
Enteric Nervous System Alterations in Inflammatory Bowel Disease: Perspectives and Implications


{kind=link}
{kind=link}
Abstract
:1. Introduction
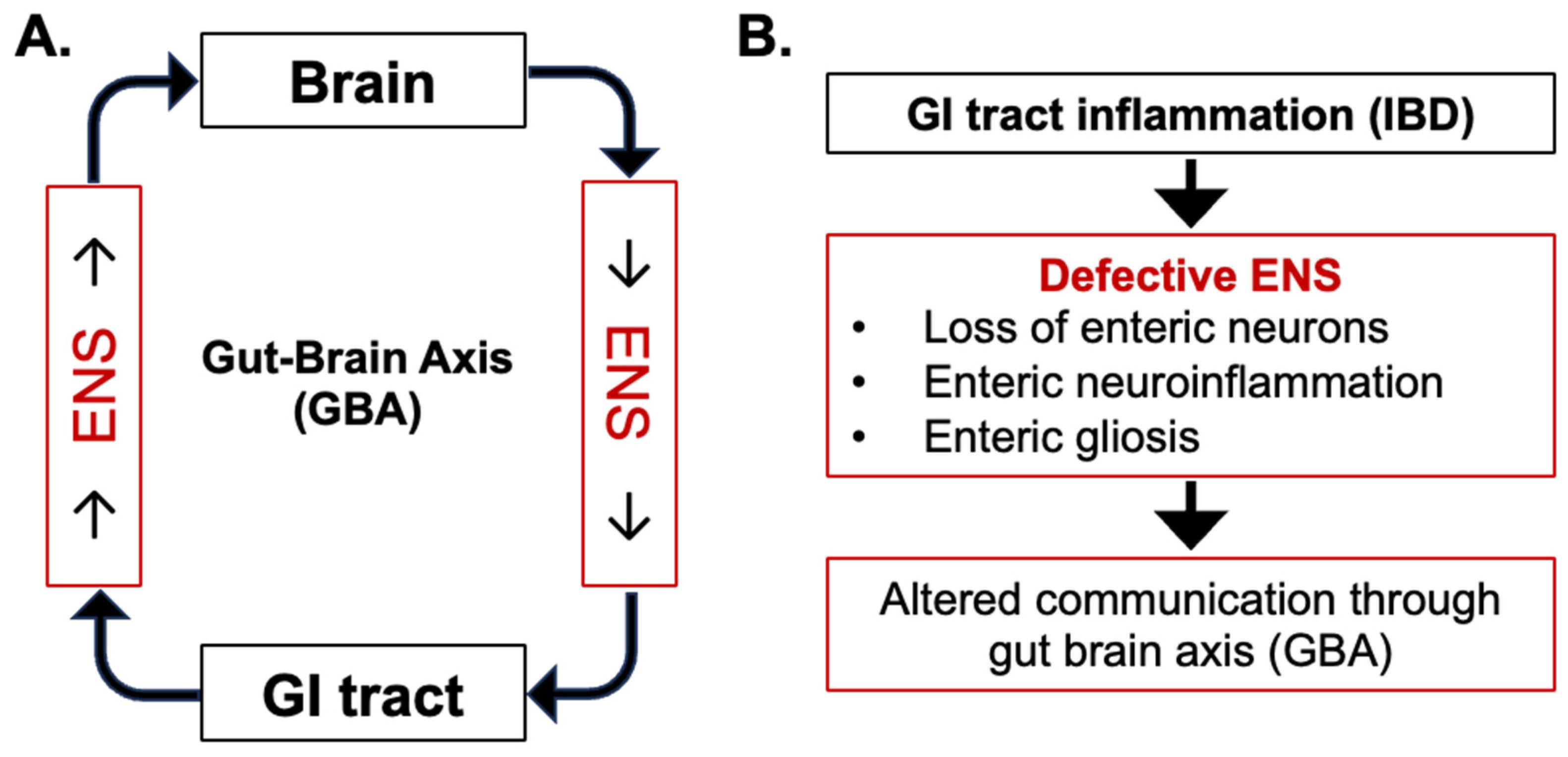
2. Central Role of ENS in Gut–Brain Axis
3. Enteric Nervous System Alterations in IBD
3.1. Pathophysiology of IBD and Alterations to Enteric Neurons
3.2. Pathophysiology of IBD and Alterations to Enteric Glial Cells (EGCs)
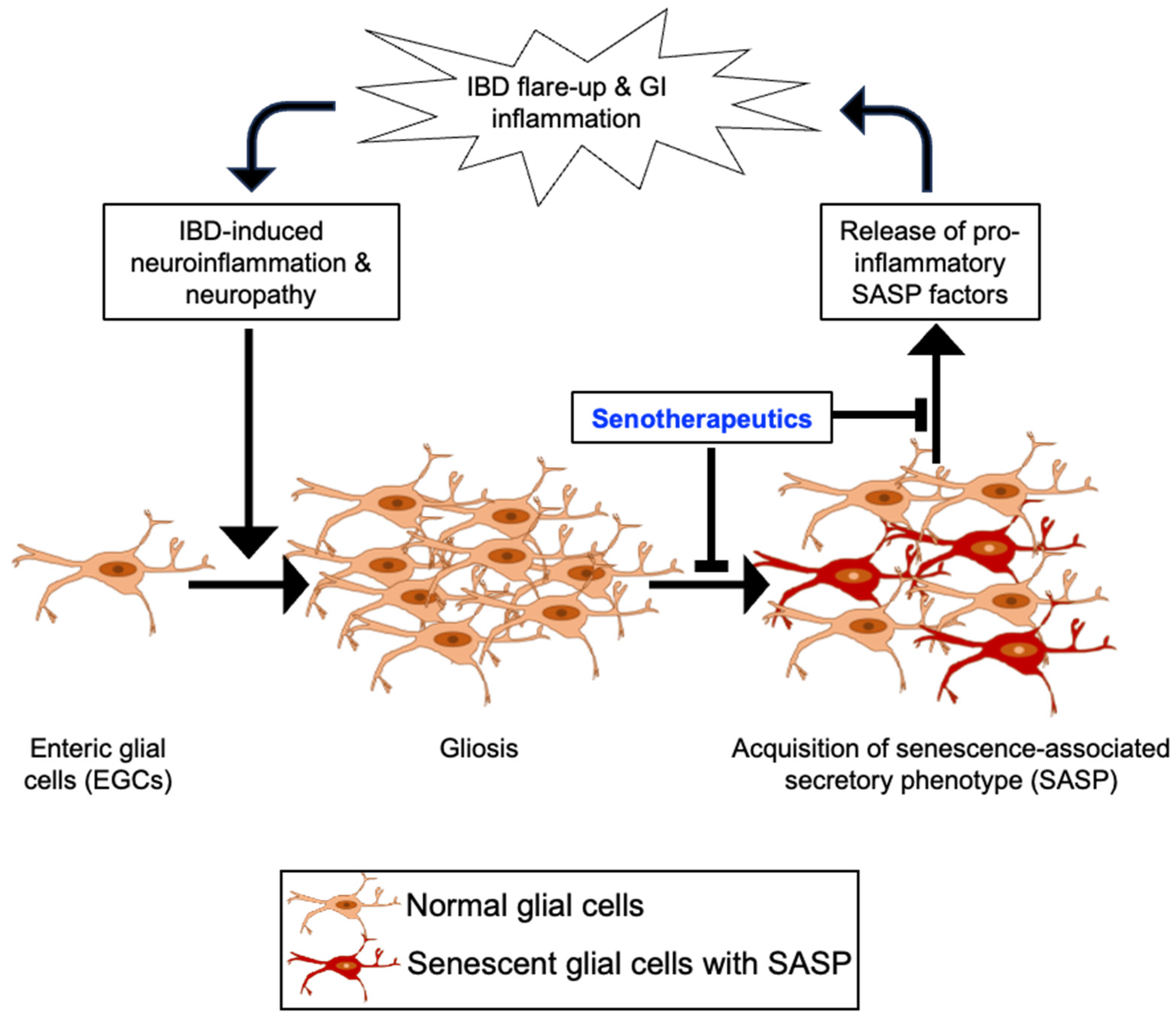
4. EGC Senescence in the Etiology of IBD
5. Plausibility of Senolytics Intervention for the Management of IBD
6. Effects of Serotherapeutic Agents on Gut Dysbiosis
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rudbaek, J.J.; Agrawal, M.; Torres, J.; Mehandru, S.; Colombel, J.F.; Jess, T. Deciphering the different phases of preclinical inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 86–100. [Google Scholar] [CrossRef]
- Oddsson, S.J.; Gunnarsdottir, T.; Johannsdottir, L.G.; Amundadottir, M.L.; Frimannsdottir, A.; Molander, P.; Ylanne, A.K.; Islind, A.S.; Oskarsdottir, M.; Thorgeirsson, T. A New Digital Health Program for Patients with Inflammatory Bowel Disease: Preliminary Program Evaluation. JMIR Form. Res. 2023, 7, e39331. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.K.; Auyeung, K.K. Inflammatory bowel disease: Etiology, pathogenesis and current therapy. Curr. Pharm. Des. 2014, 20, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Malik, T.F.; Aurelio, D.M. Extraintestinal Manifestations of Inflammatory Bowel Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Vadstrup, K.; Alulis, S.; Borsi, A.; Jørgensen, T.R.; Nielsen, A.; Munkholm, P.; Qvist, N. Extraintestinal Manifestations and Other Comorbidities in Ulcerative Colitis and Crohn Disease: A Danish Nationwide Registry Study 2003–2016. Crohns Colitis 360 2020, 2, otaa070. [Google Scholar] [CrossRef]
- Gong, W.; Guo, P.; Li, Y.; Liu, L.; Yan, R.; Liu, S.; Wang, S.; Xue, F.; Zhou, X.; Yuan, Z. Role of the Gut-Brain Axis in the Shared Genetic Etiology between Gastrointestinal Tract Diseases and Psychiatric Disorders: A Genome-Wide Pleiotropic Analysis. JAMA Psychiatry 2023, 80, 360–370. [Google Scholar] [CrossRef]
- Peppas, S.; Pansieri, C.; Piovani, D.; Danese, S.; Peyrin-Biroulet, L.; Tsantes, A.G.; Brunetta, E.; Tsantes, A.E.; Bonovas, S. The Brain-Gut Axis: Psychological Functioning and Inflammatory Bowel Diseases. J. Clin. Med. 2021, 10, 377. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Enayati, A.; Soghi, A.; Butler, A.E.; Rizzo, M.; Sahebkar, A. The Effect of Curcumin on the Gut-Brain Axis: Therapeutic Implications. J. Neurogastroenterol. Motil. 2023, 29, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, X.; Huang, S.; Yin, G.; Wang, X.; Shen, G. Targeting the gut-microbiota-brain axis in irritable bowel disease to improve cognitive function—Recent knowledge and emerging therapeutic opportunities. Rev. Neurosci. 2023, 34, 763–773. [Google Scholar] [CrossRef]
- Günther, C.; Rothhammer, V.; Karow, M.; Neurath, M.; Winner, B. The Gut-Brain Axis in Inflammatory Bowel Disease-Current and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 8870. [Google Scholar] [CrossRef]
- Fung, C.; Vanden Berghe, P. Functional circuits and signal processing in the enteric nervous system. Cell Mol. Life Sci. 2020, 77, 4505–4522. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.J.; Hu, H. Enteric nervous system: Sensory transduction, neural circuits and gastrointestinal motility. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Sternini, C. Structural and chemical organization of the myenteric plexus. Annu. Rev. Physiol. 1988, 50, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Jessen, H.; Thuneberg, L. Interstitial cells of Cajal and Auerbach’s plexus. A scanning electron microscopical study of guinea-pig small intestine. J. Submicrosc. Cytol. Pathol. 1991, 23, 195–212. [Google Scholar] [PubMed]
- Clapp, M.; Aurora, N.; Herrera, L.; Bhatia, M.; Wilen, E.; Wakefield, S. Gut microbiota’s effect on mental health: The gut-brain axis. Clin. Pract. 2017, 7, 987. [Google Scholar] [CrossRef] [PubMed]
- Heravi, F.S.; Naseri, K.; Hu, H. Gut Microbiota Composition in Patients with Neurodegenerative Disorders (Parkinson’s and Alzheimer’s) and Healthy Controls: A Systematic Review. Nutrients 2023, 15, 4365. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Ganz, J.; Bayrer, J.; Becker, L.; Bogunovic, M.; Rao, M. Advances in Enteric Neurobiology: The “Brain” in the Gut in Health and Disease. J. Neurosci. 2018, 38, 9346–9354. [Google Scholar] [CrossRef] [PubMed]
- Furness, J.B.; Callaghan, B.P.; Rivera, L.R.; Cho, H.J. The enteric nervous system and gastrointestinal innervation: Integrated local and central control. Adv. Exp. Med. Biol. 2014, 817, 39–71. [Google Scholar] [PubMed]
- Mittal, R.; Debs, L.H.; Patel, A.P.; Nguyen, D.; Patel, K.; O’Connor, G.; Grati, M.; Mittal, J.; Yan, D.; Eshraghi, A.A.; et al. Neurotransmitters: The Critical Modulators Regulating Gut-Brain Axis. J. Cell Physiol. 2017, 232, 2359–2372. [Google Scholar] [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain-Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef]
- Ye, L.; Liddle, R.A. Gastrointestinal hormones and the gut connectome. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, S.E.; Kirchgessner, A. Neuroinflammation in inflammatory bowel disease. J. Neuroinflamm. 2010, 7, 37. [Google Scholar] [CrossRef]
- Craig, C.F.; Filippone, R.T.; Stavely, R.; Bornstein, J.C.; Apostolopoulos, V.; Nurgali, K. Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease. J. Neuroinflamm. 2022, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Hey, G.E.; Vedam-Mai, V.; Beke, M.; Amaris, M.; Ramirez-Zamora, A. The Interface between Inflammatory Bowel Disease, Neuroinflammation, and Neurological Disorders. Semin. Neurol. 2023, 43, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Villanacci, V.; Bassotti, G.; Nascimbeni, R.; Antonelli, E.; Cadei, M.; Fisogni, S.; Salerni, B.; Geboes, K. Enteric nervous system abnormalities in inflammatory bowel diseases. Neurogastroenterol. Motil. 2008, 20, 1009–1016. [Google Scholar] [CrossRef]
- Cabarrocas, J.; Savidge, T.C.; Liblau, R.S. Role of enteric glial cells in inflammatory bowel disease. Glia 2003, 41, 81–93. [Google Scholar] [CrossRef]
- Cornet, A.; Savidge, T.C.; Cabarrocas, J.; Deng, W.L.; Colombel, J.F.; Lassmann, H.; Desreumaux, P.; Liblau, R.S. Enterocolitis induced by autoimmune targeting of enteric glial cells: A possible mechanism in Crohn’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 13306–13311. [Google Scholar] [CrossRef]
- De Giorgio, R.; Guerrini, S.; Barbara, G.; Stanghellini, V.; De Ponti, F.; Corinaldesi, R.; Moses, P.L.; Sharkey, K.A.; Mawe, G.M. Inflammatory neuropathies of the enteric nervous system. Gastroenterology 2004, 126, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Nezami, B.G.; Srinivasan, S. Enteric nervous system in the small intestine: Pathophysiology and clinical implications. Curr. Gastroenterol. Rep. 2010, 12, 358–365. [Google Scholar] [CrossRef]
- Holland, A.M.; Bon-Frauches, A.C.; Keszthelyi, D.; Melotte, V.; Boesmans, W. The enteric nervous system in gastrointestinal disease etiology. Cell Mol. Life Sci. 2021, 78, 4713–4733. [Google Scholar] [CrossRef]
- Brosens, E.; Burns, A.J.; Brooks, A.S.; Matera, I.; Borrego, S.; Ceccherini, I.; Tam, P.K.; García-Barceló, M.M.; Thapar, N.; Benninga, M.A.; et al. Genetics of enteric neuropathies. Dev. Biol. 2016, 417, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.D. Enteric Nervous System: Neuropathic Gastrointestinal Motility. Dig. Dis. Sci. 2016, 61, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Polin, A.; Lavergne-Slove, A.; Panis, Y.; Treton, X.; Dray, X.; Bouhnik, Y.; Valleur, P.; Marteau, P. Plexitis as a predictive factor of early postoperative clinical recurrence in Crohn’s disease. Gut 2009, 58, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Ray, K. IBD: Intestinal inflammation affected by density of enteric neurons. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 534. [Google Scholar] [CrossRef] [PubMed]
- Nestor-Kalinoski, A.; Smith-Edwards, K.M.; Meerschaert, K.; Margiotta, J.F.; Rajwa, B.; Davis, B.M.; Howard, M.J. Unique Neural Circuit Connectivity of Mouse Proximal, Middle, and Distal Colon Defines Regional Colonic Motor Patterns. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 309–337. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Wright, C.M.; Heuckeroth, R.O. Unexpected Roles for the Second Brain: Enteric Nervous System as Master Regulator of Bowel Function. Annu. Rev. Physiol. 2019, 81, 235–259. [Google Scholar] [CrossRef] [PubMed]
- Morales-Soto, W.; Gulbransen, B.D. Enteric Glia: A New Player in Abdominal Pain. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 433–445. [Google Scholar] [CrossRef]
- Delvalle, N.M.; Dharshika, C.; Morales-Soto, W.; Fried, D.E.; Gaudette, L.; Gulbransen, B.D. Communication between Enteric Neurons, Glia, and Nociceptors Underlies the Effects of Tachykinins on Neuroinflammation. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 321–344. [Google Scholar] [CrossRef]
- Lucarini, E.; Micheli, L.; Toti, A.; Ciampi, C.; Margiotta, F.; Di Cesare Mannelli, L.; Ghelardini, C. Anti-Hyperalgesic Efficacy of Acetyl L-Carnitine (ALCAR) Against Visceral Pain Induced by Colitis: Involvement of Glia in the Enteric and Central Nervous System. Int. J. Mol. Sci. 2023, 24, 14841. [Google Scholar] [CrossRef]
- Drokhlyansky, E.; Smillie, C.S.; Wittenberghe, N.V.; Ericsson, M.; Griffin, G.K.; Eraslan, G.; Dionne, D.; Cuoco, M.S.; Goder-Reiser, M.N.; Sharova, T.; et al. The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell 2020, 182, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Sanovic, S.; Lamb, D.P.; Blennerhassett, M.G. Damage to the enteric nervous system in experimental colitis. Am. J. Pathol. 1999, 155, 1051–1057. [Google Scholar] [CrossRef]
- Brierley, S.M.; Linden, D.R. Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 611–627. [Google Scholar] [CrossRef]
- Stavely, R.; Abalo, R.; Nurgali, K. Targeting Enteric Neurons and Plexitis for the Management of Inflammatory Bowel Disease. Curr. Drug Targets 2020, 21, 1428–1439. [Google Scholar] [CrossRef]
- Linden, D.R.; Couvrette, J.M.; Ciolino, A.; McQuoid, C.; Blaszyk, H.; Sharkey, K.A.; Mawe, G.M. Indiscriminate loss of myenteric neurones in the TNBS-inflamed guinea-pig distal colon. Neurogastroenterol. Motil. 2005, 17, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Lomax, A.E.; Fernández, E.; Sharkey, K.A. Plasticity of the enteric nervous system during intestinal inflammation. Neurogastroenterol. Motil. 2005, 17, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, H.I.R.; Castelucci, P. Enteric nervous system and inflammatory bowel diseases: Correlated impacts and therapeutic approaches through the P2 × 7 receptor. World J. Gastroenterol. 2021, 27, 7909–7924. [Google Scholar] [CrossRef]
- Arseneau, K.O.; Cominelli, F. Targeting leukocyte trafficking for the treatment of inflammatory bowel disease. Clin. Pharmacol. Ther. 2015, 97, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Luzentales-Simpson, M.; Pang, Y.C.F.; Zhang, A.; Sousa, J.A.; Sly, L.M. Vedolizumab: Potential Mechanisms of Action for Reducing Pathological Inflammation in Inflammatory Bowel Diseases. Front. Cell Dev. Biol. 2021, 9, 612830. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, A.; Yang, D.; Vella, M.; Chiu, I.M. The intestinal neuro-immune axis: Crosstalk between neurons, immune cells, and microbes. Mucosal Immunol. 2021, 14, 555–565. [Google Scholar] [CrossRef]
- Serafini, M.A.; Paz, A.H.; Nunes, N.S. Cholinergic immunomodulation in inflammatory bowel diseases. Brain Behav. Immun. Health 2021, 19, 100401. [Google Scholar] [CrossRef]
- Cheon, G.J.; Cui, Y.; Yeon, D.S.; Kwon, S.C.; Park, B.G. Mechanisms of motility change on trinitrobenzenesulfonic Acid-induced colonic inflammation in mice. Korean J. Physiol. Pharmacol. 2012, 16, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Siegel, R.M. Wishing away inflammation? New links between serotonin and TNF signaling. Mol. Interv. 2009, 9, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Li, D.; Yan, X.; Lan, J.; Han, D.; Fan, K.; Chang, J.; Ma, Y. Activation of GABA receptor attenuates intestinal inflammation by modulating enteric glial cells function through inhibiting Nf-kB pathway. Life Sci. 2023, 329, 121984. [Google Scholar] [CrossRef] [PubMed]
- Bubeck, M.; Becker, C.; Patankar, J.V. Guardians of the gut: Influence of the enteric nervous system on the intestinal epithelial barrier. Front. Med. 2023, 10, 1228938. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Chand, H.S.; Banerjee, S.; Agarwal, H.; Raizada, V.; Roy, S.; Sopori, M. Acetylcholinesterase Inhibitor Pyridostigmine Bromide Attenuates Gut Pathology and Bacterial Dysbiosis in a Murine Model of Ulcerative Colitis. Dig. Dis. Sci. 2020, 65, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.T.; Costa, D.V.S.; Gomes, A.S.; Martins, C.S.; Silva, A.M.H.P.; Coelho-Aguiar, J.M.; Castelucci, P.; Lima-Júnior, R.C.P.; Leitão, R.F.C.; Moura-Neto, V.; et al. The involvement of mast cells in the irinotecan-induced enteric neurons loss and reactive gliosis. J. Neuroinflamm. 2017, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Shea-Donohue, T.; Urban, J.F. Neuroimmune modulation of gut function. Handb. Exp. Pharmacol. 2017, 239, 247–267. [Google Scholar] [PubMed]
- Seguella, L.; Gulbransen, B.D. Enteric glial biology, intercellular signalling and roles in gastrointestinal disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 571–587. [Google Scholar] [CrossRef]
- Brown, I.A.; McClain, J.L.; Watson, R.E.; Patel, B.A.; Gulbransen, B.D. Enteric glia mediate neuron death in colitis through purinergic pathways that require connexin-43 and nitric oxide. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 77–91. [Google Scholar] [CrossRef]
- Schneider, R.; Leven, P.; Glowka, T.; Kuzmanov, I.; Lysson, M.; Schneiker, B.; Miesen, A.; Baqi, Y.; Spanier, C.; Grants, I.; et al. A novel P2 × 2-dependent purinergic mechanism of enteric gliosis in intestinal inflammation. EMBO Mol. Med. 2021, 13, e12724. [Google Scholar] [CrossRef]
- Turco, F.; Sarnelli, G.; Cirillo, C.; Palumbo, I.; De Giorgi, F.; D’Alessandro, A.; Cammarota, M.; Giuliano, M.; Cuomo, R. Enteroglial-derived S100B protein integrates bacteria-induced Toll-like receptor signalling in human enteric glial cells. Gut 2014, 63, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Leven, P.; Schneider, R.; Schneider, L.; Mallesh, S.; Vanden Berghe, P.; Sasse, P.; Kalff, J.C.; Wehner, S. β-adrenergic signaling triggers enteric glial reactivity and acute enteric gliosis during surgery. J. Neuroinflamm. 2023, 20, 255. [Google Scholar] [CrossRef]
- Mazzotta, E.; Grants, I.; Villalobos-Hernandez, E.; Chaudhuri, S.; McClain, J.L.; Seguella, L.; Kendig, D.M.; Blakeney, B.A.; Murthy, S.K.; Schneider, R.; et al. BQ788 reveals glial ETB receptor modulation of neuronal cholinergic and nitrergic pathways to inhibit intestinal motility: Linked to postoperative ileus. Br. J. Pharmacol. 2023, 180, 2550–2576. [Google Scholar] [CrossRef]
- Schneider, R.; Leven, P.; Mallesh, S.; Breßer, M.; Schneider, L.; Mazzotta, E.; Fadda, P.; Glowka, T.; Vilz, T.O.; Lingohr, P.; et al. IL-1-dependent enteric gliosis guides intestinal inflammation and dysmotility and modulates macrophage function. Commun. Biol. 2022, 5, 811. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Qian, W.; Hou, X.H.; Dai, C.B. Bifidobacterium bifidum and Bacteroides fragilis Induced Differential Immune Regulation of Enteric Glial Cells Subjected to Exogenous Inflammatory Stimulation. Inflammation 2022, 45, 2388–2405. [Google Scholar] [CrossRef]
- Biskou, O.; Meira de-Faria, F.; Walter, S.M.; Winberg, M.E.; Haapaniemi, S.; Myrelid, P.; Söderholm, J.D.; Keita, Å.V. Increased Numbers of Enteric Glial Cells in the Peyer’s Patches and Enhanced Intestinal Permeability by Glial Cell Mediators in Patients with Ileal Crohn’s Disease. Cells 2022, 11, 335. [Google Scholar] [CrossRef]
- Pochard, C.; Coquenlorge, S.; Freyssinet, M.; Naveilhan, P.; Bourreille, A.; Neunlist, M.; Rolli-Derkinderen, M. The multiple faces of inflammatory enteric glial cells: Is Crohn’s disease a gliopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G1–G11. [Google Scholar] [CrossRef]
- Chow, A.K.; Grubišić, V.; Gulbransen, B.D. Enteric Glia Regulate Lymphocyte Activation via Autophagy-Mediated MHC-II Expression. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 1215–1237. [Google Scholar] [CrossRef] [PubMed]
- da Silveira, A.B.; de Oliveira, E.C.; Neto, S.G.; Luquetti, A.O.; Fujiwara, R.T.; Oliveira, R.C.; Brehmer, A. Enteroglial cells act as antigen-presenting cells in chagasic megacolon. Hum. Pathol. 2011, 42, 522–532. [Google Scholar] [CrossRef]
- Rosenberg, H.J.; Rao, M. Enteric glia in homeostasis and disease: From fundamental biology to human pathology. iScience 2021, 24, 102863. [Google Scholar] [CrossRef]
- Gulbransen, B.D.; Bashashati, M.; Hirota, S.A.; Gui, X.; Roberts, J.A.; MacDonald, J.A.; Muruve, D.A.; McKay, D.M.; Beck, P.L.; Mawe, G.M.; et al. Activation of neuronal P2 × 7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat. Med. 2012, 18, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.S.; Parr, E.J.; Sharkey, K.A. Effects of inflammation on cell proliferation in the myenteric plexus of the guinea-pig ileum. Cell Tissue Res. 1997, 289, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, C.; Schick, M.A.; Wollborn, J.; Heider, A.; Scholz, C.J.; Cecil, A.; Niesler, B.; Hirrlinger, J.; Walles, H.; Metzger, M. Activation of Myenteric Glia during Acute Inflammation In Vitro and In Vivo. PLoS ONE 2016, 11, e0151335. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, C.; Sarnelli, G.; Esposito, G.; Turco, F.; Steardo, L.; Cuomo, R. S100B protein in the gut: The evidence for enteroglial-sustained intestinal inflammation. World J. Gastroenterol. 2011, 17, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Liñán-Rico, A.; Turco, F.; Ochoa-Cortes, F.; Harzman, A.; Needleman, B.J.; Arsenescu, R.; Abdel-Rasoul, M.; Fadda, P.; Grants, I.; Whitaker, E.; et al. Molecular Signaling and Dysfunction of the Human Reactive Enteric Glial Cell Phenotype: Implications for GI Infection, IBD, POI, Neurological, Motility, and GI Disorders. Inflamm. Bowel Dis. 2016, 22, 1812–1834. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Capoccia, E.; Cirillo, C.; Gigli, S.; Pesce, M.; D’Alessandro, A.; Cuomo, R.; Sarnelli, G.; Steardo, L.; Esposito, G. Enteric glia: A new player in inflammatory bowel diseases. Int. J. Immunopathol. Pharmacol. 2015, 28, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, C.; Sarnelli, G.; Turco, F.; Mango, A.; Grosso, M.; Aprea, G.; Masone, S.; Cuomo, R. Proinflammatory stimuli activates human-derived enteroglial cells and induces autocrine nitric oxide production. Neurogastroenterol. Motil. 2011, 23, e372–e382. [Google Scholar] [CrossRef] [PubMed]
- Fettucciari, K.; Macchioni, L.; Davidescu, M.; Scarpelli, P.; Palumbo, C.; Corazzi, L.; Marchegiani, A.; Cerquetella, M.; Spaterna, A.; Marconi, P.; et al. Clostridium difficile toxin B induces senescence in enteric glial cells: A potential new mechanism of Clostridium difficile pathogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1945–1958. [Google Scholar] [CrossRef]
- Korsak, K.; Dolatshad, N.F.; Silva, A.T.; Saffrey, M.J. Ageing of enteric neurons: Oxidative stress, neurotrophic factors and antioxidant enzymes. Chem. Cent. J. 2012, 6, 80. [Google Scholar] [CrossRef]
- Bassotti, G.; Fruganti, A.; Stracci, F.; Marconi, P.; Fettucciari, K. Cytotoxic synergism of Clostridioides difficile toxin B with proinflammatory cytokines in subjects with inflammatory bowel diseases. World J. Gastroenterol. 2023, 29, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Progatzky, F.; Shapiro, M.; Chng, S.H.; Garcia-Cassani, B.; Classon, C.H.; Sevgi, S.; Laddach, A.; Bon-Frauches, A.C.; Lasrado, R.; Rahim, M.; et al. Regulation of intestinal immunity and tissue repair by enteric glia. Nature 2021, 599, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Manne, S.; Treem, W.R.; Bennett, D. Prevalence of Inflammatory Bowel Disease in Pediatric and Adult Populations: Recent Estimates from Large National Databases in the United States, 2007-2016. Inflamm. Bowel Dis. 2020, 26, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Witkowski, J.M.; McElhaney, J.; Loeb, M.; Mitnitski, A.; Pawelec, G. Aging, frailty and age-related diseases. Biogerontology 2010, 11, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and aging: Signaling pathways and intervention therapies. Signal Transduct. Target. Ther. 2023, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Monaghan, T.M.; Duggal, N.A.; Tighe, P.; Peerani, F. Microbial-Immune Crosstalk in Elderly-Onset Inflammatory Bowel Disease: Unchartered Territory. J. Crohns Colitis 2023, 17, 1309–1325. [Google Scholar] [CrossRef]
- Kumar, K.; Kumar, S.; Datta, K.; Fornace, A.J.; Suman, S. High-LET-Radiation-Induced Persistent DNA Damage Response Signaling and Gastrointestinal Cancer Development. Curr. Oncol. 2023, 30, 5497–5514. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Datta, K.; Fornace, A.J.; Suman, S. Total body proton and heavy-ion irradiation causes cellular senescence and promotes pro-osteoclastogenic activity in mouse bone marrow. Heliyon 2022, 8, e08691. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Tripathi, U.; Misra, A.; Tchkonia, T.; Kirkland, J.L. Impact of Senescent Cell Subtypes on Tissue Dysfunction and Repair: Importance and Research Questions. Mech. Ageing Dev. 2021, 198, 111548. [Google Scholar] [CrossRef]
- Mebratu, Y.A.; Soni, S.; Rosas, L.; Rojas, M.; Horowitz, J.C.; Nho, R. The aged extracellular matrix and the profibrotic role of senescence-associated secretory phenotype. Am. J. Physiol. Cell Physiol. 2023, 325, C565–C579. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Munck, L.K. Drug insight: Aminosalicylates for the treatment of IBD. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Barrett, K.; Saxena, S.; Pollok, R. Using corticosteroids appropriately in inflammatory bowel disease: A guide for primary care. Br. J. Gen. Pract. 2018, 68, 497–498. [Google Scholar] [CrossRef] [PubMed]
- Ardizzone, S.; Cassinotti, A.; Manes, G.; Porro, G.B. Immunomodulators for all patients with inflammatory bowel disease. Therap Adv. Gastroenterol. 2010, 3, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wang, S.; Li, J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front. Med. 2021, 8, 765474. [Google Scholar] [CrossRef] [PubMed]
- Hindmarch, D.C.; Malashanka, S.; Shows, D.M.; Clarke, A.S.; Lord, J.D. Janus kinase inhibitors differentially inhibit specific cytokine signals in the mesenteric lymph node cells of IBD patients. J. Crohns Colitis 2023, jjad173. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, G.; Yang, Y.; Zhang, S.; Jiang, H.; Tian, K.; Arenbaoligao; Chen, D. The treatment of inflammatory bowel disease with monoclonal antibodies in Asia. Biomed. Pharmacother. 2023, 157, 114081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2023, 290, 1362–1383. [Google Scholar] [CrossRef]
- Kim, E.C.; Kim, J.R. Senotherapeutics: Emerging strategy for healthy aging and age-related disease. BMB Rep. 2019, 52, 47–55. [Google Scholar] [CrossRef]
- Popescu, I.; Deelen, J.; Illario, M.; Adams, J. Challenges in anti-aging medicine-trends in biomarker discovery and therapeutic interventions for a healthy lifespan. J. Cell Mol. Med. 2023, 27, 2643–2650. [Google Scholar] [CrossRef]
- Suman, S.; Fornace, A.J. Countermeasure development against space radiation-induced gastrointestinal carcinogenesis: Current and future perspectives. Life Sci. Space Res. 2022, 35, 53–59. [Google Scholar] [CrossRef]
- Cominelli, F.; Nast, C.C.; Duchini, A.; Lee, M. Recombinant interleukin-1 receptor antagonist blocks the proinflammatory activity of endogenous interleukin-1 in rabbit immune colitis. Gastroenterology 1992, 103, 65–71. [Google Scholar] [CrossRef]
- Saccon, T.D.; Nagpal, R.; Yadav, H.; Cavalcante, M.B.; Nunes, A.D.C.; Schneider, A.; Gesing, A.; Hughes, B.; Yousefzadeh, M.; Tchkonia, T.; et al. Senolytic Combination of Dasatinib and Quercetin Alleviates Intestinal Senescence and Inflammation and Modulates the Gut Microbiome in Aged Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, S.H.; Yang, E.J.; Kim, E.K.; Kim, J.K.; Shin, D.Y.; Cho, M.L. Metformin Ameliorates Inflammatory Bowel Disease by Suppression of the STAT3 Signaling Pathway and Regulation of the between Th17/Treg Balance. PLoS ONE 2015, 10, e0135858. [Google Scholar] [CrossRef] [PubMed]
- Shaul, E.; Conrad, M.A.; Dawany, N.; Patel, T.; Canavan, M.C.; Baccarella, A.; Weinbrom, S.; Aleynick, D.; Sullivan, K.E.; Kelsen, J.R. Canakinumab for the treatment of autoinflammatory very early onset- inflammatory bowel disease. Front. Immunol. 2022, 13, 972114. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.P.; Ooi, J.D.; Goldberg, R. The interplay between the microbiota, diet and T regulatory cells in the preservation of the gut barrier in inflammatory bowel disease. Front. Microbiol. 2023, 14, 1291724. [Google Scholar]
- Wu, R.; Xiong, R.; Li, Y.; Chen, J.; Yan, R. Gut microbiome, metabolome, host immunity associated with inflammatory bowel disease and intervention of fecal microbiota transplantation. J. Autoimmun. 2023, 141, 103062. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, J. Gut microbiota and inflammatory bowel disease. WIREs Mech. Dis. 2022, 14, e1540. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Lee, M.; Chang, E.B. The Gut Microbiome and Inflammatory Bowel Diseases. Annu. Rev. Med. 2022, 73, 455–468. [Google Scholar] [CrossRef]
- Kawamoto, S.; Hara, E. Crosstalk between gut microbiota and cellular senescence: A vicious cycle leading to aging gut. Trends Cell Biol. 2024, S0962-8924. [Google Scholar] [CrossRef]
- Sharma, R. Emerging Interrelationship Between the Gut Microbiome and Cellular Senescence in the Context of Aging and Disease: Perspectives and Therapeutic Opportunities. Probiotics Antimicrob. Proteins 2022, 14, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Petakh, P.; Kamyshna, I.; Kamyshnyi, A. Effects of metformin on the gut microbiota: A systematic review. Mol. Metab. 2023, 77, 101805. [Google Scholar] [CrossRef] [PubMed]
- Induri, B.N.R.; Kansara, P.; Thomas, S.C.; Xu, F.; Saxena, D.; Li, X. The Gut Microbiome, Metformin, and Aging. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Ashiqueali, S.A.; Chaudhari, D.; Zhu, X.; Noureddine, S.; Siddiqi, S.; Garcia, D.N.; Gostynska, A.; Stawny, M.; Rubis, B.; Zanini, B.M.; et al. Fisetin modulates the gut microbiota alongside biomarkers of senescence and inflammation in a DSS-induced murine model of colitis. Geroscience 2024. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suman, S. Enteric Nervous System Alterations in Inflammatory Bowel Disease: Perspectives and Implications. Gastrointest. Disord. 2024, 6, 368-379. https://doi.org/10.3390/gidisord6020025
Suman S. Enteric Nervous System Alterations in Inflammatory Bowel Disease: Perspectives and Implications. Gastrointestinal Disorders. 2024; 6(2):368-379. https://doi.org/10.3390/gidisord6020025
Chicago/Turabian StyleSuman, Shubhankar. 2024. "Enteric Nervous System Alterations in Inflammatory Bowel Disease: Perspectives and Implications" Gastrointestinal Disorders 6, no. 2: 368-379. https://doi.org/10.3390/gidisord6020025
APA StyleSuman, S. (2024). Enteric Nervous System Alterations in Inflammatory Bowel Disease: Perspectives and Implications. Gastrointestinal Disorders, 6(2), 368-379. https://doi.org/10.3390/gidisord6020025