
The Rearrangement of Alkylallenes to 1,3-Dienes


1
Department of Chemistry, College of Education, Mosul University, Mosul 41002, Iraq
2
Department of Chemistry, School of Science, Loughborough University, Leicestershire LE11 3TU, UK
*
Authors to whom correspondence should be addressed.
Reactions 2022, 3(1), 70-86; https://doi.org/10.3390/reactions3010006
Submission received: 1 December 2021
/
Revised: 16 December 2021
/
Accepted: 19 December 2021
/
Published: 5 January 2022
(This article belongs to the Special Issue Feature Papers in Reactions in 2021)
Abstract
:1,3-Dienes are vital building blocks in organic synthesis. They underpin many fundamental synthetic transformations and are present in numerous natural products and drug candidate molecules. The rearrangement of an alkylallene to a 1,3-diene is an atom efficient, redox neutral, transformation that provides a straightforward synthetic route to functionalized 1,3-dienes. Herein, we provide an account of this transformation using allenes that are not predisposed by the presence of heteroatoms or electron-withdrawing groups directly attached to the allene. Early reports of this skeletal rearrangement are acid-mediated approaches, with limited substrate scope, but they provide valuable mechanistic insights. More recent transition metal-mediated approaches that exhibit improved substrate scope are described, together with isolated examples that have utilized this rearrangement.
1. Introduction
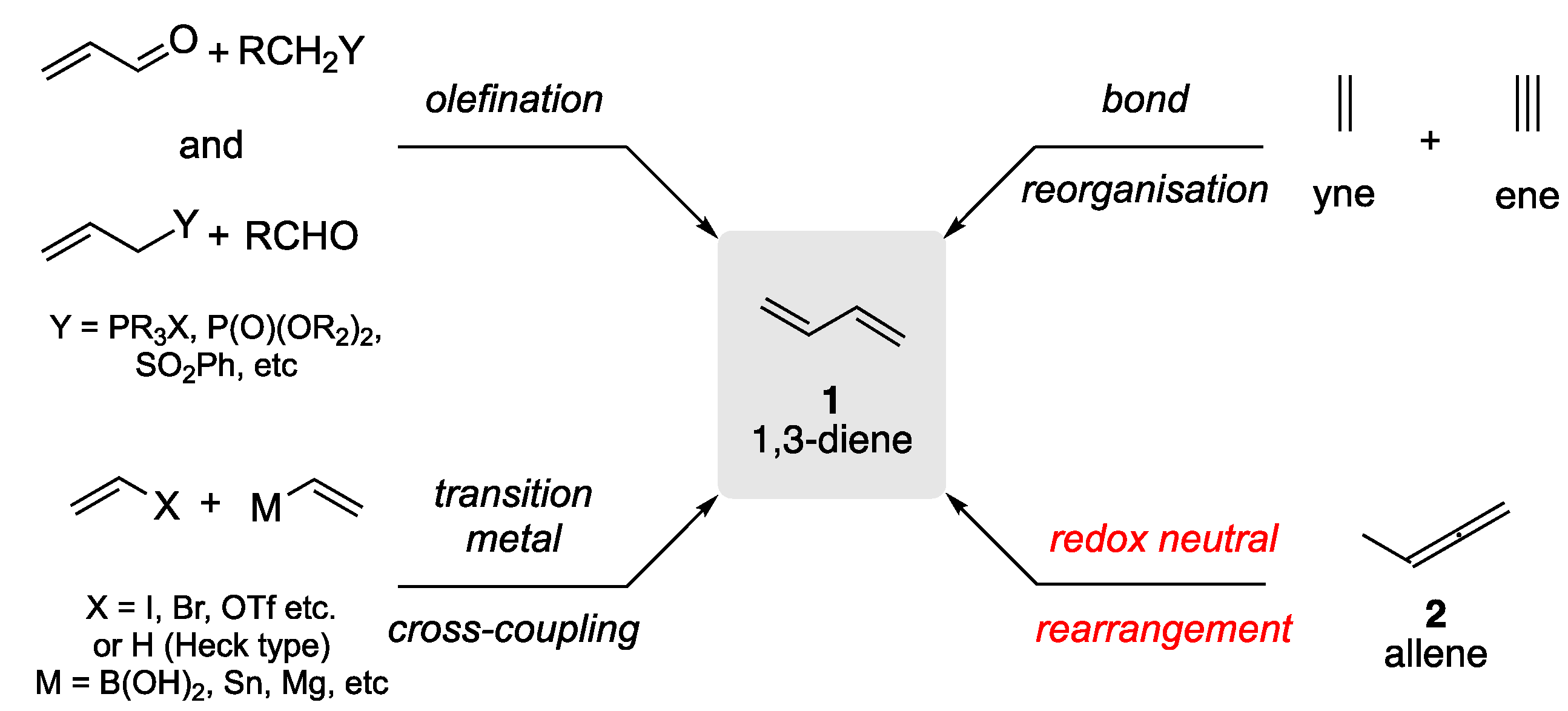
1,3-Dienes (1) are essential building blocks in organic synthesis and materials chemistry. They have been crucial in the development of fundamental synthetic reactions such as the Diels-Alder and related pericyclic transformations [1,2,3,4,5]. The 1,3-diene (1) structural motif can be found across numerous natural product classes, as well as in several high-profile on-market drugs and drug candidates; their inherent electronic characteristics have made them indispensable in the development of innovative photoactive organic materials [6,7,8,9]. Given their significance, the synthesis of 1,3-dienes (1) is rich in history and innovation, and a recent extensive review, centered on stereoselective methods provides an excellent account of current synthetic approaches to the 1,3-diene motif [10]. The application of olefination strategies has dominated their synthesis [11,12,13,14,15,16]; this was subsequently augmented by transition metal cross-coupling approaches [17,18,19,20,21], and the bond reorganization of appropriately substituted alkene and alkyne substrates [22,23,24,25], giving rise to 1,3-dienes (1) with unique connectivity’s that can be tailored to the synthetic target of choice (Scheme 1).
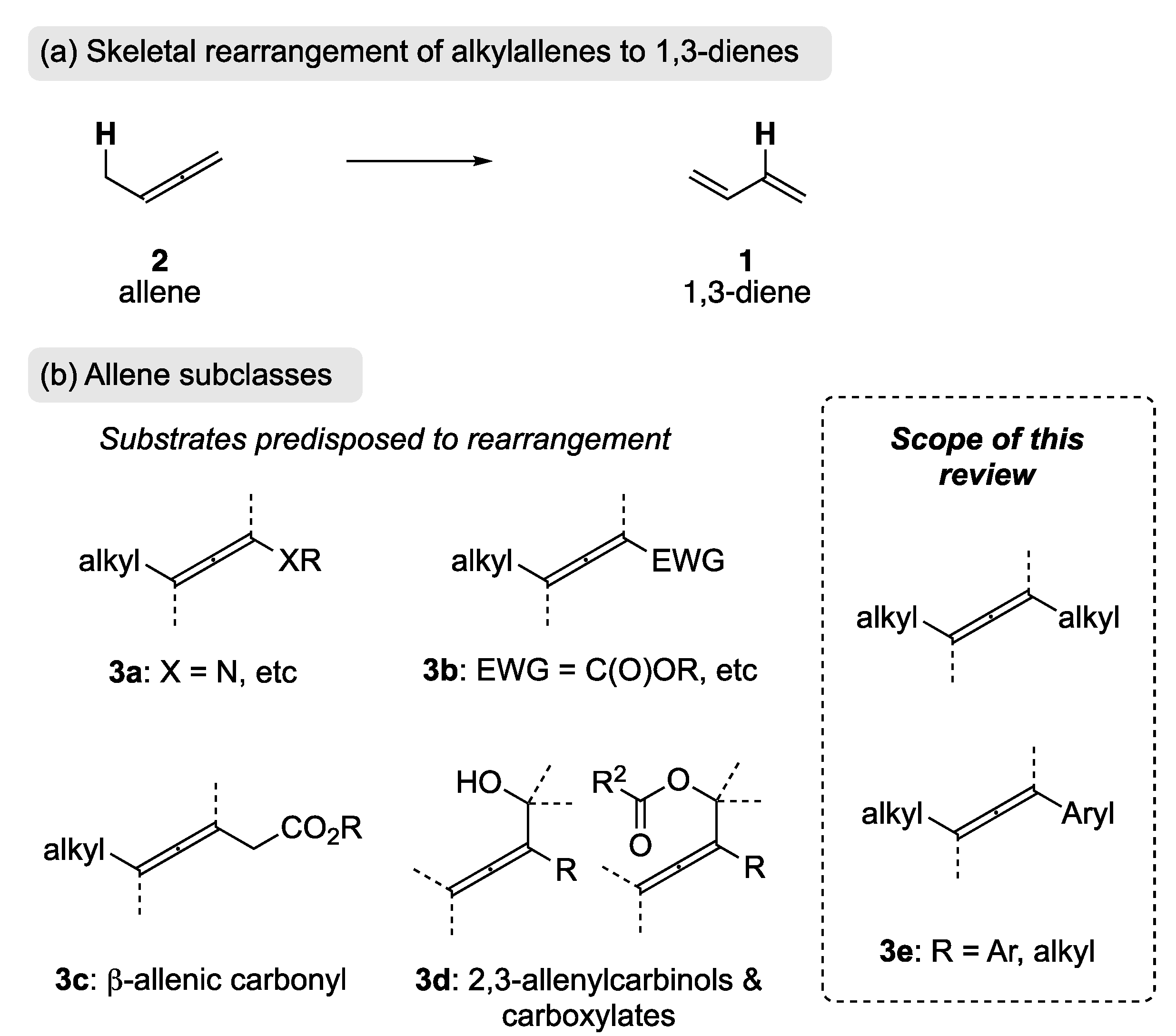
The rearrangement of an alkyl-allene (2) is an appealing approach to the synthesis 1,3-dienes 1, as it is redox neutral and can be viewed as a formal 1,3-hydrogen migratory process (Scheme 2). Unlike olefination strategies, transition metal cross-coupling, and bond-reorganization approaches, this approach can utilize the inherent thermodynamic stability of the product 1,3-diene to promote the rearrangement.
The rearrangement of allenes activated by a heteroatom attached directly to the allene (3a) has been reported by Hsung et al. [26,27], and an extensive review has been published by the same authors [28]. An electron-withdrawing group attached directly to the allene will bias it toward rearrangement (e.g., 3b); this has been realized through phosphine-catalyzed conjugate addition [29,30,31] and via palladium catalysis [32]. β-Allenic carbonyl substrates (3c) are also predisposed to rearrangement, and these substrates can be readily re-conjugated via base catalysis thereby providing dienoic esters, which are present in several natural occurring pheromones and flavors [33,34,35]. Additionally, while the rearrangement of 2,3-allenylcarbinols and their corresponding carboxylates (e.g., 3d) can be achieved through a [3,3]-sigmatropic rearrangement [36,37,38], SN2′ pathways [39,40,41], and metal-mediated processes [42,43,44], it does not fall within the scope of this review, as it is not a formal 1,3-hydrogen migratory process. With the emergence of allenes as building blocks in organic synthesis [45,46], the rearrangement of 2 to 1 (Scheme 2) has the potential to be a valuable, complementary tool, in the synthesis of 1,3-dienes. The focus of this review will therefore be the rearrangement of allenes not predisposed to rearrangement through direct attachment of heteroatoms or electron-withdrawing groups (e.g., 3e).
The rearrangement of this allene class has been a focus of interest since the early 1960s, and it is only recently that its synthetic potential has been improved. The review of Soengas and Rodriguez-Solla [10] provides an excellent account of stereoselective methods for 1,3-diene synthesis, and we hope that this review can provide the reader with an appreciation of the origins of the rearrangement and future challenges in improving substrate scope and utility. Therefore, this review begins with an initial account of this rearrangement, centering on thermal and acidic methods. These earlier reports must be examined in context, as they are important in defining scope and generality. Within the literature, isolated examples of this rearrangement have been observed, and frequently, these examples are very substrate specific, or the rearrangement has been observed as a side reaction. Transition-metal-promoted methods are also analyzed, with the substrate scope being greatly enlarged, thereby providing some of the first synthetically useful methodologies. In this review, the reaction scope for each synthetic approach is highlighted, including substrate bias, the role of electron-rich substituents that may facilitate the rearrangement, and how this impacts the underlying mechanistic rationale for each transformation.
2. Acid-Mediated Rearrangements
Acid-mediated rearrangements are primarily realized through the protonation of the central sp-hybridized carbon of the allene, where stabilization of a transient carbocation facilitates the formation of a thermodynamically stable 1,3-diene. This method, therefore, limits the scope of the allene substrates that be effectively rearranged to their 1,3-diene counterparts.
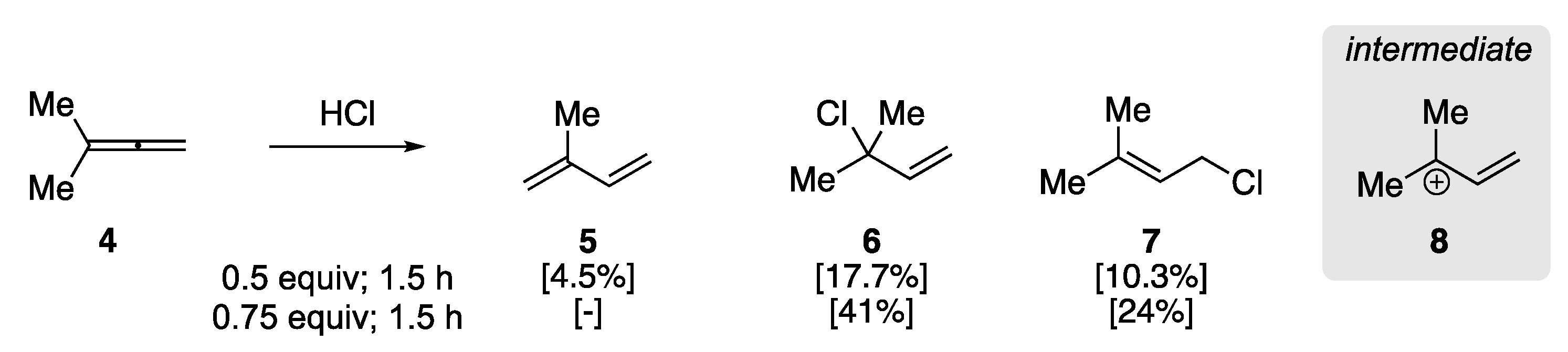
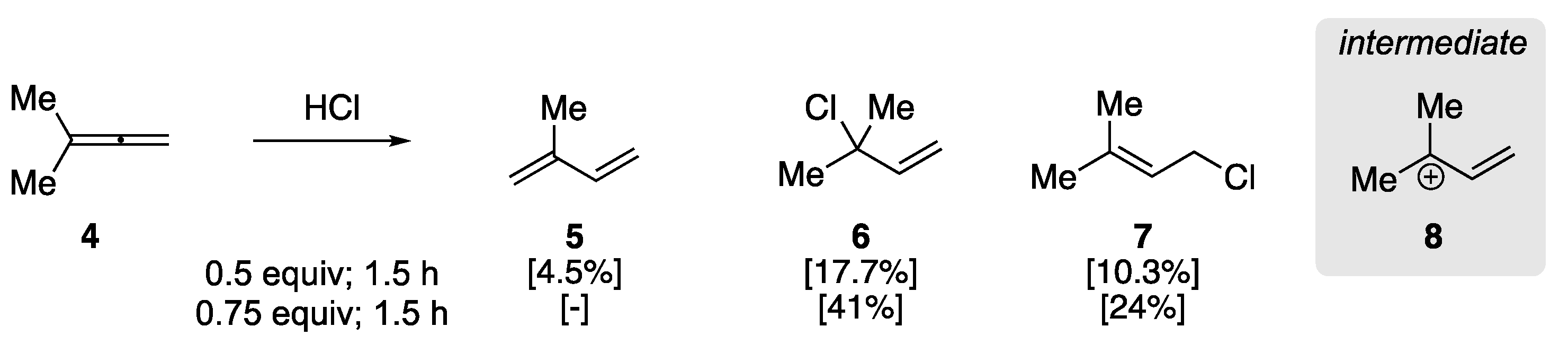
In 1960, Johnson et al. examined the addition of hydrogen chloride to aliphatic allenes to elucidate the regiochemistry of the initial protonation [47]. This early report observed the rearrangement of 3-methyl-1,2-butadiene (4) to isoprene in the presence of HCl (Scheme 3), clarifying previous studies on the addition of HX to allenes. They found that the treatment of 4 with HCl (0.5 equiv) at −78 °C for 0.5 h yielded a mixture of isoprene 5 and allylic chloride addition products (6/7), as determined by GC analysis. The ratio of 5 to 6/7 could be affected through HCl stoichiometry and reaction time, where an increase in the amount of HCl and reaction time resulted in primarily allylic chlorides 6 and 7, respectively. The authors rationalized the formation of the products by the protonation of the central sp-hybridized carbon yielding carbocation 8, with the formation of 1,3-diene 5 resulting from subsequent elimination.
Wenkert et al. found that allenes 9 and 11 could be rearranged to 1,3-dienes 10 and 12, respectively (Scheme 4). [48]. Analogous to the work of Johnson, protonation occurs through the central sp-hybridized allenic carbon to provide an intermediate such as 13, which can be further stabilized by the electron-donating 4-methoxy aryl substituent. Consequently, both allenes 9 and 11 can be considered as being activated.
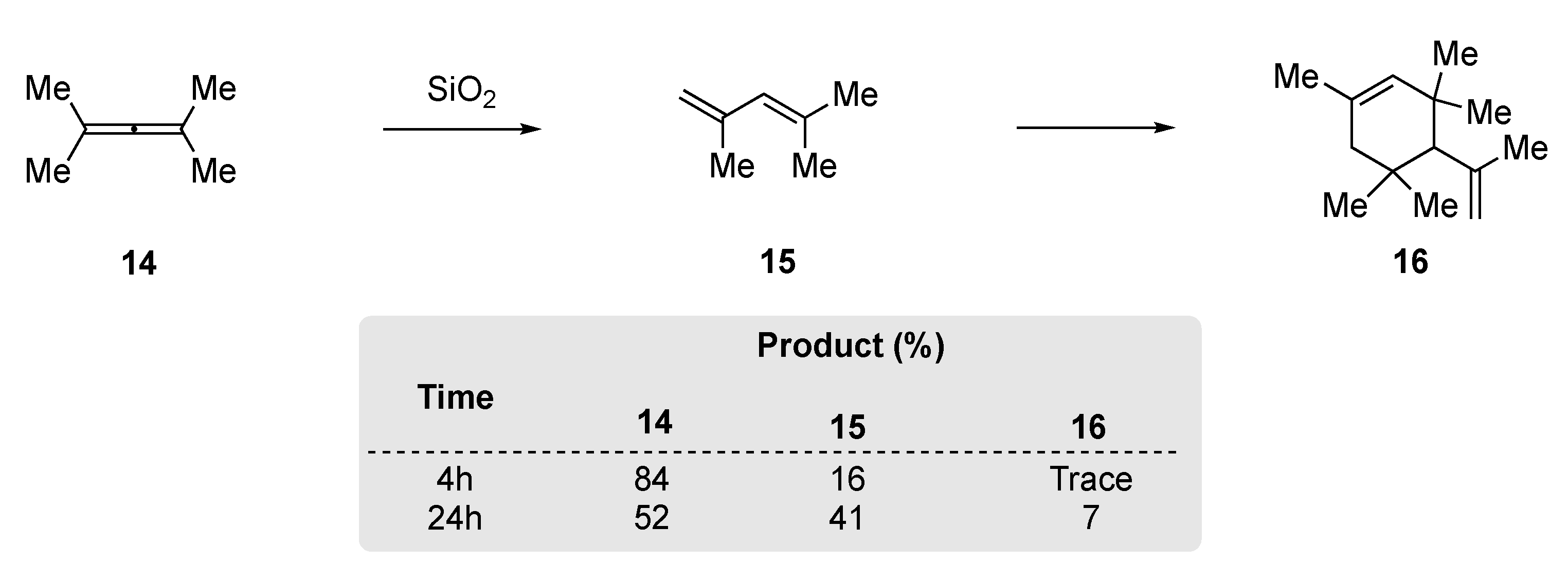
Kropp et al. had demonstrated that silica gel and alumina facilitated the addition of HX to alkenes and alkynes [49]. In a related study, they observed that tetramethylallene 14 could be rearranged to 2,4-dimethylpenta-1,3-diene (15) through treatment with silica gel (Scheme 5). However, they found that extended reactions times (24 h) were required to achieve modest conversion and that 15 would undergo further dimerization to produce 16.
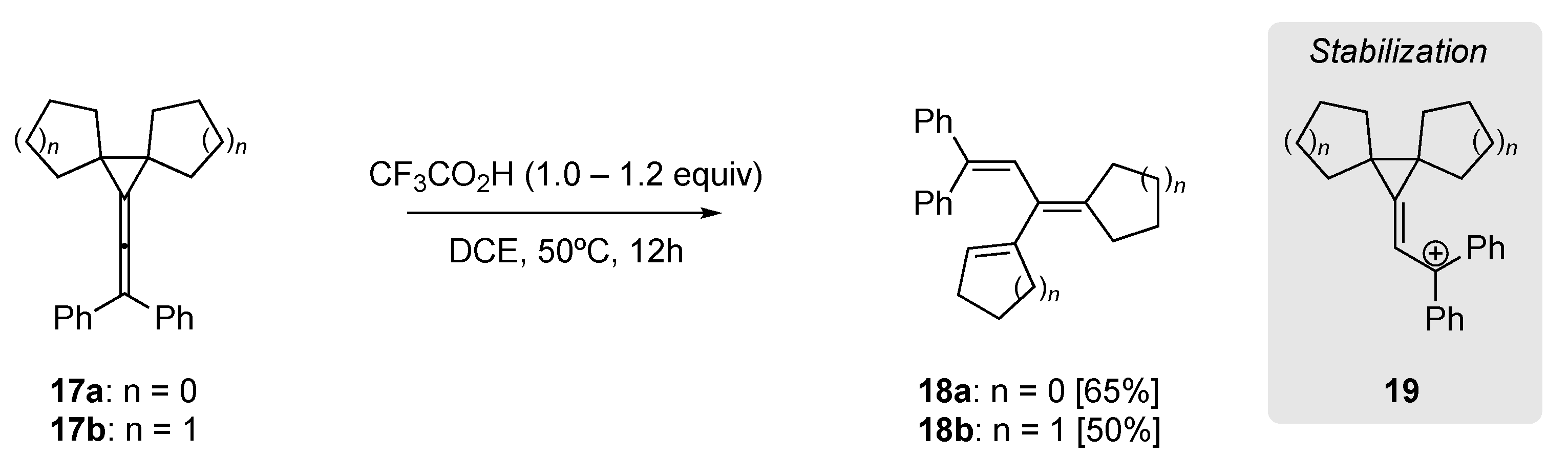
Li et al. utilized the intrinsic strain of diarylvinylidenecyclopropanes in their acid mediate rearrangement to yield trienes (Scheme 6) [50]. For example, the dicyclopentyl (17a) and dicyclohexyl (17b) diarylvinylidenecyclopropanes could be effectively rearranged to provide trienes 18a and 18b, in 65% and 50% isolated yield, respectively. Additionally, using DFT calculations they identified the formation of the carbocation 19 as key within this rearrangement.
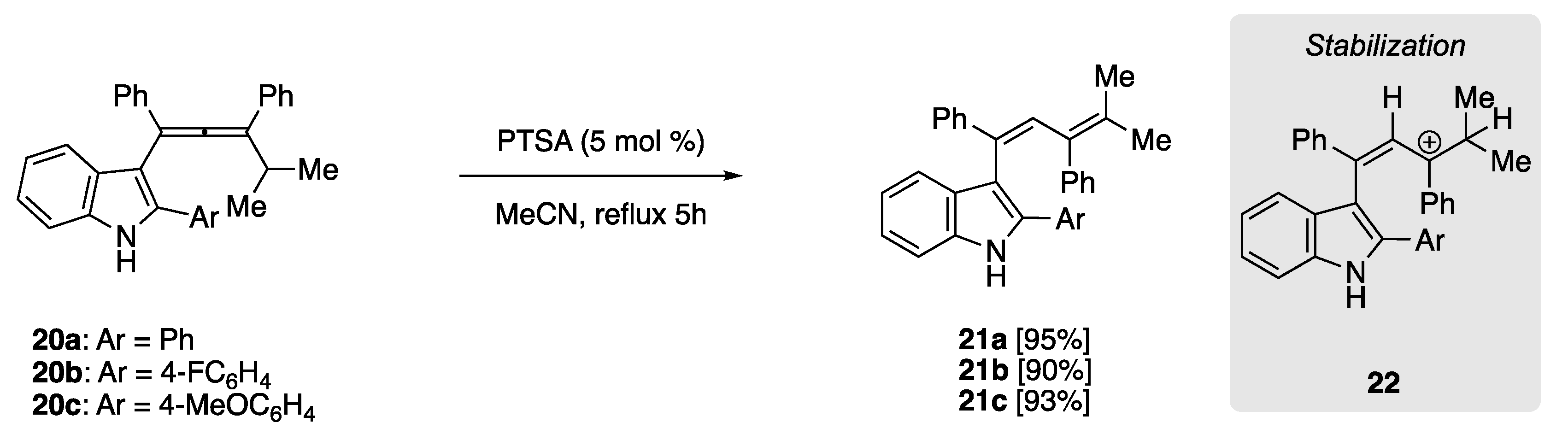
In 2010, Sanz et al. treated indolyl allenes (20) in refluxing MeCN and catalytic PTSA, yielding 1,3-dienes 21 in high isolated yield (Scheme 7) [51]. The rearrangement of this allene subgroup benefited from subsequent stabilization of carbocation 22, formed upon protonation with PTSA. Stabilization is afforded through the participation of electron-rich indolyl substituent, as well as the flanking phenyl groups, although there appears to be no perceived benefit in isolated chemical yield when using electron-poor (20b, Ar = 4F-C6H4) or electron-rich (20c, Ar = 4MeOC6H4) aryl substituents.
Titov et al., in a study examining the synthesis of potential bioactive tetrahydrobenzazecines, observed that cyclic allene 23 underwent rearrangement to 1,3-diene 24 upon microwave irradiation in acetic acid (Scheme 8) [52]. The yields were found to be good to excellent, depending on the substitution. Again, this type of rearrangement was believed to occur by the benzylic carbocation 25. This intermediate also benefits from stabilization via the N-heteroatom of the enamine. Another key driving force for the formation of diene 24, could well be the release of the inherent ring strain within cyclic allene 23.
3. Thermal-Mediated Rearrangements
The rearrangement of strained allenic systems, such as the cyclopropyl allenes 26 and 28, to 1,3-dienes has been known since the late 1960s (Scheme 9). Seminal works by Crandall and Paulson [53], Conia et al. [54], and Maitland Jones et al. [55] demonstrated that thermal rearrangements of strained exo-cyclic allenyl cyclopropanes, employing pyrolysis through a flow system at temperatures in excess of 300 °C, gave access to hitherto unknown 1,3-dienyl cyclopropanes through a homolytic ring breakage and subsequent ring-closure events.
The synthetic utility of this transformation is limited. Rearrangement in these strained allenic systems (e.g., 28a–d) benefits from aryl substitution on the cyclopropane, and this aligns well with the proposed mechanism to provide added stabilization in the radical process.
In 1968, Taylor and Wright re-examined the thermal rearrangement of tetramethylallene 14, as prior reports had shown that 14 undergoes dimerization at 150 °C [56]. They observed that heating of 14 in a glass vessel that had been pre-treated with base gave the dimer, tetramethyl-1,2-di-isopropylidenecyclobutane. However, in an unwashed or acid-treated vessel, rapid rearrangement to 1,3-diene, 2,4-dimethylpenta-1,3-diene (15) was observed; subsequently, 15 underwent further dimerization to yield a complex mixture. This led to a synthetically useful procedure being established, where the allene 14 could be successfully rearranged to 2,4-dimethylpenta-1,3-diene (15) through heating a diluted solution in a polar aprotic solvent at elevated temperatures (Scheme 10).
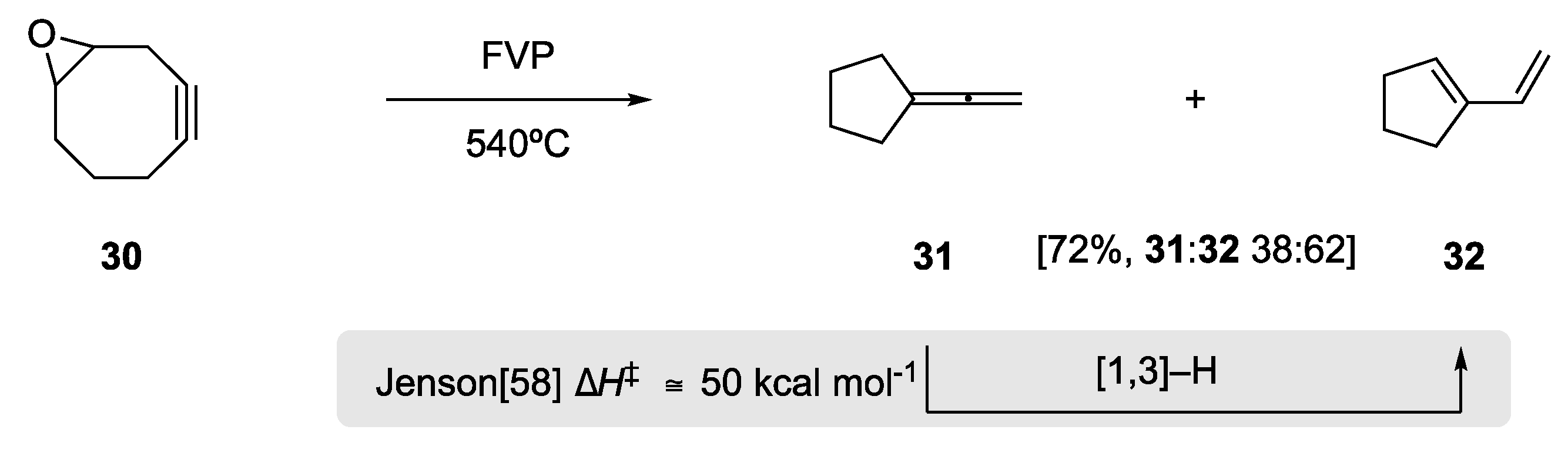
In 1989, Meier and Schmitt observed a formal 1,3-hydrogen shift in the exo-cyclic allene 31 (Scheme 11) [57]. Upon Flash Vacuum Pyrolysis of the cyclooctynyl epoxide 30, they observed two major products in 72% yield, exo-cyclic allene 31 and 1,3-diene 32 in a 38:62 ratio. The 1,3-diene was postulated to occur through formal [1,3]-hydrogen migration. Experimentally this reaction occurs at 540 °C under the FVP conditions; Jenson, in a very insightful theoretical study on the effects of allenes in sigmatropic H shift, subsequently calculated an energy barrier of ΔH‡ of ≅50 kcal mol−1 for this thermal [1,3]-H rearrangement [58].
4. Transition-Metal-Promoted Rearrangements
4.1. Gold
Due to their carbophilic character, gold catalysts are ideal for the selective activation of allenes to nucleophilic attack [59]. An Au(III)-catalyzed rearrangement of aryl-substituted allenes to 1,3-dienes was accomplished by Liu et al. (Scheme 12) [60,61]. This report is important, as it provides a catalytic method to achieve this rearrangement at ambient temperature, and importantly, it exhibited excellent substrate scope.
Reaction optimization revealed that AuCl3 (5 mol%) with 10 mol% of PhNO at 28 °C for 2 h, was essential to achieve full rearrangement; Au(I)-catalysts such as AuCl provided only partial 1,3-diene product 34 and reduced chemical yields of 34 were observed when reaction times were in excess of 2 h. When the additive PhNO was replaced by tertiary amines such as DBU or Et3N, no rearrangement was observed, and replacing the solvent with CH3CN or DCE resulted in a marginal reduction in the yield of 34. However, solvents such as 1,4-dioxane or toluene resulted in little-to-no observed 1,3-diene product 34. The scope of the transformation was examined on di-, tri-, and tetrasubstituted allenes.
For tri- and tetrasubstituted allenes, all reactions were performed with AuCl3 (5 mol%) and PhNO (10 mol%) at 28 °C, but the duration of each was substrate dependent and ranged from 0.5 h to 72 h (Scheme 13). For example, the trisubstituted 1,3-dienes 37a and 37b required significantly more time to achieve reaction completion, compared with 37a. 1,3-Dienes 38b and 38c were obtained as mixtures of geometric isomers, with each isomer being isolated as the internal olefin through an extended reaction time of 9 h and 5 h, respectively. Isolated chemical yields were good to excellent, and the double bond geometry of the isolated 1,3-dienes reflected thermodynamic equilibrium; this also accounts for the extended reaction times particularly in the formation of 1,3-dienes 38b and 38c, respectively. The isomerization was shown to be applicable to heteroaromatic allenes, providing 1,3-dienes 40–43 in very good yields and short reaction times. Tetrasubstituted allenes 44, 46, and 48 performed well, providing 1,3-dienes 45, 47, and 49, respectively. In the case of 44, the product 1,3-dienes were isolated as a mixture of 45a and 45b in approximately 2:1 ratio. Under the reaction conditions, 1,1-disubstituted allenes required heating to 50 °C and extended reaction times (Scheme 13c). Under the reactions conditions 1,2-disubstituted allene 52 failed to undergo any rearrangement with only starting material being returned in 43% yield. Significantly, this result implies that the formation of a tertiary carbocation is vital to this Au(III)-catalyzed transformation (see Scheme 14).
A mechanism for this transformation is presented in Scheme 14. Following allene activation by Au(III), the authors proposed the formation of an initial tertiary carbocation 54. Nitrosobenzene then facilitates a cooperative intramolecular proton transfer ultimately providing 56 that undergoes protodeauration to yield the observed 1,3-diene (34). Importantly, deuterium-labeling experiments using 33′ provided evidence of the mechanism with deuterium incorporation at C1 (0.11%) and C2 (0.21%) within 34′.
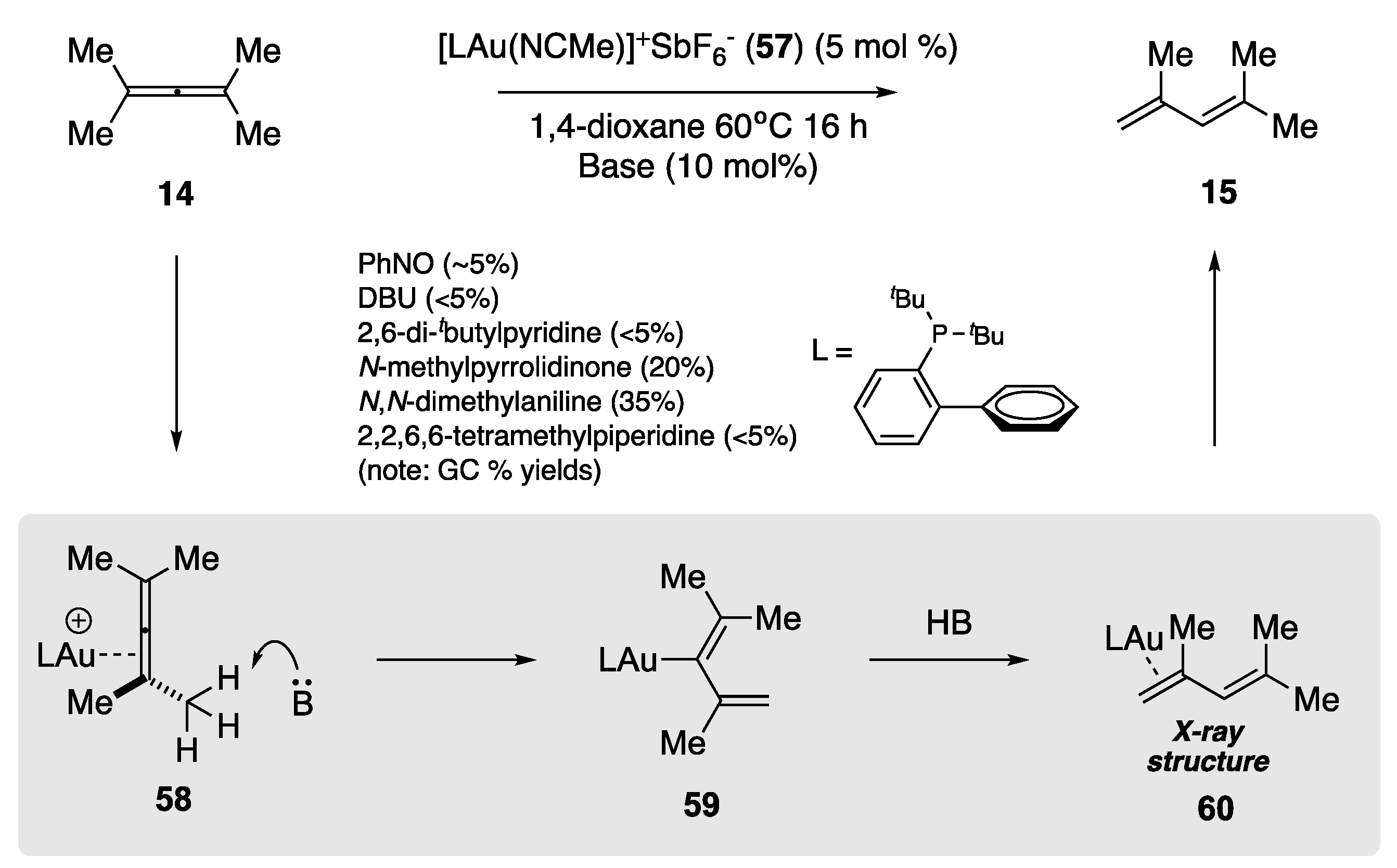
The Au-catalyzed rearrangement described by Lui et al. [60,61] works solely for aryl-substituted allenes. There are limited examples of the rearrangement of alkyl-substituted allenes within the literature. The rearrangement of a cationic tetramethylallene complex Au(I) complex has been established by Widenhoefer et al. (Scheme 15) [62]. Treatment of tetramethylallene (14) with Au(I) complex 57 generates the π-tetramethylallene complex 58 in a 96% isolated yield. Attempts at the crystallization of this Au(I) complex led to rearrangement, via a formal 1,3-hydrogen migration, to the π-1,3-diene complex 60, whose structure was confirmed by X-ray analysis. The authors were able to estimate the enthalpy for the conversion of 58 to 60 to be −8.6 kcal/mol based on the calculated heats of formation and binding affinities of 2,4-dimethyl-2,3-pentadiene (14) and 2,4-dimethyl-1,3-pentadiene (15), respectively. The authors also probed a deprotonation mechanism to account for the transformation and found that heating a solution of 58 with 1 equiv. of triethylamine yielded approx. 50% rearrangement and quantitative ligand displacement to form a 2:1:1 mixture of 57. NEt3+SbF6−, 2,4-dimethyl-2,3-pentadiene (14) and 2,4-dimethyl-1,3-pentadiene (15). The mechanism of the rearrangement was postulated to occur through deprotonation of the allenylmethyl, providing the Au σ-dienyl intermediate 59 that subsequently underwent protodeauration. This mechanism was reinforced by the work of Das et al., who performed an in-depth DFT study on the Au(III)/PhNO-catalyzed rearrangement of substituted allenes to 1,3-dienes [63]. Attempts by Widenhoefer et al. at improving the efficiency of this rearrangement by using weakly coordinating bases revealed N,N-dimethylaniline to be the most effective, providing 2,4-dimethyl-1,3-pentadiene (15) in an improved 35% GC yield.
As seen above, the rearrangement of an alkyl-substituted allene to a 1,3-diene is still limited in reaction scope. However, the detailed work by Widenhoefer et al. [62], together with computational studies [63], may provide opportunities to expand this scope and complement the methodology reported by Lui et al. [60,61].
4.2. Palladium
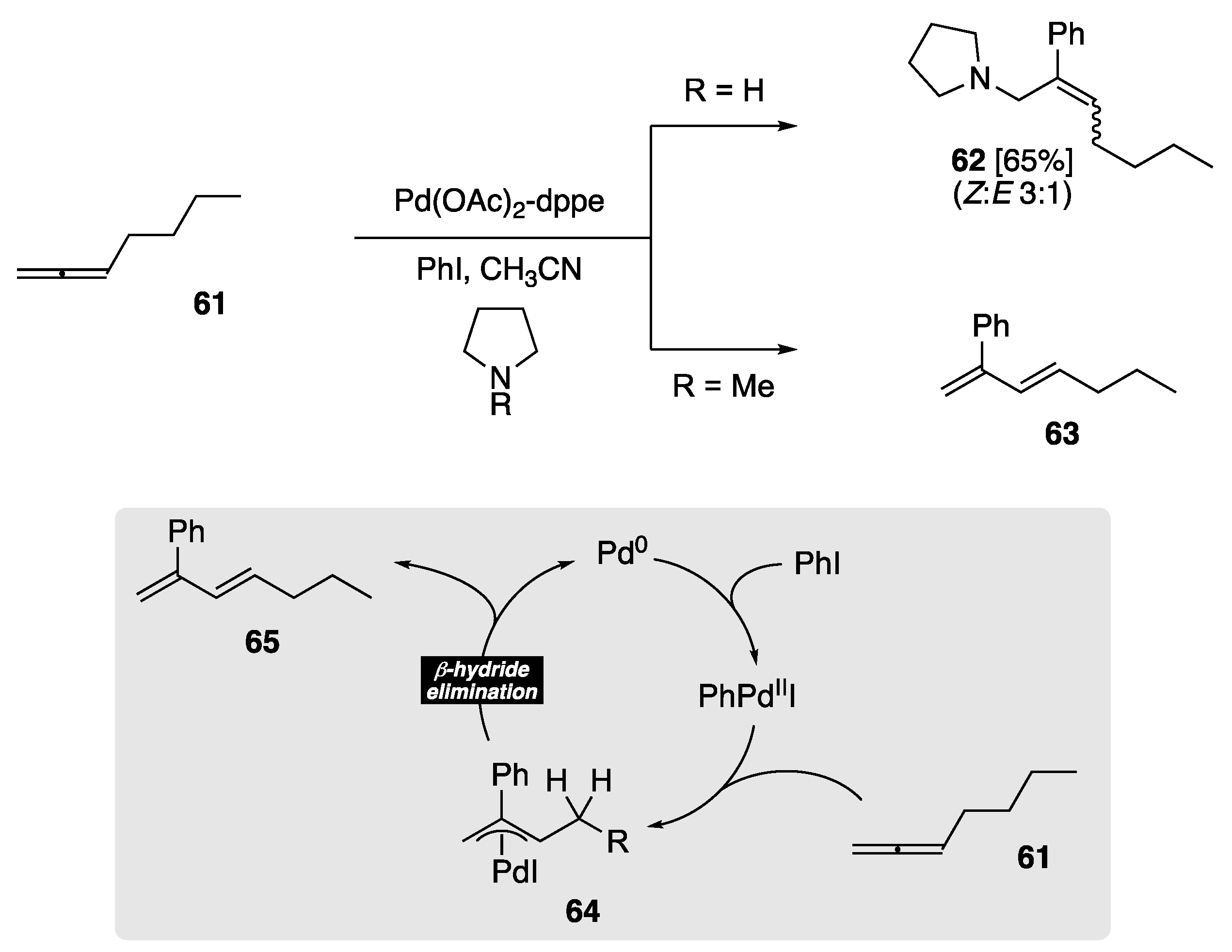
The reaction of Pd(0) with 1,2-dienes has been extensively studied and provides a convenient method to access synthetically useful π-allylpalladium intermediates [64]. Prior to the work of Tsuji et al. [65,66], the fate of this intermediate was oligomerization or polymerization. A key study by Tsuji found that this π-allylpalladium species could be intercepted by secondary amine nucleophiles such as pyrrolidine to generate allylamines (Scheme 16). However, the authors also observed that the π-allylpalladium species derived from 1,2-heptadiene, in the presence of N-methylpyrrolidine, generated 1,3-diene 65, exclusively. The authors postulated that in the absence of a suitable nucleophile, in this case, N-methylpyrrolidine, the intermediate π-allylpalladium species 64 instead undergoes elimination of HPd, yielding the observed 1,3-diene 65. While this is not formally a direct rearrangement of an allene to a 1,3-diene, given the intermediate π-allylpalladium species 64 is a consequence of the syn arylpalladium addition, it does represent the mechanistic basis of subsequent direct allene to 1,3-diene rearrangements via Pd catalysis.
The first direct rearrangement of 1,2-diene to 1,3-diene through palladium catalysis was reported by Al-Masum and Yamamoto [67]. Building on previous reports on the addition of nucleophiles to 1,2-dienes, they found that the palladium-catalyzed hydrocarboxylation of aliphatic allenes provided substantial amounts of the 1,3-diene products instead of the anticipated allyl esters (Scheme 17a). For example, 1,1-disubstituted allene 66, when treated with Pd2dba3 in the presence of a slight excess of acetic acid, resulted in a moderate yield of 1,3-diene 67 as a 1:1 mixture of E:Z isomers; similarly, the disubstituted aryl allenes 68, under the same reaction conditions, resulted in 1,3-dienes 69 in 50% isolated yield.
The authors postulated a plausible mechanism involving the initial formation of hydridopalladium species HPd(II)OAc, through the insertion of Pd(0) into HOAc; hydropalladiation of allenes 66 and 68 then provides π-allylpalladium intermediates 70 and 71, respectively (Scheme 17b,c). In the case of π-allylpalladium intermediate 70, β-hydride elimination of the allylic hydrogen can be accomplished to yield two possible intermediates, therefore producing 1,3-diene 67 as a 1:1 mixture of E:Z isomers; β-hydride elimination via the allylic hydrogen on adjacent methyl group was not observed. π-Allylpalladium intermediate 71 also undergoes β-hydride elimination of the allylic hydrogen, but this results in the formation of a single 1,3-diene 69. This disclosure by Yamamoto of using an in situ formed H-Pd(II)OAc species to undertake a hydropalladiation-β-hydride elimination of an allene to generate 1,3-diene is significant. It demonstrated the potential of the Pd-catalyzed reaction pathway to achieve the rearrangement; however, it did suffer from the limited scope and, importantly, reaction selectivity over the more desired hydrocarboxylation pathway.
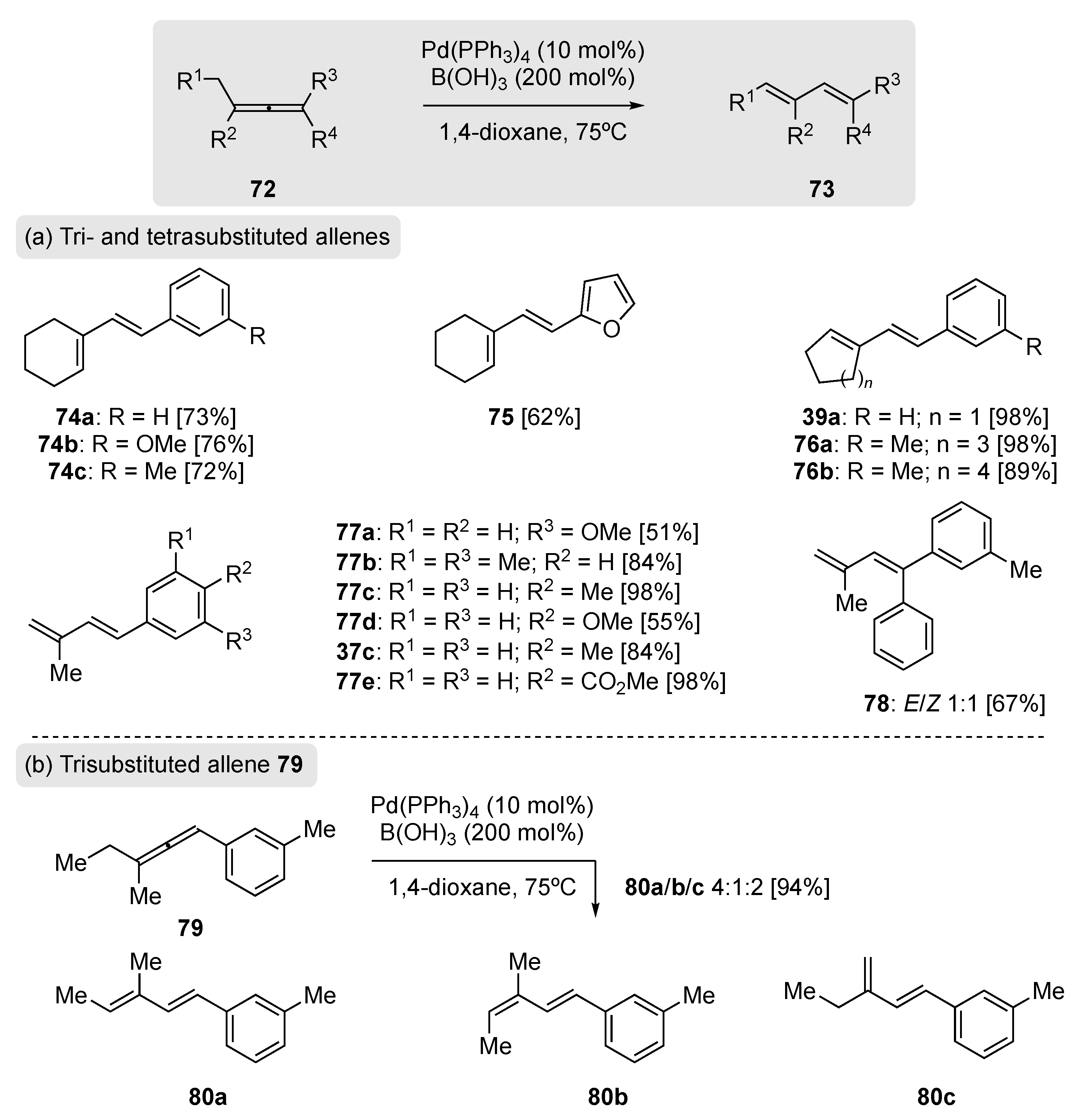
In 2018, Al-Jawaheri and Kimber disclosed a direct conversion of aryl allene 72 to 1,3-diene 73 using an H-Pd(II)-OB(OH)2-mediated reaction manifold (Scheme 18) [68]. This transformation followed a previous report by the same authors on a base-free Pd coupling of arylboronic acid with propargylic alcohols, giving direct access to substituted 1,3-dienes (Scheme 19) [69]. After optimization, they found 200 mol% of boric acid in the presence of 10 mol% of Pd(0) could effectively rearrange a number of arylallenyl substrates to their 1,3-diene counterparts in good-to-excellent isolated yields, including tri- and tetra-substituted allenes (Scheme 18a). When tri-substituted allene 79 was exposed to the rearrangement conditions, three 1,3-diene products were isolated, 80a, 80b, and 80c, respectively, in a 4:1:2 ratio (Scheme 18b).
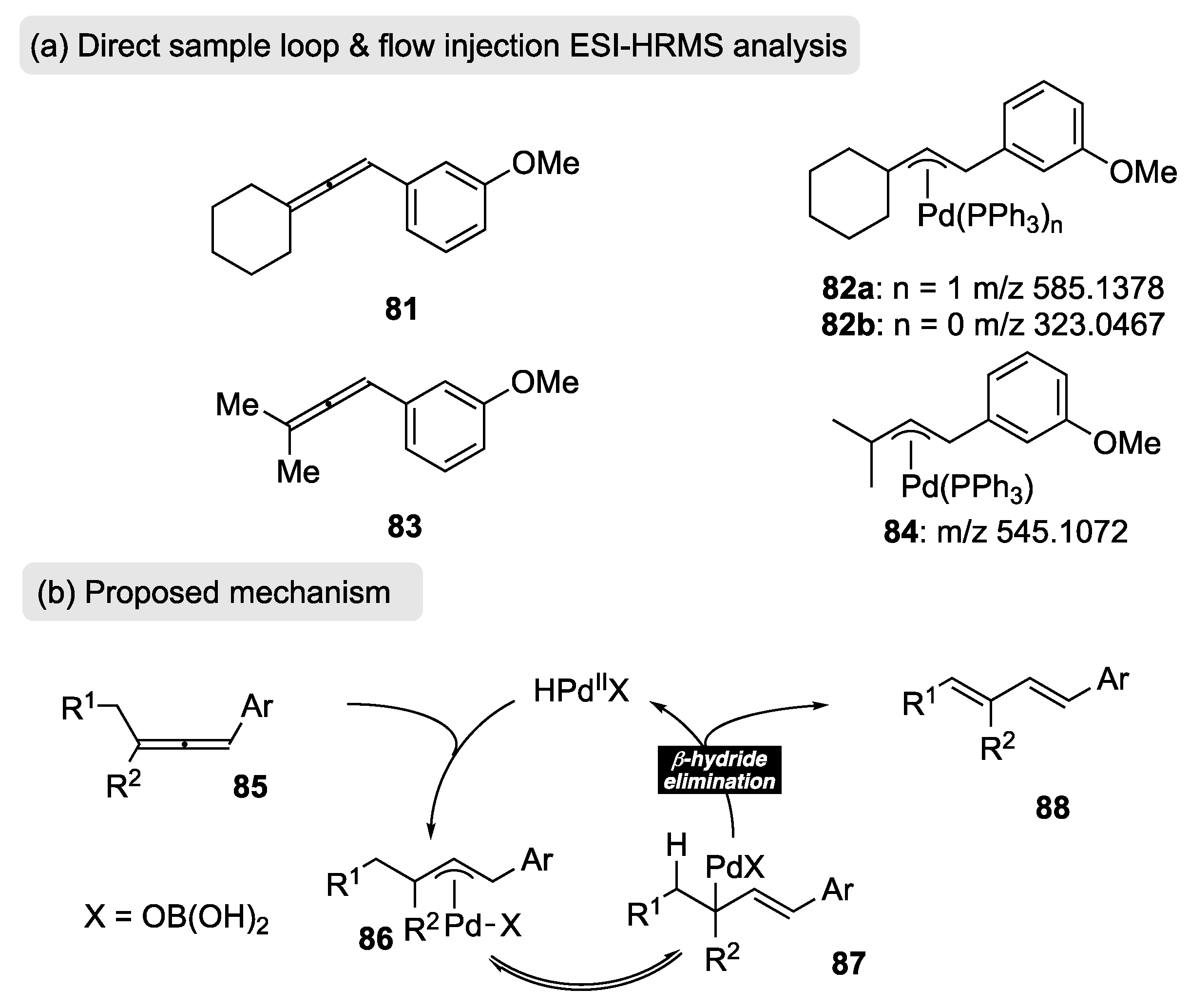
The authors were also able to follow the course of the rearrangement using direct sample loop and flow injection ESI-HRMS analysis (Scheme 19a). Using this very sensitive technique, they analyzed the rearrangement of two arylallenes (81 and 83) whose selection was based on the presence of a meta-functional group (OMe) for adequate ionization. They were subsequently able to identify π-allylpalladium species 82 and 84 to further support the mechanism proposed in Scheme 19b. Accordingly, it was postulated that allene 85 undergoes hydropalladation with the Pd species H-PdIIOB(OH)2 to yield π-allylpalladium 86, whose ultimate fate is to undergo a subsequent β-hydride elimination through intermediate 87, resulting in the observed 1,3-diene 88.
As with the Au-catalyzed method of Liu et al. [60,61], the scope of this rearrangement was limited to aryl-substituted allenes, illustrating the importance of the aryl substituent in stabilizing the intermediate π-allylpalladium species.
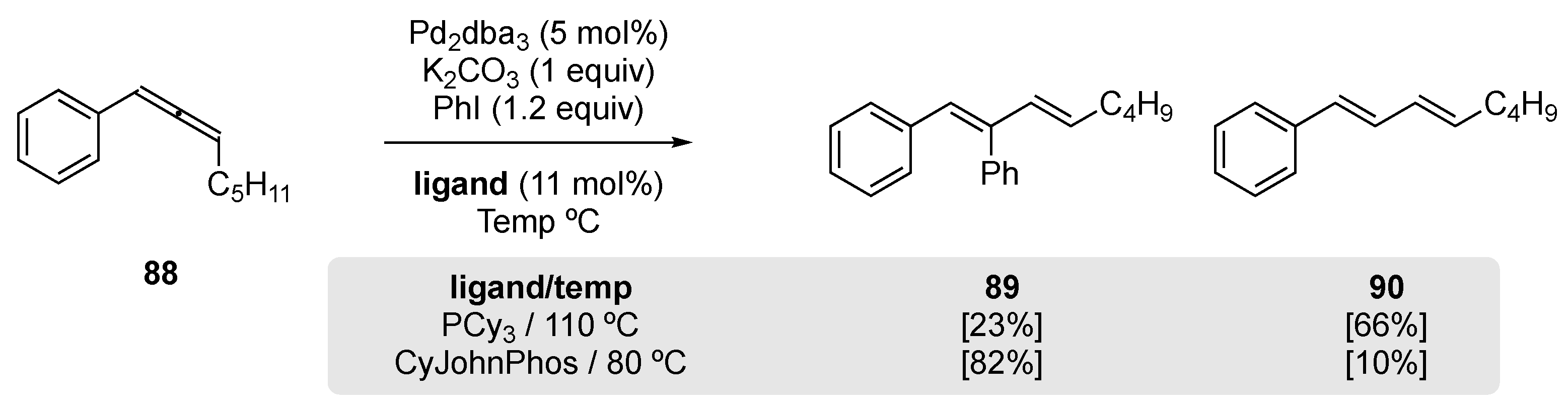
In a recent study by Vine and Schomaker [70], investigating Pd-catalyzed couplings between allenes and arylhalides, the authors observed significant amounts of 1,3-diene 90 instead of the desired Heck addition product 89 when exposing disubstituted allene 88 to the conditions shown in Scheme 20. During an optimization study, they observed the formation of 90 when undertaking the reaction with the electron-rich phosphine PCy3; this could be suppressed when the ligand was exchanged for the more hindered phosphine CyJohnPhos, together with lowering of the reaction temperature and extending the reaction time. The authors invoked the formation of a π-allylpalladium species in the formation of the undesired 1,3-diene 90.
4.3. Iron
Pettit et al. observed that the reaction of tetramethylallene 14 with Fe2(CO)9 provided a mixture of two complexes, 91 and 92, with 92 being the predominant product (Scheme 21) [71]. Furthermore, upon prolonged heating, or treatment with excess Fe2(CO)9, complex 91 could be converted to complex 92. Oxidative degradation of each complex was then performed, with 91 providing the starting tetramethylallene (14), while complex 92 yielded 2,4-dimethyl-1,3-pentadiene 15. This study established the effectiveness of Fe2(CO)9 to facilitate the allene to 1,3-diene rearrangement albeit with a stoichiometric amount of the metal complex.
4.4. Nickel
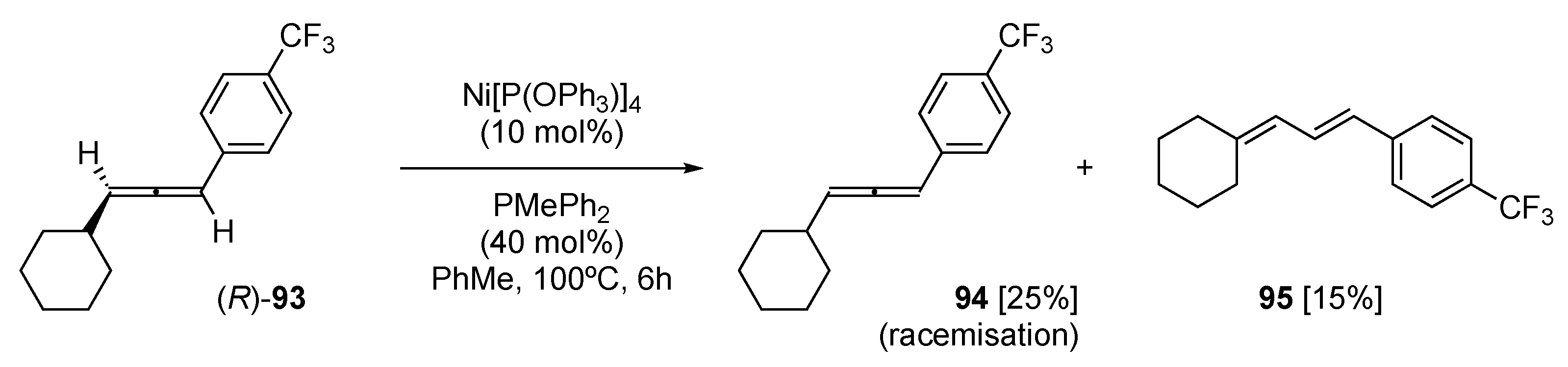
Nickel catalysis has been exploited to great effect to achieve the stereoselective hydrocyanation of allenes, with a π-allylnickel intermediate being key to this transformation [72]. This transformation can also be performed with enantiopure allenes with a good-to-excellent transfer of chiral information to the resultant products. When investigating the mechanistic basis of their Ni-catalyzed hydrocyanation of chiral allenes, Arai et al. performed their hydrocyanation reaction in the absence of a cyano source on enantiopure allene 93 (Scheme 22) [73]. As anticipated, the predominant product was racemic allene 94, together with 15% of 1,3-diene 95. The rationalization of the formation of 95 was through initial oxidative addition of Ni(0) to the allene followed by β-H elimination and reductive elimination to give the observed 1,3-diene 95. In this study, the allene-to-1,3-diene rearrangement was only observed for a single substrate but does illustrate the potential for Ni(0) catalysis to achieve this transformation.
5. Radical
Jones and Pattenden demonstrated that allene 96, either as a mixture or as separate diastereoisomers, could undergo arrangement to 1,3-diene 97 using a catalytic amount of AIBN under reflux (Scheme 23) [74]. The authors postulate that allene 97 undergoes initial hydrogen abstraction to yield an allylic radical center that is propagated by an intermediate vinylic radical. This radical rearrangement manifold is reminiscent of the previously described thermal rearrangements of cyclopropylalkylidenes. Although this is an isolated example on one specific allenic substrate, it does illustrate the potential of a homolytic pathway to facilitate this allene-to-1,3-diene rearrangement.
6. Summary
This review discussed the rearrangement of alkylallenes to 1,3-dienes. The 1,3-diene motif continues to be an essential structural motif in organic chemistry, and therefore, innovative synthetic methods that can achieve its construction are essential. Contemporary synthetic methodologies continue to deliver structurally diverse allenes. Therefore, having the synthetic capability to rearrange them to their respective 1,3-dienes opens opportunities to access structurally diverse 1,3-dienes, hitherto inaccessible using traditional synthetic methods.
The early reports were acid or thermally mediated and are often substrate dependent, harnessing inherent ring strain or the participation of a remote heteroatom to achieve the rearrangement. Of the methods discussed to achieve this rearrangement, transition metals such as gold and palladium have displayed the greatest promise. They have good substrate scope for aryl-substituted allenes, where stabilization of the intermediate p-allyl is required. There are exceptions, such as the performance of 1,2-disubstituted allenes in the gold-mediated pathway and the inability of haloboronic acid to participate cleanly in the palladium-mediated pathway. These two exceptions highlight the differing mechanistic pathways of each manifold. The gold(III)-mediated rearrangement of Lui et al. is predicated on the formation of a sufficiently stable carbocation, reflecting early acid-mediated reports of the rearrangement and subsequent DFT studies, whereas the palladium(0)-mediated methods are reliant on the formation of stable p-allylpalladium intermediates, with a subsequent β-hydride elimination of the allylic hydrogen to generate the thermodynamic 1,3-diene. The iron-mediated rearrangement of simple tetramethylallene requires further investigation regarding the scope and, importantly, needs to be rendered catalytic. In contrast, the nickel-mediated method can be achieved catalytically, but the scope of the reaction requires expansion. The AIBN-mediated rearrangement of a single tetraalkylallene shows promise given the recent development of mild photoredox methods for the generation of carbon-centered radicals.
More generally, the scope and generality of allenes that now can undergo rearrangement to their 1,3-dienes have been established. However, the successful application of this methodology requires further exploration, conceivably through target synthesis of natural products and functional materials. Additionally, it may be feasible to rearrange appropriately substituted allene substrates to provide new synthetic routes to much-sought-after polyene substrates, such as trienes and tetraenes.
Author Contributions
Writing—original draft preparation, writing—review and editing, M.C.K. and Y.A.-J. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors wish to acknowledge support from the Higher Education of Iraq (Y.A.-J.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Deagostino, A.; Prandi, C.; Zavattaro, C.; Venturello, P. Functionalized 1-Alkoxy-1,3-dienes: Their Preparation and Applications in Synthetic Organic Chemistry. Eur. J. Org. Chem. 2006, 2006, 2463–2483. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels-Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Negishi, E.; Huang, Z.; Wang, G.; Mohan, S.; Wang, C.; Hattori, H. Recent Advances in Efficient and Selective Synthesis of Di-, Tri-, and Tetrasubstituted Alkenes via Pd-Catalyzed Alkenylation-Carbonyl Olefination Synergy. Acc. Chem. Res. 2008, 41, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- de Paolis, M.; Chataigner, I.; Maddaluno, J. Recent Advances in Stereoselective Synthesis of 1,3-dienes. In Stereoselective Alkene Synthesis. Topics in Current Chemistry; Wang, J., Ed.; Springer: Berlin/Heidleberg, Germany, 2012; Volume 327, pp. 87–146. [Google Scholar]
- Ghogare, A.A.; Greer, A. Using Singlet Oxygen to Synthesize Natural Products and Drugs. Chem. Rev. 2016, 116, 9994–10034. [Google Scholar] [CrossRef]
- Kobayashi, M.; Higuchi, K.; Murakami, N.; Tajima, H.; Aoki, S. Total synthesis of callystatin A, a potent cytotoxic polyketide from the marine sponge, Callyspongia truncata. Tetrahedron Lett. 1997, 38, 2859–2862. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Kupchan, S.M.; Komoda, Y.; Court, W.A.; Thomas, G.J.; Smith, R.M.; Karim, A.; Gilmore, C.J.; Haltiwanger, R.C.; Bryan, R.F. Maytansine, a Novel Antileukemic Ansa Macrolide from Maytenus ovatus. J. Am. Chem. Soc. 1972, 94, 1354–1356. [Google Scholar] [CrossRef] [PubMed]
- Madden, K.S.; Mosa, F.A.; Whiting, A. Non-isoprenoid polyene natural products – structures and synthetic strategies. Org. Biomol. Chem. 2014, 12, 7877–7899. [Google Scholar] [CrossRef]
- Soengas, R.G.; Rodriguez-Solla, H. Modern Synthetic Methods for the Stereoselective Construction of 1,3-Dienes. Molecules 2021, 26, 249–289. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Reitz, A.B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Ager, D.J. Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; pp. 1–223. [Google Scholar]
- Dong, D.-J.; Li, H.-H.; Tian, S.-K. A Highly Tunable Stereoselective Olefination of Semistabilized Triphenylphosphonium Ylides with N-Sulfonyl Imines. J. Am. Chem. Soc. 2010, 132, 5018–5020. [Google Scholar] [CrossRef] [PubMed]
- Borg, T.; Tuzina, P.; Somfai, P. Lewis Acid-Promoted Addition of 1,3-Bis(silyl)propenes to Aldehydes: A Route to 1,3-Dienes. J. Org. Chem. 2011, 76, 8070–8075. [Google Scholar] [CrossRef] [PubMed]
- Billard, F.; Robiette, R.; Pospíšil, J. Julia–Kocienski Reaction-Based 1,3-Diene Synthesis: Aldehyde-Dependent (E,E/E,Z)-Selectivity. J. Org. Chem. 2012, 77, 6358–6364. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Wang, C.; Song, H.; He, Z. Wittig Olefination between Phosphine, Aldehyde, and Allylic Carbonates: A General Method for Stereoselective Synthesis of Trisubstituted 1,3-Dienes with Highly Variable Substituents. Org. Lett. 2010, 12, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Lemhadri, M.; Battace, A.; Berthiol, F.; Zair, T.; Doucet, H.; Santelli, M. Palladium-Tetraphosphine Complex Catalysed Heck Reaction of Vinyl Bromides with Alkenes: A Powerful Access to Conjugated Dienes. Synthesis 2008, 7, 1142–1152. [Google Scholar]
- Molander, G.A.; Felix, L.A. Stereoselective Suzuki−Miyaura Cross-Coupling Reactions of Potassium Alkenyltrifluoroborates with Alkenyl Bromides. J. Org. Chem. 2005, 70, 3950–3956. [Google Scholar] [CrossRef]
- Zheng, C.; Wang, D.; Stahl, S.S. Catalyst-Controlled Regioselectivity in the Synthesis of Branched Conjugated Dienes via Aerobic Oxidative Heck Reactions. J. Am. Chem. Soc. 2012, 134, 16496–16499. [Google Scholar] [CrossRef] [Green Version]
- Delcamp, J.H.; Gormisky, P.E.; White, M.C. Oxidative Heck Vinylation for the Synthesis of Complex Dienes and Polyenes. J. Am. Chem. Soc. 2013, 135, 8460–8463. [Google Scholar] [CrossRef] [Green Version]
- Diver, S.T.; Giessert, A.J. Enyne Metathesis (Enyne Bond Reorganization). Chem. Rev. 2004, 104, 1317–1382. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, J.; Kozmin, S.A. Gold and Platinum Catalysis of Enyne Cycloisomerization. Adv. Synth. Catal. 2006, 348, 2271–2296. [Google Scholar] [CrossRef]
- Poulsen, C.S.; Madsen, R. Enyne Metathesis Catalyzed by Ruthenium Carbene Complexes. Synthesis 2003, 1, 1–18. [Google Scholar] [CrossRef]
- Trost, B.M.; Frederiksen, M.U.; Rudd, M.T. Ruthenium-Catalyzed Reactions—A Treasure Trove of Atom-Economic Transformations. Angew. Chem. Int. Ed. 2005, 44, 6630–6666. [Google Scholar] [CrossRef]
- Hayashi, R.; Hsung, R.P.; Feltenberger, J.B.; Lohse, A.G. Regio- and Stereoselective Isomerization of Allenamides: Synthesis of 2-Amido-Dienes and Their Tandem Isomerization-Electrocyclic Ring-Closure. Org. Lett. 2009, 11, 2125–2128. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, R.; Feltenberger, J.B.; Lohse, A.G.; Walton, M.C.; Hsung, R.P. An efficient and practical entry to 2-amido-dienes and 3-amido-trienes from allenamides through stereoselective 1,3-hydrogen shifts. Beilstein J. Org. Chem. 2011, 7, 410–420. [Google Scholar] [CrossRef]
- Lu, T.; Lu, Z.; Ma, Z.-X.; Zhang, Y.; Hsung, R.P. Allenamides: A Powerful and Versatile Building Block in Organic Synthesis. Chem. Rev. 2013, 113, 4862–4904. [Google Scholar] [CrossRef] [Green Version]
- Trost, B.M.; Kazmaier, U. Internal redox catalyzed by triphenylphosphine. J. Am. Chem. Soc. 1992, 114, 7933–7935. [Google Scholar] [CrossRef]
- Hampton, C.S.; Harmata, M. Mechanistic Aspects of the Phosphine-Catalyzed Isomerization of Allenic Sulfones to 2-Arylsulfonyl 1,3-Dienes. J. Org. Chem. 2015, 80, 12151–12158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, X.; Kwon, O. Phosphine catalysis of allenes with electrophiles. Chem. Soc. Rev. 2014, 43, 2927–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, C.S.; Harmata, M. Mechanistic Aspects of the Palladium-Catalyzed Isomerization of Allenic Sulfones to 1-Arylsulfonyl 1,3-Dienes. J. Org. Chem. 2016, 81, 4807–4822. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, S.; Masuda, T.; Takeda, A. Highly stereocontrolled synthesis of (2E,4Z)-dienoic esters with alumina catalyst. Its application to total syntheses of flavor components and insect pheromones. J. Org. Chem. 1982, 47, 4478–4482. [Google Scholar] [CrossRef]
- Amos, R.A.; Katzenellenbogen, J.A. Reaction of copper enolates of esters with propargylic systems. Facile preparation of 3,4-dienoic esters, stereoselective rearrangement to (2E,4Z)- and (2E,4E)-dienoic esters, and stereoselective synthesis of a fragrance from the Bartlett pear. J. Org. Chem. 1978, 43, 555–560. [Google Scholar] [CrossRef]
- Hendrick, C.A.; Willy, W.E.; McKean, D.R.; Baggiolini, E.; Siddall, J.B. Approaches to the synthesis of the insect juvenile hormone analog ethyl 3,7,11-trimethyl-2,4-dodecadienoate and its photochemistry. J. Org. Chem. 1975, 40, 8–15. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Aragoncilli, C.; Redondo, M.C. Stereoselective Synthesis of 1,2,3-Trisubstituted 1,3-Dienes through Novel [3,3]-Sigmatropic Rearrangements in α-Allenic Methanesulfonates: Application to the Preparation of Fused Tricyclic Systems by Tandem Rearrangement/Diels-Alder Reaction. Eur. J. Org. Chem. 2005, 1, 98–106. [Google Scholar] [CrossRef]
- Matsumoto, K.; Mizushina, N.; Yoshida, M.; Shindo, M. Stereocontrolled Synthesis of Multisubstituted 1,3-dienes via an Allene-Claisen Rearrangement. Synlett 2017, 28, 2340–2344. [Google Scholar] [CrossRef] [Green Version]
- Krafft, M.E.; Hallal, K.M.; Vidhani, D.V.; Cran, J.W. Gold(I)-catalysed Claisen rearrangement of allenyl vinyl ethers; synthesis of substituted 1,3-dienes. Org. Biomol. Chem. 2011, 9, 7535–7538. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jin, X.; Fu, C.; Ma, S. Efficient Highly Selective Synthesis of Methyl 2-(Ethynyl)alk-2-(E)-enoates and 2-(1’-Chlorovinyl)alk-2(Z)-enoates from 2-(Methoxycarbonyl)2,3-allenols. Org. Lett. 2009, 11, 2169–2172. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jin, X.; Ma, S. Studies on Highly Stereoselective Addition-Elimination Reactions of 3-(Methoxycarbonyl)-1,2-allen-4-ols with MX. An Efficient Synethsis of 3-(Methoxycarbonyl)-2-1,3(Z)-dienes. J. Org. Chem. 2007, 72, 5901–5904. [Google Scholar] [CrossRef]
- Horvárth, A.; Backväll, J.E. Palladium(II)-Catalyzed SN2’ Reactions of α-Allenic Acetates. Stereoconvergent Synthesis of (Z,E)-2-Bromo-1,3-dienes. J. Org. Chem. 2001, 66, 8120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fu, C.; Ma, S. Highly Stereoselective Facile Synthesis of 2-Acetoxy-1,3(E)-alkadienes via Rh(I)-Catalyzed Isomerization of 2,3-Allenyl Carboxylates. Org. Lett. 2011, 13, 1920–1923. [Google Scholar] [CrossRef]
- Buzas, A.K.; Istrate, F.M.; Gagosz, F. Gold(I)-Catalyzed Isomerization of Allenyl Carbinol Esters: An Efficient Access to Functionalized 1,3-Butadien-2-ol Esters. Org. Lett. 2007, 9, 985. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, L. Gold-Catalyzed Efficient Formation of Alkenyl Enol Esters/Carbonates from Trimethylsilylmethyl-Substituted Propargyl Esters/Carbonates. Org. Lett. 2006, 8, 4585–4587. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ma, S. Allenes in Catalytic Asymmetric Synthesis and Natural Product Syntheses. Angew. Chem. Int. Ed. 2012, 51, 3074–3112. [Google Scholar] [CrossRef] [PubMed]
- Neff, R.K.; Frantz, D.E. Recent Advances in the Catalytic Syntheses of Allenes: A Critical Assessment. ACS Catal. 2014, 4, 519–528. [Google Scholar] [CrossRef]
- Jacobs, T.L.; Johnson, R.N. The Addition of Hydrogen Chloride to Aliphatic Allenic Hydrocarbons. J. Am. Chem. Soc. 1960, 82, 6397–6404. [Google Scholar] [CrossRef]
- Wenkert, E.; Leftin, M.H.; Michelotti, E.L. Synthesis of Allenes by Nickel-Catalyzed Grignard Reactions with Silylpropargyl Alcohols. J. Org. Chem. 1985, 50, 1122–1124. [Google Scholar] [CrossRef]
- Kropp, P.J.; Breton, G.W.; Craig, S.L.; Crawford, S.D.; Durland, W.F.; Jones, J.E.; Raleigh, J.S. Surface-Mediated Reactions. 6. Effects of Silica Gel and Alumina on Acid-Catalyzed Reactions. J. Org. Chem. 1995, 60, 4146–4152. [Google Scholar] [CrossRef]
- Li, W.; Shi, M.; Li, Y. Brønsted Acid Mediated Novel Rearrangements of Diarylvinylidenecyclopropanes and Mechanistic Investigations Based on DFT Calculations. Chem. Eur. J. 2009, 15, 8852–8860. [Google Scholar] [CrossRef]
- Sanz, R.; Miguel, D.; Martínez, A.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M.A.; Álvarez, E.; Rodríguez, F. Brønsted Acid Catalyzed Alkylation of Indoles with Tertiary Propargylic Alcohols: Scope and Limitations. Eur. J. Org. Chem. 2010, 36, 7027–7039. [Google Scholar] [CrossRef] [Green Version]
- Titov, A.A.; Kobzev, M.S.; Borisova, T.N.; Listratova, A.V.; Evenko, T.V.; Varlamov, A.V.; Voskressensky, L.G. Facile Methods for the Synthesis of 8-Ylidene-1,2,3,8-tetrahydrobenzazecines. Eur. J. Org. Chem. 2020, 20, 3041–3049. [Google Scholar] [CrossRef]
- Crandall, J.K.; Paulson, D.R. The Pyrolysis of Alkenylidenecyclopropanes. A Convenient Synthesis of Dimethylenecyclopropanes. J. Am. Chem. Soc. 1966, 88, 4302–4303. [Google Scholar] [CrossRef]
- Bloch, R.; le Perchec, P.; Conia, J.-M. Dimethylenecyclopropane. Angew. Chem. Int. Ed. Engl. 1970, 9, 798–799. [Google Scholar] [CrossRef]
- Jones, M.; Hendrick, M.E.; Hardie, J.A. Pyrolysis of Phenylalkenylidenecyclopropanes. J. Org. Chem. 1971, 36, 3061–3062. [Google Scholar] [CrossRef]
- Taylor, D.R.; Wright, D.B. Thermal Cycloadditions and Isomerisation of Tetramethylallene. Chem. Commun. 1968, 8, 434–435. [Google Scholar] [CrossRef]
- Meier, H.; Schmitt, M. Cycloalkin-vinylidnencycloalkan-Umlagerungen. Tetrahedron Lett. 1989, 30, 5873–5876. [Google Scholar] [CrossRef]
- Jensen, F. A Theoretical Study of the Allene Effect in [1,n] Sigmatropic Hydrogen Shifts. J. Am. Chem. Soc. 1995, 117, 7487–7492. [Google Scholar] [CrossRef]
- Krause, N.; Winter, C. Gold-Catalyzed Nucleophilic Cyclization of Functionalized Allenes: A Powerful Access to Carbo- and Heterocycles. Chem. Rev. 2011, 111, 1994–2009. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.-M.; Hsu, Y.-L.; Liu, R.-S. Gold-catalysed isomerisation of unactivated allenes into 1,3-dienes under ambient conditions. Chem. Commun. 2012, 48, 6577–6579. [Google Scholar] [CrossRef]
- Chen, J.-M.; Chang, C.-J.; Ke, Y.-J.; Liu, R.-S. Catalytic Formal [4+2] Cycloadditions between Unactivated Allenes and N-Hydroxyaniline Catalyzed by AlCl3/CuCl2/O2. J. Org. Chem. 2014, 79, 4306–4311. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Robertson, B.D.; Widenhoefer, R.A. Synthesis and X-ray crystal structure of a cationic gold (I) π-(1,3-diene) complex generated via isomerization of a gold π-allene complex. J. Organomet. Chem. 2014, 758, 25–28. [Google Scholar] [CrossRef]
- Basak, A.; Chakrabarty, K.; Ghosh, A.; Das, G.K. Mechanism of the Gold(III)-Catalyzed Isomerization of Substituted Allenes to Conjugated Dienes: A DFT Study. J. Org. Chem. 2013, 78, 9715–9724. [Google Scholar] [CrossRef]
- Lechel, T.; Pfrengle, F.; Reissig, H.-U.; Zimmer, R. Three Carbons for Complexity! Recent Developments of Palladium-Catalyzed Reactions of Allenes. ChemCatChem. 2013, 5, 2100–2130. [Google Scholar] [CrossRef]
- Shimizu, I.; Tsuji, J. Palladium-Catalyzed Synthesis of 2,3-Disubstituted Allylamines by Regioselective Aminophenylation or Aminoalkenylation of 1,2-Dienes. Chem. Lett. 1984, 13, 233–236. [Google Scholar] [CrossRef]
- Shimizu, I.; Sugiura, T.; Tsuji, J. Facile Synthesis of β-Aryl- or β-Alkenyl-β-methyl-α,β-unsaturated Carbonyl Compounds by Palladium-Catalyzed Reaction of 1,2-Dien-4-ols with Aryl or Alkenyl Halides. J. Org. Chem. 1985, 50, 537–539. [Google Scholar] [CrossRef]
- Al-Masum, M.; Yamamoto, Y. Palladium-Catalyzed Hydrocarboxylation of Allenes. J. Am. Chem. Soc. 1998, 120, 3809–3810. [Google Scholar] [CrossRef]
- Al-Jawaheri, Y.; Turner, M.; Kimber, M.C. Enabling the Rearrangement of Unactivated Allenes to 1,3-Dienes by Used of a Palladium (0) / Boric acid System. Synthesis 2018, 50, 2329–2336. [Google Scholar]
- Al-Jawaheri, Y.; Kimber, M.C. Synthesis of 1,3-Dienes via a Sequential Suzuki-Miyaura Coupling/Palladium-Mediated Allene Isomerization Sequence. Org. Lett. 2016, 18, 3502–3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vine, L.E.; Schomaker, J.M. Pd-Catalyzed Heck-Type Reactions of Allenes for Stereoselective Synthesis of Substituted 1,3-Dienes. Chem. Eur. J. 2021. [Google Scholar] [CrossRef]
- Ben-Shoshan, R.; Pettit, R. A Novel Type of Valence Tautomerism in Organometallic Complexes. J. Am. Chem. Soc. 1967, 89, 2231–2232. [Google Scholar] [CrossRef]
- Arai, S.; Hori, H.; Amako, Y.; Nishida, A. A new protocol for nickel-catalysed regio- and stereoselective hydrocyanation of allenes. Chem. Commun. 2015, 51, 7493–7496. [Google Scholar] [CrossRef] [PubMed]
- Amako, Y.; Arai, S.; Nishida, A. Transfer of axial chirality through the nickel-catalysed hydrocyanation of chiral allenes. Org. Biomol. Chem. 2017, 15, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.R.; Pattenden, G. Alkene-allenecyclopropane radical cyclisations promooted by tris-(trimethylsilyl)silane. Tetrahedron Lett. 2009, 50, 3527–3529. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of 1,3-dienes (1).
Scheme 2.
(a) Rearrangement of an alkylallene to a 1,3-diene. (b) Different allene subclasses.
Scheme 3.
Acid-catalyzed rearrangement of 3-methyl-1,2-butadiene 4 [47].
Scheme 3.
Acid-catalyzed rearrangement of 3-methyl-1,2-butadiene 4 [47].
Scheme 4.
HCl-promoted allene rearrangement. The rearrangement was facilitated by the presence of an electron-rich 4MeO-arene substituent [48].
Scheme 4.
HCl-promoted allene rearrangement. The rearrangement was facilitated by the presence of an electron-rich 4MeO-arene substituent [48].
Scheme 5.
SiO2 rearrangement of tetramethylallene 14 [49].
Scheme 5.
SiO2 rearrangement of tetramethylallene 14 [49].
Scheme 6.
Trifluoroacetic acid-mediated rearrangement of diaryl-vinylidenecyclopropanes [50].
Scheme 6.
Trifluoroacetic acid-mediated rearrangement of diaryl-vinylidenecyclopropanes [50].
Scheme 7.
PTSA-catalyzed rearrangement of indolylallenes to yield penta-substituted 1,3-dienes. The intermediate tertiary carbocation is stabilized by the electron-rich indolyl substituent [51].
Scheme 7.
PTSA-catalyzed rearrangement of indolylallenes to yield penta-substituted 1,3-dienes. The intermediate tertiary carbocation is stabilized by the electron-rich indolyl substituent [51].
Scheme 8.
Acetic acid- and microwave-mediated rearrangement of allenes 23 [52].
Scheme 8.
Acetic acid- and microwave-mediated rearrangement of allenes 23 [52].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme 10.
Thermal rearrangement of tetramethylallene in polar aprotic solvents [56].
Scheme 10.
Thermal rearrangement of tetramethylallene in polar aprotic solvents [56].

Scheme 11.
Flash vacuum pyrolysis of the epoxides provides a mixture of the allene and 1,3-diene [57]. A subsequent theoretical study by Jenson provided an energy barrier for this rearrangement [58].
Scheme 12.
Accomplishing the rearrangement of allenes at ambient temperature through the action of AuCl3 and nitrosobenzene (PHNO) [60].
Scheme 12.
Accomplishing the rearrangement of allenes at ambient temperature through the action of AuCl3 and nitrosobenzene (PHNO) [60].
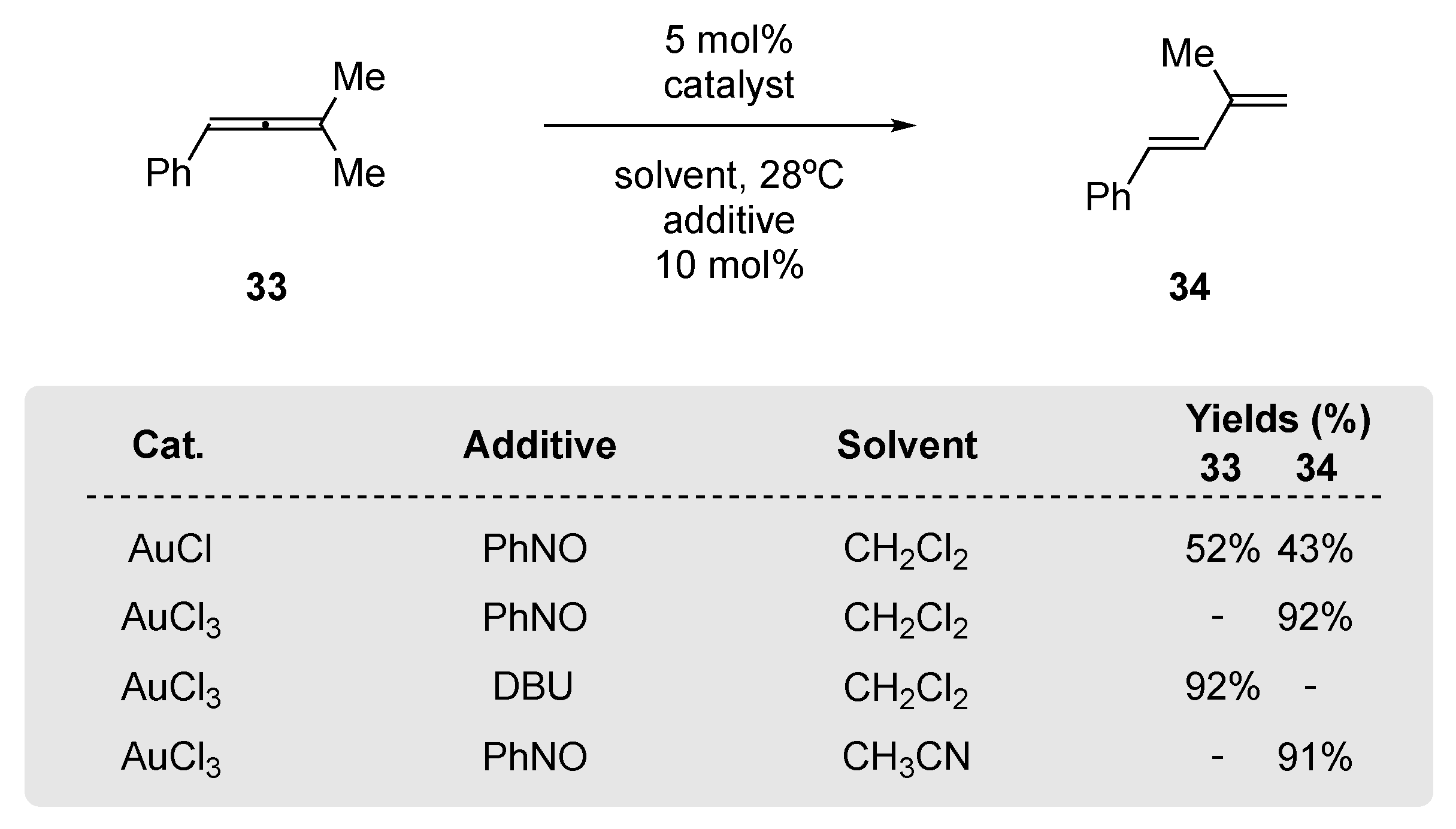
Scheme 13.
(a,b) Tri- and tetra-allene reaction scope of Lui’s Au(III)/PhNO rearrangement. (c) 1,1-Disubstituted allene rearrangement can be accomplished using Au(III)/PhNO protocol; 1,2-disubstituted allenes do not participate. For comparison, the reaction times (h) for each rearrangement are given in italics [60].
Scheme 13.
(a,b) Tri- and tetra-allene reaction scope of Lui’s Au(III)/PhNO rearrangement. (c) 1,1-Disubstituted allene rearrangement can be accomplished using Au(III)/PhNO protocol; 1,2-disubstituted allenes do not participate. For comparison, the reaction times (h) for each rearrangement are given in italics [60].

Scheme 14.
A proposed mechanism for the Au(III)/PhNO-assisted rearrangement [60].
Scheme 14.
A proposed mechanism for the Au(III)/PhNO-assisted rearrangement [60].
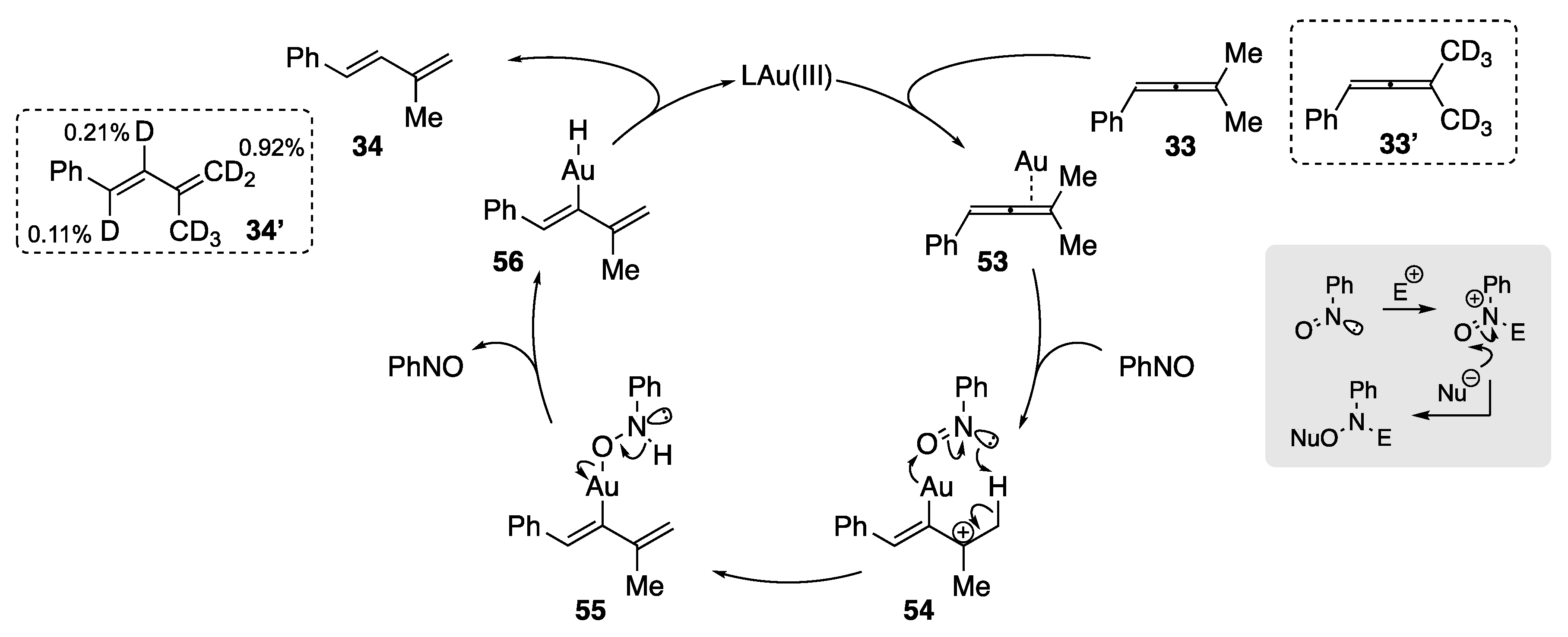
Scheme 15.
Widenhoefer’s Au(I)-facilitated rearrangement and the effect of different bases on the product outcome [62].
Scheme 15.
Widenhoefer’s Au(I)-facilitated rearrangement and the effect of different bases on the product outcome [62].
Scheme 16.
Early work by Tsuji provided the basis of the β-hydride elimination pathway yielding 1,3-diene 65 [65,66].
Scheme 17.
(a) Yamamoto’s observed rearrangement of allenes to 1,3-dienes through hydropalladation; (b,c) plausible mechanism for Yamamoto’s rearrangement of allenes 66 and 68 via hydropalladation [67].
Scheme 17.
(a) Yamamoto’s observed rearrangement of allenes to 1,3-dienes through hydropalladation; (b,c) plausible mechanism for Yamamoto’s rearrangement of allenes 66 and 68 via hydropalladation [67].

Scheme 18.
(a) Substrate scope of the Pd(0)/boric acid mediated rearrangement; (b) product distribution for allene 79 [68].
Scheme 18.
(a) Substrate scope of the Pd(0)/boric acid mediated rearrangement; (b) product distribution for allene 79 [68].
Scheme 19.
(a) ESI-HRMS analysis using allenes 81 and 83; (b) proposed mechanism.
Scheme 20.
Observed rearrangement of allene 88 to 1,3-diene 90 under Heck-type coupling conditions [70].
Scheme 20.
Observed rearrangement of allene 88 to 1,3-diene 90 under Heck-type coupling conditions [70].
Scheme 21.
Pettit’s observed rearrangement of tetramethylallene via oxidative degradation of iron complex 92 [71].
Scheme 21.
Pettit’s observed rearrangement of tetramethylallene via oxidative degradation of iron complex 92 [71].

Scheme 22.
The Ni(0)-facilitated rearrangement of enantio-enrich allene 93 [73].
Scheme 22.
The Ni(0)-facilitated rearrangement of enantio-enrich allene 93 [73].
Scheme 23.
Pattenden’s radical-mediated rearrangement of tetrasubstituted allene 96 [74].
Scheme 23.
Pattenden’s radical-mediated rearrangement of tetrasubstituted allene 96 [74].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Al-Jawaheri, Y.; Kimber, M.C. The Rearrangement of Alkylallenes to 1,3-Dienes. Reactions 2022, 3, 70-86. https://doi.org/10.3390/reactions3010006
AMA Style
Al-Jawaheri Y, Kimber MC. The Rearrangement of Alkylallenes to 1,3-Dienes. Reactions. 2022; 3(1):70-86. https://doi.org/10.3390/reactions3010006
Chicago/Turabian StyleAl-Jawaheri, Yassir, and Marc Colin Kimber. 2022. "The Rearrangement of Alkylallenes to 1,3-Dienes" Reactions 3, no. 1: 70-86. https://doi.org/10.3390/reactions3010006