Immunomodulatory Effects of TGF-? Family Signaling within Intestinal Epithelial Cells and Carcinomas


{kind=link}
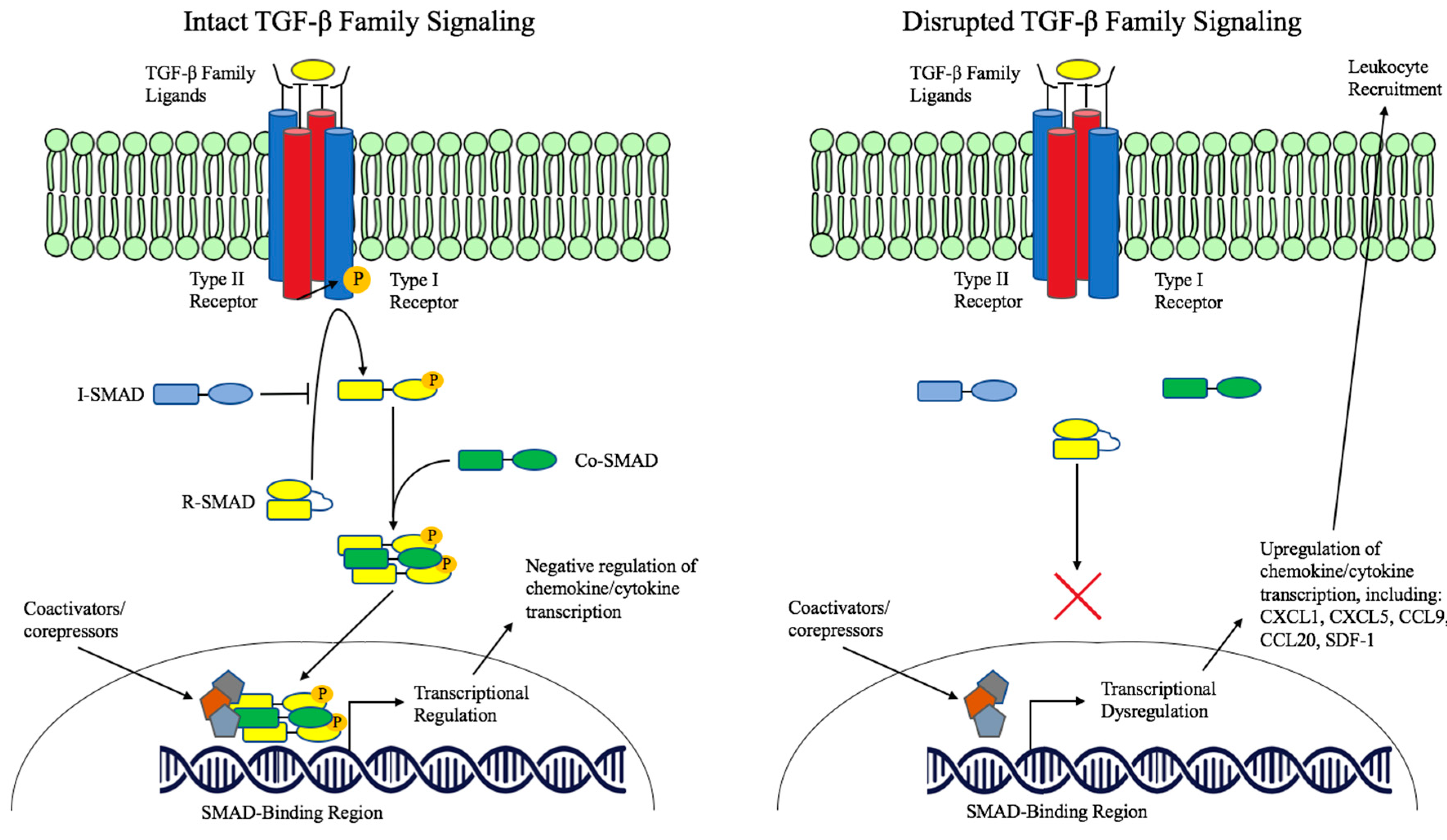
Abstract
:1. TGF-β Family Signaling
2. TGF-β Pathway Dysregulation in Cancer
3. TGF-β in Immune Cell Regulation
4. TGF-β in Epithelial Homeostasis
4.1. The Immunomodulatory Role of TGF-β in Epithelial Cells and Epithelial Cancers
4.2. TGF-β Dysregulation in Inflammatory Bowel Disease
5. Conclusions and Unanswered Questions
Author Contributions
Funding
Conflicts of Interest
References
- Weiss, A.; Attisano, L. The TGFbeta Superfamily Signaling Pathway. WIREs Dev. Biol. 2012, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Moses, H.L.; Serra, R. Regulation of differentiation by TGF-beta. Curr. Opin. Genet. Dev. 1996, 6, 581–586. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Samanta, D.; Datta, P.K. Alterations in the Smad pathway in human cancers. Front Biosci. 2015, 17, 1281–1293. [Google Scholar] [CrossRef]
- Moustakas, A.; Heidin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Morikawa, M.; Koinuma, D.; Miyazono, K.; Heldin, C.-H. Genome-wide mechanisms of Smad binding. Oncogene 2012, 32, 1609–1615. [Google Scholar] [CrossRef]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A.; Wrana, J.L.; et al. The MAD-Related Protein Smad7 Associates with the TGFbeta Receptor and Functions as an Antagonist of TGFbeta Signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Aloysius, A.; DasGupta, R.; Dhawan, J. The transcription factor Lef1 switches partners from beta-catenin to Smad3 during muscle stem cell quiescence. Sci. Signal. 2018, 11, 1–15. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Zent, R.; Ghiassi, M.; McDonnell, M.; Moses, H.L. Integrin β 1Signaling Is Necessary for Transforming Growth Factor-β Activation of p38MAPK and Epithelial Plasticity. J. Biol. Chem. 2001, 276, 46707–46713. [Google Scholar] [CrossRef]
- Engel, M.E.; McDonnell, M.A.; Law, B.K.; Moses, H.L. Interdependent SMAD and JNK Signaling in Transforming Growth Factor-beta-mediated Transcription. J. Biol. Chem. 1999, 274, 37413–37420. [Google Scholar] [CrossRef]
- Hanafusa, H.; Ninomiya-Tsuji, J.; Masuyama, N.; Nishita, M.; Fujisawa, J.; Shibuya, H.; Matsumoto, K.; Nishida, E. Involvement of the p38 Mitogen-activated Protein Kinase Pathway in Transforming Growth Factor-beta-induced Gene Expression. J. Biol. Chem. 1999, 274, 27161–27167. [Google Scholar] [CrossRef] [PubMed]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-β induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming Growth Factor-beta1 Mediates Epithelial to Mesenchymal Transdifferentiation threough a RhoA-dependent Mechanism. Mol. Biol. Cell. 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Edlund, S.; Landstrom, M.; Heldin, C.-H.; Aspenstrom, P. Transforming Growth Factor-beta-induced Mobilization of Actin Cytoskeleton Requires Signaling by Small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, M.C.; Mitchell, H.; Penheiter, S.G.; Dore, J.J.; Suzuki, K.; Edens, M.; Sharma, D.K.; Pagano, R.E.; Leof, E.B. Transforming Growth Factor-β Activation of Phosphatidylinositol 3-Kinase Is Independent of Smad2 and Smad3 and Regulates Fibroblast Responses via p21-Activated Kinase-2. Cancer Res. 2005, 65, 10431–10440. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I Transforming Growth Factor β Receptor Binds to and Activates Phosphatidylinositol 3-Kinase. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Bakin, A.V.; Rodeck, U.; Brunet, A.; Arteaga, C.L. Transforming Growth Factor Beta Enhances Epithelial Cell Survival via Akt-dependent Regulation of FKHRL1. Mol. Biol. Cell 2001, 12, 3328–3339. [Google Scholar] [CrossRef]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-Kinase Function Is Required for Transforming Growth Factor β-mediated Epithelial to Mesenchymal Transition and Cell Migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2008, 19, 128–139. [Google Scholar] [CrossRef]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J. Inactivation of the type II TGF-Beta receptor in colon cancer cells with microsatellite instability. Science 2019, 268, 1336–1338. [Google Scholar] [CrossRef]
- Parsons, R.; Myeroff, L.L.; Liu, B.; Willson, J.K.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Microsatellite Instability and Mutations of the Transforming Growth Factor Beta Type II Receptor Gene in Colorectal Cancer. Cancer Res. 1995, 55, 5548–5550. [Google Scholar]
- Grady, W.M.; Myeroff, L.L.; Swinler, S.E.; Rajput, A.; Thiagalingam, S.; Lutterbaugh, J.D.; Neumann, A.; Brattain, M.G.; Chang, J.; Kim, S.J.; et al. Mutational Inactivation of Transforming Growth Factor Beta Receptor Type II in Microsatellite Stable Colon Cancers. Cancer Res. 1999, 59, 320–324. [Google Scholar] [PubMed]
- Levy, L.; Hill, C. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006, 17, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Hinshelwood, R.A.; Huschtscha, L.I.; Melki, J.; Stirzaker, C.; Abdipranoto, A.; Vissel, B.; Ravasi, T.; Wells, C.A.; Hume, D.A.; Reddel, R.R.; et al. Concordant epigenetic silencing of transforming growth factor-beta signaling pathway genes occurs early in breast carcinogenesis. Cancer Res. 2007, 67, 11517–11527. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, A.A.; Veerakumarasivam, A.; Vias, M.; Kumar, R.; Hamdy, F.C.; Neal, D.E.; Mills, I.G. Promoter methylation correlates with reduced Smad4 expression in advanced prostate cancer. Prostate 2008, 68, 661–674. [Google Scholar] [CrossRef]
- Monteleone, G.; Kumberova, A.; Croft, N.M.; McKenzie, C.; Steer, H.W.; MacDonald, T.T. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J. Clin. Invest. 2001, 108, 601–609. [Google Scholar] [CrossRef]
- Aytac, E.; Sulu, B.; Heald, B.; O’Malley, M.; LaGuardia, L.; Remzi, F.H.; Kalady, M.F.; Burke, C.A.; Church, J.M. Genotype-defined cancer risk in juvenile polyposis syndrome. Br. J. Surg. 2014, 102, 114–118. [Google Scholar] [CrossRef]
- Pasche, B.; Kolachana, P.; Nafa, K.; Satagopan, J.; Chen, Y.G.; Lo, R.S.; Brener, D.; Yang, D.; Kirstein, L.; Oddoux, C.; et al. TbetaR-I Is a Candidate Tumor Susceptibility Allele. Cancer Res. 1999, 59, 5678–5682. [Google Scholar]
- Pasche, B.; Kaklamani, V.; Hou, N.; Young, T.; Rademaker, A.; Peterlongo, P.; Ellis, N.; Offit, K.; Caldes, T.; Reiss, M.; et al. TGFBR1*6A and Cancer: A Meta-Analysis of 12 Case-Control Studies. JCO 2004, 22, 754–756. [Google Scholar] [CrossRef]
- Liao, R.-Y.; Mao, C.; Qiu, L.-X.; Ding, H.; Chen, Q.; Pan, H.-F. TGFBR1*6A/9A polymorphism and cancer risk: A meta-analysis of 13,662 cases and 14,147 controls. Mol. Biol. Rep. 2009, 37, 3227–3232. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFβ signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-beta: Duality of Function Between Tumor Prevention and Carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Ehata, S.; Koinuma, D. Tumor-promoting functions of transforming growth factor-β in progression of cancer. Upsala J. Med. Sci. 2012, 117, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. KrasG12D and Smad4/Dpc4 Haploinsufficiency Cooperate to Induce Mucinous Cystic Neoplasms and Invasive Adenocarcinoma of the Pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef]
- Teng, Y.; Sun, A.-N.; Pan, X.-C.; Yang, G.; Yang, L.L.; Wang, M.R.; Yang, X. Synergistic Function of Smad4 and PTEN in Suppressing Forestomach Squamous Cell Carcinoma in the Mouse. Cancer Res. 2006, 66, 6972–6981. [Google Scholar] [CrossRef]
- Xu, X.; Brodie, S.G.; Yang, X.; Im, Y.H.; Parks, W.T.; Chen, L.; Zhou, Y.X.; Weinstein, M.; Kim, S.J.; Deng, C.X. Haploid loss of the tumor suppressor Smad4/Dpc4 initiates gastric polyposis and cancer in mice. Oncogene 2000, 19, 1868–1874. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Kobayashi, S.; Qiao, W.; Li, C.; Xiao, C.; Radaeva, S.; Stiles, B.; Wang, R.H.; Ohara, N.; Yoshino, T.; et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption ofSmad4 andPten in mice. J. Clin. Invest. 2006, 116, 1843–1852. [Google Scholar] [CrossRef]
- Qiao, W.; Li, A.G.; Owens, P.; Xu, X.; Wang, X.J.; Deng, C.-X. Hair follicle defects and squamous cell carcinoma formation in Smad4 conditional knockout mouse skin. Oncogene 2005, 25, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W. Squamous cell carcinoma and mammary abscess formation through squamous metaplasia in Smad4/Dpc4 conditional knockout mice. Development 2003, 130, 6143–6153. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Mao, C.; Teng, Y.; Li, W.; Zhang, J.; Cheng, X.; Li, X.; Han, X.; Xia, Z.; Deng, H.; et al. Targeted Disruption of Smad4 in Mouse Epidermis Results in Failure of Hair Follicle Cycling and Formation of Skin Tumors. Cancer Res. 2005, 65, 8671–8678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal Tumorigenesis in Compound Mutant Mice of both Dpc4 (Smad4) and Apc Genes. Cell 1998, 92, 645–656. [Google Scholar] [CrossRef]
- Means, A.L.; Freeman, T.J.; Zhu, J.; Woodbury, L.G.; Marincola-Smith, P.; Wu, C.; Meyer, A.R.; Weaver, C.J.; Padmanabhan, C.; An, H.; et al. Epithelial Smad4 Deletion Up-Regulates Inflammation and Promotes Inflammation-Associated Cancer. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 257–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, T.J.; Smith, J.J.; Chen, X.; Washington, M.K.; Roland, J.T.; Means, A.L.; Eschrich, S.A.; Yeatman, T.J.; Deane, N.G.; Beauchamp, R.D. Smad4-Mediated Signaling Inhibits Intestinal Neoplasia by Inhibiting Expression of β-Catenin. YGAST 2012, 142, 562–571.e562. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; DeCant, B.; Stauacher, J.; Vitello, D.; Mangan, R.J.; Wayne, E.A.; Mascarinas, E.; Diaz, A.M.; Bauer, J.; McKinney, R.D.; et al. Loss of TGFβ signaling promotes colon cancer progression and tumor-associated inflammation. Oncotarget 2017, 8, 3826–3829. [Google Scholar] [CrossRef]
- Hahm, K.; Im, Y.; Parks, T.W.; Park, S.H.; Markowitz, S.; Jung, H.Y.; Green, J.; Kim, S.J. Loss of transforming growth factor beta signalling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut 2001, 49, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.L.; Herrington, H.; Reh, D.; Weber, S.; Bornstein, S.; Wang, D.; Li, A.G.; Tang, C.F.; Siddiqui, Y.; Nord, J.; et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamos cell carcinoma. Genes Dev. 2006, 20, 1331–1342. [Google Scholar] [CrossRef]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, C.P.; et al. Abrogation of TGFβ Signaling in Mammary Carcinomas Recruits Gr-1+CD11b+ Myeloid Cells that Promote Metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine Growth Factor Rev. 2010, 21, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Flavell, R.A. Abrogation of TGFbeta Signaling in T Cells Leads to Spontaneous T Cell Differentiation and Autoimmune Disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef]
- Wrzesinski, S.H.; Wan, Y.Y.; Flavell, R.A. Transforming growth factor-beta and the immune response: Implications for anticancer therapy. Clin. Cancer Res. 2007, 13, 5262–5270. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Flavell, R.A. Transforming growth factor-β in T-cell biology. Nat. Rev. Immunol. 2002, 2, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. Transforming Growth Factor-β Regulation of Immune Responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Kontani, K.; Kajino, K.; Huangi, C.-L.; Fujino, S.; Taguchi, O.; Yamauchi, A.; Yokomise, H.; Ogasawara, K. Spontaneous elicitation of potent antitumor immunity and eradication of established tumors by administration of DNA encoding soluble transforming growth factor-β II receptor without active antigen-sensitization. Cancer Immunol. Immunother. 2005, 55, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marcais, A.; Guimaraes, F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-b inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, 1–15. [Google Scholar] [CrossRef]
- Brandes, M.E.; Mai, U.; Ohura, K.; Wahl, S.M. Type I transforming growth factor-beta receptors on neutrophils mediate chemotaxis to transforming growth factor-beta. J. Immunol. 1991, 147, 1600–1606. [Google Scholar]
- Chen, J.J.; Sun, Y.; Nabel, G.J. Regulation of the Proinflammatory Effects of Fas Ligand (CD95L). Science 1998, 282, 1714–1717. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Otegbeye, F.; Ojo, E.; Moreton, S.; Mackowski, N.; Lee, D.A.; de Lima, M.; Wald, D.N. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE 2018, 13, e0191358-13. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, H.; Wang, X.; Guanmin, J.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [PubMed]
- Hofmann, T.G.; Stollberg, N.; Schmitz, M.L.; Will, H. HIPK2 Regulates Transforming Growth Factor-beta-Induced c-Jun NH2-Terminal Kinase Activation and Apoptosis in Human Hepatoma Cells. Cancer Res. 2003, 63, 8271–8277. [Google Scholar] [PubMed]
- Gottfried, Y.; Rotem, A.; Lotan, R.; Stellar, H.; Larisch, S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. 2004, 23, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Chytil, A.; Washington, M.K.; Romero-Gallo, J.; Gorska, A.E.; Wirth, P.S.; Gautum, S.; Moses, H.L.; Grady, W.M. Transforming Growth Factor beta Receptor Type II Inactivation Promotes the Establishment and Progression of Colon Cancer. Cancer Res. 2004, 64, 4687–4692. [Google Scholar] [CrossRef]
- Yan, M.; Rerko, R.M.; Platzer, P.; Dawson, D.; Willis, J.; Tong, M.; Lawrence, E.; Lutterbaugh, J.; Lu, S.; Willson, J.K.V.; et al. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 17468–17473. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Fujishita, T.; Kakizaki, F.; Hirai, H.; Matsumoto, T.; Iwamoto, M.; Inamoto, S.; Hatano, E.; Hasegawa, S.; et al. Loss of SMAD4 From Colorectal Cancer Cells Promotes CCL15 Expression to Recruit CCR1+ Myeloid Cells and Facilitate Liver Metastasis. Gastroenterology 2013, 145, 1064–1075.e11. [Google Scholar] [CrossRef] [Green Version]
- Inamoto, S.; Itatani, Y.; Yamamoto, T.; Minamiguchi, S.; Hirai, H.; Iwamoto, M.; Hasegawa, S.; Taketo, M.M.; Sakai, Y.; Kawada, K. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin. Cancer Res. 2016, 22, 492–501. [Google Scholar] [CrossRef]
- Kitamura, T.; Kometani, K.; Hashida, H.; Matsunaga, A.; Miyoshi, H.; Hosogi, H.; Aoki, M.; Oshima, M.; Hattori, M.; Takabayashi, A.; et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat. Genet. 2007, 39, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, I.; Lee, L.H.; Ogino, S.; Marco, M.R.; Wu, C.; Chen, X.; Datta, J.; Sadot, E.; Szeglin, B.; Guillem, J.G.; et al. SMAD4 Loss in Colorectal Cancer Patients Correlates with Recurrence, Loss of Immune Infiltrate, and Chemoresistance. Clin. Cancer Res. 2019, 25, 1948–1956. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.; White, R.; Malkoski, S.; Oka, M.; Han, G.; Cleaver, T.; Reh, D.; Andersen, P.; Gross, N.; Olson, S.; et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J. Clin. Invest. 2009, 119, 3408–3419. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Chung, C.H.; Parker, J.S.; Stover, D.G.; Cheng, N.; Chytil, A.; Aakre, M.; Shyr, Y.; Moses, H.L. Abrogation of TGF-β signaling enhances chemokine production and correlates with prognosis in human breast cancer. J. Clin. Invest. 2009, 119, 1571–1582. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-Z.; Li, Y.-Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Del Vecchio Blanco, G.; Monteleone, I.; Fina, D.; Caruso, R.; Gioia, V.; Ballerini, S.; Federici, G.; Bernardini, S.; Pallone, F.; et al. Post-transcriptional Regulation of Smad7 in the Gut of Patients With Inflammatory Bowel Disease. Gastroenterology 2005, 129, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Fantini, M.C.; Onali, S.; Zorzi, F.; Sancesario, G.; Bernardini, S.; Calabrese, E.; Viti, F.; Monteleone, I.; Biancone, L.; et al. Phase I Clinical Trial of Smad7 Knockdown Using Antisense Oligonucleotide in Patients With Active Crohn’s Disease. Mol. Therapy 2016, 20, 870–876. [Google Scholar] [CrossRef]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Struniolo, G.C.; et al. Mongersen, an Oral SMAD7Antisense Oligonucleotide, and Crohn’s Disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marincola Smith, P.; Means, A.L.; Beauchamp, R.D. Immunomodulatory Effects of TGF-? Family Signaling within Intestinal Epithelial Cells and Carcinomas. Gastrointest. Disord. 2019, 1, 290-300. https://doi.org/10.3390/gidisord1020024
Marincola Smith P, Means AL, Beauchamp RD. Immunomodulatory Effects of TGF-? Family Signaling within Intestinal Epithelial Cells and Carcinomas. Gastrointestinal Disorders. 2019; 1(2):290-300. https://doi.org/10.3390/gidisord1020024
Chicago/Turabian StyleMarincola Smith, Paula, Anna L. Means, and R. Daniel Beauchamp. 2019. "Immunomodulatory Effects of TGF-? Family Signaling within Intestinal Epithelial Cells and Carcinomas" Gastrointestinal Disorders 1, no. 2: 290-300. https://doi.org/10.3390/gidisord1020024
APA StyleMarincola Smith, P., Means, A. L., & Beauchamp, R. D. (2019). Immunomodulatory Effects of TGF-? Family Signaling within Intestinal Epithelial Cells and Carcinomas. Gastrointestinal Disorders, 1(2), 290-300. https://doi.org/10.3390/gidisord1020024