
The Innate Immune System Surveillance Biomarker p87 in African Americans and Caucasians with Small High-Grade Dysplastic Adenoma [SHiGDA] and Right-Sided JAK3 Colon Mutations May Explain the Presence of Multiple Cancers Revealing an Important Minority of Patients with JAK3 Mutations and Colorectal Neoplasia

 , ,
, ,
Abstract
:1. Introduction
2. Results
3. Materials and Methods
3.1. Patient Populations
3.2. Ion Torrent™ PGM Sequencing
3.3. PCR Assays
- Left primer: JAK3exon4F 5’-AAGGTACAAGCTGGGCTCTG-3´ Integrated DNA Technologies
- Right primer: JAK3exon4R 5´-TGAGGCCACCCAACTTCAAG-3´ Integrated DNA Technologies
3.4. Microbiome Determination
3.5. ELISA Testing and Immunohistochemistry
3.6. Data Analysis
4. Conclusions
Pitfalls and Promise
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levin, B.; Lieberman, D.A.; McFarland, B.; Andrews, K.S.; Brooks, D.; Bond, J.; Dash, C.; Giardiello, F.M.; Glick, S.; Johnson, D.; et al. Screening and Surveillance for the Early Detection of Colorectal Cancer and Adenomatous Polyps, 2008: A Joint Guideline From the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 2008, 134, 1570–1595. [Google Scholar] [CrossRef] [PubMed]
- Tobi, M.; Antaki, F.; Rambus, M.A.; Yang, Y.-X.; Kaplan, D.; Rodriguez, R.; Maliakkal, B.; Majumdar, A.; Demian, E.; Tobi, Y.Y.; et al. The Non-Invasive Prediction of Colorectal Neoplasia (NIPCON) Study 1995–2022: A Comparison of Guaiac-Based Fecal Occult Blood Test (FOBT) and an Anti-Adenoma Antibody, Adnab-9. Int. J. Mol. Sci. 2023, 24, 17257. [Google Scholar] [CrossRef] [PubMed]
- Bretthauer, M.; Loberg, M.; Wieszczy, P.; Kalager, M.; Emilsson, L.; Garborg, K.; Rupinski, M.; Dekker, E.; Spaander, M.; Bugajski, M.; et al. Effect of Colonoscopy Screening on Risks of Colorectal Cancer and Related Death. New Engl. J. Med. 2022, 387, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, D.A.; Rex, D.K.; Winawer, S.J.; Giardiello, F.M.; Johnson, D.A.; Levin, T.R. Guidelines for Colonoscopy Surveillance After Screening and Polypectomy: A Consensus Update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2012, 143, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Hilsden, R.J.; McGregor, E.; Murray, A.; Khoja, S.; Bryant, H. Coloretal cancer screening: Practices and attitudes of gastroenterologists, internists and surgeons. Can. J. Surg. 2006, 48, 434–440. [Google Scholar]
- Singh, H.; Turner, D.; Xue, L.; Targownik, L.E.; Bernstein, C.N. Risk of Developing Colorectal Cancer Following a Negative Colonoscopy Examination. Jama 2006, 295, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Baxter, N.N.; Goldwasser, M.A.; Paszat, L.F.; Saskin, R.; Urbach, D.R.; Rabeneck, L. Association of colonoscopy and death from colorectal cancer. Ann. Intern. Med. 2009, 150, 1–8. [Google Scholar] [CrossRef]
- Brenner, H.; Hoffmeister, M.; Arndt, V.; Stegmaier, C.; Altenhofen, L.; Haug, U. Protection from Right- and Left-Sided Colorectal Neoplasms After Colonoscopy: Population-Based Study. J. Natl. Cancer Inst. 2010, 102, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; White, P.; Nieto, J.; Vieira, D.; Francois, F.; Hamilton, F. Colorectal Cancer in African Americans: An Update. Clin. Transl. Gastroenterol. 2016, 7, e185. [Google Scholar] [CrossRef]
- Shimoda, T.; Ikegami, M.; Fujisaki, J.; Matsui, T.; Aizawa, S.; Ishikawa, E. Early colorectal carcinoma with special reference to its development de novo. Cancer 1989, 64, 1138–1147. [Google Scholar] [CrossRef]
- Baxter, N.N.; Warren, J.L.; Barrett, M.J.; Stukel, T.A.; Doria-Rose, V.P. Association Between Colonoscopy and Colorectal Cancer Mortality in a US Cohort According to Site of Cancer and Colonoscopist Specialty. J. Clin. Oncol. 2012, 30, 2664–2669. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, C.R. AGA Future trends committee report: CRC: A Qualitative review of emerging screening and diagnostic technologies. Gastroenterology 2005, 129, 1083–1103. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.E.; Baron, J.A.; Lieberman, D.A.; Schatzkin, A.; Lanza, E.; Winawer, S.J.; Zauber, A.G.; Jiang, R.; Ahnen, D.J.; Bond, J.H.; et al. A Pooled Analysis of Advanced Colorectal Neoplasia Diagnoses After Colonoscopic Polypectomy. Gastroenterology 2009, 136, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Ahnen, D.J. The ACG Emily Couric Lecture- The adenoma-carcinoma sequence revisited: Has the era of genetic tailoring finally arrived? Am. J. Gastroenterol. 2011, 106, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.; Chang-Claude, J.; Seiler, C.M.; Rickert, A.; Hoffmeister, M. Protection from Colorectal Cancer After Colonoscopy: A Population-Based, Case–Control Study. Ann. Intern. Med. 2011, 154, 22–30. [Google Scholar] [CrossRef]
- Ullah, N.; Qureshi, K.; Hatfield, J.; Sochacki, P.; David, D.; Albataineh, H.; Mejia, L.; Kenkre, C.; Lawson, M.; Tobi, M. Small early tubular adenomas and mixed colonic polyps found on screening flexible sigmoidoscopy do not predict proximal neoplasia in males. Clin. Gastroenterol. Hepatol. 2004, 2, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; and Obrien, M. Hyperplastic polyps, serrated adenomas, and the serrated polyp neoplasia pathway. Am. J. Gastroenterol. 2004, 99, 2242–2255. [Google Scholar] [CrossRef]
- O’Brien, M.J.; Yang, S.; Mack, C.; Xu, H.; Huang, C.S.; Mulcahy, E.; Amorosino, M.; Farraye, F.A. Comparison of Microsatellite Instability, CpG Island Methylation Phenotype, BRAF and KRAS Status in Serrated Polyps and Traditional Adenomas Indicates Separate Pathways to Distinct Colorectal Carcinoma End Points. Am. J. Surg. Pathol. 2006, 30, 1491–1501. [Google Scholar] [CrossRef]
- East, J.; Saunders, B.; Jass, J. Sporadic and syndromic hyperplastic polyps and serrated adenomas of the colon: Classification, molecular genetics, natural history, and clinical management. Gastroenterol. Clin. N. Am. 2008, 37, 25–46. [Google Scholar] [CrossRef]
- Boland, C.R.; Sato, J.; Appelman, H.D.; Bresalier, R.S.; Feinberg, A.P. Microallelotyping defines the sequence and tempo of allelic losses at tumour suppressor gene loci during colorectal cancer progression. Nat. Med. 1995, 9, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Doffe, F.; Carbonnier, V.; Tissier, M.; Leroy, B.; Martins, I.; Mattsson, J.S.M.; Micke, P.; Pavlova, S.; Pospisilova, S.; Smardova, J.; et al. Identification and functional characterization of new missense SNPs in the coding region of the TP53 gene. Cell Death Differ. 2020, 28, 1477–1492. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Screening for Colorectal Cancer in African Americans: Determinants and Rationale for an Earlier Age to Commence Screening. Dig. Dis. Sci. 2014, 60, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Nguyen, H.-N.; Van Hoang, M.; Bui, T.T.T.; Vu, B.-Q.; Dinh, T.H.T.; Vo, H.T.M.; Blaydon, D.C.; Eldirany, S.A.; Bunick, C.G.; et al. Altered skin microbiome, inflammation, and JAK/STAT signaling in Southeast Asian ichthyosis patients. Hum. Genom. 2024, 18, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Drew, D.A.; Markowitz, A.; Lloyd-Price, J.; Abu-Ali, G.; Nguyen, L.H.; Tran, C.; Chung, D.C.; Gilpin, K.K.; Meixell, D.; et al. Structure of the Mucosal and Stool Microbiome in Lynch Syndrome. Cell Host Microbe 2020, 27, 585–600.e4. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, Y.J.; Oh, G.M.; Jung, W.; Park, S.J. How is gut microbiome of patients with familial adenomatous polyposis different from healthy people? Medicine 2022, 101, e32194. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tobi, M.; Prabhu, S.; Gage, R.E.; Orr, T.; Lawson, M.J. Colorectal cancer risk: The impact of evidence of a field effect of carcinogenesis on blinded diagnosis using an anti-adenoma antibody test performed on colonoscopic effluent. Dig. Dis. Sci. 2002, 47, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Xhang, X.; Leu, Y.; Xu, Y.; Ullah, N.; Lawson, M.; Tobi, M. Fecal Adnab-9 binding as a risk marker for colorectal neoplasia. Cancer Lett. 2006, 235, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, R.A.; Min, B.H.; Yasmeen, S.; Teplitz, R.; Tesluk, H.; Ruebner, B.H.; Tobi, M.; Hatfield, J.; Fligiel, S.; Lawson, M.J. Correlation of Ki-67, p53, and Adnab-9 immunohistochemical staining and ploidy with clinical and histopathologic features of severely dysplastic colorectal adenomas. Dig. Dis. Sci. 2003, 48, 223–229. [Google Scholar] [CrossRef]
- Buchert, M.; Burns, C.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939–951. [Google Scholar] [CrossRef]
- Bhatavdekar, J.M.; Patel, D.D.; Chikhlikar, P.R.; Shah, N.G.; Vora, H.H.; Ghosh, N.; Trivedi, T.I. Molecular markers are predictors of recurrence and survival in patients with Dukes B and Dukes C colorectal adenocarcinoma. Dis. Colon. Rectum. 2001, 44, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Tobi, M.; Hatfield, J.; Adsay, V.; Galagan, K.; Kozarek, R.; Inagaki, M.; Kasai, S.; Tokusashi, Y.; Obara, T.; Hruban, R.H.; et al. Prognostic Significance of the Labeling of Adnab-9 in Pancreatic Intraductal Papillary Mucinous Neoplasms. J. Gastrointest. Cancer 2001, 29, 141–150. [Google Scholar] [CrossRef]
- Tobi, M.; Kim, M.; Weinstein, D.H.; Rambus, M.A.; Hatfield, J.; Adsay, N.V.; Levi, E.; Evans, D.; Lawson, M.J.; Fligiel, S. Prospective Markers for Early Diagnosis and Prognosis of Sporadic Pancreatic Ductal Adenocarcinoma. Dig. Dis. Sci. 2012, 58, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Welch, H.G.; Robertson, D.J. Colorectal Cancer on the Decline Why Screening Can’t Explain It All. N. Engl. J. Med. 2016, 374, 1605–1607. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Ji, P.; Ouyang, N.; Zhang, Y.; Wang, X.Y.; Rubin, D.C.; Davidson, N.O.; Bergamaschi, R.; Shroyer, K.R.; Burke, S.; et al. Differential expression of miRNAs in colon cancer between African and Caucasian Americans: Implications for cancer racial health disparities. Int. J. Oncol. 2014, 45, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Weige, C.C.; Birtwistle, M.R.; Mallick, H. Transcriptomes and shRNA Suppressors in a TP53 Allele specific Model of Early-onset Colon Cancer in African Americans. Mol. Cancer Res. 2014, 12, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Kuracha, M.R.; Govindarajan, V.; Loggie, B.W.; Tobi, M.; McVicker, B.L. Pictilisib-Induced Resistance Is Mediated through FOXO1-Dependent Activation of Receptor Tyrosine Kinases in Mucinous Colorectal Adenocarcinoma Cells. Int. J. Mol. Sci. 2023, 24, 12331. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jovov, B.; Araujo-Perez, F.; Sigel, C.S.; Stratford, J.K.; McCoy, A.N.; Yeh, J.J.; Keku, T. Differential Gene Expression between African American and European American Colorectal Cancer Patients. PLoS ONE 2012, 7, e30168. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Lee, S.-J.; Jung, Y.S. Blocking of p53-Snail binding, promoted by oncogenic K-Ras, recovers p53 expression and function. Neoplasia 2009, 11, 22–31. [Google Scholar] [CrossRef]
- Ashktorab, H.; Paydar, M.; Yazdi, S.; Namin, H.H.; Sanderson, A.; Begum, R.; Semati, M.; Etaati, F.; Lee, E.; Brim, H.; et al. BMI and the risk of colorectal adenoma in African-Americans. Obesity 2014, 22, 1387–1391. [Google Scholar] [CrossRef]
- Uckun, F.M.; Erbeck, D.; Qazi, S.; Venkatachalam, T.; Tibbies, H.; Vassilev, A. Effect of Targeting Janus Kinase 3 on the Development of Intestinal Tumors in the Adenomatous Polyposis Colimin Mouse Model of Familial Adenomatous Polyposis. Arzneimittelforschung 2007, 57, 320–329. [Google Scholar] [CrossRef]
- Dammeijer, F.; van Gulijk, M.; Klaase, L.; van Nimwegen, M.; Bouzid, R.; Hoogenboom, R.; Joosse, M.E.; Hendriks, R.W.; van Hall, T.; Aerts, J.G. Low-Dose JAK3 Inhibition Improves Antitumor T-Cell Immunity and Immunotherapy Efficacy. Mol. Cancer Ther. 2022, 21, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ma, X.; Sun, X.; Wu, T.; Yu, J.; Wang, C.; Jin, Y.; Zheng, X. Novel Potent EGFR-JAK3 Dual-Target Inhibitor that Overcomes KRAS Mutation Resistance in Colorectal Cancer. Anti-Cancer Agents Med. Chem. 2023, 23, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Akintola-Ogunremi, O.; Pfeifer, J.D.; Tan, B.R.; Yan, Y.; Zhu, X.; Hart, J.; Goldblum, J.R.; Burgart, L.; Lauwers, G.Y.; Montgomery, E.; et al. Analysis of Protein Expression and Gene Mutation of c-kit in Colorectal Neuroendocrine Carcinomas. Am. J. Surg. Pathol. 2003, 27, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Quek, R.; Farid, M.; Kanjanapan, Y.; Lim, C.; Tan, I.B.; Kesavan, S.; Lim, T.K.H.; Oon, L.L.-E.; Goh, B.K.; Chan, W.H.; et al. Prognostic significance of KITexon 11 deletion mutation in intermediate-risk gastrointestinal stromal tumor. Asia-Pacific J. Clin. Oncol. 2016, 13, 115–124. [Google Scholar] [CrossRef]
- Choudhary, S.; Pardo, A.; Rosinke, R.; Batra, J.K.; Barth, S.; Verma, R.S. Targeting c-kit receptor in neuroblastomas and colorectal cancers using stem cell factor (SCF)-based recombinant bacterial toxins. Appl. Microbiol. Biotechnol. 2016, 100, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Küçükköse, E.; Peters, N.A.; Ubink, I.; van Keulen, V.A.M.; Daghighian, R.; Verheem, A.; Laoukili, J.; Kranenburg, O. KIT promotes tumor stroma formation and counteracts tumor-suppressive TGFβ signaling in colorectal cancer. Cell Death Dis. 2022, 13, 1–10. [Google Scholar] [CrossRef]

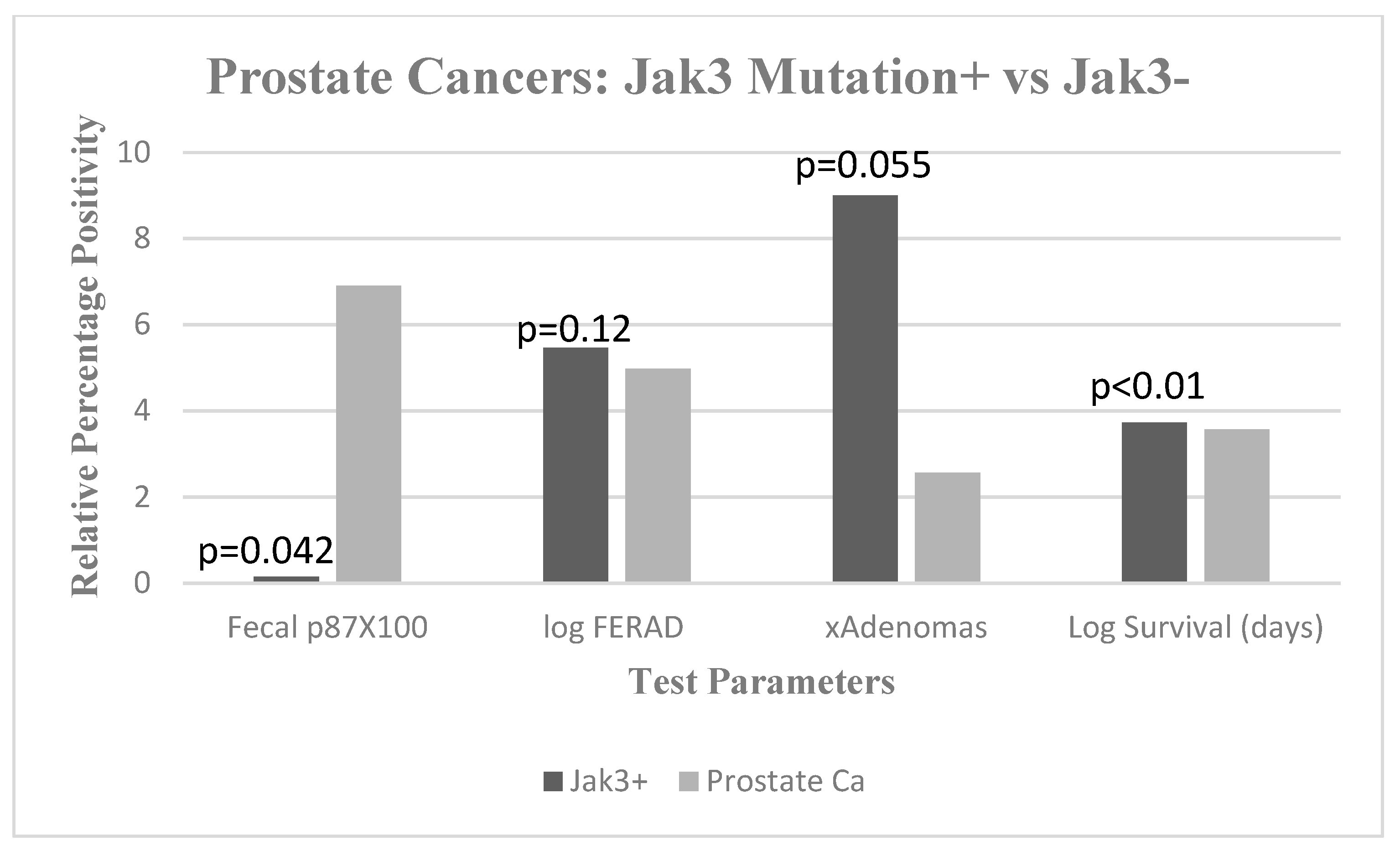
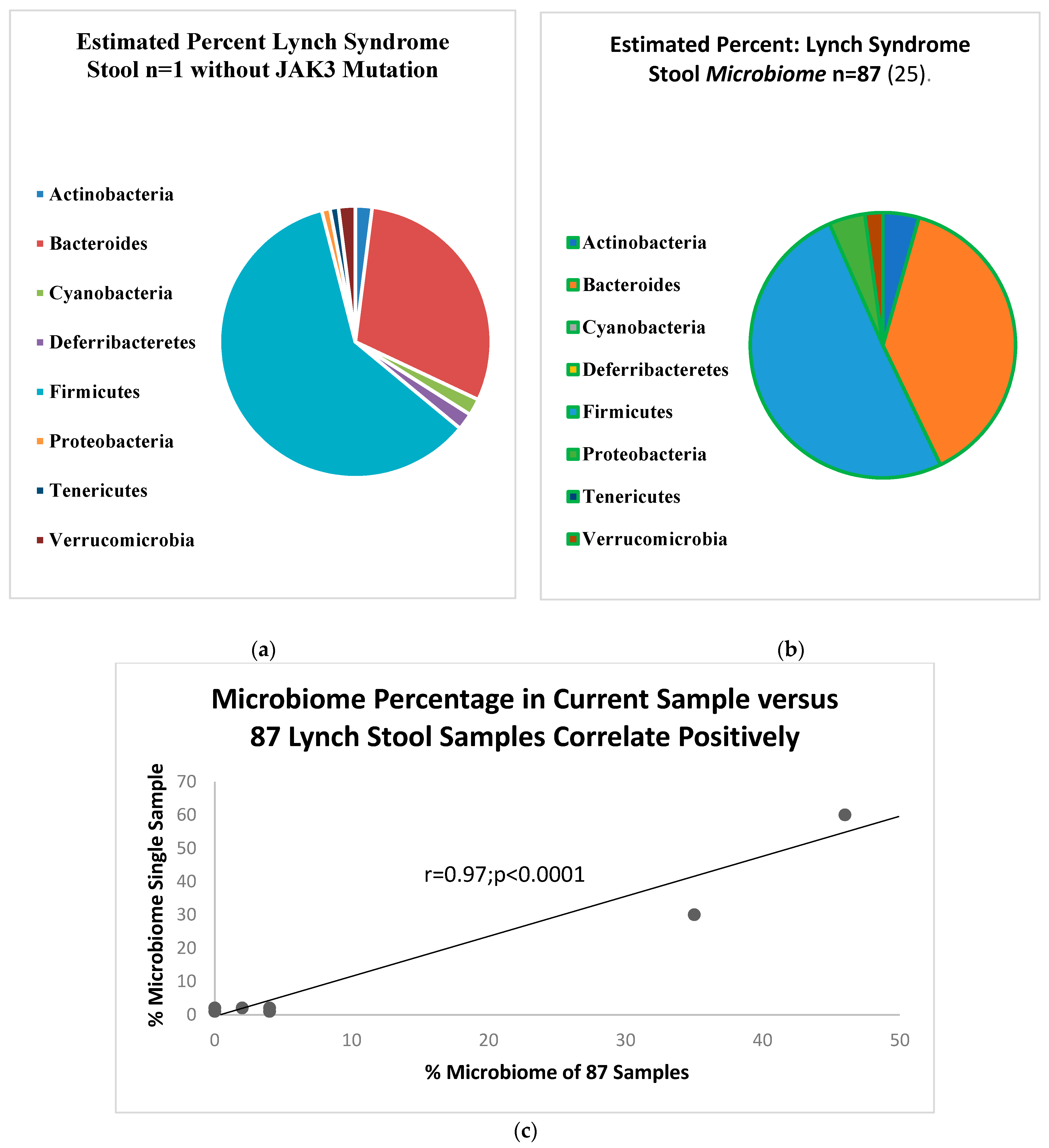
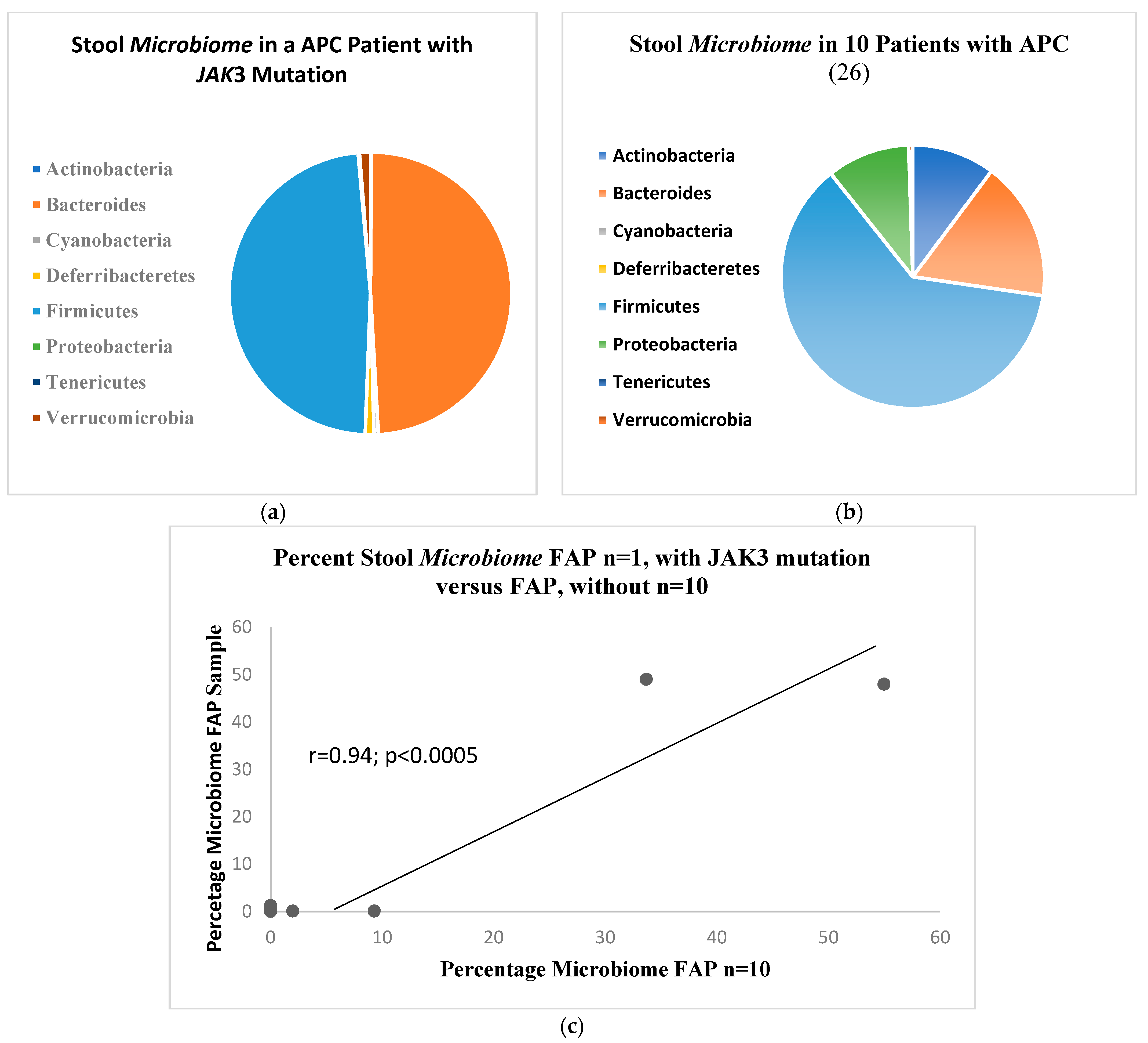


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | African American n = 119 | Caucasian n = 114 | p-Values |
|---|---|---|---|
| Age mean ± sd [yrs] | 60.73 ± 10.61 | 59.98 ± 12.87 | 0.62 |
| % Male | 88.1 | 84.5 | 0.46 |
| BMI | 29.25 ± 6.08 | 29.21 ± 5.98 | 0.96 |
| % GI symptoms | 60.5 | 46.6 | OR1.76 [CI 1.06–2.9] < 0.04 |
| Average Adenoma # | 1.91 ± 3.29 | 1.93 ± 2.80 | 0.97 |
| %Family History CRC | 16.8 | 10.5 | 0.19 |
| % Smokers (%FE + FE-) | 51.1 (35.3 vs. 47.5) | 47.1 (71.4 vs. 30) | 0.5 (AA0.075:Cau < 0.00005) * |
| HGD | N | Age yr | %AA | %Left | %TA | Size mm | %Smoke | #Syn | FOBT + % |
|---|---|---|---|---|---|---|---|---|---|
| Large | 28 | 66 ± 8.9 | 70 | 21 | 44 | 20.2 ± 8.1 | 67 | 1.4 ± 1.3 | 67 |
| Small | 21 | 63 ± 10.1 | 29 | 33 | 79 | 7.4 ± 2.7 | 30 | 2.5 ± 2.1 | 20 |
| p value | N/A | 0.33 | <0.01 | 0.54 | <0.04 | N/A | 0.11 | 0.43 | <0.05 |
| Group | Family Cancer Syndrome | High Risk and Others | High Grade Dysplasia |
|---|---|---|---|
| Number | 3 | 6 | 10 |
| Age mean ± sd (yrs) | 43.7 ± 7.0 * | 58.2 ± 8.7 | 55.7 ± 11.4 |
| % Male | 33.3 | 85.7 | 100 |
| % African American | 0 | 16.7 | 66.7 |
| % JAK3 c.394C > A | 14.3 | 0 | 16.7 |
| IonXpress Code | Mapped Reads | On Target | Mean Depth | Uniformity | Variants | Hotspot Variants |
|---|---|---|---|---|---|---|
| NipCon_001 Large | 98,792 | 95.95% | 423.4 | 99.88% | 20 | 7 |
| NipCon_002 | 476,704 | 87.74% | 1932 | 100.00% | 16 | 5 |
| NipCon_003 | 515,585 | 87.35% | 2079 | 100.00% | 16 | 5 |
| NipCon_004 | 283,449 | 89.56% | 1169 | 99.57% | 18 | 6 |
| NipCon_005 | 619,529 | 92.56% | 2651 | 99.53% | 14 | 4 |
| NipCon_006 | 271,188 | 91.03% | 1129 | 100.00% | 19 | 6 |
| NipCon_007 | 260,021 | 91.85% | 1089 | 100.00% | 18 | 6 |
| NipCon_008 | 194,255 | 79.3% | 712.8 | 100.00% | 17 | 5 |
| NipCon_009 | 212,251 | 90.40% | 995.2 | 100.00% | 17 | 5 |
| IonXpressCode | Size HGD | Location | Distinct Mutation | #Mut | %ACS * | ACS Phase |
|---|---|---|---|---|---|---|
| NipCon_001 * | Large lesion | Descending | APC, KRAS, KIT, SMO | 4 | 50 | Early, mid |
| NipCon_002 | Large | Cecum | TP53, KIT | 2 | 50 | Late |
| NipCon_003 | Large | Ascending | APC, TP53 | 2 | 100 | Early, late |
| NipCon_004 | Large | Descending | TP53 | 1 | 100 | Late |
| NipCon_005 | Large | Sigmoid | TP53 | 1 | 100 | Late |
| NipCon_006 | Small | Cecum | APC, TP53, PIK3CA, JAK3 | 4 | 50 | Early, late |
| NipCon_007 | Small | Ascending | APC, TP53, PIK3CA, JAK3 | 4 | 50 | Early, late |
| NipCon_008 | Small | Descending | APC, TP53, PIK3CA, JAK3, KIT | 5 | 40 | Early, late |
| NipCon_009 | Small | Sigmoid | APC, KIT, PIKCA, JAK3 | 4 | 25 | Early |
| Sample | Gene | Variant | Result | AA | Zygosity | rsID | MAF AF | MAF EUR |
|---|---|---|---|---|---|---|---|---|
| Patient A | ||||||||
| HGDpol | APC | c.3950_3956delAAGATCC | frameshift | nonsense | HET | novel | N/A | N/A |
| HGDpol | KIT | c.1672A > G | missense | pLys558Glu | HET | novel | N/A | N/A |
| HGDpol | KRAS | c.35G > T | missense | p.Gly12Val | HET | rs121913529 | 0.0001 | 1.87 × 10−5 |
| HGDpol | SMO | c.1886G > A | missense | p.Arg629Lys | HET | novel | N/A | N/A |
| Cecum | APC | c.4744G > A | missense | p.Ala1582Thr | HET | novel | N/A | N/A |
| Descend | KIT | c.4744G > A | missense | p.Lys558Glu | HET | novel | N/A | N/A |
| Patient B | ||||||||
| Cecum | APC | c.4744G > A | missense | p.Ala1582Thr | HET | novel | N/A | N/A |
| Cecum | APC | c.4479_4480delGG | frameshift | nonsense | HOM | novel | N/A | N/A |
| Cecum | JAK3 | c.394C > A | missense | p.Pro132Thr | HET | rs3212723 | 0.14 | 0.0001 |
| Ascend | APC | c.4479_4480delGG | frameshift | nonsense | HOM | novel | N/A | N/A |
| Ascend | JAK3 | c.394C > A | missense | p.Pro132Thr | HET | rs3212723 | 0.14 | 0.0001 |
| Ascend | KIT | c.1672A > G | missense | p.Lys558Glu | HET | novel | N/A | N/A |
| Descend | APC | c.4479_4480delGG | frameshift | nonsense | HOM | novel | N/A | N/A |
| Descend | JAK3 | c.394C > A | missense | p.Pro132Thr | HET | rs3212723 | 0.14 | 0.0001 |
| Sigmoid | APC | c.4479_4480delGG | frameshift | nonsense | HOM | novel | N/A | N/A |
| Sigmoid | JAK3 | c.394C > A | missense | p.Pro132Thr | HET | rs3212723 | 0.14 | 0.0001 |
| Sigmoid | KIT | c.1672A > G | missense | p.Lys558Glu | HET | novel | N/A | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tobi, M.; Zhao, X.; Rodriquez, R.; Tobi, Y.Y.; Ganguly, T.; Kuhn, D.; McVicker, B.; Lawson, M.J.; Lieb, J., II; Lopes, J.L. The Innate Immune System Surveillance Biomarker p87 in African Americans and Caucasians with Small High-Grade Dysplastic Adenoma [SHiGDA] and Right-Sided JAK3 Colon Mutations May Explain the Presence of Multiple Cancers Revealing an Important Minority of Patients with JAK3 Mutations and Colorectal Neoplasia. Gastrointest. Disord. 2024, 6, 497-512. https://doi.org/10.3390/gidisord6020034
Tobi M, Zhao X, Rodriquez R, Tobi YY, Ganguly T, Kuhn D, McVicker B, Lawson MJ, Lieb J II, Lopes JL. The Innate Immune System Surveillance Biomarker p87 in African Americans and Caucasians with Small High-Grade Dysplastic Adenoma [SHiGDA] and Right-Sided JAK3 Colon Mutations May Explain the Presence of Multiple Cancers Revealing an Important Minority of Patients with JAK3 Mutations and Colorectal Neoplasia. Gastrointestinal Disorders. 2024; 6(2):497-512. https://doi.org/10.3390/gidisord6020034
Chicago/Turabian StyleTobi, Martin, Xiaoqing Zhao, Rebecca Rodriquez, Yosef Y. Tobi, Tapan Ganguly, Donald Kuhn, Benita McVicker, Michael J. Lawson, John Lieb, II, and Jaime L. Lopes. 2024. "The Innate Immune System Surveillance Biomarker p87 in African Americans and Caucasians with Small High-Grade Dysplastic Adenoma [SHiGDA] and Right-Sided JAK3 Colon Mutations May Explain the Presence of Multiple Cancers Revealing an Important Minority of Patients with JAK3 Mutations and Colorectal Neoplasia" Gastrointestinal Disorders 6, no. 2: 497-512. https://doi.org/10.3390/gidisord6020034
APA StyleTobi, M., Zhao, X., Rodriquez, R., Tobi, Y. Y., Ganguly, T., Kuhn, D., McVicker, B., Lawson, M. J., Lieb, J., II, & Lopes, J. L. (2024). The Innate Immune System Surveillance Biomarker p87 in African Americans and Caucasians with Small High-Grade Dysplastic Adenoma [SHiGDA] and Right-Sided JAK3 Colon Mutations May Explain the Presence of Multiple Cancers Revealing an Important Minority of Patients with JAK3 Mutations and Colorectal Neoplasia. Gastrointestinal Disorders, 6(2), 497-512. https://doi.org/10.3390/gidisord6020034