Electrochemical Behavior of Al–Al9Co2 Alloys in Sulfuric Acid



Abstract
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
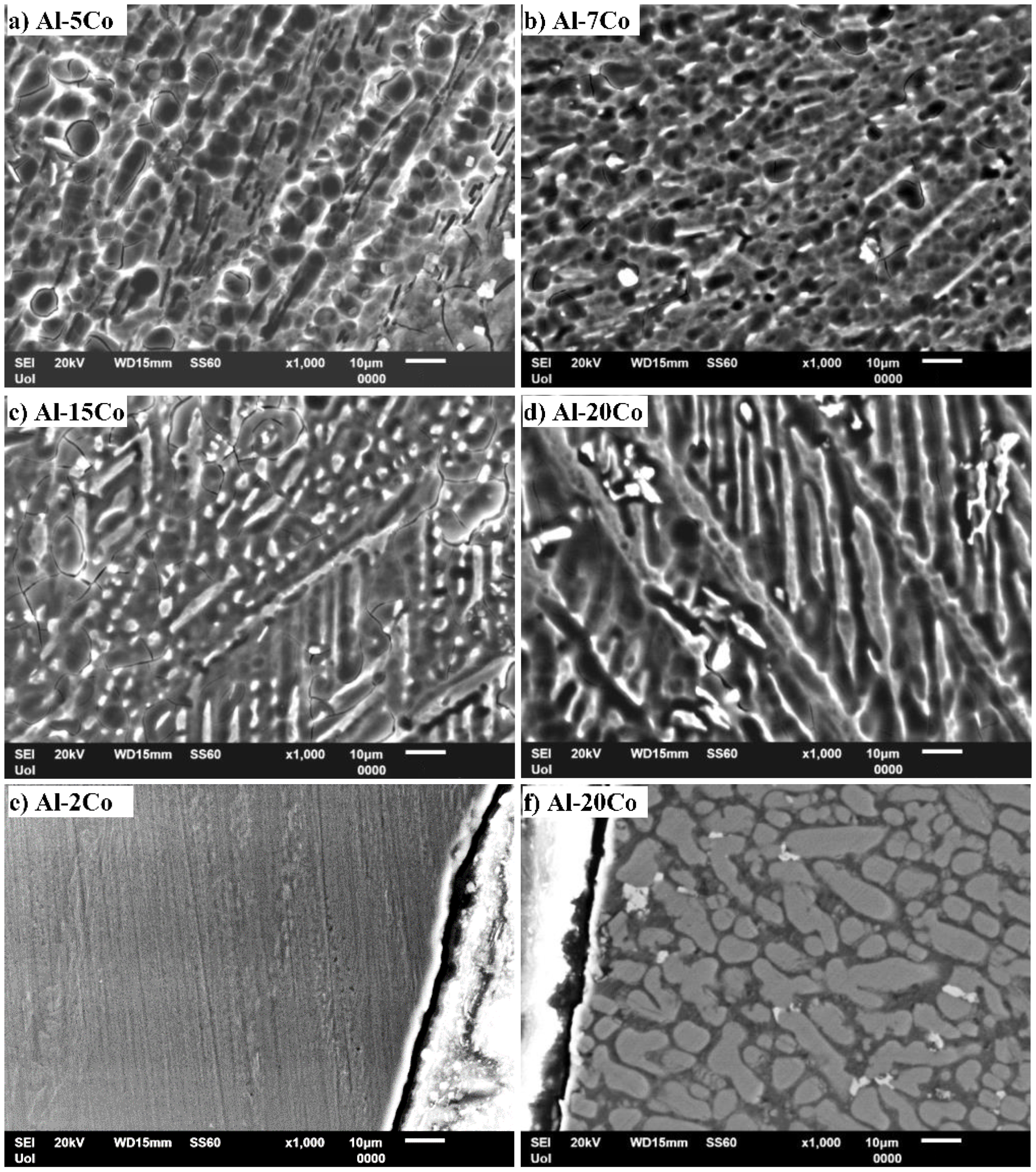
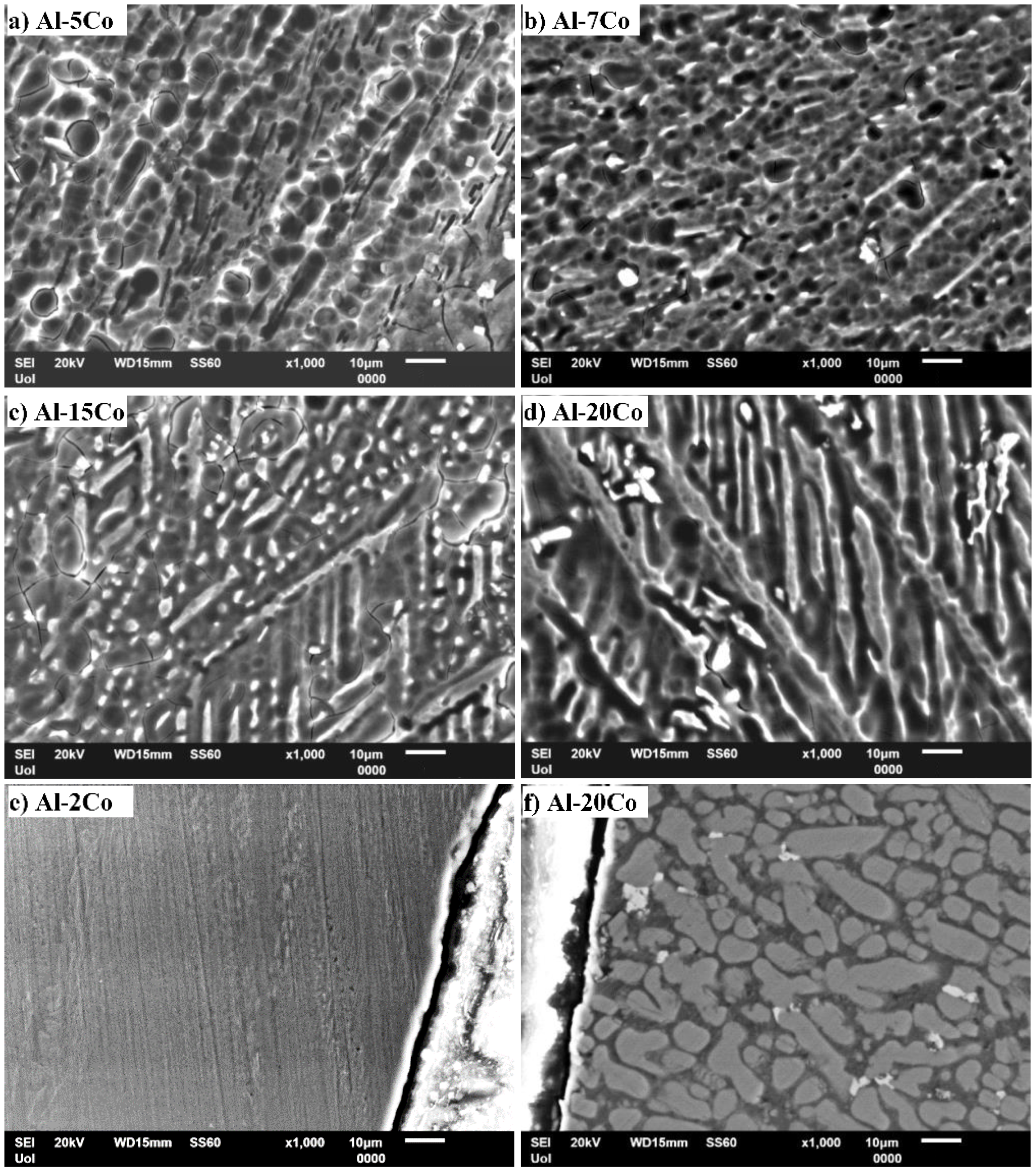
3.1. Microstructural Analysis
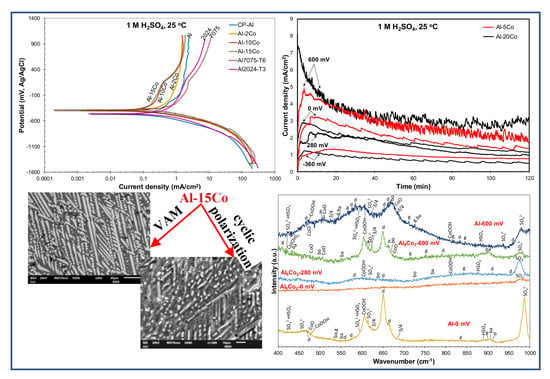
3.2. Electrochemical Performance in 1 M H2SO4
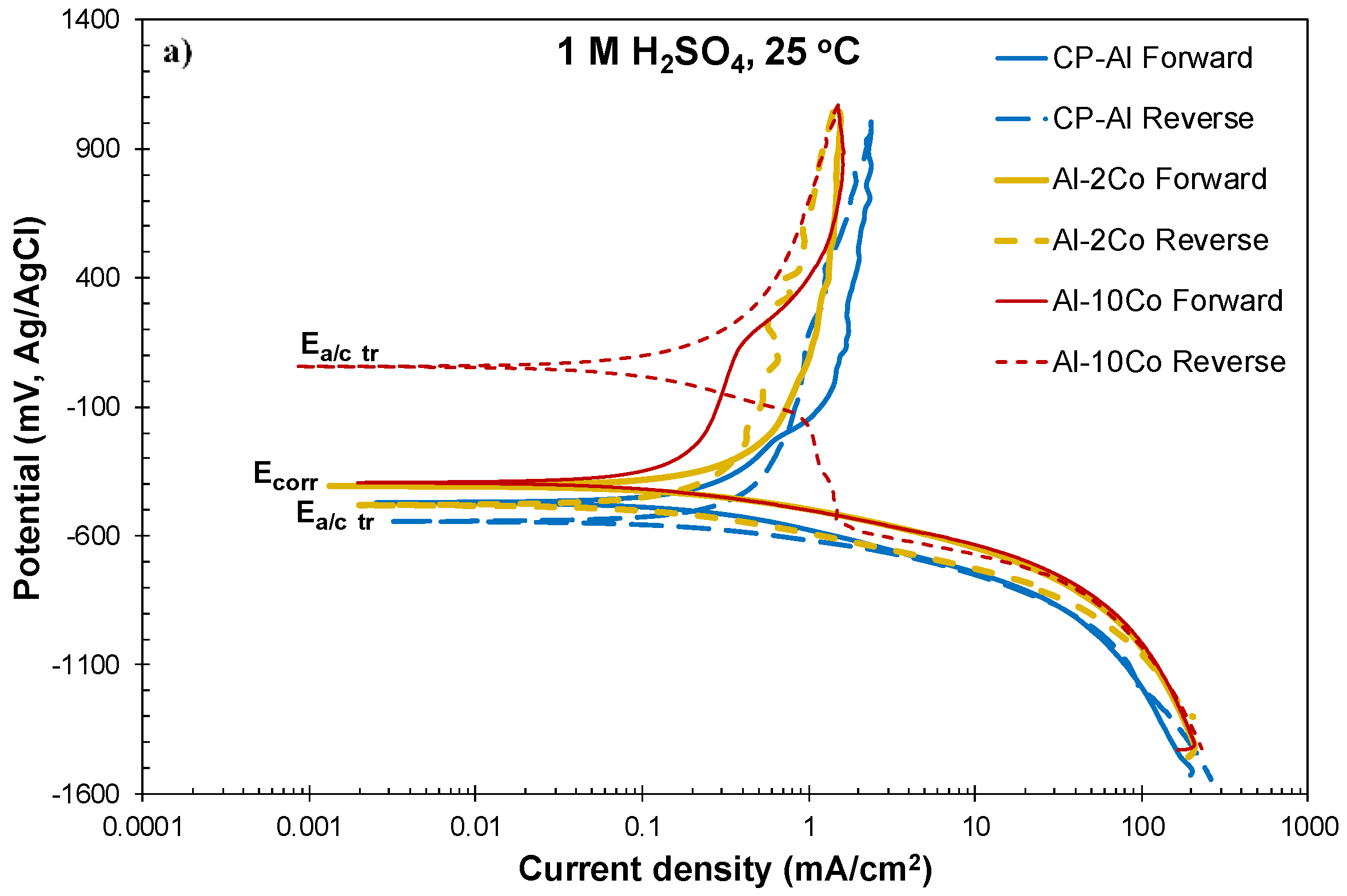
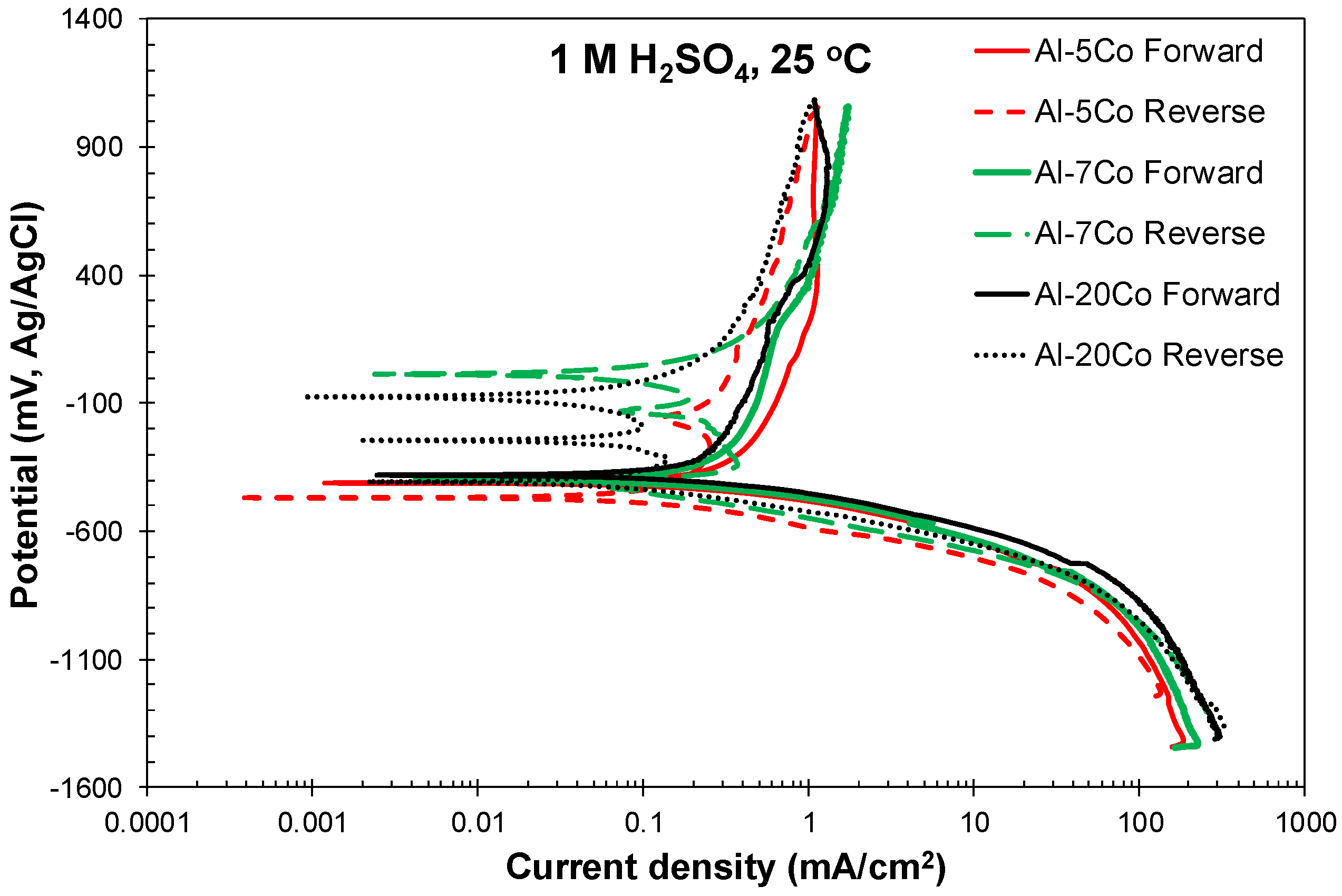
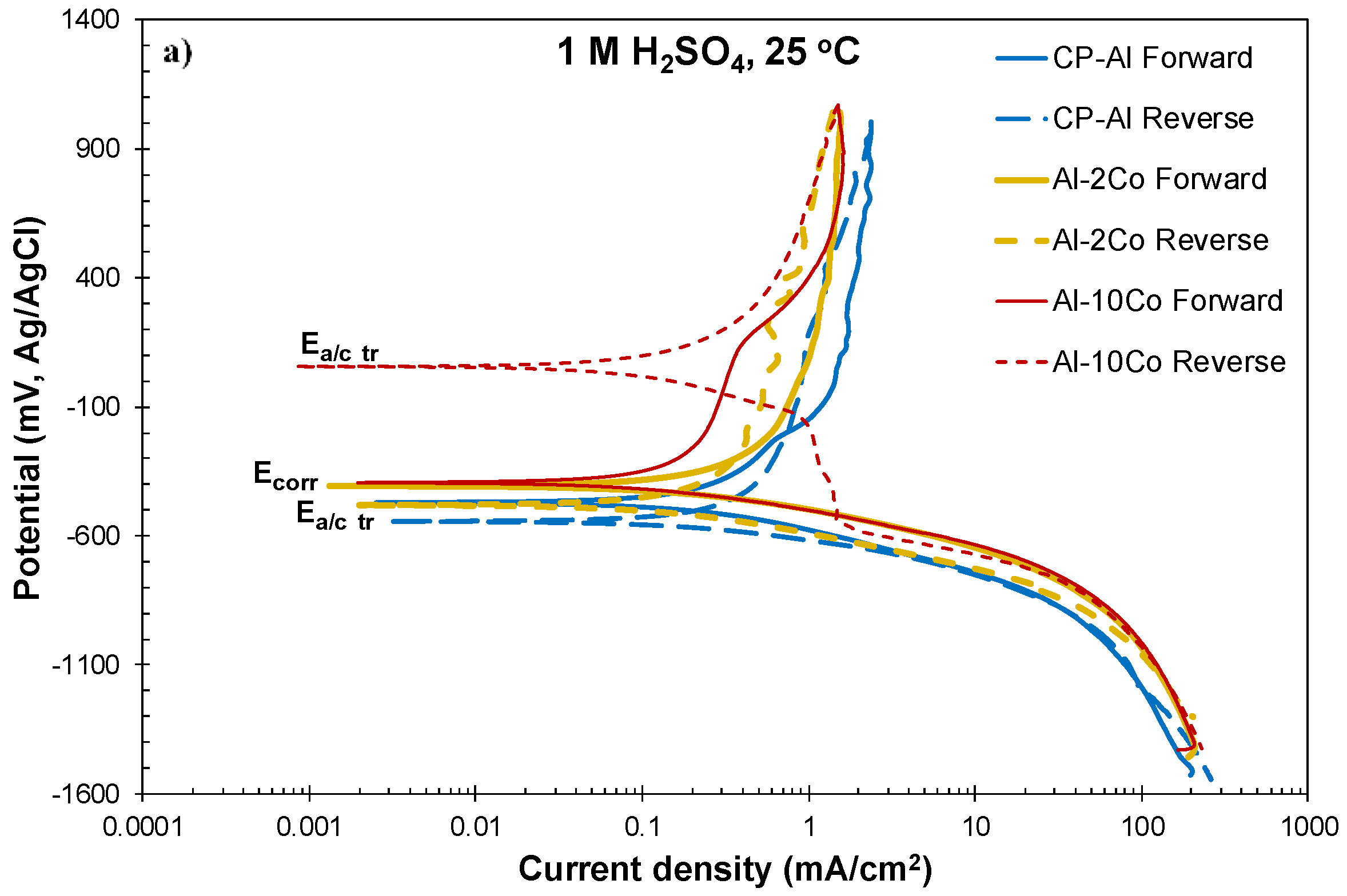
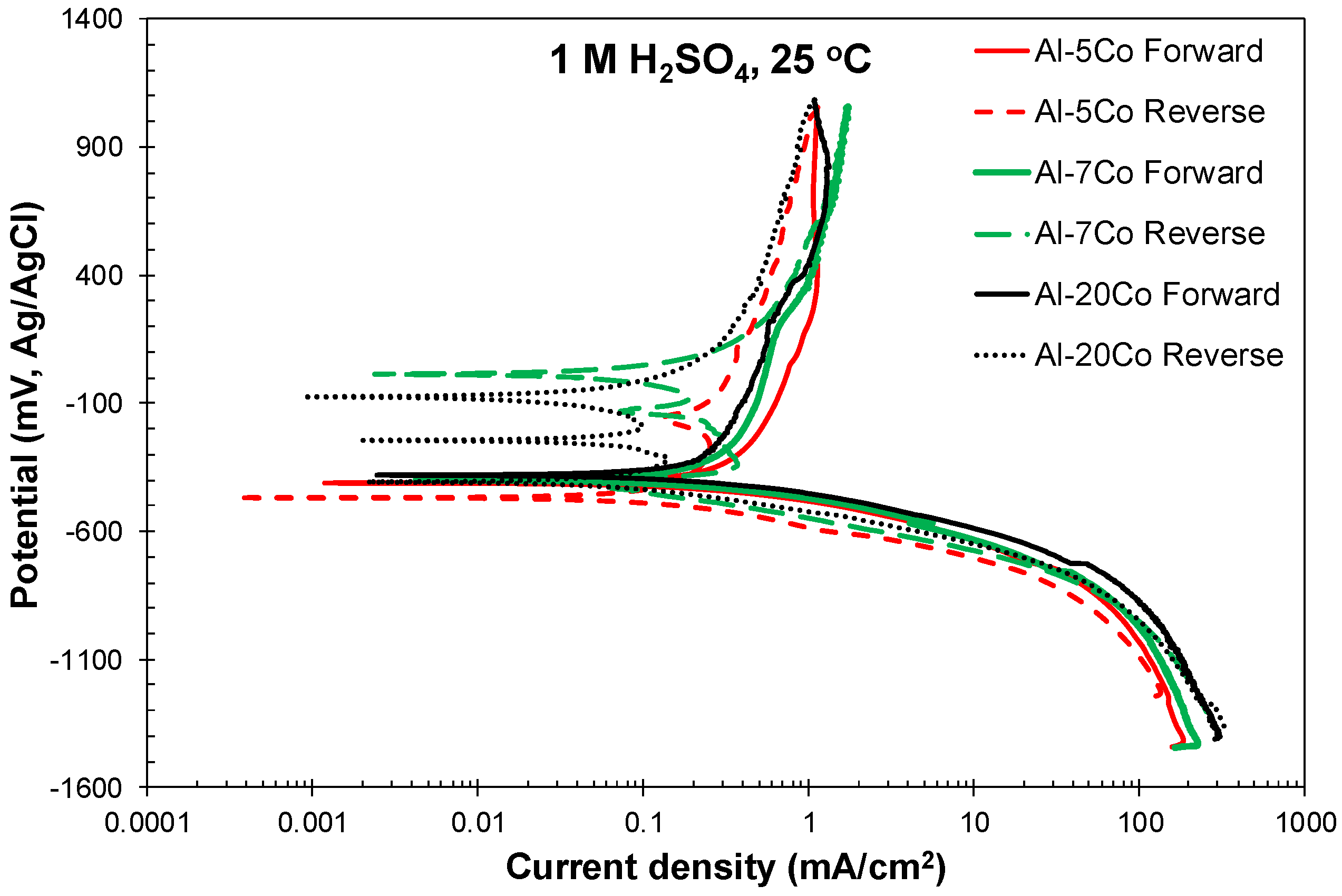
3.2.1. Cyclic Polarization
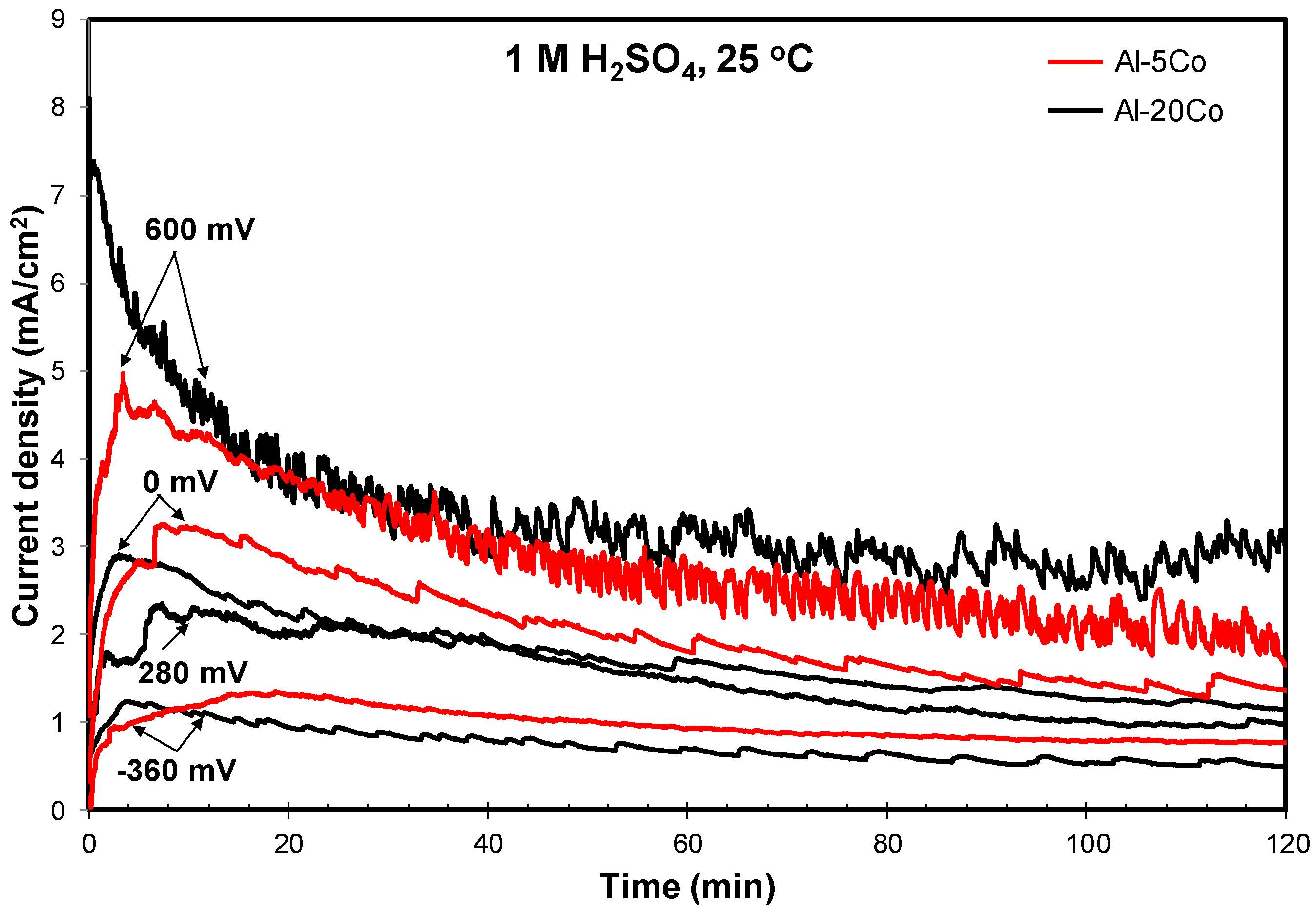
3.2.2. Chronoamperometry
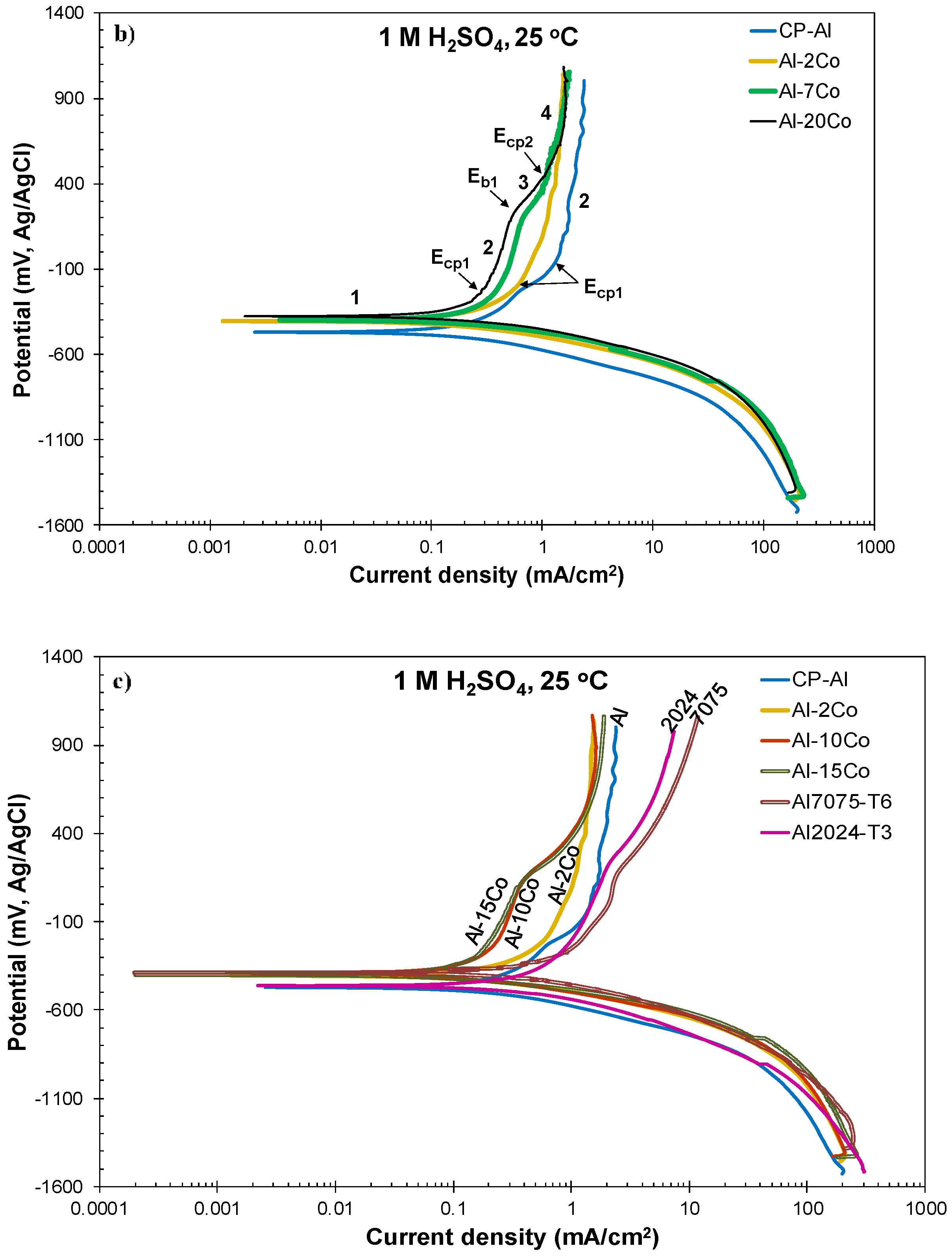
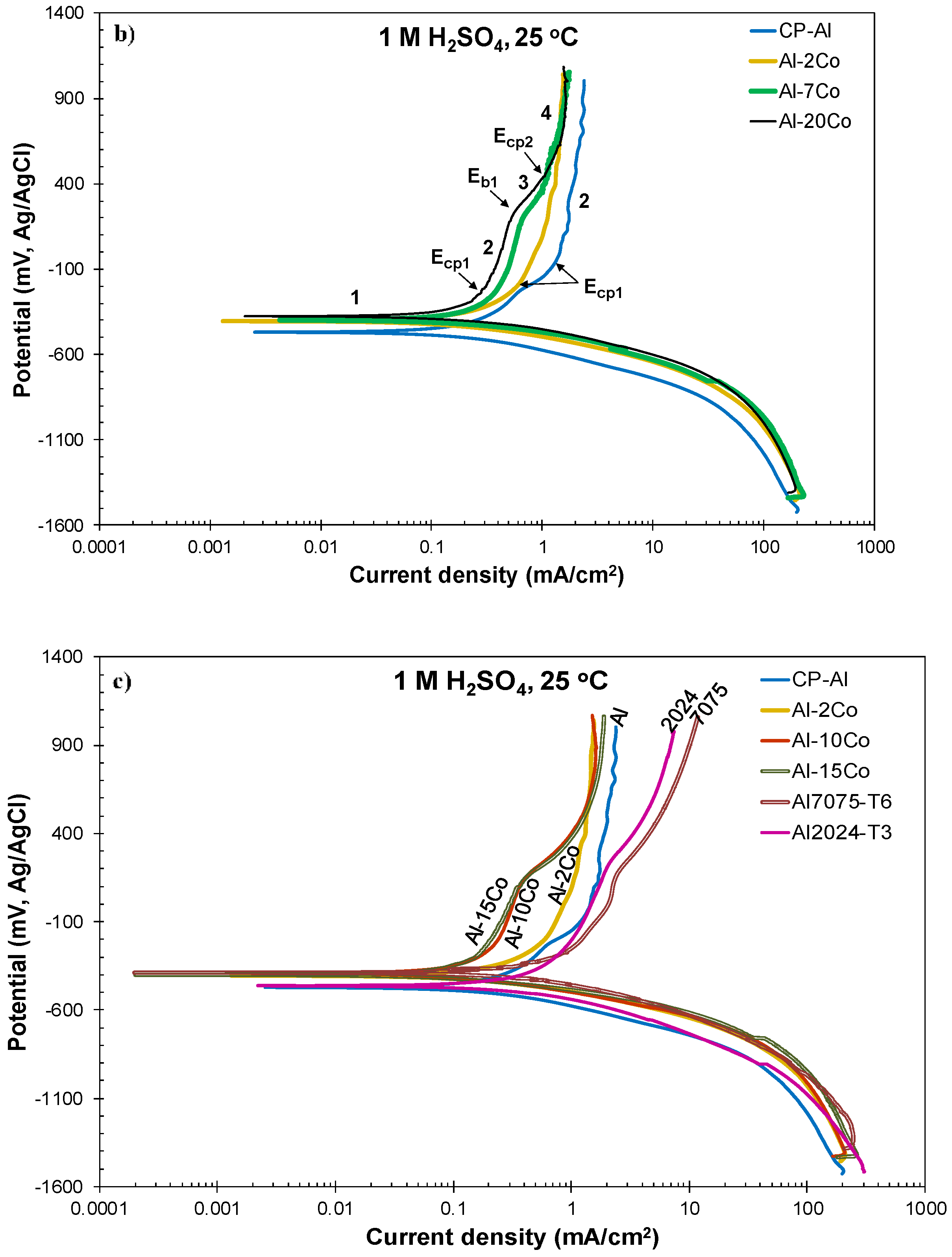
3.2.3. Polarization Performance in 1 M H2SO4 Versus Co Content
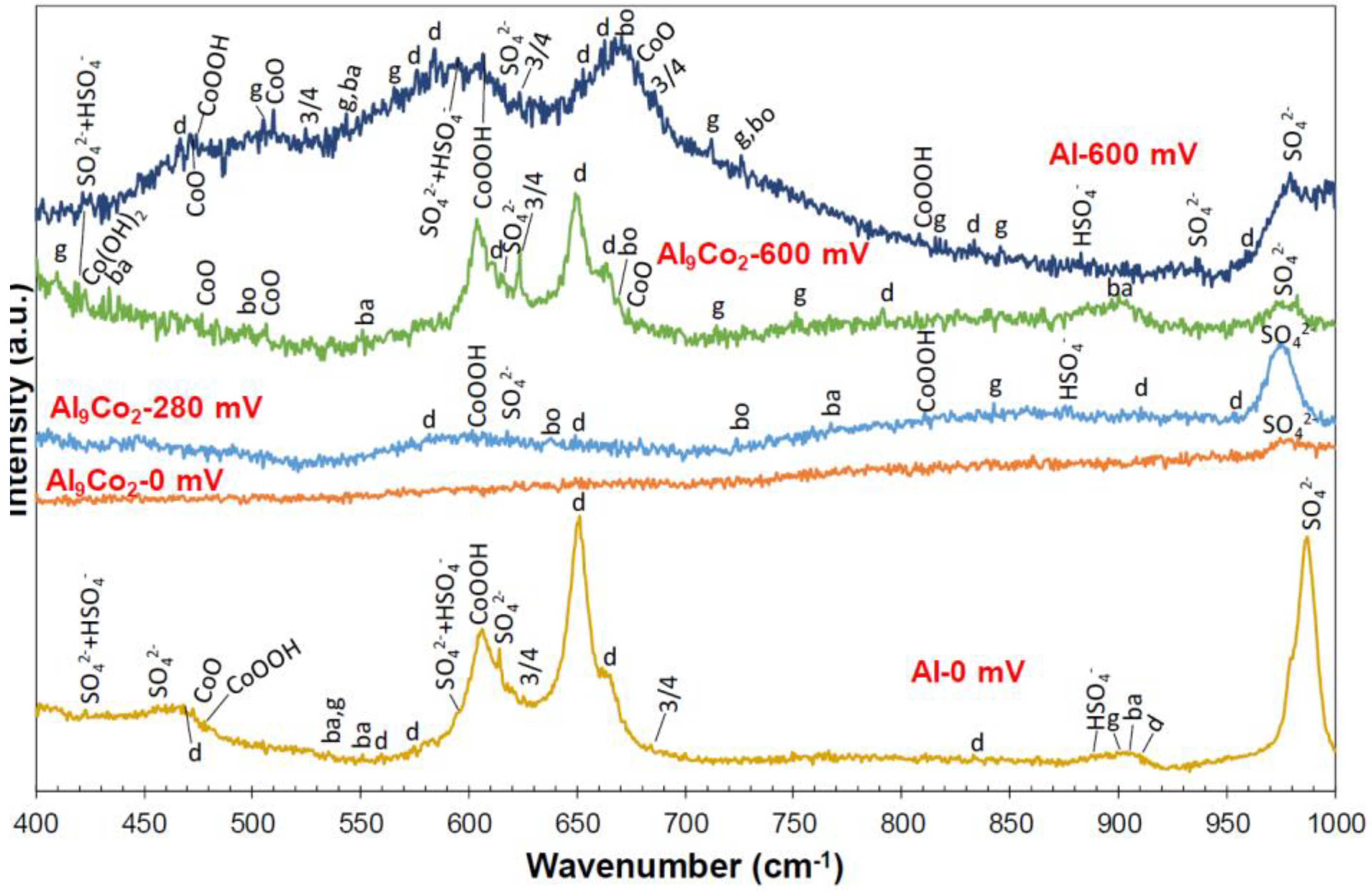
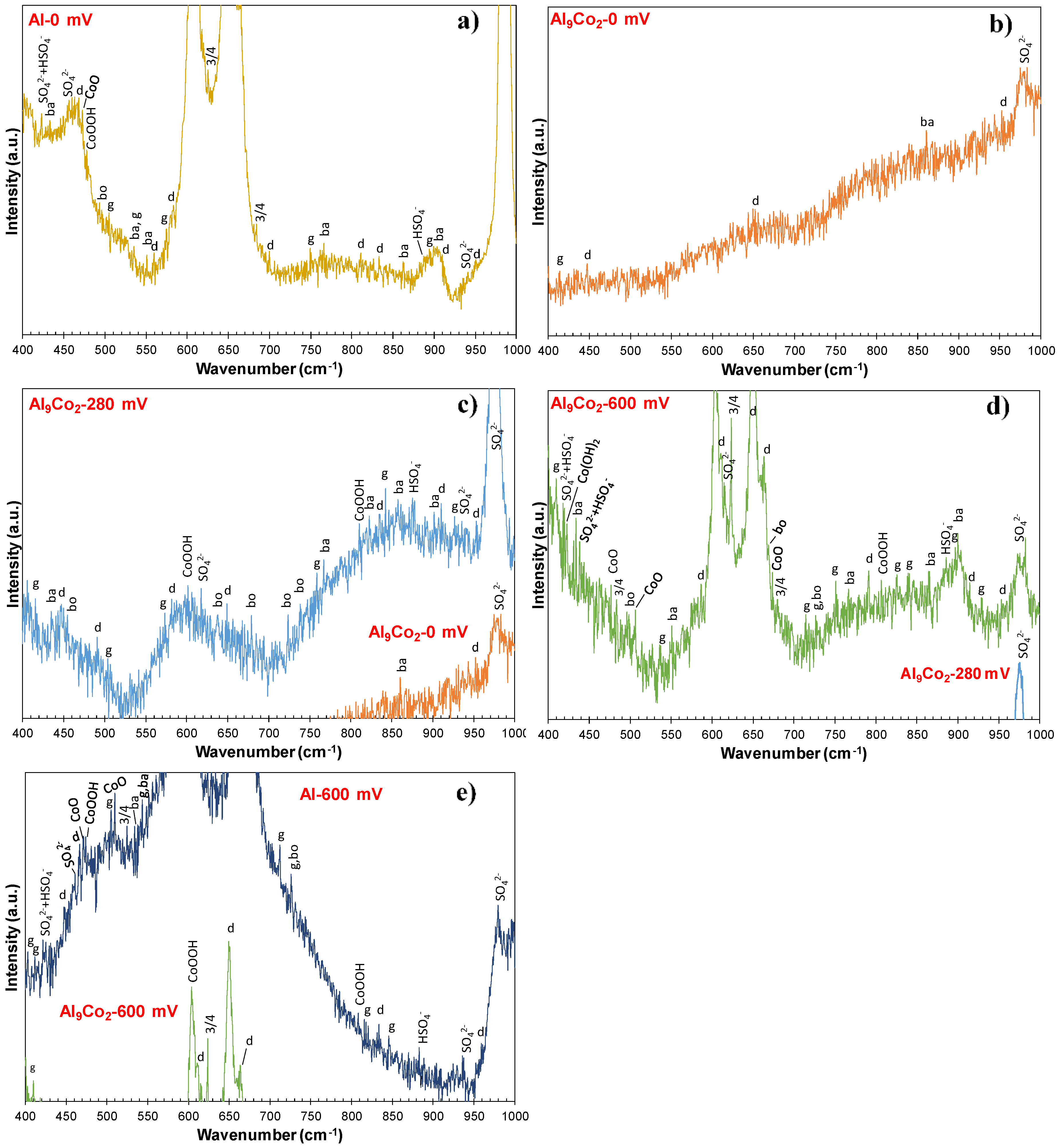
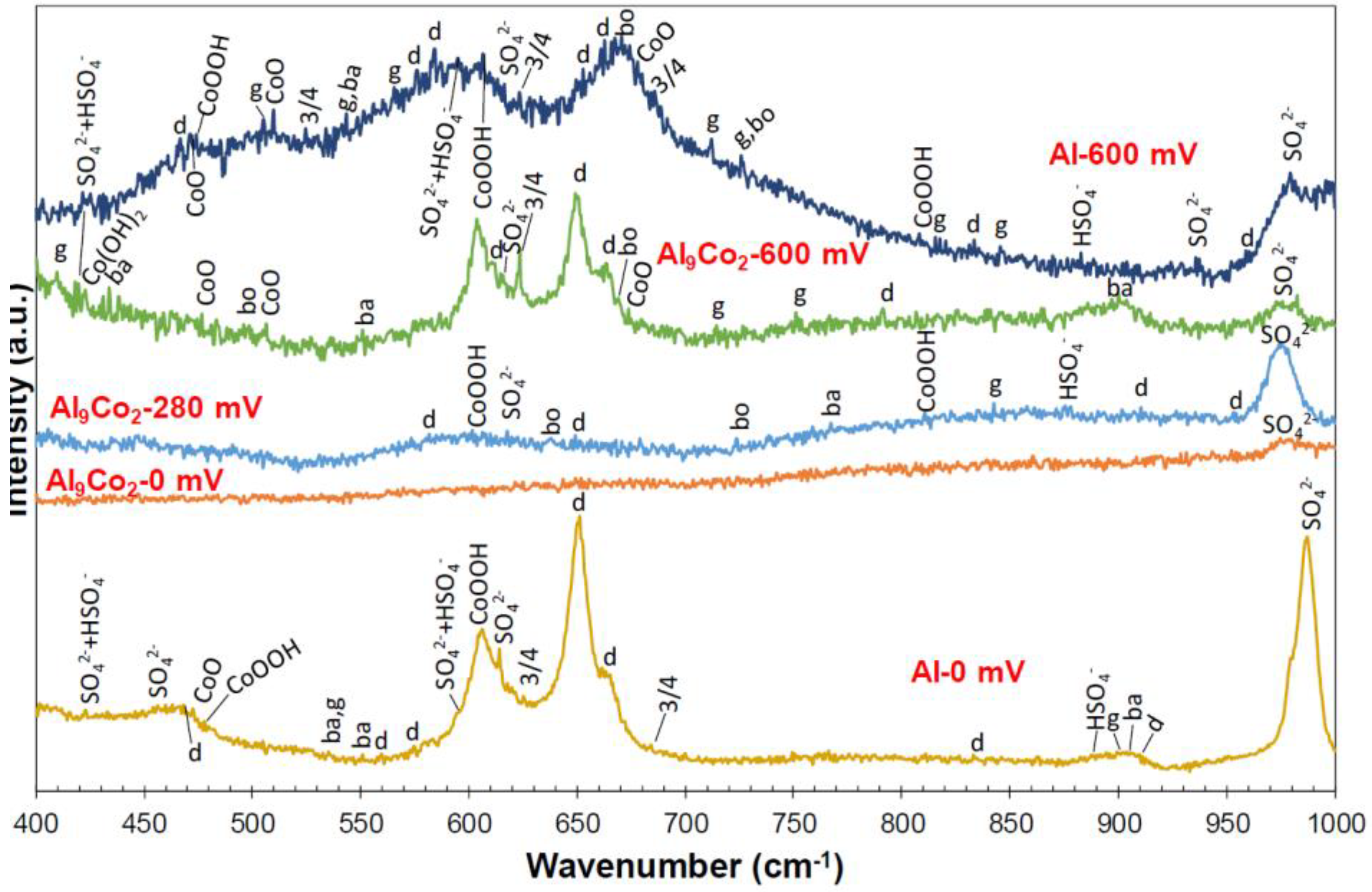
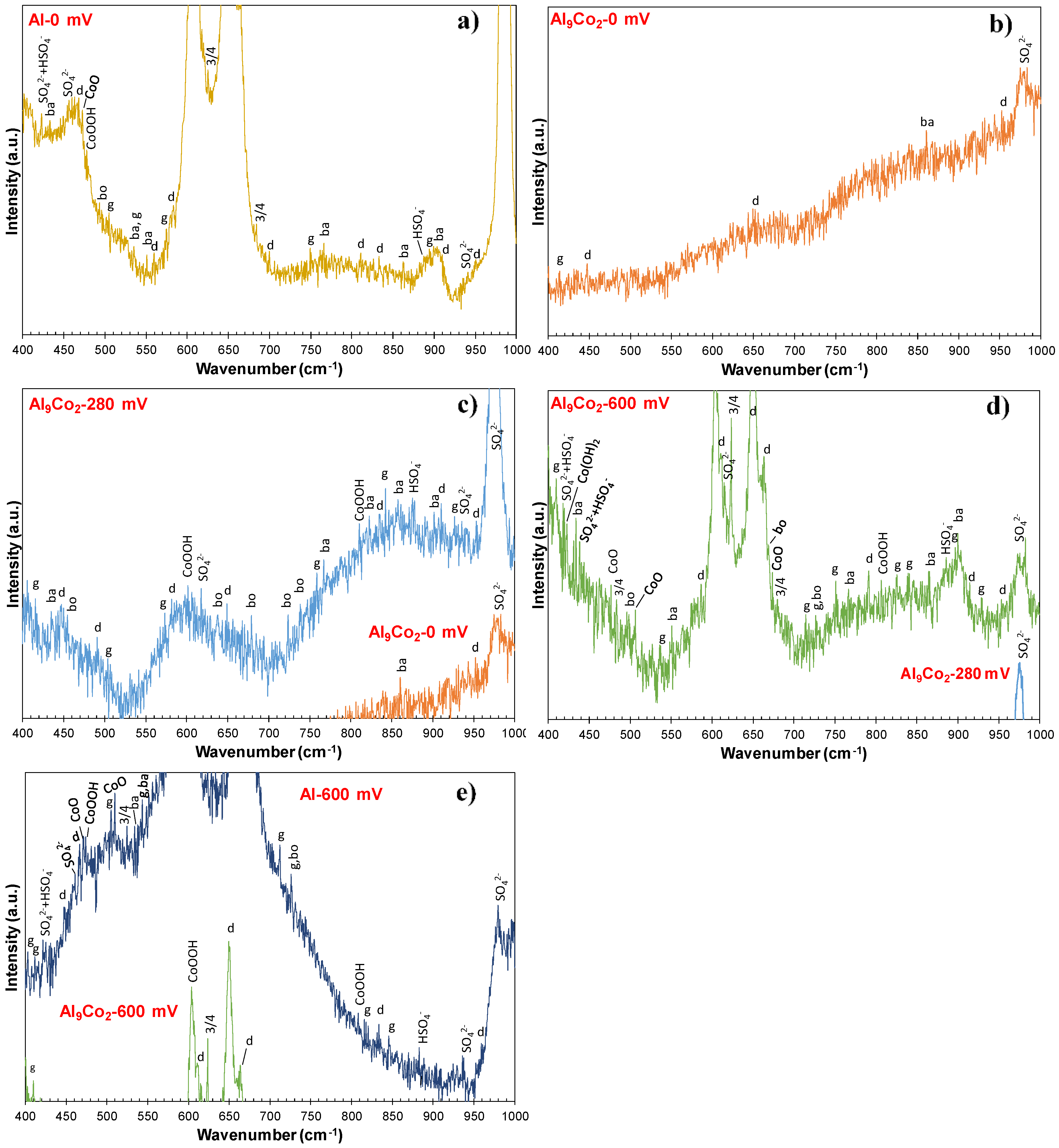
3.2.4. Nature of Surface Films in 1 M H2SO4
3.2.5. Some Comments on the Objectives of This Work
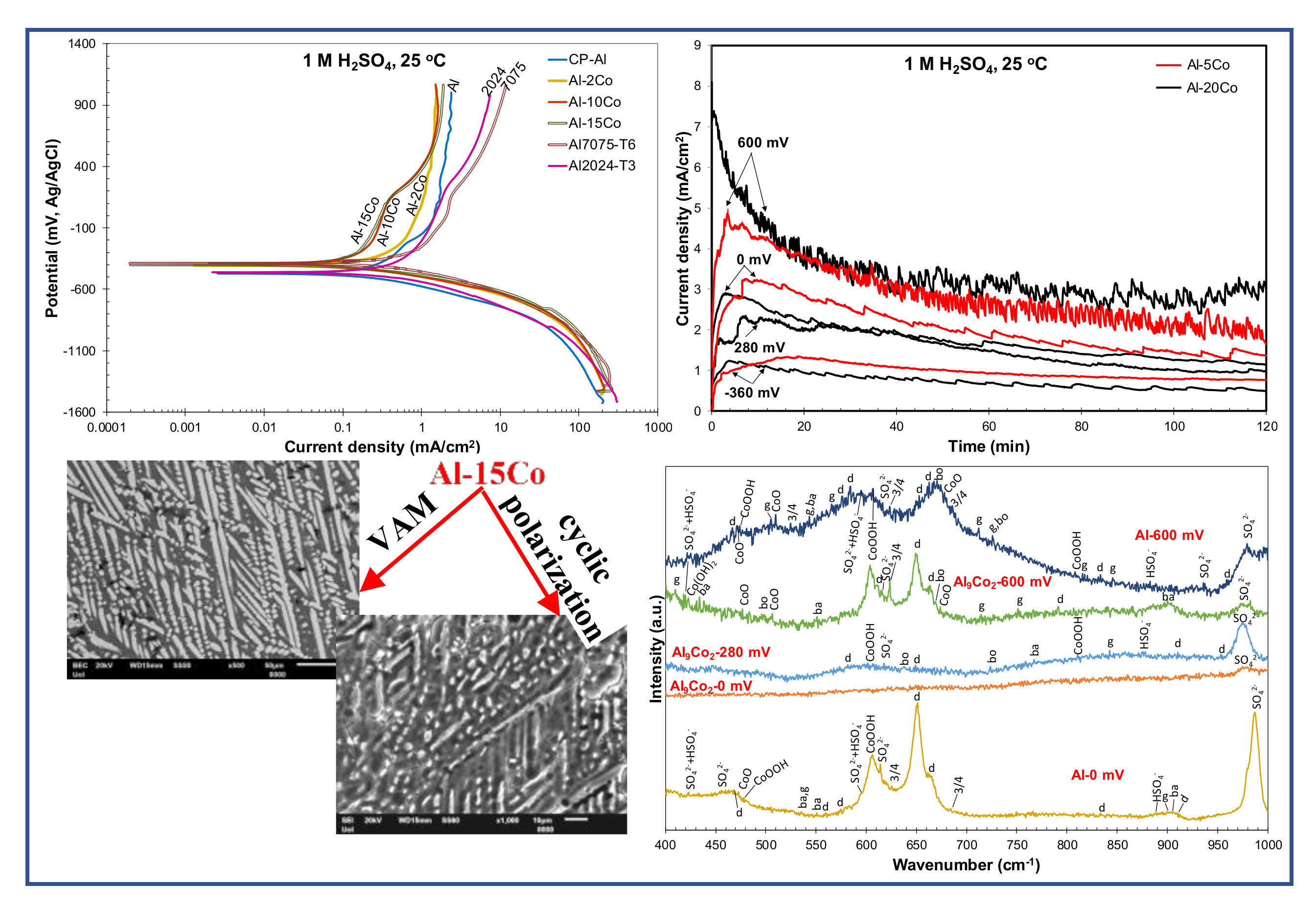
4. Conclusions
- Al–Co alloys of various Co contents in the 2–20 wt.% Co range were produced by vacuum arc melting. The alloys were composed of Al9Co2 particles uniformly distributed in an Al matrix in a directional pattern. As the Co content of the alloy increased, the volume fraction and coarseness of Al9Co2 increased.
- Both potentiodynamic polarization and chronoamperometry experiments showed that the anodic polarization of Al–Co in 1 M H2SO4 at 25 °C proceeded via the stages: (1) Active dissolution of the Al-matrix by ionic conduction across the imperfections of the preexistent surface film; (2) pseudo-passivation of the Al-matrix by film formation at the metal/oxide interface and film dissolution at the film/electrolyte interface; (3) oxidation of the surface of the Al9Co2 particles; and (4) extension of pseudo-passivation to the surface of the Al9Co2 particles. Stage 3 was distinct only in the case of the high-Co alloys (7–20 wt.% Co).
- Different passivation behaviors were observed for the 2–5 wt.% Co and 7–20 wt.% Co alloys in 1 M H2SO4. The low-Co alloys presented one stage passivation governed by the passivation of the (Al) matrix; this stage included passivation of Al9Co2 at high overpotentials. The high-Co alloys presented passivation of two distinct stages. The first stage was governed by the passivation of the (Al) matrix. The second stage was governed by the passivation of Al9Co2. A transitional active stage separated the two passive stages; it corresponded to the oxidation of Al9Co2.
- Alloying Al with Co did not decrease the rate of uniform corrosion. Nevertheless, increasing the Co content led to a decrease in the passivation current density (during the first stage) in 1 M H2SO4, due to the increasing participation of Co in the surface layer of the (Al) matrix. Even low percentages of Co led to accountable decreases in the passive current density of Al (Al–2 wt.% Co: 1.4 times, Al–5 wt.% Co: 2 times, Al–7 wt.% Co: 3.5 times).
- All Al–Co compositions displayed greater passivation ability in 1 M H2SO4 than commercially pure Al, Al7075-T6, and Al2024-T3, in terms of passive current density and initiation of passivation at lower potentials with reference to the corrosion potential.
- All Al–Co compositions showed high resistance to localized corrosion. High Co alloys (Co ≥ 7 wt. %) did not show any susceptibility to localized corrosion.
- Mixtures of crystalline phases (Al2O3-hydrates, CoO, Co3O4, CoOOH, sulfates, bisulfates) and amorphous phases (dispersed Co-oxide species, Al2O3 underlayer to Al2O3-hydrate overlayers) composed the passive films.
- Taking into account that: (a) High Co alloys are more brittle than low Co alloys due to the high amounts of Co-aluminides, (b) addition of 7 wt.% Co to Al has previously led to higher localized and uniform corrosion resistances in 3.5 wt% NaCl, as compared to richer Co compositions, (c) Co additions raise the cost of raw materials, and (d) low and high Co containing alloys have herein shown comparable resistances to localized corrosion and uniform corrosion in 1 M H2SO4, the present work tends to support that alloying Al with 7 wt.% Co could be a quite suitable option, towards the threefold: Ductility-corrosion resistance-cost of raw materials. A planned publication on the effect of the Co content on the tribological performance of Al will further elucidate this matter.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karakose, E.; Keskin, M. Structural investigations of mechanical properties of Al based rapidly solidified alloys. Mater. Des. 2011, 32, 4970–4979. [Google Scholar] [CrossRef]
- Liu, S.; Cui, C.; Wang, X.; Li, N.; Shi, J.; Cui, S.; Chen, P. Effect of cooling rate on microstructure and grain refining behavior of in-situ CeB6/Al composite inoculant in aluminum. Metals 2017, 7, 204. [Google Scholar] [CrossRef] [Green Version]
- Władysiak, R.; Kozuń, A.; Dębowska, K.; Pacyniak, T. Analysis of crystallization process of intensive cooled AlSi20CuNiCoMg alloy. Arch. Foundry Eng. 2017, 17, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Gogebakan, M.; Uzun, O.; Karaaslan, T.; Keskin, M. Rapidly solidified Al-6.5 wt.% Ni alloy. J. Mater. Process. Technol. 2003, 142, 87–92. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, B.; Li, S.; Mao, S.; Liu, X.; Zhang, Y.; Wang, L. The quantitative relationship between microstructure and mechanical property of a melt spun Al–Mg alloy. Mater. Sci. Eng. A 2015, 621, 212–217. [Google Scholar] [CrossRef]
- Lichioiu, I.; Peter, I.; Varga, B.; Rosso, M. Preparation and structural characterization of rapidly solidified Al–Cu alloys. J. Mater. Sci. Technol. 2014, 30, 394–400. [Google Scholar] [CrossRef]
- Menon, J.; Suryanarayana, C. Metallography of a melt-quenched aluminum-cobalt alloy. Metallography 1988, 21, 179–197. [Google Scholar] [CrossRef]
- Stan, K.; Litynska-Dobrzynska, L.; Ochin, P.; Garzeł, G.; Wierzbicka-Miernik, A.; Wojewoda-Budka, J. Effect of Ti, Zr and Hf addition on microstructure and properties of rapidly solidified Al–Mn–Fe alloy. J. Alloy. Compd. 2014, 615, S607–S611. [Google Scholar] [CrossRef]
- Donnadieu, P.; Ochin, P. Amorphous and crystalline phases in rapidly solidified Al-Ta and Al-Ta-V alloys. J. Alloys Compd. 2007, 434–435, 255–258. [Google Scholar] [CrossRef]
- Dorin, T.; Stanford, N.; Birbilis, N.; Gupta, B.K. Influence of cooling rate on the microstructure and corrosion behaviour of Al-Fe alloys. Corros. Sci. 2015, 100, 396–403. [Google Scholar] [CrossRef]
- Sameljuk, A.V.; Neikov, O.D.; Krajnikov, A.V.; Milman, Y.V.; Thompson, G.E.; Zhou, X. Effect of rapid solidification on the microstructure and corrosion behaviour of Al–Zn–Mg based material. Corros. Sci. 2007, 49, 276–286. [Google Scholar] [CrossRef]
- Mozhi, T.A.; Jha, S.C.; Ray, R. Technical note: Corrosion behavior of rapidly solidified Al-Ti-base alloys in chloride environment. Corrosion 1989, 45, 811–813. [Google Scholar] [CrossRef]
- Fass, M.; Itzhak, D.; Eliezer, D.; Froes, F.H. Corrosion behaviour of rapidly solidified Al-Er binary and ternary alloys in NaCl solution at room temperature. J. Mater. Sci. Lett. 1987, 6, 1227–1228. [Google Scholar] [CrossRef]
- Yoshioka, H.; Yoshida, S.; Kawasima, A.; Asami, K.; Hashimoto, K. The pitting corrosion behavior of rapidly solidified aluminum alloys. Corros. Sci. 1986, 26, 795–812. [Google Scholar] [CrossRef]
- Kim, Y.; Buchheit, R.G. A characterization of the inhibiting effect of Cu on metastable pitting in dilute Al–Cu solid solution alloys. Electrochim. Acta 2007, 52, 2437–2446. [Google Scholar] [CrossRef]
- Sundararajan, G.; Gigliotti, M.; Subramanian, P.R. Corrosion performances of rapidly solidified aluminum (RSA) alloys in chloride media. In Corrosion 2014; NACE International: Huston, TX, USA, 2014. [Google Scholar]
- Valencia, J.J.; McCullough, C.; Levi, C.G.; Mehrabian, R. Solidification microstructure of supercooled Ti-Al alloys containing intermetallic phases. Acta Metall. 1989, 37, 2517–2530. [Google Scholar] [CrossRef]
- Sakuma, T.; Yoshizawa, Y.I.; Suto, H. The microstructure and mechanical properties of yttria-stabilized zirconia prepared by arc-melting. J. Mater. Sci. 1985, 20, 2399–2407. [Google Scholar] [CrossRef]
- Osório, W.R.; Spinelli, J.E.; Ferreira, I.L.; Garcia, A. The roles of macrosegregation and of dendritic array spacings on the electrochemical behavior of an Al–4.5 wt.% Cu alloy. Electrochim. Acta 2007, 52, 3265–3273. [Google Scholar] [CrossRef]
- Lekatou, A.; Sfikas, A.K.; Petsa, C.; Karantzalis, A.E. Al-Co alloys prepared by vacuum arc melting: Correlating microstructure evolution and aqueous corrosion behaviour with Co content. Metals 2016, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Zuo, M.; Dong, Y.; Zhao, D.; Wang, Y.; Teng, X. Study on the anti-poison performance of Al–Y–P master alloy for impurity Ca in aluminum alloys. Materials 2017, 10, 1356. [Google Scholar] [CrossRef] [Green Version]
- Karantzalis, A.E.; Lekatou, A.; Evaggelidou, M. Microstructure and sliding wear assessment of Co–TiC composite materials. Int. J. Cast Metal. Res. 2014, 27, 73–79. [Google Scholar] [CrossRef]
- Vizureanu, P.; Minciună, M.G.; Achiţei, D.C.; Sandu, A.V.; Hussin, K. Mechanical Behaviour of CoCrMo Alloy with Si Content. Appl. Mech. Mater. 2015, 754–755, 1017–1022. [Google Scholar] [CrossRef]
- Yan, H.Y.; Vorontsov, V.A.; Dye, D. Alloying effects in polycrystalline γ΄ strengthened Co-Al-W base alloys. Intermetallics 2014, 48, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Palcut, M.; Priputen, P.; Salgo, K.; Janovec, J. Phase constitution and corrosion resistance of Al-Co alloys. Mater. Chem. Phys. 2015, 166, 95–104. [Google Scholar] [CrossRef]
- Priputen, P.; Palcut, M.; Babinec, M.; Misik, J.; Cernickova, I.; Janovec, J. Correlation between microstructure and corrosion behavior of near-equilibrium Al-Co alloys in various environments. J. Mater. Eng. Perform. 2017, 26, 3970–3976. [Google Scholar] [CrossRef]
- Heggen, M.; Feuerbacher, M. Core structure and motion of the dislocations in the orthorhombic structurally complex alloy Al13Co4. Mater. Res. Lett. 2014, 2, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Kandaskalov, D.; Fournée, V.; Ledieu, J.; Gaudry, E. Adsorption properties of the o-Al13Co4(100) surface toward molecules involved in the semihydrogenation of acetylene. J. Phys. Chem. C 2014, 118, 23032–23041. [Google Scholar] [CrossRef]
- Krajčí, M.; Hafner, J. Catalytic properties of five-fold surfaces of quasicrystal approximants. In Aperiodic Crystals; Schmid, S., Withers, R., Lifshitz, R., Eds.; Springer: Dordrecht, Germany, 2013; pp. 269–274. [Google Scholar]
- Soler, L.; Macanas, J.; Munoz, M.; Casado, J. Synergistic hydrogen generation from aluminum, aluminum alloys and sodium borohydride in aqueous solutions. Int. J. Hydrogen Energy 2007, 32, 4702–4710. [Google Scholar] [CrossRef]
- Geng, K.; Yang, Y.; Li, S.; Misra, R.D.K.; Zhu, Q. Enabling high-performance 3D printing of Al powder by decorating with high laser absorbing Co phase. Addit. Manuf. 2020, 32, 101012. [Google Scholar]
- Hung, C.J.; Nayak, S.K.; Sun, Y.; Fennessy, C.; Vedula, V.K.; Tulyani, S.; Lee, S.W.; Alpay, S.P.; Hebert, R.J. Novel Al-X alloys with improved hardness. Mater. Des. 2020, 192, 108699. [Google Scholar] [CrossRef]
- Men‘Shikova, S.G.; Shirinkina, I.G.; Brodova, I.G.; Lad‘yanov, V.I.; Suslov, A.A. Structures of thin ribbons from an Al-Co alloy under rapid cooling. Met. Sci. Heat Treat. 2016, 58, 393–399. [Google Scholar] [CrossRef]
- Adam, A.M. Dendrite refinement of Al9Co2 compound by a continuous increase of the cooling rate during solidification. UPB Sci. Bull. Ser. B 2012, 74, 289–300. [Google Scholar]
- Lekatou, A.; Sfikas, A.K.; Karantzalis, A.E.; Sioulas, D. Microstructure and corrosion performance of Al-32%Co alloys. Corros. Sci. 2012, 63, 193–209. [Google Scholar] [CrossRef]
- Palcut, M.; Priputen, P.; Kusy, M.; Janovec, J. Corrosion behaviour of Al-29%atCo alloy in aqueous NaCl. Corros. Sci. 2013, 75, 461–466. [Google Scholar] [CrossRef]
- Sui, H.X.; Zhu, M.; Qi, M.; Li, G.B.; Yang, D.Z. The enhancement of solid solubility limits of AlCo intermetallic compound by high energy ball milling. J. Appl. Phys. 1992, 71, 2945–2949. [Google Scholar] [CrossRef]
- Gille, P.; Bauer, B. Single crystal growth of Al13Co4 and Al13Fe4 from Al-rich solutions by the Czochralski method. Cryst. Res. Technol. 2008, 43, 1161–1167. [Google Scholar] [CrossRef]
- Scully, J.R.; Presuel-Moreno, F.; Goldman, M.; Kelly, R.G.; Tailleart, N. User-selectable barrier, sacrificial anode, and active corrosion inhibiting properties of Al-Co-Ce alloys for coating applications. Corrosion 2008, 64, 210–229. [Google Scholar] [CrossRef]
- Lekatou, A.G.; Sfikas, A.K.; Karantzalis, A.E. The influence of the fabrication route on the microstructure and surface degradation properties of Al reinforced by Al9Co2. Mater. Chem. Phys. 2017, 200, 33–49. [Google Scholar] [CrossRef]
- Bakoulis, G.; Lekatou, A.G.; Poulia, A.; Sfikas, A.K.; Lentzaris, K.; Karantzalis, A.E. Al-(Al9Co2- Al13Co4) powder metallurgy processed composite materials: Analysis of microstructure, sliding wear and aqueous corrosion. Mater. Sci. Eng. Adv. Res. 2017. [Google Scholar] [CrossRef]
- Lekatou, A.; Sioulas, D.; Karantzalis, A.E.; Grimanelis, D. A comparative study on the microstructure and surface property evaluation of coatings produced from nanostructured and conventional WC-Co powders HVOF-sprayed on Al 7075. Surf. Coat. Tech. 2015, 276, 539–556. [Google Scholar] [CrossRef]
- Silverman, D.C. Tutorial on cyclic potentiodynamic polarization technique. In Proceedings of the CORROSION 98 Research Topical Symposium, San Diego, CA, USA, 22–27 March 1998. [Google Scholar]
- Beavers, J.A.; Durr, C.L.; Thompson, N.G. Unique interpretations of potentiodynamic polarization technique. In Proceedings of the CORROSION 98 Research Topical Symposium, San Diego, CA, USA, 22–27 March 1998. [Google Scholar]
- Davies, R.H.; Dinsdale, A.T.; Gisby, J.A.; Robinson, J.A.J.; Martin, S.M. MTDATA—Thermodynamics and phase equilibrium software from the National Physical Laboratory. Calphad 2002, 26, 229–271. [Google Scholar] [CrossRef]
- Karantzalis, A.E.; Arni, Z.; Tsirka, K.; Evangelou, A.; Lekatou, A.; Dracopoulos, V. Fabrication of TiC-reinforced composites by vacuum arc melting: TiC mode of reprecipitation in different molten metals and alloys. J. Mater. Eng. Perform. 2016, 25, 3161–3172. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.M. Rapidly solidified MC carbide morphologies of a pulsed laser surface alloyed γ-TiAl intermetallic with carbon. Scr. Mater. 2004, 50, 507–510. [Google Scholar] [CrossRef]
- Karantzalis, A.E.; Lekatou, A.; Tsirka, K. Solidification observations and sliding wear behaviour of vacuum arc melting processed Ni–Al–TiC composites. Mater. Charact. 2012, 69, 97–107. [Google Scholar] [CrossRef]
- Maciá, E. Thermal conductivity in complex metallic alloys: Beyond Wiedemann-Franz law. Phys. Rev. B 2009, 79, 245112. [Google Scholar] [CrossRef] [Green Version]
- Mercier, D.; Herinx, M.; Barthes-Labrousse, M.G. Influence of 1,2-diaminoethane on the mechanism of aluminum corrosion in sulfuric acid solutions. Corros. Sci. 2010, 52, 3405–3412. [Google Scholar] [CrossRef]
- Arellanes-Lozada, P.; Olivares-Xometl, O.; Guzmán-Lucero, D.; Likhanova, N.V.; Domínguez-Aguilar, M.A.; Lijanova, I.V.; Arce-Estrada, E. The inhibition of aluminum corrosion in sulfuric acid by poly(1-vinyl-3-alkyl-imidazolium Hexafluorophosphate). Materials 2014, 7, 5711–5734. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Chiba, M. Role of anodic oxide films in the corrosion of aluminum and its alloys. Corros. Rev. 2017, 36, 35–54. [Google Scholar] [CrossRef]
- Curioni, M.; Skeldon, P.; Thompson, G.E. Anodizing of aluminum under nonsteady conditions. J. Electrochem. Soc. 2009, 156, C407–C413. [Google Scholar] [CrossRef]
- Curioni, M.; Scenini, F. The mechanism of hydrogen evolution during anodic polarization of aluminium. Electrochim. Acta 2015, 180, 712–721. [Google Scholar] [CrossRef]
- Kelly, R.G.; Scully, J.R.; Shoesmith, D.; Buchheit, R.G. Electrochemical Techniques in Corrosion Science and Engineering; CRC Press: Boca Raton, FL, USA, 2002; pp. 59–65. [Google Scholar]
- Rajan, S.S.S. Sulfate adsorbed on hydrous alumina, ligands displaced, and changes in surface charge. Soil Sci. Soc. Am. J. 1978, 42, 39–44. [Google Scholar] [CrossRef]
- Ledieu, J.; Gaudry, É.; Fournée, V. Surfaces of Al-based complex metallic alloys: Atomic structure, thin film growth and reactivity. Sci. Technol. Adv. Mater. 2014, 15, 034802. [Google Scholar] [CrossRef] [PubMed]
- Wille, G.; Bourrat, X.; Maubec, N.; Guegan, R.; Lahfid, A. Raman-in-SEM Studies of inorganic materials. In Spectroscopic Properties of Inorganic and Organometallic Compounds: Techniques, Materials and Applications; Yarwood, J., Douthwaite, R., Duckett, S.B., Eds.; The Royal Society of Chemistry: London, UK, 2014; Volume 45, pp. 79–116. [Google Scholar]
- Bockris, J.O’.M.; Kang, Y. The protectivity of aluminum and its alloys with transition metals. J. Solid State Electrochem. 1997, 1, 17–35. [Google Scholar] [CrossRef]
- Shankar Rao, V. Repassivation behaviour and surface analysis of Fe3Al based iron aluminide in 0.25 M H2SO4. Corros. Sci. 2005, 47, 183–194. [Google Scholar] [CrossRef]
- Kutz, T.N.; Zander, D. The influence of chromium on the passivation of Fe3Al iron aluminides, investigated via potentiodynamic polarization in 0.25 M H2SO4. Corrosion 2017, 73, 648–654. [Google Scholar] [CrossRef]
- Ura-Binczyk, E.; Homazava, N.; Ulrich, A.; Hauert, R.; Lewandowska, M.; Kurzydlowski, K.J.; Schmutz, P. Passivation of Al-Cr-Fe and Al-Cu-Fe-Cr complex metallic alloys in 1 M H2SO4 and 1 M NaOH solutions. Corros. Sci. 2011, 53, 1825–1837. [Google Scholar] [CrossRef]
- Esquivel, J.; Gupta, R.K. Review—Corrosion-resistant metastable Al alloys: An overview of corrosion mechanisms. J. Electrochem. Soc. 2020, 167, 081504. [Google Scholar] [CrossRef]
- Kharitonov, D.S.; Örnek, C.; Claesson, P.M.; Sommertune, J.; Zharskii, I.M.; Kurilo, I.I.; Pan, J. Corrosion inhibition of aluminum alloy AA6063-T5 by vanadates: Microstructure characterization and corrosion analysis. J. Electrochem. Soc. 2018, 165, C116–C126. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Nilsson, J.-A.; Pan, J. In Situ and Operando AFM and EIS Studies of Anodization of Al 6060: Influence of Intermetallic Particles. J. Electrochem. Soc. 2016, 163, C609–C618. [Google Scholar] [CrossRef]
- Tang, C.W.; Wang, C.; Chien, S.H. Characterization of cobalt oxides studied by FT-IR, Raman, TPR and TG-MS. Thermochim. Acta 2008, 473, 68–73. [Google Scholar] [CrossRef]
- Demichelis, R.; Noel, Y.; Civalleri, B.; Roetti, C.; Ferrero, M.; Dovesi, R. The vibrational spectrum of r-AlOOH diaspore: An ab initio study with the CRYSTAL Code. J. Phys. Chem. B 2007, 111, 9337–9346. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.D.; Frost, R.L.; Kloprogge, J.T.; Schulze, D.G.; Duong, L. FT-Raman spectroscopy and SEM of gibbsite, bayerite, boehmite and diaspore in relation to the characterization of bauxite. 2001. In A Clay Odyssey; Domínguez, E., Mas, G., Cravero, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 545–552. [Google Scholar]
- Lund Myhre, C.E.; Christensen, D.H.; Nicolaisen, F.M.; Nielsen, C.J. Spectroscopic study of aqueous H2SO4 at different temperatures and compositions: Variations in dissociation and optical properties. J. Phys. Chem. A 2003, 107, 1979–1991. [Google Scholar] [CrossRef]
- Hope, G.A.; Woods, R. Transient adsorption of sulfate ions during copper electrodeposition. J. Electrochem. Soc. 2004, 151, C550–C553. [Google Scholar] [CrossRef]
- Knopf, D.A.; Luo, B.P.; Krieger, U.K.; Koop, T. Thermodynamic dissociation constant of the bisulfate ion from Raman and ion interaction modeling studies of aqueous sulfuric acid at low temperatures. J. Phys. Chem. A 2003, 107, 4322–4332. [Google Scholar] [CrossRef]
- Lekatou, A.; Zois, D.; Karantzalis, A.E.; Grimanelis, D. Electrochemical behaviour of cermet coatings with a bond coat on Al7075: Pseudopassivity, localized corrosion and galvanic effect considerations in a saline environment. Corros. Sci. 2010, 52, 2616–2635. [Google Scholar] [CrossRef]
- Vuurman, M.A.; Stufkens, D.J.; Oskam, A.; Deo, G.L.; Wachs, I.E. Combined Raman and IR study of MOX–V2O5/Al2O3 (MOx = MoO3, WO3, NiO, CoO) catalysts under dehydrated conditions. J. Chem. Soc. Faraday Trans. 1996, 92, 3259–3265. [Google Scholar] [CrossRef]
- Bernard, W.J.; Randall, J.J., Jr. The reaction between anodic aluminum oxide and water. J. Electrochem. Soc. 1961, 108, 822–825. [Google Scholar] [CrossRef]
- Cox, R.A.; Haldna, U.L.; Idler, K.L.; Yate, K. Resolution of Raman spectra of aqueous sulfuric acid mixtures using principal factor analysis. Can. J. Chem. 1981, 59, 2591–2598. [Google Scholar] [CrossRef]
- Mabrouk, K.B.; Kauffmann, T.H.; Arouic, H.; Marc, D.; Fontana, M.D. Raman study of cation effect on sulfate vibration modes in solid state and in aqueous solutions. J. Raman Spectrosc. 2013, 44, 1603–1608. [Google Scholar] [CrossRef]
- Qiu, J.; Li, X.; Qi, X. Raman spectroscopic investigation of sulfates using mosaic grating spatial heterodyne raman spectrometer. IEEE Photonics J. 2019, 11, 1–12. [Google Scholar] [CrossRef]
- Omori, K. Infrared diffraction and the far infrared spectra of anhydrous sulfates. Mineral. J. 1968, 5, 334–354. [Google Scholar] [CrossRef] [Green Version]
- Kloprogge, T.; Frost, R.L. Raman microscopy study of basic aluminum sulfate. J. Mater. Sci. 1999, 34, 4199–4202. [Google Scholar] [CrossRef]
- Buzgar, N.; Buzatu, A.; Sanislav, J.V. The Raman study of certain sulfates. An. Stiin. U Al I-Mat. 2009, 55, 5–23. [Google Scholar]
- Sobron, P.; Sobron, F.; Eide, U.M.; Nielsen, C.J.; Rull, F. Model-based measurements of diffusion of sulfuric acid into water using Raman spectroscopy. Appl. Spectrosc. 2009, 63, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Pinhero, P.J.; Sordelet, D.J.; Anderegg, J.W.; Brunet, P.; Dubois, J.M.; Thiel, P.A. Room temperature oxidation of Al-Cu-Fe and Al-Cu-Fe-Cr quasicrystals. Mater. Res. Soc. Symp. Proc. 1999, 553, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Beni, A.; Ott, N.; Pawelkiewicz, M.; Wardé, M.; Young, K.; Bauer, B.; Rajput, P.; Detlefs, B.; Zegenhagen, J.; McGrath, R.; et al. Hard X-ray Photoelectron Spectroscopy (HAXPES) characterisation of electrochemical passivation oxide layers on Al–Cr–Fe complex metallic alloys (CMAs). Electrochem. Comm. 2014, 46, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Davis, G.D.; Shaw, B.A.; Rees, B.J.; Pecile, C.A. Electrochemical behavior and surface chemistry of nonequilibrium aluminum-tantalum alloys: Solute-rich interphase model. Surf. Interf. Anal. 1995, 23, 609–617. [Google Scholar] [CrossRef]
- Frangini, S. Corrosion rate and anodic dissolution behavior of a B2-iron aluminide alloy in sulfuric acid. Corrosion 1999, 55, 89–95. [Google Scholar] [CrossRef]
- El-Sherbini, E.E.F.; Abd-El-Wahab, S.M.; Deyab, M.A. Studies on corrosion inhibition of aluminum in 1.0 M HCl and 1.0 M H2SO4 solutions by ethoxylated fatty acids. Mater. Chem. Phys. 2003, 82, 631–637. [Google Scholar] [CrossRef]
- Dumitrascu, V.; Benea, L.; Danaila, E. Corrosion behavior of aluminum oxide film growth by controlled anodic oxidation. IOP Conf. Ser. Mater. Sci. Eng. 2016, 209, 012016. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alloy | Ecorr (mV vs Ag/AgCl) | Ecp1 (mV vs Ag/AgCl) | Ecp1 − Ecorr (mV vs Ag/AgCl) | Eb1 (mV vs Ag/AgCl) | Ecp2 (mV vs Ag/AgCl) | Ea/c tr_1 (mV vs Ag/AgCl) | Ea/c tr_2 (mV vs Ag/AgCl) | ip1 (mA/cm2) | ip2 (mA/cm2) |
|---|---|---|---|---|---|---|---|---|---|
| CP-Al | −463 ± 8 | −101 ± 18 | 363 ± 29 | – | – | – | −530 ± 21 | 1.69 ± 0.04 | 1.96 ± 0.07 |
| Al–2Co | −406 ± 7 | −193 ± 9 | 213 ± 13 | – | – | – | −436 ± 52 | 1.20 ± 0.02 | 1.62 ± 0.21 |
| Al–5Co | −412 ± 2 | −226 ± 18 | 187 ± 20 | – | – | – | −467 ± 5 | 0.85 ± 0.07 | 1.09 ± 0.15 |
| Al–7Co | −394 ± 10 | −218 ± 23 | 166 ± 19 | 213 ± 46 | 374 ± 35 | 25 ± 10 | −426 ± 16 | 0.48 ± 0.06 | 1.23 ± 0.03 |
| Al–10Co | −396 ± 5 | −227 ± 10 | 169 ± 12 | 157 ± 21 | 421 ± 21 | 9 ± 51 | −444 ± 32 | 0.36 ± 0.09 | 1.45 ± 0.24 |
| Al–15Co | −387 ± 18 | −227 ± 25 | 159 ± 16 | 179 ± 28 | 439 ± 33 | −4 ± 39 | −391 ± 23 | 0.35 ± 0.08 | 1.50 ± 0.25 |
| Al–20Co | −378 ± 7 | −248 ± 16 | 130 ± 15 | 184 ± 46 | 401 ± 48 | 22 ± 64 | −413 ± 20 | 0.34 ± 0.06 | 1.34 ± 0.19 |
| Al7075-T6 | −406 ± 25 | −191 ± 24 | 214 ± 47 | 239 ± 68 | 431 ± 17 | −50 ± 65 | −418 ± 18 | 2.42 ± 0.25 | 7.20 ± 1.56 |
| Al2024-T3 | −457± 4 | −134 ± 20 | 322 ± 16 | 193 ± 16 | 456 ± 35 | −46 ± 71 | −433 ± 60 | 1.32 ± 0.20 | 5.34 ± 1.48 |
| Alloy | icorr (mA/cm2) | βc (mV/d) | αc (mV) | rc2 | ΔΕ (mV vs. Ag/AgCl) | Δi (mA/cm2) |
|---|---|---|---|---|---|---|
| CP-Al | 0.21 ± 0.01 | −159 ± 4 | −572 ± 20 | 0.998 ± 0.001 | (−687±9)–(−532±11) | (0.58±0.08)–(5.87±0.71) |
| Al–2Co | 0.19 ± 0.03 | −128 ± 16 | −500 ± 18 | 0.996 ± 0.002 | (−610±9)–(−476±7) | (0.64±0.08)–(6.45±0.89) |
| Al–5Co | 0.36 ± 0.06 | −143 ± 12 | −484 ± 15 | 0.991 ± 0.004 | (−651±21)–(−492±12) | (0.94±0.40)–(9.49±0.43) |
| Al–7Co | 0.26 ± 0.06 | −137 ± 6 | −480 ± 10 | 0.996 ± 0.002 | (−598±10)–(−464±4) | (0.75±0.17)–(7.65±1.64) |
| Al–10Co | 0.19 ± 0.06 | −121 ± 9 | −485 ± 24 | 0.995 ± 0.004 | (−585±8)–(−465±3) | (0.75±0.18)–(7.67±1.84) |
| Al–15Co | 0.31 ± 0.08 | −119 ± 7 | −448 ± 22 | 0.992 ± 0.003 | (−575±11)–(−456±17) | (1.05±0.23)–(10.56±2.26) |
| Al–20Co | 0.29 ± 0.04 | −137 ± 6 | −452 ± 10 | 0.997 ± 0.001 | (−583±9)–(−448±7) | (0.85±0.09)–(8.54±0.95) |
| Al7075-T6 | 0.66 ± 0.18 | −185 ± 12 | −438 ± 12 | 0.986 ± 0.007 | (−656±37)–(−475±24) | (1.35±0.40)–(13.41±3.74) |
| Al2024-T3 | 0.36 ± 0.08 | −183 ± 6 | −539 ± 38 | 0.994 ± 0.001 | (−707±10)–(−527±4) | (0.71±0.19)–(7.18±1.85) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sfikas, A.K.; Lekatou, A.G. Electrochemical Behavior of Al–Al9Co2 Alloys in Sulfuric Acid. Corros. Mater. Degrad. 2020, 1, 249-272. https://doi.org/10.3390/cmd1020012
Sfikas AK, Lekatou AG. Electrochemical Behavior of Al–Al9Co2 Alloys in Sulfuric Acid. Corrosion and Materials Degradation. 2020; 1(2):249-272. https://doi.org/10.3390/cmd1020012
Chicago/Turabian StyleSfikas, Athanasios K., and Angeliki G. Lekatou. 2020. "Electrochemical Behavior of Al–Al9Co2 Alloys in Sulfuric Acid" Corrosion and Materials Degradation 1, no. 2: 249-272. https://doi.org/10.3390/cmd1020012
APA StyleSfikas, A. K., & Lekatou, A. G. (2020). Electrochemical Behavior of Al–Al9Co2 Alloys in Sulfuric Acid. Corrosion and Materials Degradation, 1(2), 249-272. https://doi.org/10.3390/cmd1020012