test found that the Langmuir isotherm model yielded the lower χ2 value for V sorption onto both saprolites (Table S2). The maximum sorption capacities predicted by the Langmuir model were 5.8 and 6.8 mmol V/kg saprolite for the UC and RL samples, respectively (Figure 3B).
3.4. Chemical Extractions of Native and Cr- and V-Loaded Saprolites
Solid-phase Cr and V concentrations from chemical extractions of Cr and V-loaded samples and native saprolites are shown in Figure 4. The CBD extraction targeting Cr and V bound to crystalline oxides released the most Cr and V from both the native UC (0.7 mmol Cr/kg and 0.32 mmol V/kg) and RL (0.12 mmol Cr/kg and 1.57 mmol V/kg) saprolites (Figure 4A,C). In the UC saprolite, CBD-extractable Cr and V were ~7 and 5% of the total Cr and V, whereas in the RL saprolite, CBD-extractable V was 37% of the total V, and CBD-extractable Cr was 23% of the environmentally available Cr for the RL saprolite. Ammonium oxalate-targeting ions coprecipitated with amorphous oxyhydroxides released the second highest amount of Cr and V from both UC (0.017 mmol Cr/kg and 0.063 mmol V/kg) and RL native saprolites (0.017 mmol Cr/kg and 0.31 mmol V/kg). MgCl2 extraction targeting ionically bound Cr and V released no detectable Cr from the native UC and RL saprolites, whereas 0.0050 and 0.027 mmol V/kg were released from the native UC and RL saprolites, respectively.
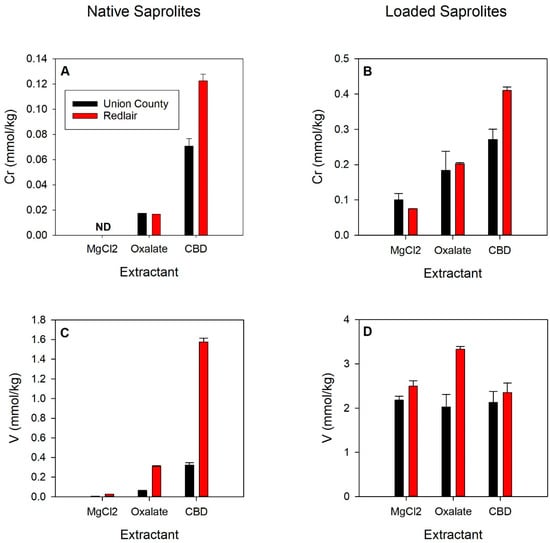
Figure 4.
Concentrations of Cr (A,B) and V (C,D) desorbed from native (A,C) and Cr- (B) or V-loaded (D) Union County (black bars) and Redlair (red bars) saprolites via MgCl2, oxalate, and CBD extractions. Elemental release from Cr- and V-loaded saprolites was corrected for the measured oxalate-, CBD-, and MgCl2-extracted Cr and V from the native saprolites. Error bars represent the standard error of replicates. Reaction conditions: ionic strength (I) = 0.02 M NaCl; pH = 6.5; initial [Cr] = 40 µM; initial [V] = 300 µM; solid solution ratio = 0.5 g/10 mL.
In the Cr-loaded saprolites, the most Cr (0.27 and 0.52 mmol Cr/kg for UC and RL) was released from the CBD extraction (bound to crystalline oxides), followed by the oxalate (0.18 and 0.20 mmol Cr/kg; amorphous oxides) and MgCl2 (0.10 and 0.075 mmol Cr/kg; ionically bound) extractions (Figure 4B). CBD-extracted Cr from the Cr-loaded samples was ~3.8 and 3.4 times greater than it was from the native saprolites, whereas oxalate extractions mobilized ~10.5 and 12 times more Cr in the UC and RL Cr-loaded saprolites than for the native saprolites.
In V-loaded saprolites, extracted V was relatively comparable across all parallel extractants (Figure 4D). The most V was released by the MgCl2 (2.18 mmol V/kg; ionically bound) and oxalate extractions (3.33 mmol V/kg amorphous oxides) for the UC and RL saprolites, respectively. The second greatest amounts of V were released by the CBD (2.13 mmol V/kg; crystalline oxides) and MgCl2 (2.49 mmol V/kg; ionically bound) extractions for the UC and RL saprolites, respectively. The least amount of V was released by the oxalate (2.02 mmol V/kg; amorphous oxides) and CBD extractions (2.35 mmol V/kg; crystalline oxides) for the UC and RL saprolites, respectively.
3.5. Bulk XANES and µ-XRF Analysis
XANES analysis of the Cr- and V-loaded samples showed Cr and V in reduced oxidation states (Figure 5 and Figure 6). Due to concentrations and mineral–host variability, linear-combination fits (LCFs) of the Cr XANES spectra for Cr-loaded samples were accomplished using native (unloaded) saprolite (denoted UC sap or RL sap in Figure 5) and representative Cr(III) and Cr(VI) mineral standards, Cr(OH)3 for Cr(III) and K2Cr2O7 for Cr(VI). Results indicate that little additional Cr(VI) was detected in Cr-loaded samples, as LCFs were comprised primarily of native saprolite spectra (78.1% for UC, 69.3% for RL) and Cr(OH)3 standard spectra (20.8% for UC, 29% for RL), but only negligible (<2%) amounts of the K2Cr2O7 standard in the final fits (Figure 5). Although there may have been small amounts of Cr(VI) in the native, unreacted saprolite, particularly considering the weak pre-edge feature at 5993 eV that is indicative of Cr(VI), the addition of Cr(VI) in the sorption experiments did not appreciably grow that feature in the Cr-loaded saprolite spectra.
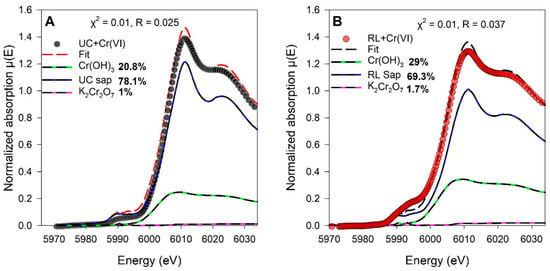
Figure 5.
Linear combination fits of Cr K-edge XANES normalized spectra of (A) Cr-loaded (60 µM) Union County saprolite and (B) Cr-loaded (60 µM) Redlair saprolite. Native saprolite, K2Cr2O7, and Cr(OH)3 standard spectra that were used in the LCF are shown as references. Black and red dots show experimental data, and red and black dashed lines represent the LCF fits for Cr-loaded UC and RL saprolites, respectively. Final LCF results are noted as percentages next to the reference standards in the legends.
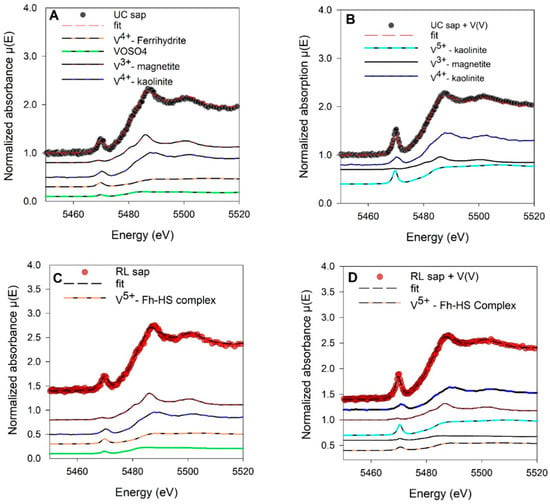
Figure 6.
Linear combination fits of V K-edge XANES normalized spectra of (A) native Union County saprolite materials, (B) V-loaded (450 µM) Union County, (C) native Redlair saprolite, and (D) V-loaded (450 µM) Redlair saprolite. Black and red dots show experimental data, and red and black dashed lines represent the LCF fits for native and V-loaded UC and RL saprolites, respectively.
Vanadium XANES spectra of native and V-loaded saprolites were able to be fit with representative V(III), V(IV), and V(V) mineral standards. The average oxidation state of V was 4.2 ± 0.3 and 4.2 ± 1.4 in the UC and RL V-loaded saprolites, respectively (Figure 6). Vanadium XANES spectra LCF showed that V4+-substituted kaolinite and V5+-sorbed kaolinite were important components of our UC saprolite spectra fit. On the other hand, in addition to V4+ substituted in or V5+ sorbed on well-ordered kaolinite, RL V-loaded XANES fits were improved by ~10% with a standard of V5+ sorbed on a ferrihydrite-humic substance complex.
µ-XRF results show that Cr and V were associated with Fe-bearing phases. However, the distribution of Cr and V across the UC sample was diffuse (Figure 7 and Figure 8). Due to this, three hotspots were selected for microscale elemental spatial correlation analysis in some samples. Correlation analysis for the Cr-loaded Union County (UC-1) sample showed that Cr was positively correlated with Fe and Mn at the different spots (R2 = 0.84 and 0.73 for Cr-Fe and Cr-Mn, respectively, in spot A; 0.72 and 0.75, respectively, in spot B; and 0.93 and 0.89, respectively, in spot C) (Figure 7). Cr was highly positively correlated with Fe and Mn-bearing in Cr-loaded RL saprolites (Figure S8). Vanadium was correlated with Fe at spots A (R2 = 0.94), B (R2 = 0.71), and C (R2 = 0.61) and with Mn at spots A (R2 = 0.97), B (R2 = 0.56), and C (R2 = 0.48) in V-loaded Union County saprolite (UC-2) (Figure 8).
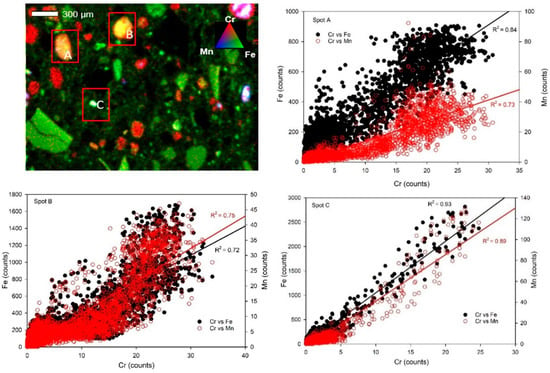
Figure 7.
Solid-phase Cr, Fe, and Mn spatial associations. (Upper left): Tricolor µ-XRF maps of Cr-loaded (60 µM) Union County saprolite (UC-1). Alphabetical labels represent areas chosen for elemental correlation analysis. Red, green, and blue colors represent Cr, Fe, and Mn counts, respectively. (Other panels): Correlation plots of Cr, Fe, and Mn are at spots A, B, and C, respectively.
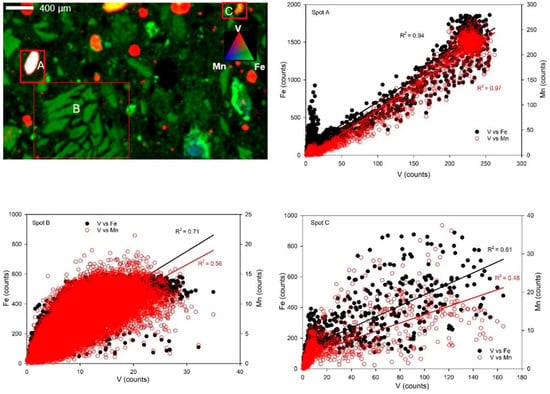
Figure 8.
Solid-phase V, Fe, and Mn spatial associations. (Upper left): Tricolor µ-XRF maps of V-loaded (450 µM) Union County saprolite (UC-2). Alphabetical labels represent areas chosen for elemental correlation analysis. Red, green, and blue colors represent V, Fe, and Mn counts, respectively. (Other panels): Correlation plots of V, Fe, and Mn are at spots A, B, and C, respectively.
4. Discussion
4.1. Controls on Cr Binding to Saprolites
Previous studies have shown the sorption of Cr(VI) on the surface of synthetic Fe and Mn(oxyhydr)oxide surfaces [15,37,40]. Here the results of our sorption experiments on chemically variable natural UC and RL saprolites showed extensive Cr sorption. Both saprolites sorbed similar amounts of Cr, with Langmuir-modeled maximum sorption capacities of 0.35 mmol Cr/kg and 0.30 mmol Cr/kg for the UC and RL saprolites, respectively. While both saprolites sorbed similar amounts of Cr, kinetic differences may be due to differences in the proportions of amorphous Fe and Mn(oxyhydroxide) surfaces of the saprolites, a supposition that is in concert with other authors who have found that natural and synthetic Fe oxyhydroxides could increase the rates and extent of Cr sorption [20,37,41,42,43,44]. In addition, the weak Cr(VI) peak at 5993 eV in our Cr K-edge XANES spectra and the minimal role of Cr(VI) in the LCF fits suggest that reduction of Cr took place when Cr(VI) was added to the saprolites (Figure 5). Added Cr may have been reduced by the Fe(II)-bearing phases detected in the samples (Table S7), a hypothesis in agreement with a number of reports of Fe(II) minerals participating in the abiotic reduction of Cr(VI) to Cr(III) [37,45,46,47,48].
Although we could not distinguish primary vs. secondary Fe and Mn phases with our spectroscopic imaging technique, µ-XRF analysis showed that Fe- and Mn-bearing minerals are important host phases for added Cr. This was highlighted by the positive correlation between Cr, Fe, and Mn in the Cr-loaded (UC-1 and RL-1) samples (Figure 7 and Figure S8). Although Mn is <0.15 wt.% in the saprolites, the correlation between Cr and Mn may be because the surface function group on the mixed-valence variably charged Mn-bearing phase was slightly deprotonated at the pH of our experiment; the electrostatic attraction could have resulted in the sorption of Cr(III) on Mn-bearing phases, and/or Mn was simply associated with the same Fe phases that retained Cr [49]. Nevertheless, spectroscopic imaging confirms that Fe- and Mn-bearing minerals are important sorbents for Cr in both the UC and RL saprolites, findings also supported by the chemical extractions of Cr-loaded saprolites (Figure 4).
Overall, our analytical data suggest that incorporation and sorption of Cr on amorphous and crystalline (oxyhydr)oxide phases are the main mechanisms for Cr retention. Immobilization of added Cr(VI) is further enhanced through its reduction to Cr(III) phases, which may be promoted when abundant Fe(II) is present as an oxidant, as was the case for our sediments whose Fe speciation via XAS indicated some mixed valence and reduced Fe phases (e.g., chlorite, magnetite, and hornblende; Table S7). Resulting Cr(III) was associated with Fe and Mn (oxyhydr)oxide phases.
4.2. Controls on V Binding to Saprolites
Vanadium has been shown to strongly adsorb on the surface of Fe, Mn, Al-bearing, and clay minerals [5,6,40,50,51]. Here, maximum sorption capacities from our Langmuir adsorption isotherm model show that the UC (5.8 mmol V/kg) and RL saprolites (6.8 mmol V/kg) could sorb similar amounts of V. Interestingly, both saprolites’ affinity for V was over an order of magnitude greater than for Cr (on a molar basis), consistent with the relative abundances of V vs. Cr in the native samples (Table 1). This could be a result of the binding mechanism of Cr and V on Fe oxide phases. Cr is known to form both inner- and outer-sphere complexes with Fe oxides, while V has been shown to exclusively form strong covalent, inner-sphere polymeric complexes with Fe oxides, especially at higher V concentrations such as those used in our study [22,52]. The reason for the slightly higher maximum sorption capacity of V on RL than UC remains unclear, though the greater amount of Fe (oxyhydr)oxides in RL is likely a factor. Previous studies have shown that due to high surface area, site density, and abundant singly and doubly coordinated -OH groups, Fe (oxyhydr)oxides can display high adsorption capacities for V [25,51,52,53,54].
Chemical extractions revealed that for any of our extractants, CBD released the most amounts of total V from the native UC (5% of total V released) and RL (37% of total V released) saprolites, highlighting the occurrence of V in crystalline oxide mineral phases. This is comparable with the study by Wisawapipat and Kretzschmar [25], which showed that ~38% of V was extracted from crystalline Fe oxides in well-weathered soils. In contrast, CBD extractions released the least V in the V-loaded RL saprolite, while the highest V was released by amorphous phases in UC saprolite. Our collective data suggest that V was preferentially substituted into vacant sites in crystalline Fe phases in the native RL saprolites, but when these sites became saturated due to increased V sorption in V-loaded RL saprolites, V was either sorbed onto amorphous surfaces or ionically bound on surfaces of other minerals in the saprolite [53,54]. MgCl2 extractions that targeted ionically sorbed metals showed that ~250 and 100 times the initial native V concentration was released in the V-loaded UC and RL saprolites, indicating a dominant electrostatic interaction mechanism for V(V) retention. To further highlight this, our follow-up experiment showed that increasing ionic strength increased V adsorption in UC saprolite (Figure S9).
Results from XANES analysis suggest that V in native and V-loaded UC and RL saprolites may be associated with Fe oxide phases. In addition, XANES LCF also indicated that up to 34–83% of V in both native and loaded saprolite samples were likely associated with kaolinite phases. This is unsurprising because XRD showed the presence of 1:1 crystalline clay minerals (nacrite and kaolinite) in our saprolites. Lastly, µ-XRF results also corroborated the association of V with Fe and Mn phases in UC V-loaded saprolites, with strongly positive correlations of up to 0.94 and 0.97 recorded for V-Fe and V-Mn, respectively.
In our studies, although the average redox state of V in native saprolites was V(IV), XANES analyses of V-loaded samples suggested that mixed species of V(IV,V) may coexist (Figure 6B,D). The presence of small amounts of Fe(II)-bearing minerals in UC and RL saprolites could have reduced V(V), producing mixed-valence V species and modulating the retention and reduction of V [25,55]; however, our analysis of vanadium speciation in V-loaded samples was also influenced by native V in the saprolite.
Collectively, we identify that sorption dominates retention of dissolved V added to the saprolites. Although most native V was found as V(IV), the addition of V(V) did not appear to lead to an appreciable reduction of this added V, resulting in loaded saprolites with mixed V speciation. The saprolites had large capacities for sorbing added V, substantially through binding to Fe (oxyhdry)oxides, although the strength of this binding needs further investigation as much of the added V was able to be extracted by MgCl2, indicating electrostatic V retention and suggesting that added V could be remobilized under flowing groundwater conditions.
4.3. Significance for Groundwater Quality
Our data indicate that the proposed possible mechanism of retention in both Cr- and V-loaded saprolites is a combination of sorption by surface complexation, electrostatic interactions, and ion exchange, while structural incorporation is likely the dominant process in native saprolites. The presence of exchangeable Cr and V in Cr- and V-loaded saprolites indicates a potential for easy release of these sorbed elements from solids to the aqueous phase. In our native saprolites, minimal amounts of Cr and V were associated with ionically active species. However, in Cr- and V-loaded saprolites, a small fraction of Cr and a large fraction of V were found to be environmentally labile (Figure 4B,D). This finding holds significant environmental implications, especially in the presence of chloride and phosphate introduced into waste streams, which could outcompete with weakly sorbed Cr and V for adsorption sites, facilitating their release into the environment.
The release of naturally occurring Cr and V from aquifer solids into groundwater would pose health and environmental risks. Our study demonstrates the potential for elemental release from various host phases in both native and Cr- and V-sorbed chemically variable Piedmont saprolites. Particularly in settings with complex and evolving groundwater chemistry and in the presence of abundant reactive Mn oxides, interactions with saprolites may oxidatively dissolve Cr and V. These contaminants, once mobilized, could easily be transported along groundwater flow paths to wells. However, in the absence of anthropogenic influences, amorphous and crystalline phases like Fe and Mn(oxyhydr)oxides and aluminosilicate clays might serve as temporary repositories of remobilized Cr and V.
5. Conclusions
We examined the phase associations and sorption behavior of Cr and V in two saprolite samples from a region of the North Carolina Piedmont where elevated Cr and V have been detected in well water. Chromium and V in both native and loaded saprolite samples were associated with iron (oxyhydr)oxides and Mn-bearing minerals. Across all samples and treatments, Cr primarily existed as Cr(III), likely precipitating as Cr(OH)3 even after being introduced as Cr(VI) in solution. Vanadium, however, existed as a combination of V(III), V(IV), and V(V) species, and introduced V(V) was associated with Fe oxides, kaolinite, and other clay minerals. Overall, this study has shown that the mechanisms controlling Cr and V retention include sorption onto clay minerals, Fe and Mn oxides through surface complexation, and electrostatic interactions.
Although both Cr and V were strongly retained onto NC Piedmont saprolites, higher sorption capacities were observed for V over Cr, consistent with the higher natural abundance of V on the saprolites. Finally, although Cr and V were primarily strongly sorbed to mineral phases, the presence of exchangeable forms of Cr and V in native and loaded saprolites was also observed, indicating a repository of easily mobilizable forms of these contaminants. More studies are needed to determine how land use and management strategies may impact the release of both Cr and V from their host phases, compromising groundwater quality.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/soilsystems8040127/s1. Supplementary Material includes Additional Methods and the following tables and figures: Table S1: Chemical extraction procedure for native and Cr- and V-loaded saprolites; Table S2: Statistical analysis of goodness of fit from Langmuir and Freundlich models; Table S3: Standard spectra used in the V K-edge XANES LCF; Table S4: V K-edge XANES linear combination fits for native saprolites; Table S5: V K-edge XANES linear combination fits (LCFs) for vanadium-loaded saprolites; Table S6: Standard spectra used in the Fe K-edge XANES LCF; Table S7: Fe XANES linear combination fits; and Table S8: R2 values for kinetic models. Figure S1: X-ray diffraction pattern of Union County saprolite; Figure S2: X-ray diffraction pattern of Redlair saprolite; Figure S3: Fe K-edge XANES spectra of Union County and Redlair saprolites; Figure S4: Normalized V pre-edge peak intensity plotted against pre-edge energy position for standards and native UC and Redlair saprolites; Figure S5: Mn K-edge XANES first derivatives of native saprolites and standards; Figure S6: Cr K-edge XANES first derivatives of native saprolites and standards; Figure S7: Freundlich isotherm models for sorption of Cr and V onto saprolites; Figure S8: Solid-phase Cr, Fe, and Mn spatial associations in Cr-loaded Redlair saprolite; and Figure S9: Effect of ionic strength on Cr and V sorption on Union County saprolite.
Author Contributions
Conceptualization, F.O.B., O.W.D. and M.L.P.; methodology, F.O.B., H.R.P., O.W.D. and M.L.P.; investigation, F.O.B., H.R.P., R.E.A., I.G.O. and D.S.V.; data curation, F.O.B., H.R.P., R.E.A., I.G.O. and M.L.P.; writing—original draft preparation, F.O.B. and M.L.P.; writing—review and editing, F.O.B., D.S.V., O.W.D. and M.L.P.; supervision, O.W.D. and M.L.P.; funding acquisition, O.W.D., D.S.V., R.E.A. and M.L.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Institute of Health through the National Institute of Environmental Sciences under grant No. NIH—5P42ES031007-02 and by the Geologic Society of America (GSA).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. The raw data supporting the conclusions of this article are available from the corresponding author on request.
Acknowledgments
The spectroscopic analysis portion of this study was performed at the Stanford Synchrotron Radiation Light Source (SSRL), a Directorate of SLAC National Laboratory operated for the U.S. Department of Energy office of Science by Stanford University. We thank Sadeya Ulfat Tashnia, Macon Abernathy, Alireza Namayandeh, Edward Oloughlin, Maxim Boyanov, and Kenneth Kemner for their assistance in sample collection, GIS analysis, and provision of databases and standards for XRD and XANES analyses. We also thank Chris Russo and Jesse Muratli of the Keck Laboratory for Plasma Spectrometry, Oregon State University, for the analytical support. We thank Sam Webb for assistance at SSRL. We acknowledge Redlair Observatory, a partnership including the North Carolina Plant Conservation Program, Catawba Lands Conservancy, and UNC Charlotte, for sample site access. Finally, our unreserved appreciation also goes to every member of the Soil and Water Laboratory, University of Oregon, for their support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, Z.Q.; Yu, Y.; Zhang, X.H.; Komorowski, J. Chromium-insulin reduces insulin clearance and enhances insulin signaling by suppressing hepatic insulin-degrading enzyme and proteasome protein expression in KKAy mice. Front. Endocrinol. 2014, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Jardine, P.M.; Fendorf, S.E.; Mayes, M.A.; Larsen, I.L.; Brooks, S.C.; Bailey, W.B. Fate and transport of hexavalent chromium in undisturbed heterogeneous soil. Environ. Sci. Technol. 1999, 33, 2939–2944. [Google Scholar] [CrossRef]
- Peel, H.R.; Balogun, F.O.; Bowers, C.A.; Miller, C.T.; Obeidy, C.S.; Polizzotto, M.L.; Tashnia, S.U.; Vinson, D.S.; Duckworth, O.W. Towards Understanding Factors Affecting Arsenic, Chromium, and Vanadium Mobility in the Subsurface. Water 2022, 14, 3687. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Gong, Y.; Gao, J.; Sun, T.; Liu, Y.; Oturan, N.; Oturan, M.A. The reduction of Cr(VI) to Cr(III) mediated by environmentally relevant carboxylic acids: State-of-the-art and perspectives. J. Hazard. Mater. 2019, 365, 205–226. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.M.; Alessi, D.S.; Tack, F.M.; Ok, Y.S.; Kim, K.-H.; Gustafsson, J.P.; Sparks, D.L.; Rinklebe, J. Redox chemistry of vanadium in soils and sediments: Interactions with colloidal materials, mobilization, speciation, and relevant environmental implications—A review. Adv. Colloid Interface Sci. 2019, 265, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.A.; Hadialhejazi, G.; Gustafsson, J.P. Vanadium sorption by mineral soils: Development of a predictive model. Chemosphere 2017, 168, 925–932. [Google Scholar] [CrossRef]
- Schlesinger, W.H.; Klein, E.M.; Vengosh, A. Global biogeochemical cycle of vanadium. Proc. Natl. Acad. Sci. USA. 2017, 114, E11092–E11100. [Google Scholar] [CrossRef]
- Vengosh, A.; Coyte, R.; Karr, J.; Harkness, J.S.; Kondash, A.J.; Ruhl, L.S.; Merola, R.B.; Dywer, G.S. Origin of Hexavalent Chromium in Drinking Water Wells from the Piedmont Aquifers of North Carolina. Environ. Sci. Technol. Lett. 2016, 3, 409–414. [Google Scholar] [CrossRef]
- Coyte, R.M.; McKinley, K.L.; Jiang, S.; Karr, J.; Dwyer, G.S.; Keyworth, A.J.; Davis, C.C.; Kondash, A.J.; Vengosh, A. Occurrence and distribution of hexavalent chromium in groundwater from North Carolina, USA. Sci. Total Environ. 2019, 711, 135135. [Google Scholar] [CrossRef]
- Coyte, R.M.; Vengosh, A. Factors Controlling the Risks of Co-occurrence of the Redox-Sensitive Elements of Arsenic, Chromium, Vanadium, and Uranium in Groundwater from the Eastern United States. Environ. Sci. Technol. 2020, 54, 4367–4375. [Google Scholar] [CrossRef]
- Gillispie, E.C.; Austin, R.E.; Rivera, N.A.; Bolich, R.; Duckworth, O.W.; Bradley, P.; Amoozegar, A.; Hesterberg, D.; Polizzotto, M.L. Soil Weathering as an Engine for Manganese Contamination of Well Water. Environ. Sci. Technol. 2016, 50, 9963–9971. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, T.R.; Austin, R.E.; Bradley, P.J.; Eaves, L.A.; Fry, R.C.; George, A.; Gray, K.M.; Osborne, J.A.; Stýblo, M.; Vinson, D.S.; et al. Geologic predictors of drinking water well contamination in North Carolina. PLoS Water 2024, 3, e0000194. [Google Scholar] [CrossRef]
- Wright, M.T.; Belitz, K. Factors controlling the regional distribution of vanadium in groundwater. Ground Water 2010, 48, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yan, X.; Peacock, C.L.; Zhang, S.; Li, W.; Zhang, J.; Feng, X.; Liu, F.; Yin, H. Adsorption of Cr(VI) on Al-substituted hematites and its reduction and retention in the presence of Fe2+ under conditions similar to subsurface soil environments. J. Hazard. Mater. 2020, 390, 122014. [Google Scholar] [CrossRef]
- Landrot, G.; Ginder-Vogel, M.; Livi, K.; Fitts, J.P.; Sparks, D.L. Chromium(III) oxidation by three poorly-crystalline manganese(IV) oxides. 1. chromium(III)-oxidizing capacity. Environ. Sci. Technol. 2012, 46, 11594–11600. [Google Scholar] [CrossRef]
- Pan, C.; Liu, H.; Catalano, J.G.; Wang, Z.; Qian, A.; Giammar, D.E. Understanding the Roles of Dissolution and Diffusion in Cr(OH)3 Oxidation by Î-MnO2. ACS Earth Sp. Chem. 2019, 3, 357–365. [Google Scholar] [CrossRef]
- Oze, C.; Bird, D.K.; Fendorf, S. Genesis of hexavalent chromium from natural sources in soil and groundwater. Proc. Natl. Acad. Sci. USA 2007, 104, 6544–6549. [Google Scholar] [CrossRef]
- Liang, J.; Huang, X.; Yan, J.; Li, Y.; Zhao, Z.; Liu, Y.; Ye, J.; Wei, Y. A review of the formation of Cr(VI) via Cr(III) oxidation in soils and groundwater. Sci. Total Environ. 2021, 774, 145762. [Google Scholar] [CrossRef]
- Abernathy, J.; Schaefer, V.; Vessey, J.; Liu, H.; Ying, C. Oxidation of V(IV) by Birnessite: Kinetics and Surface Complexation. Environ. Sci. Technol. 2021, 55, 11703–11712. [Google Scholar] [CrossRef]
- Johnston, C.P.; Chrysochoou, M. Investigation of Chromate Coordination on Ferrihydrite by in Situ ATR-FTIR Spectroscopy and Theoretical Frequency Calculations. Environ. Sci. Technol. 2012, 46, 5851–5858. [Google Scholar] [CrossRef]
- Ding, Z.; Sun, G.; Fu, F.; Ye, C. Phase transformation of Cr(VI)-adsorbed ferrihydrite in the presence of Mn(II): Fate of Mn(II) and Cr(VI). J. Environ. Sci. 2022, 113, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, M.J.; Schaefer, M.V.; Ramirez, R.; Garniwan, A.; Lee, I.; Zaera, F.; Polizzotto, M.L.; Ying, S.C. Vanadate Retention by Iron and Manganese Oxides. ACS Earth Sp. Chem. 2022, 6, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J.P. Vanadium geochemistry in the biogeosphere–speciation, solid-solution interactions, and ecotoxicity. Appl. Geochem. 2019, 102, 1–25. [Google Scholar] [CrossRef]
- Oze, C.; Fendorf, S.; Coleman, R.G. Chromium Geochemistry in Serpentinized Ultramafic Rocks and Serpentine Soils from the Franciscan Formation of California Organic Matter Mineralization and Metal Cycling During Flood Plain Evolution View Project Uranium Sequestration View Project. 2002. Available online: https://www.researchgate.net/publication/252099396 (accessed on 21 November 2024).
- Wisawapipat, W.; Kretzschmar, R. Solid Phase Speciation and Solubility of Vanadium in Highly Weathered Soils. Environ. Sci. Technol. 2017, 51, 8254–8262. [Google Scholar] [CrossRef]
- Scheinost, A.C.; Kretzschmar, R.; Pfister, S.; Roberts, D.R. Combining Selective Sequential Extractions, X-ray Absorption Spectroscopy, and Principal Component Analysis for Quantitative Zinc Speciation in Soil. Environ. Sci. Technol. 2002, 36, 5021–5028. [Google Scholar] [CrossRef]
- Eaves, L.A.; Keil, A.P.; Rager, J.E.; George, A.; Fry, R.C. Analysis of the novel NCWELL database highlights two decades of co-occurrence of toxic metals in North Carolina private well water: Public health and environmental justice implications. Sci. Total Environ. 2022, 812, 151479. [Google Scholar] [CrossRef]
- Eaves, L.; Keil, A.P.; Henry, A.; Fry, R.C. A Dataset Describing Well Water Metal/Metalloid Contamination of Private Wells in North Carolina, 1998–2019, V1 ed.; UNC Dataverse: Chapel Hill, NC, USA, 2021. [Google Scholar]
- Smith, S.M. National Geochemical Database—Reformatted Data from the National Uranium Resource Evaluation (NURE) Hydrogeochemical and Stream Sediment Reconnaissance (HSSR) Program [Version 1.40]: US Geological Survey Open-File Report OF 97-492. 2006. Available online: http://pubs.usgs.gov/of/1997/ofr-97-0492/ (accessed on 3 June 2024).
- Vinson, D.S.; Vengosh, A.; Hirschfeld, D.; Dwyer, G.S. Relationships between radium and radon occurrence and hydrochemistry in fresh groundwater from fractured crystalline rocks, North Carolina (USA). Chem. Geol. 2009, 260, 159–171. [Google Scholar] [CrossRef]
- LaFayette, G.N.; Knappett, P.S.K.; Li, Y.; Loza-Aguirre, I.; Polizzotto, M.L. Geogenic sources and chemical controls on fluoride release to groundwater in the Independence Basin, Mexico. Appl. Geochem. 2020, 123, 104787. [Google Scholar] [CrossRef]
- Keon, N.E.; Swartz, C.H.; Brabander, D.J.; Harvey, C.; Hemond, H.F. Validation of an arsenic sequential extraction method for evaluating mobility in sediments. Environ. Sci. Technol. 2001, 35, 2778–2784. [Google Scholar] [CrossRef]
- Strawn, D.G. Sorption mechanisms of chemicals in soils. Soil Syst. 2021, 5, 13. [Google Scholar] [CrossRef]
- Balogun, F.O.; Aiken, M.L.; Namayandeh, A.; Duckworth, O.W.; Polizzotto, M.L. Dissolved Organic Carbon Diminishes Manganese Oxide-Driven Oxidation of Chromium. Chemosphere 2023, 344, 140424. [Google Scholar] [CrossRef] [PubMed]
- Polizzotto, M.L.; Harvey, C.F.; Li, G.; Badruzzman, B.; Ali, A.; Newville, M.; Sutton, S.; Fendorf, S. Solid-phases and desorption processes of arsenic within Bangladesh sediments. Chem. Geol. 2006, 228, 97–111. [Google Scholar] [CrossRef]
- Duckworth, O.W.; Akafia, M.M.; Andrews, M.Y.; Bargar, J.R. Siderophore-promoted dissolution of chromium from hydroxide minerals. Environ. Sci. Process. Impacts. 2014, 16, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, A.H.; Peña, J.; Amor, M.; Duckworth, O.W. Cr(VI) uptake and reduction by biogenic iron (oxyhydr)oxides. Environ. Sci. Proc. Impacts 2018, 20, 1056–1068. [Google Scholar] [CrossRef]
- Manceau, A.; Marcus, M.A.; Grangeon, S. Determination of Mn valence states in mixed-valent manganates by XANES spectroscopy. Am. Mineral. 2012, 97, 816–827. [Google Scholar] [CrossRef]
- Ho, Y.S.; McKay, G. Pseudo-second order model for sorption processes. Process Biochem. 1999, 34, 451–465. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, S.; Fernandez-Marcos, M.L. Sorption and desorption of vanadate, arsenate and chromate by two vol-canic soils of equatorial africa. Soil Syst. 2021, 5, 22. [Google Scholar] [CrossRef]
- Lilli, M.A.; Nikolaidis, N.P.; Karatzas, G.P.; Kalogerakis, N. Identifying the controlling mechanism of geogenic origin chromium release in soils. J. Hazard. Mater. 2019, 366, 169–176. [Google Scholar] [CrossRef]
- Choppala, G.; Kunhikrishnan, A.; Seshadri, B.; Park, J.H.; Bush, R.; Bolan, N. Comparative sorption of chromium species as influenced by pH, surface charge and organic matter content in contaminated soils. J. Geochem. Explor. 2018, 184, 255–260. [Google Scholar] [CrossRef]
- Fendorf, S.E. Surface reactions of chromium in soils and waters. Geoderma 1995, 67, 55–71. [Google Scholar] [CrossRef]
- Johnston, C.P.; Chrysochoou, M. Mechanisms of chromate adsorption on hematite. Geochim. Cosmochim. Acta 2014, 138, 146–157. [Google Scholar] [CrossRef]
- Fendorf, S.; Wielinga, B.W.; Hansel, C.M. Chromium transformations in natural environments: The role of biological and abiological processes in chromium(vi) reduction. Int. Geol. Rev. 2000, 42, 691–701. [Google Scholar] [CrossRef]
- Agrawal, S.G.; Fimmen, R.L.; Chin, Y.P. Reduction of Cr(VI) to Cr(III) by Fe(II) in the presence of fulvic acids and in lacustrine pore water. Chem. Geol. 2009, 262, 328–335. [Google Scholar] [CrossRef]
- Joe-Wong, C.; Brown, G.E.J.; Maher, K. Kinetics and Products of Chromium(VI) Reduction by Iron(II/III)-Bearing Clay Minerals. Environ. Sci. Technol. 2017, 51, 9817–9825. [Google Scholar] [CrossRef]
- Liao, W.; Ye, Z.; Yuan, S.; Cai, Q.; Tong, M.; Qian, A.; Cheng, D. Effect of Coexisting Fe(III) (oxyhydr)oxides on Cr(VI) Reduction by Fe(II)-Bearing Clay Minerals. Environ. Sci. Technol. 2019, 53, 13767–13775. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, F.; Xue, J.; Chen, S.; Wang, J.; Yang, Y. Enhanced removal of heavy metal ions from aqueous solution using manganese dioxide-loaded biochar: Behavior and mechanism. Sci. Rep. 2020, 10, 6067. [Google Scholar] [CrossRef]
- Larsson, M.A.; D’Amato, M.; Cubadda, F.; Raggi, A.; Öborn, I.; Kleja, D.B.; Gustafsson, J.P. Long-term fate and transformations of vanadium in a pine forest soil with added converter lime. Geoderma 2015, 259–260, 271–278. [Google Scholar] [CrossRef]
- Gäbler, H.-E.; Glüh, K.; Bahr, A.; Utermann, J. Quantification of vanadium adsorption by German soils. J. Geochem. Explor. 2009, 103, 37–44. [Google Scholar] [CrossRef]
- Vessey, C.J.; Lindsay, M.B.J. Aqueous Vanadate Removal by Iron(II)-Bearing Phases under Anoxic Conditions. Environ. Sci. Technol. 2020, 54, 4006–4015. [Google Scholar] [CrossRef]
- Kajjumba, G.W.; Aydın, S.; Güneysu, S. Adsorption isotherms and kinetics of vanadium by shale and coal waste. Adsorpt. Sci. Technol. 2018, 36, 936–952. [Google Scholar] [CrossRef]
- Baldermann, A.; Stamm, F.M. Effect of kinetics, pH, aqueous speciation and presence of ferrihydrite on vanadium (V) uptake by allophanic and smectitic clays. Chem. Geol. 2022, 607, 121022. [Google Scholar] [CrossRef]
- O’Loughlin, E.; Boyanov, M.; Kemner, K. Reduction of Vanadium (V) by Iron (II)-Bearing Minerals. Minerals 2021, 11, 316. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).