
Recent Progresses in the Preparation of Chlorinated Molecules: Electrocatalysis and Photoredox Catalysis in the Spotlight



{kind=link}
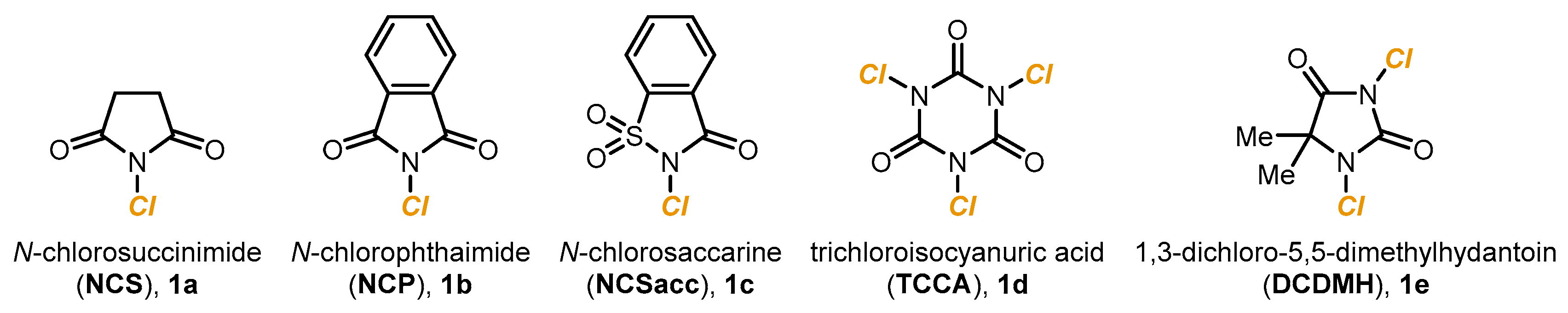{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
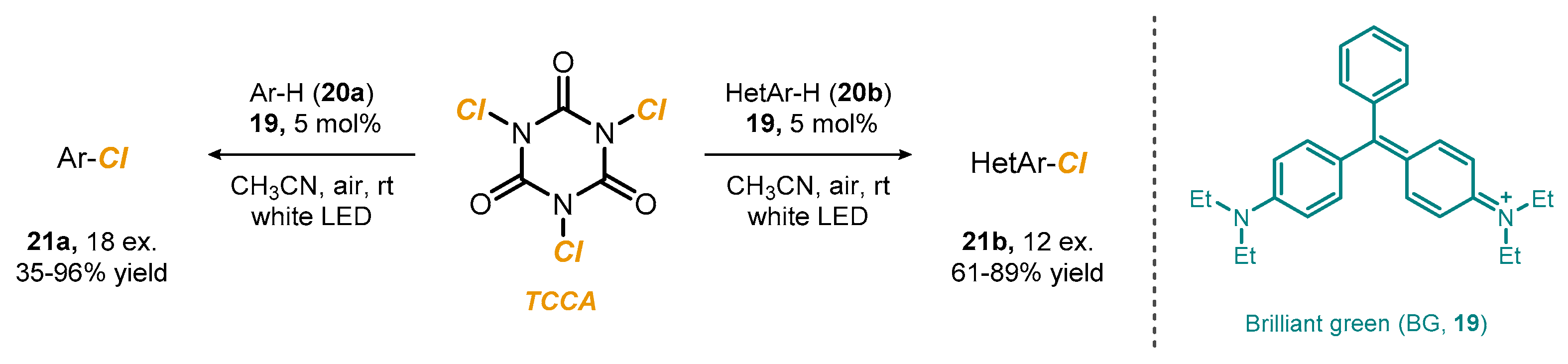{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
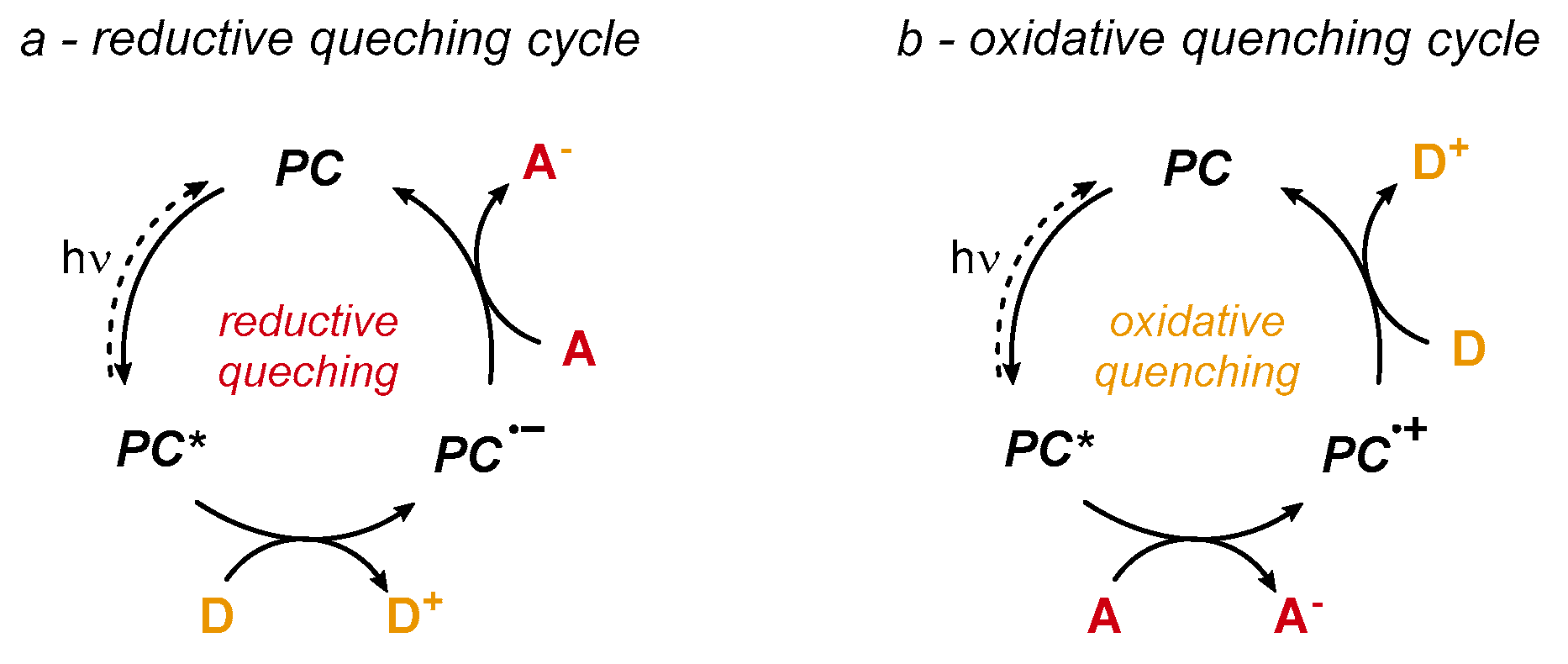
2. Chlorination Enabled by Visible Light Photoredox Catalysis
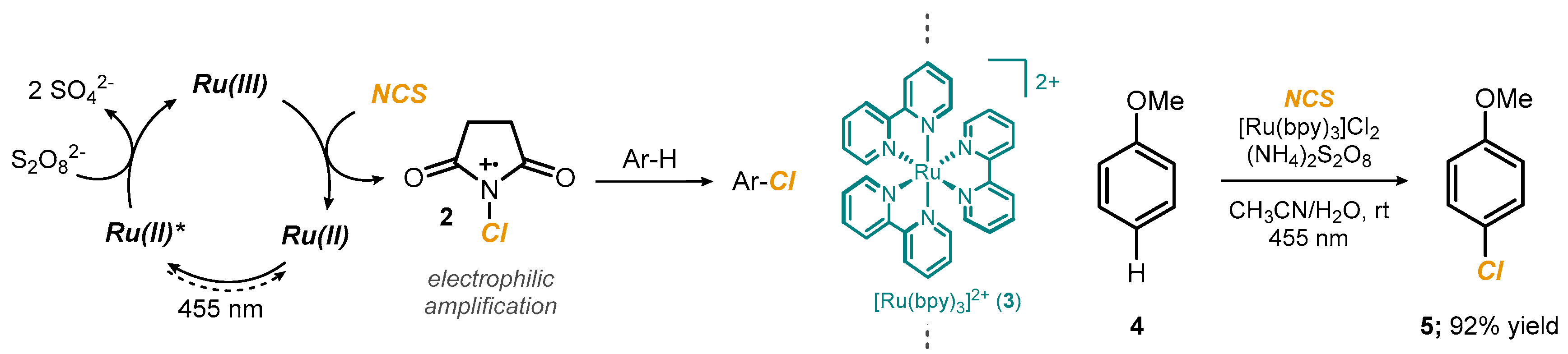
2.1. Photoredox Electrophilic Amplification of NCS and Related Reagents
2.2. Photoredox Chlorination Using Alternative Chlorinating Reagents
2.3. Photoredox Chlorination of Nitrogen-Containing Aliphatic Molecules
3. Chlorination Enabled by Electrocatalysis
3.1. Electrocatalyzed Dichlorination of Alkenes
3.2. Electrocatalyzed Heterofunctionalization of Alkenes
3.3. Electrocatalyzed Arene Chorination
3.4. Electrocatalyzed Miscellaneous Reactions
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
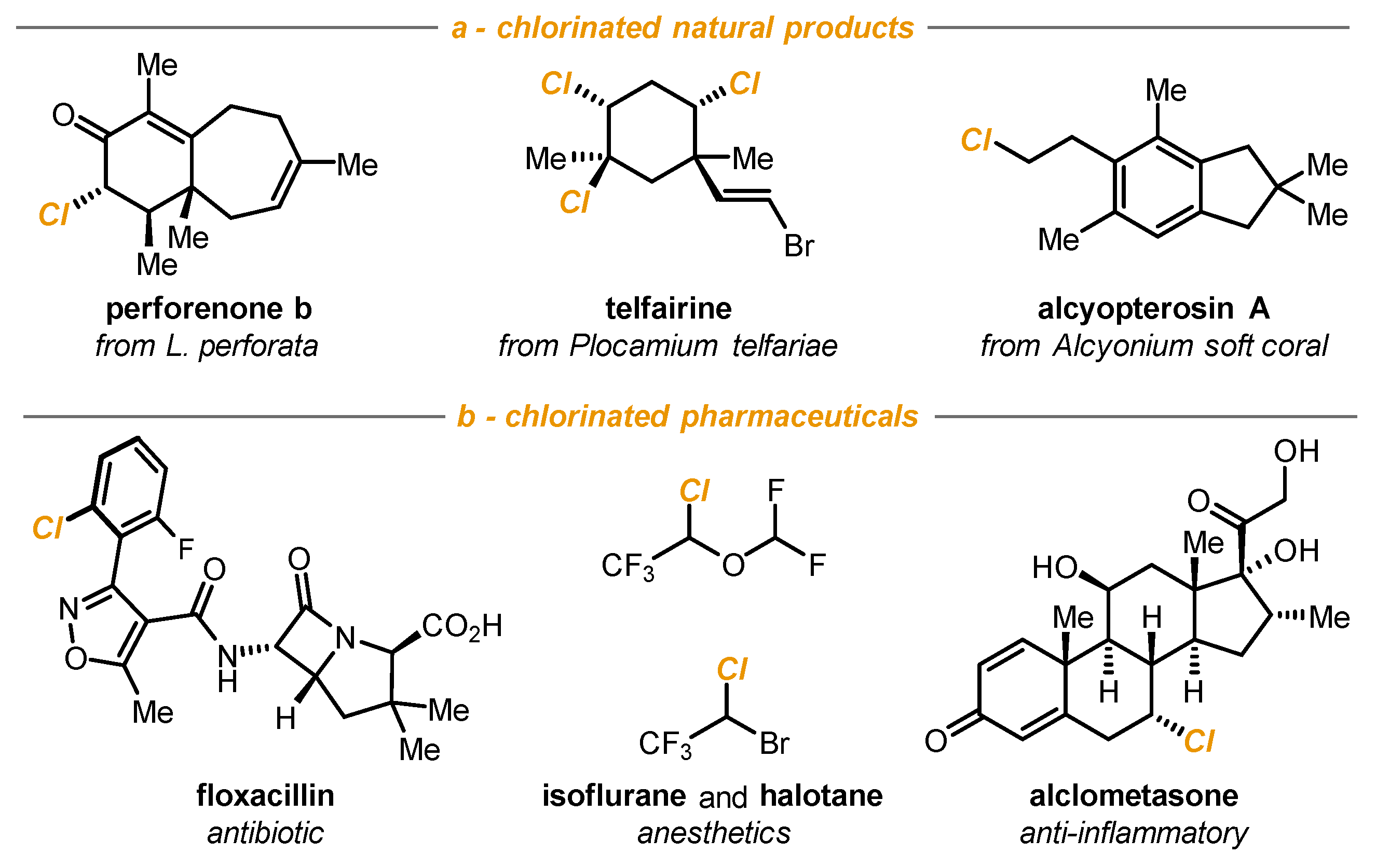
- Zhukova, N.V.; Gloriozova, T.A.; Poroikov, V.V.; Dembitsky, V.M. Halogenated (Cl, Br and I) marine steroids and their biological activities: A brief review. Pharma Innov. J. 2017, 6, 456–462. [Google Scholar]
- Wang, B.-G.; Gloer, J.B.; Ji, N.-Y.; Zhao, J.-C. Halogenated organic molecules of Rhodomelaceae origin: Chemistry and biology. Chem. Rev. 2013, 113, 3632–3685. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Naturally occurring organohalogen compounds. Acc. Chem. Res. 1998, 31, 141–152. [Google Scholar] [CrossRef]
- Finkelstein, H. Darstellung organischer Jodide aus den entsprechenden bromiden und chloriden. Ber. Dtsch. Chem. Ges. 1910, 43, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Juliá, F.; Constantin, T.; Leonori, D. Applications of halogen-atom transfer (XAT) for the generation of carbon radicals in synthetic photochemistry and photocatalysis. Chem. Rev. 2022, 122, 2292–2352. [Google Scholar] [CrossRef]
- Fang, W.-Y.; Ravindar, L.; Rakesh, K.P.; Manukumar, H.M.; Shantharam, C.S.; Alharbi, N.S.; Qin, H.-L. Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 173, 117–153. [Google Scholar] [CrossRef]
- Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking back, looking forward at halogen bonding in drug discovery. Molecules 2017, 22, 1397. [Google Scholar] [CrossRef]
- Gál, B.; Bucher, C.; Burns, N.Z. Chiral alkyl halides: Underexplored motifs in medicine. Mar. Drugs 2016, 14, 206. [Google Scholar] [CrossRef] [Green Version]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Dreher, E.-L.; Torkelson, T.R.; Beutel, K.K. Chlorethanes and Chlorethylenes. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar] [CrossRef]
- Sakhri, A.; Perrin, F.; Aragon, E.; Lamouric, S.; Benaboura, A. Chlorinated rubber paints for corrosion prevention of mild steel: A comparison between zinc phosphate and polyaniline pigments. Corros. Sci. 2010, 52, 901–909. [Google Scholar] [CrossRef]
- Lahimer, M.C.; Ayed, N.; Horriche, J.; Belgaied, S. Characterization of plastic packaging additives: Food contact, stability and toxicity. Arab. J. Chem. 2017, 10, S1938–S1954. [Google Scholar] [CrossRef] [Green Version]
- Baughman, T.W.; Sworen, J.C.; Wagener, K.B. The facile preparation of alkenyl metathesis synthons. Tetrahedron 2004, 60, 10943–10948. [Google Scholar] [CrossRef]
- Appel, R. Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P–N Linkage. Angew. Chem. Int. Ed. 1975, 14, 801–811. [Google Scholar] [CrossRef]
- Luu, T.G.; Jung, Y.; Kim, H.-K. Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst. Molecules 2021, 26, 7380. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Zhu, S.; Qing, F.-L.; Chu, L. Visible-light-induced halogenation of aliphatic CH bonds. Tetrahedron Lett. 2018, 59, 173–179. [Google Scholar] [CrossRef]
- Scheide, M.R.; Nicoleti, C.R.; Martins, G.M.; Braga, A.L. Electrohalogenation of organic compounds. Org. Biomol. Chem. 2021, 19, 2578–2602. [Google Scholar] [CrossRef]
- Tay, N.E.S.; Lehnherr, D.; Rovis, T. Photons or electrons? A critical comparison of electrochemistry and photoredox catalysis for organic synthesis. Chem. Rev. 2022, 122, 2487–2649. [Google Scholar] [CrossRef]
- Hering, T.; König, B. Photocatalytic activation of N-chloro compounds for the chlorination of arenes. Tetrahedron 2016, 72, 7821–7825. [Google Scholar] [CrossRef]
- Rogers, D.A.; Gallegos, J.M.; Hopkins, M.D.; Lignieres, A.A.; Pitzel, A.K.; Lamar, A.A. Visible-light photocatalytic activation of N-chlorosuccinimide by organic dyes for the chlorination of arenes and heteroarenes. Tetrahedron 2019, 75, 130498. [Google Scholar] [CrossRef]
- Rogers, D.A.; Hopkins, M.D.; Rajagopal, N.; Varshney, D.; Howard, H.A.; LeBlanc, G.; Lamar, A.A. US Food and Drug Administration-Certified Food Dyes as Organocatalysts in the Visible Light-Promoted Chlorination of Aromatics and Heteroaromatics. ACS Omega 2020, 5, 7693–7704. [Google Scholar] [CrossRef] [Green Version]
- Rogers, D.A.; Bensalah, A.T.; Espinosa, A.T.; Hoerr, J.L.; Refai, F.H.; Pitzel, A.K.; Alvarado, J.J.; Lamar, A.A. Amplification of Trichloroisocyanuric Acid (TCCA) Reactivity for Chlorination of Arenes and Heteroarenes via Catalytic Organic Dye Activation. Org. Lett. 2019, 21, 4229–4233. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.A.; Pan, C.-M.; Yabe, Y.; Kawamata, Y.; Eastgate, M.D.; Baran, P.S. Palau’chlor: A practical and reactive chlorinating reagent. J. Am. Chem. Soc. 2014, 136, 6908–6911. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Zhou, C.; Yang, X.-L.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Visible light-catalyzed benzylic C–H bond chlorination by a combination of organic dye (Acr+-Mes) and N-chlorosuccinimide. J. Org. Chem. 2020, 85, 9080–9087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, X. Room temperature C (sp 2)–H oxidative chlorination via photoredox catalysis. Chem. Sci. 2017, 8, 7009–7013. [Google Scholar] [CrossRef]
- Düsel, S.J.S.; König, B. Oxidative Photochlorination of Electron-Rich Arenes via in situ Bromination. Eur. J. Org. Chem. 2020, 2020, 1491–1495. [Google Scholar] [CrossRef]
- Hering, T.; Mühldorf, B.; Wolf, R.; König, B. Halogenase-inspired oxidative chlorination using flavin photocatalysis. Angew. Chem. Int. Ed. 2016, 55, 5342–5345. [Google Scholar] [CrossRef] [Green Version]
- Seel, C.J.; Králík, A.; Hacker, M.; Frank, A.; König, B.; Gulder, T. Atom-Economic Electron Donors for Photobiocatalytic Halogenations. ChemCatChem 2018, 10, 3960–3963. [Google Scholar] [CrossRef]
- Patil, D.V.; Kim, H.Y.; Oh, K. Visible Light-Promoted Friedel–Crafts-Type Chloroacylation of Alkenes to β-Chloroketones. Org. Lett. 2020, 22, 3018–3022. [Google Scholar] [CrossRef]
- Wolff, M.E. Cyclization of N-Halogenated Amines (The Hofmann-Löffler Reaction). Chem. Rev. 1963, 63, 55–64. [Google Scholar] [CrossRef]
- Dauncey, E.M.; Morcillo, S.P.; Douglas, J.J.; Sheikh, N.S.; Leonori, D. Photoinduced remote functionalisations by iminyl radical promoted C−C and C−H bond cleavage cascades. Angew. Chem. Int. Ed. 2018, 57, 744–748. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, J.-J.; Wu, D.; Yu, W. Visible-light-driven remote C−H chlorination of aliphatic sulfonamides with sodium hypochlorite. Asian J. Chem. 2020, 9, 1650–1654. [Google Scholar] [CrossRef]
- Wang, F.; Liu, X.; Wang, L. Visible-light-induced C (sp 3)–H functionalizations of piperidines to 3, 3-dichloro-2-hydroxy-piperidines with N-chlorosuccinimide. Org. Biomol. Chem. 2021, 19, 6141–6146. [Google Scholar] [CrossRef] [PubMed]
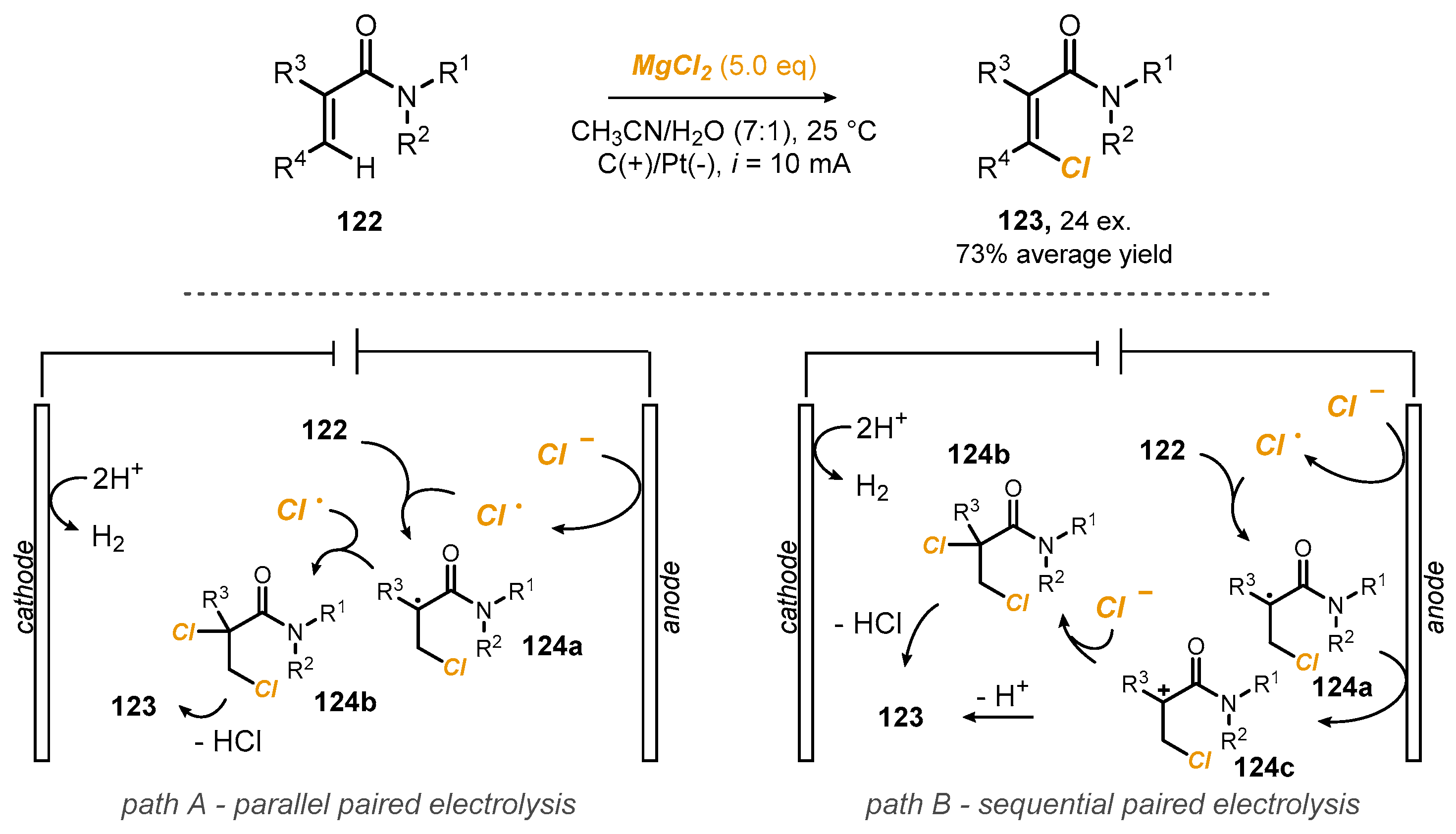
- Fu, N.; Sauer, G.S.; Lin, S. Electrocatalytic radical dichlorination of alkenes with nucleophilic chlorine sources. J. Am. Chem. Soc. 2017, 139, 15548–15553. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Roeckl, J.L.; Waldvogel, S.R.; Morandi, B. Merging shuttle reactions and paired electrolysis for reversible vicinal dihalogenations. Science 2021, 371, 507–514. [Google Scholar] [CrossRef]
- Strehl, J.; Fastie, C.; Hilt, G. The Electrochemical cis-Chlorination of Alkenes. Chem. Eur. J. 2021, 27, 17341–17345. [Google Scholar] [CrossRef]
- Ye, K.-Y.; Pombar, G.; Fu, N.; Sauer, G.S.; Keresztes, I.; Lin, S. Anodically coupled electrolysis for the heterodifunctionalization of alkenes. J. Am. Chem. Soc. 2018, 140, 2438–2441. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.; Shen, Y.; Allen, A.R.; Song, L.; Ozaki, A.; Lin, S. Mn-catalyzed electrochemical chloroalkylation of alkenes. ACS Catal. 2019, 9, 746–754. [Google Scholar] [CrossRef]
- Tian, S.; Lv, S.; Jia, X.; Ma, L.; Li, B.; Zhang, G.; Gao, W.; Wei, Y.; Chen, J. CV-driven Optimization: Cobalt-Catalyzed Electrochemical Expedient Oxychlorination of Alkenes via ORR. Adv. Synth. Catal. 2019, 361, 5626–5633. [Google Scholar] [CrossRef]
- Yu, M.; Wang, H.; Gao, Y.; Bu, F.; Cong, H.; Lei, A. Manganese-catalyzed chlorosulfonylation of terminal alkene and alkyne via convergent paired electrolysis. Cell Rep. Phys. Sci. 2021, 2, 100476. [Google Scholar] [CrossRef]
- Lin, X.; Zeng, C.; Liu, C.; Fang, Z.; Guo, K. C-5 selective chlorination of 8-aminoquinoline amides using dichloromethane. Org. Biomol. Chem. 2021, 19, 1352–1357. [Google Scholar] [CrossRef]
- Konishi, M.; Tsuchida, K.; Sano, K.; Kochi, T.; Kakiuchi, F. C-5 selective chlorination of 8-aminoquinoline amides using dichloromethane. J. Org. Chem. 2017, 82, 8716–8724. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Lin, F.; Adeli, Y.; Jin, R.; Jiao, N. Efficient electrocatalysis for the preparation of (hetero) aryl chlorides and vinyl chloride with 1, 2-dichloroethane. Angew. Chem. Int. Ed. 2019, 58, 4566–4570. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.D.W.; Hareram, M.D.; Seastram, A.C.; McBride, T.; Wirth, T.; Browne, D.L.; Morrill, L.C. Manganese-catalyzed electrochemical deconstructive chlorination of cycloalkanols via alkoxy radicals. Org. Lett. 2019, 21, 9241–9246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Deng, Y.; Song, W.; Song, C.; Lei, A. Electrochemical dearomative halocyclization of tryptamine and tryptophol derivatives. Chin. J. Chem. 2020, 38, 1070–1074. [Google Scholar] [CrossRef]
- Yu, D.; Ji, R.; Sun, Z.; Li, W.; Liu, Z.-Q. Electrochemical chlorination and bromination of electron-deficient CH bonds in quinones, coumarins, quinoxalines and 1, 3-diketones. Tetrahedron Lett. 2021, 86, 153514. [Google Scholar] [CrossRef]
- Lin, Y.; Jin, J.; Wang, C.; Wan, J.-P.; Liu, Y. Electrochemical CH Halogenations of Enaminones and Electron-Rich Arenes with Sodium Halide (NaX) as Halogen Source for the Synthesis of 3-Halochromones and Haloarenes. J. Org. Chem. 2021, 86, 12378–12385. [Google Scholar] [CrossRef]
- Harnedy, J.; Hareram, M.D.; Tizzard, G.J.; Coles, S.J.; Morrill, L.C. Electrochemical oxidative Z-selective C (sp 2)–H chlorination of acrylamides. Chem. Commun. 2021, 57, 12643–12646. [Google Scholar] [CrossRef]
































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parisotto, S.; Azzi, E.; Lanfranco, A.; Renzi, P.; Deagostino, A. Recent Progresses in the Preparation of Chlorinated Molecules: Electrocatalysis and Photoredox Catalysis in the Spotlight. Reactions 2022, 3, 233-253. https://doi.org/10.3390/reactions3020018
Parisotto S, Azzi E, Lanfranco A, Renzi P, Deagostino A. Recent Progresses in the Preparation of Chlorinated Molecules: Electrocatalysis and Photoredox Catalysis in the Spotlight. Reactions. 2022; 3(2):233-253. https://doi.org/10.3390/reactions3020018
Chicago/Turabian StyleParisotto, Stefano, Emanuele Azzi, Alberto Lanfranco, Polyssena Renzi, and Annamaria Deagostino. 2022. "Recent Progresses in the Preparation of Chlorinated Molecules: Electrocatalysis and Photoredox Catalysis in the Spotlight" Reactions 3, no. 2: 233-253. https://doi.org/10.3390/reactions3020018
APA StyleParisotto, S., Azzi, E., Lanfranco, A., Renzi, P., & Deagostino, A. (2022). Recent Progresses in the Preparation of Chlorinated Molecules: Electrocatalysis and Photoredox Catalysis in the Spotlight. Reactions, 3(2), 233-253. https://doi.org/10.3390/reactions3020018