

Effective One-Component Organocatalysts for Eco-Friendly Production of Cyclic Carbonates
 , , ,
, , ,  and
and 
Abstract

1. Introduction
2. Materials and Methods
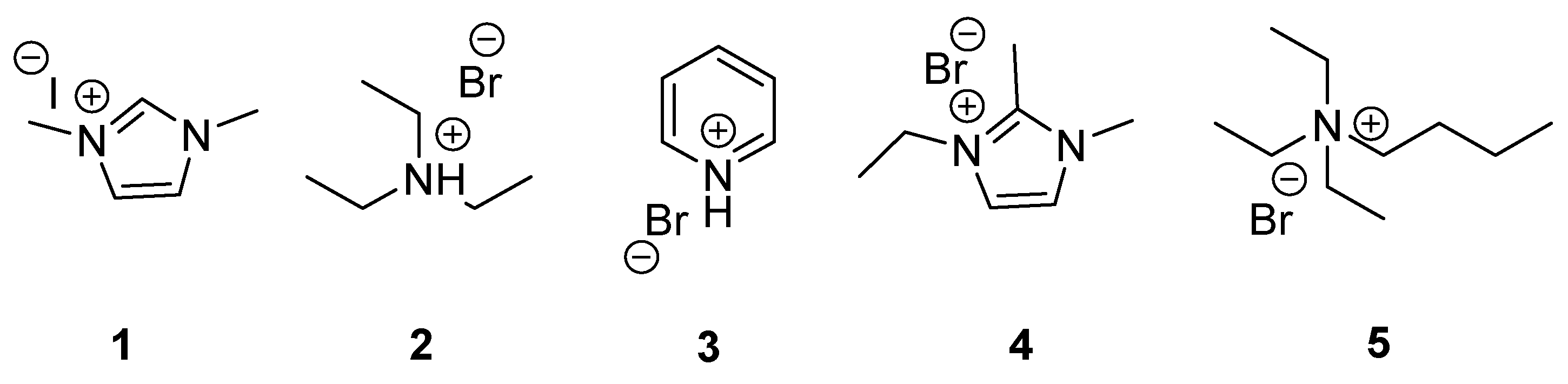
2.1. General Procedure for the Synthesis of Organocatalysts 1–5
2.2. General Procedure to Optimize Reaction Conditions for the Preparation of Styrene Carbonate
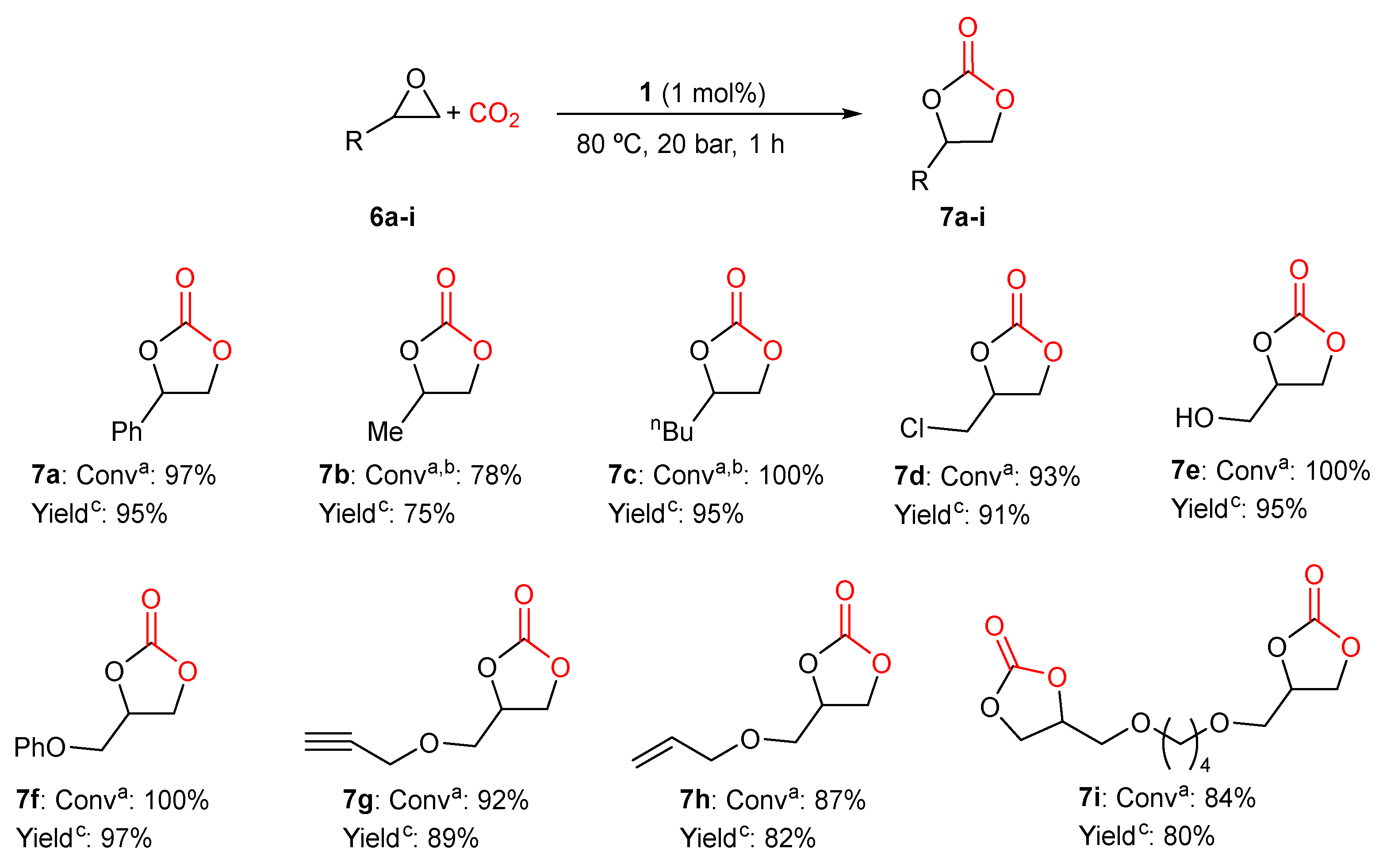
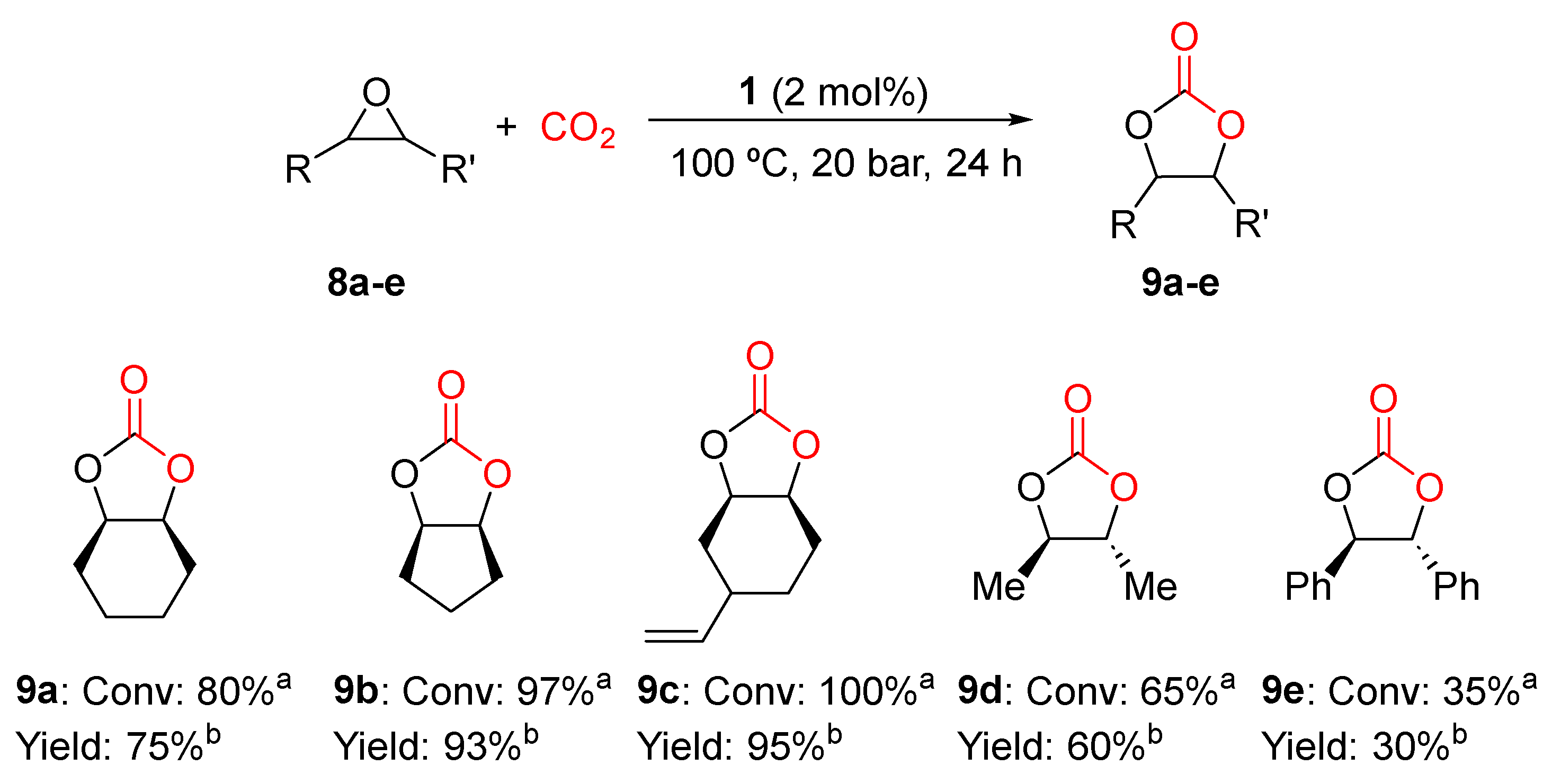
2.3. General Procedure for the Synthesis of Cyclic Carbonates
2.4. Computational Details
3. Results and Discussions
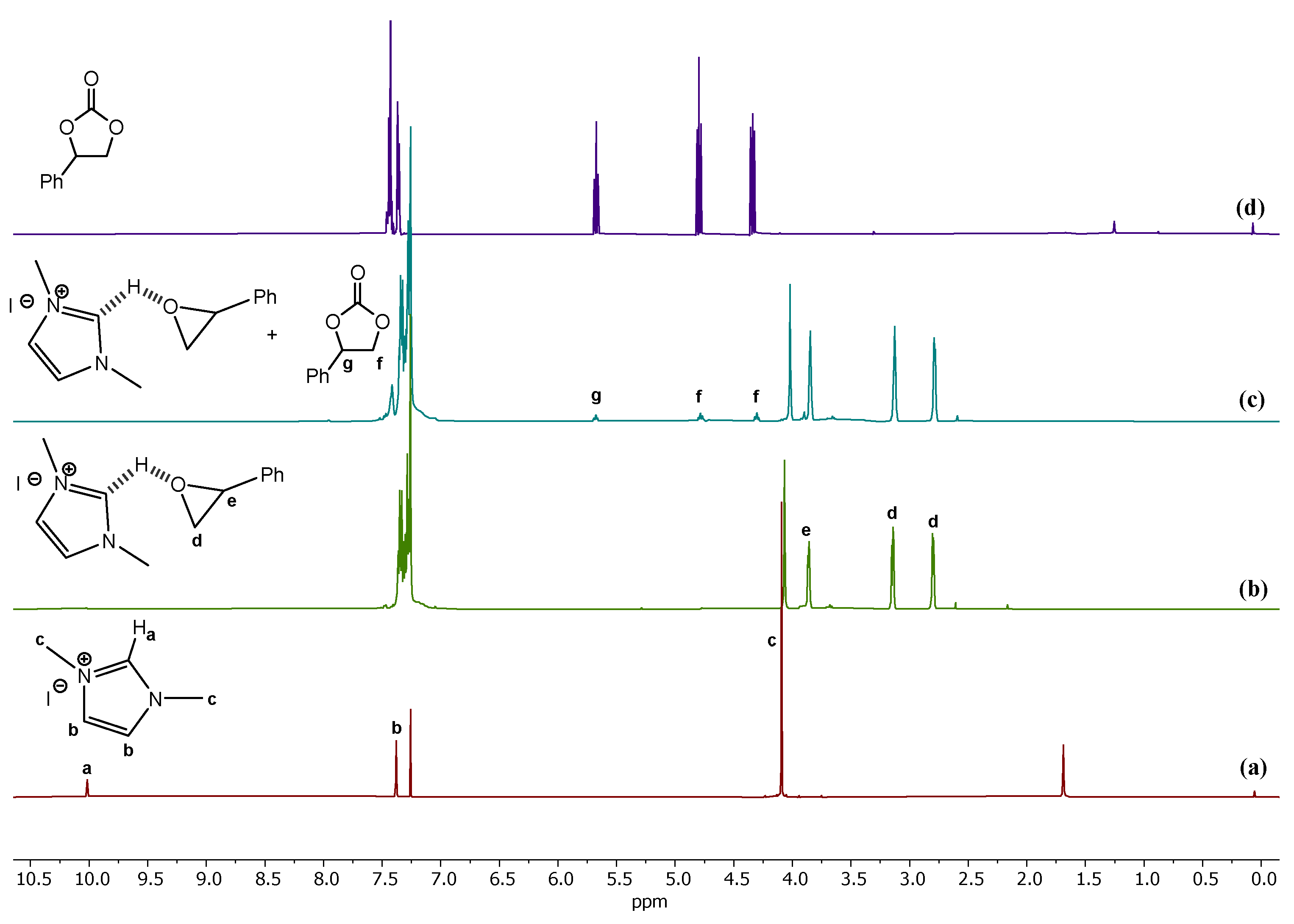
3.1. Catalytic Yield Results for the Preparation of Different Cyclic Carbonates
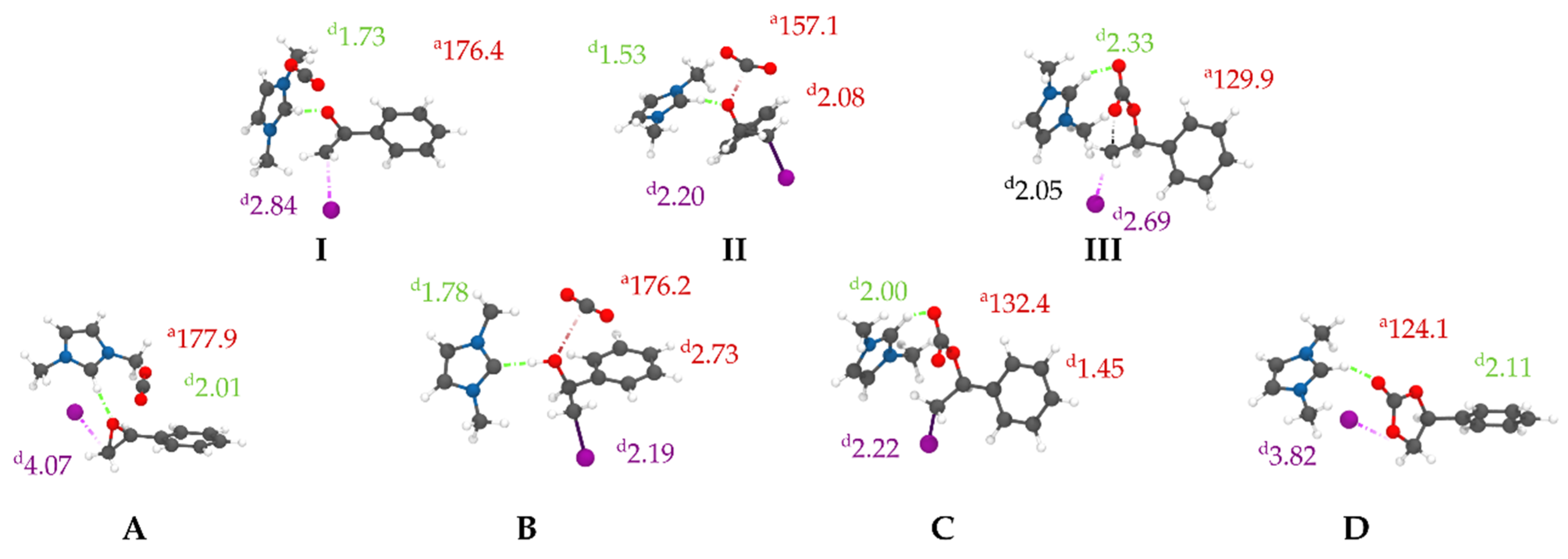
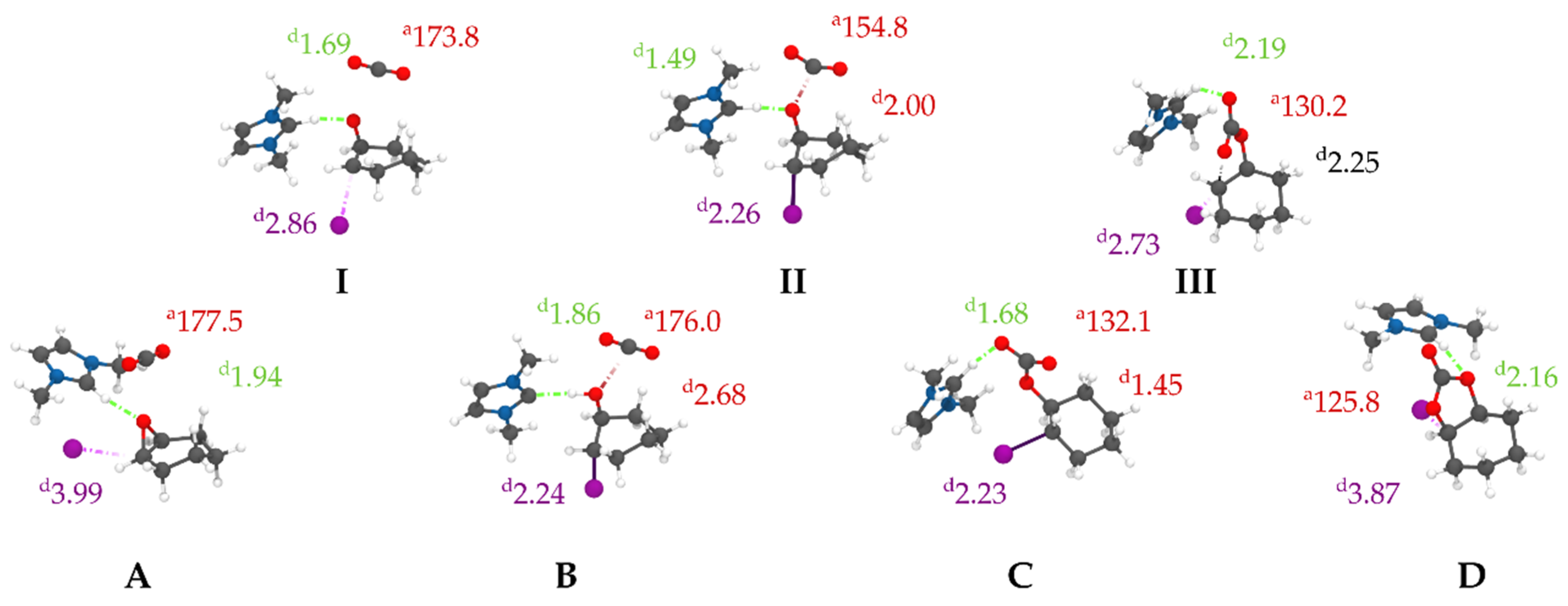
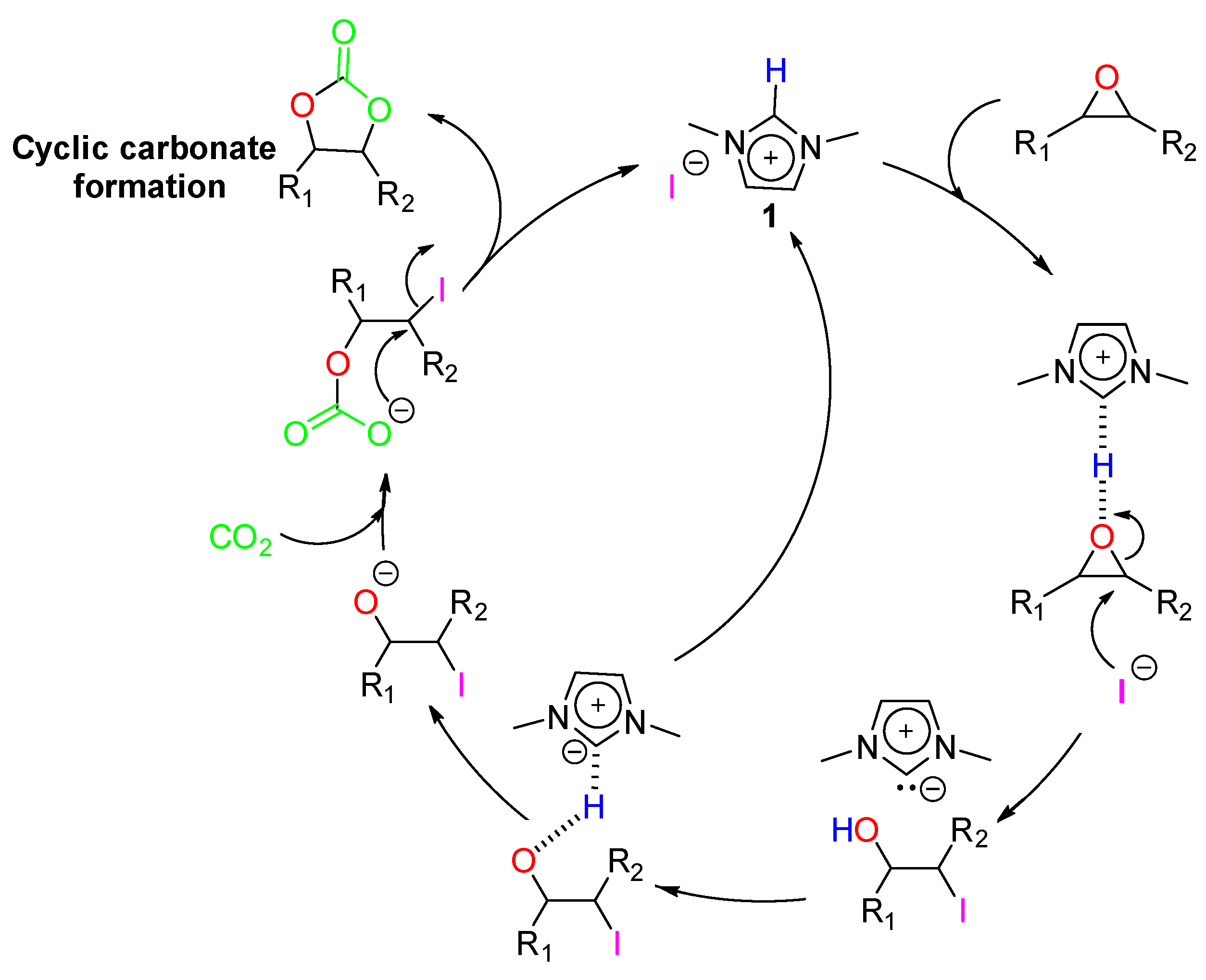
3.2. Reactive Path Computed at DFT Level
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Crippa, M.; Guizzardi, D.; Solazzo, E.; Muntean, M.; Schaaf, E.; Monforti-Ferrario, F.; Banja, M.; Olivier, J.; Grassi, G.; Rossi, S. GHG Emissions of All World Countries; Publications Office of the European Union: Ispra, Italy, 2024. [Google Scholar]
- NOAA’s Global Monitoring Lab National Oceanic and Atmospheric Administration Global Monitoring Lab. Available online: https://www.noaa.gov/climate (accessed on 6 August 2024).
- Nasirov, F.; Nasirli, E.; Ibrahimova, M. Cyclic Carbonates Synthesis by Cycloaddition Reaction of CO2 with Epoxides in the Presence of Zinc-Containing and Ionic Liquid Catalysts. J. Iran. Chem. Soc. 2022, 19, 353–379. [Google Scholar] [CrossRef]
- Nunes, L.J.R. The Rising Threat of Atmospheric CO2: A Review on the Causes, Impacts, and Mitigation Strategies. Environments 2023, 10, 66. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Bobbink, F.D.; Dyson, P.J. Synthesis of Carbonates and Related Compounds Incorporating CO2 Using Ionic Liquid-Type Catalysts: State-of-the-Art and Beyond. J. Catal. 2016, 343, 52–61. [Google Scholar] [CrossRef]
- Shu, D.Y.; Deutz, S.; Winter, B.A.; Baumgärtner, N.; Leenders, L.; Bardow, A. The Role of Carbon Capture and Storage to Achieve Net-Zero Energy Systems: Trade-Offs between Economics and the Environment. Renew. Sustain. Energy Rev. 2023, 178, 113246. [Google Scholar] [CrossRef]
- Bahman, N.; Al-Khalifa, M.; Al Baharna, S.; Abdulmohsen, Z.; Khan, E. Review of Carbon Capture and Storage Technologies in Selected Industries: Potentials and Challenges. Rev. Environ. Sci. Bio/Technol. 2023, 22, 451–470. [Google Scholar] [CrossRef]
- Ewis, D.; Arsalan, M.; Khaled, M.; Pant, D.; Ba-Abbad, M.M.; Amhamed, A.; El-Naas, M.H. Electrochemical Reduction of CO2 into Formate/Formic Acid: A Review of Cell Design and Operation. Sep. Purif. Technol. 2023, 316, 123811. [Google Scholar] [CrossRef]
- Hussain, I.; Alasiri, H.; Khan, W.U.; Alhooshani, K. Advanced Electrocatalytic Technologies for Conversion of Carbon Dioxide into Methanol by Electrochemical Reduction: Recent Progress and Future Perspectives. Coord. Chem. Rev. 2023, 482, 215081. [Google Scholar] [CrossRef]
- Wiranarongkorn, K.; Eamsiri, K.; Chen, Y.-S.; Arpornwichanop, A. A Comprehensive Review of Electrochemical Reduction of CO2 to Methanol: Technical and Design Aspects. J. CO2 Util. 2023, 71, 102477. [Google Scholar] [CrossRef]
- Faria, A.C.; Trujillano, R.; Rives, V.; Miguel, C.V.; Rodrigues, A.E.; Madeira, L.M. Cyclic Operation of CO2 Capture and Conversion into Methane on Ni-Hydrotalcite Based Dual Function Materials (DFMs). J. CO2 Util. 2023, 72, 102476. [Google Scholar] [CrossRef]
- Ren, J.; Lou, H.; Xu, N.; Zeng, F.; Pei, G.; Wang, Z. Methanation of CO/CO2 for Power to Methane Process: Fundamentals, Status, and Perspectives. J. Energy Chem. 2023, 80, 182–206. [Google Scholar] [CrossRef]
- Tlili, A.; Blondiaux, E.; Frogneux, X.; Cantat, T. Reductive Functionalization of CO2 with Amines: An Entry to Formamide, Formamidine and Methylamine Derivatives. Green Chem. 2015, 17, 157–168. [Google Scholar] [CrossRef]
- Tao, D.-J.; Qu, F.; Li, Z.-M.; Zhou, Y. Promoted Absorption of CO at High Temperature by Cuprous-Based Ternary Deep Eutectic Solvents. AIChE J. 2021, 67, e17106. [Google Scholar] [CrossRef]
- Yan, T.; Liu, H.; Zeng, Z.X.; Pan, W.G. Recent Progress of Catalysts for Synthesis of Cyclic Carbonates from CO2 and Epoxides. J. CO2 Util. 2023, 68, 102355. [Google Scholar] [CrossRef]
- Martínez, J.; de la Cruz-Martínez, F.; Martínez de Sarasa Buchaca, M.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; North, M.; Castro-Osma, J.A.; Lara-Sánchez, A. Efficient Synthesis of Cyclic Carbonates from Unsaturated Acids and Carbon Dioxide and Their Application in the Synthesis of Biobased Polyurethanes. Chempluschem 2021, 86, 460–468. [Google Scholar] [CrossRef]
- Andrea, K.A.; Kerton, F.M. Iron-Catalyzed Reactions of CO2 and Epoxides to Yield Cyclic and Polycarbonates. Polym. J. 2021, 53, 29–46. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, Y.; Zhang, X.; Zhang, X.; Liu, H.; Han, B. Recent Advances in the Coupling of CO2 and Epoxides into Cyclic Carbonates under Halogen-Free Condition. Green Chem. Eng. 2020, 1, 82–93. [Google Scholar] [CrossRef]
- Guo, L.; Lamb, K.J.; North, M. Recent Developments in Organocatalysed Transformations of Epoxides and Carbon Dioxide into Cyclic Carbonates. Green Chem. 2021, 23, 77–118. [Google Scholar] [CrossRef]
- Claver, C.; Yeamin, M.B.; Reguero, M.; Masdeu-Bultó, A.M. Recent Advances in the Use of Catalysts Based on Natural Products for the Conversion of CO2 into Cyclic Carbonates. Green Chem. 2020, 22, 7665–7706. [Google Scholar] [CrossRef]
- Martínez, J.; de la Cruz-Martínez, F.; de Sarasa Buchaca, M.M.; Caballero, M.P.; Ojeda-Amador, R.M.; Salvador, M.D.; Fregapane, G.; Tejeda, J.; Castro-Osma, J.A.; Lara-Sánchez, A. Valorization of Agricultural Waste and CO2 into Bioderived Cyclic Carbonates. J. Environ. Chem. Eng. 2021, 9, 105464. [Google Scholar] [CrossRef]
- Werlinger, F.; Caballero, M.P.; Trofymchuk, O.S.; Flores, M.E.; Moreno-Villoslada, I.; de la Cruz-Martínez, F.; Castro-Osma, J.A.; Tejeda, J.; Martínez, J.; Lara-Sánchez, A. Turning Waste into Resources. Efficient Synthesis of Biopolyurethanes from Used Cooking Oils and CO2. J. CO2 Util. 2024, 79, 102659. [Google Scholar] [CrossRef]
- Limburg, B.; Cristòfol, À.; Della Monica, F.; Kleij, A.W. Unlocking the Potential of Substrate-Directed CO2 Activation and Conversion: Pushing the Boundaries of Catalytic Cyclic Carbonate and Carbamate Formation. ChemSusChem 2020, 13, 6056–6065. [Google Scholar] [CrossRef] [PubMed]
- Bhat, G.A.; Darensbourg, D.J. Progress in the Catalytic Reactions of CO2 and Epoxides to Selectively Provide Cyclic or Polymeric Carbonates. Green Chem. 2022, 24, 5007–5034. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, G.; Xie, R.; Yang, L.; Li, B.; Wu, G. Scalable, Durable, and Recyclable Metal-free Catalysts for Highly Efficient Conversion of CO2 to Cyclic Carbonates. Angew. Chemie 2020, 132, 23491–23498. [Google Scholar] [CrossRef]
- Pescarmona, P.P. Cyclic Carbonates Synthesised from CO2: Applications, Challenges and Recent Research Trends. Curr. Opin. Green Sustain. Chem. 2021, 29, 100457. [Google Scholar] [CrossRef]
- Myri, L.; Cooper, J.F. Catalytic Process for Producing Alkylene Carbonates. U.S. Patent 2,773,070, 4 December 1956. [Google Scholar]
- Rollin, P.; Soares, L.K.; Barcellos, A.M.; Araujo, D.R.; Lenardão, E.J.; Jacob, R.G.; Perin, G. Five-Membered Cyclic Carbonates: Versatility for Applications in Organic Synthesis, Pharmaceutical, and Materials Sciences. Appl. Sci. 2021, 11, 5024. [Google Scholar] [CrossRef]
- Zhang, W.; Dai, J.; Wu, Y.-C.; Chen, J.-X.; Shan, S.-Y.; Cai, Z.; Zhu, J.-B. Highly Reactive Cyclic Carbonates with a Fused Ring toward Functionalizable and Recyclable Polycarbonates. ACS Macro Lett. 2022, 11, 173–178. [Google Scholar] [CrossRef]
- Peppel, W.J. Preparation and Properties of the Alkylene Carbonates. Ind. Eng. Chem. 1958, 50, 767–770. [Google Scholar] [CrossRef]
- Schaffner, B.; Schaffner, F.; Verevkin, S.P.; Borner, A. Organic Carbonates as Solvents in Synthesis and Catalysis. Chem. Rev. 2010, 110, 4554–4581. [Google Scholar] [CrossRef]
- Prasad, D.; Patil, K.N.; Chaudhari, N.K.; Kim, H.; Nagaraja, B.M.; Jadhav, A.H. Paving Way for Sustainable Earth-Abundant Metal Based Catalysts for Chemical Fixation of CO2 into Epoxides for Cyclic Carbonate Formation. Catal. Rev. 2022, 64, 356–443. [Google Scholar] [CrossRef]
- Della Monica, F.; Capacchione, C. Recent Advancements in Metal-catalysts Design for CO2/Epoxide Reactions. Asian J. Org. Chem. 2022, 11, e202200300. [Google Scholar] [CrossRef]
- Tyagi, P.; Singh, D.; Malik, N.; Kumar, S.; Singh Malik, R. Metal Catalyst for CO2 Capture and Conversion into Cyclic Carbonate: Progress and Challenges. Mater. Today 2023, 65, 133–165. [Google Scholar] [CrossRef]
- Al-Qaisi, F.M.; Qaroush, A.K.; Okashah, I.K.; Eftaiha, A.F.; Vasko, P.; Alsoubani, F.; Repo, T. The Use of Sustainable Transition Metals for the Cycloaddition of Epoxides and CO2 under Mild Reaction Conditions. Eur. J. Inorg. Chem. 2023, 26, e202200357. [Google Scholar] [CrossRef]
- Tong, H.; Qu, Y.; Li, Z.; He, J.; Zou, X.; Zhou, Y.; Duan, T.; Liu, B.; Sun, J.; Guo, K. Halide-Free Pyridinium Saccharinate Binary Organocatalyst for the Cycloaddition of CO2 into Epoxides. Chem. Eng. J. 2022, 444, 135478. [Google Scholar] [CrossRef]
- Zhang, F.; Bulut, S.; Shen, X.; Dong, M.; Wang, Y.; Cheng, X.; Liu, H.; Han, B. Halogen-Free Fixation of Carbon Dioxide into Cyclic Carbonates via Bifunctional Organocatalysts. Green Chem. 2021, 23, 1147–1153. [Google Scholar] [CrossRef]
- Ravi, S.; Kim, J.; Choi, Y.; Han, H.H.; Wu, S.; Xiao, R.; Bae, Y.-S. Metal-Free Amine-Anchored Triazine-Based Covalent Organic Polymers for Selective CO2 Adsorption and Conversion to Cyclic Carbonates Under Mild Conditions. ACS Sustain. Chem. Eng. 2023, 11, 1190–1199. [Google Scholar] [CrossRef]
- Pal, T.K.; De, D.; Bharadwaj, P.K. Metal–Organic Frameworks for the Chemical Fixation of CO2 into Cyclic Carbonates. Coord. Chem. Rev. 2020, 408, 213173. [Google Scholar] [CrossRef]
- Bondarenko, G.N.; Ganina, O.G.; Lysova, A.A.; Fedin, V.P.; Beletskaya, I.P. Cyclic Carbonates Synthesis from Epoxides and CO2 over NIIC-10 Metal-Organic Frameworks. J. CO2 Util. 2021, 53, 101718. [Google Scholar] [CrossRef]
- Xu, J.-H.; Peng, S.-F.; Shi, Y.-K.; Ding, S.; Yang, G.-S.; Yang, Y.-Q.; Xu, Y.-H.; Jiang, C.-J.; Su, Z.-M. A Novel Zirconium-Based Metal–Organic Framework Covalently Modified by Methyl Pyridinium Bromide for Mild and Co-Catalyst Free Conversion of CO2 to Cyclic Carbonates. Dalt. Trans. 2023, 52, 659–667. [Google Scholar] [CrossRef]
- Zhai, G.; Liu, Y.; Lei, L.; Wang, J.; Wang, Z.; Zheng, Z.; Wang, P.; Cheng, H.; Dai, Y.; Huang, B. Light-Promoted CO2 Conversion from Epoxides to Cyclic Carbonates at Ambient Conditions over a Bi-Based Metal–Organic Framework. Acs Catal. 2021, 11, 1988–1994. [Google Scholar] [CrossRef]
- Norouzi, F.; Abdolmaleki, A. CO2 Conversion into Carbonate Using Pyridinium-Based Ionic Liquids under Mild Conditions. Fuel 2023, 334, 126641. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, D.; Guo, B.; Zhao, J.; Zhou, Z.; Jin, L.; Lei, Y. Chemical Fixation of Carbon Dioxide into Cyclic Carbonates by Multi-Hydroxyl Carboxylic Acid Ionic Liquids under Atmospheric Pressure. Mol. Catal. 2023, 551, 113664. [Google Scholar] [CrossRef]
- Zhao, T.; Long, G.; Liang, H.; Xiong, W.; Hu, X. One-Pot Synthesis of Cyclic Carbonates from Olefins and CO2 Catalyzed by Silica-Supported Imidazolium Hydrogen Carbonate Ionic Liquids. Microporous Mesoporous Mater. 2023, 356, 112576. [Google Scholar] [CrossRef]
- Appaturi, J.N.; Ramalingam, R.J.; Gnanamani, M.K.; Periyasami, G.; Arunachalam, P.; Adnan, R.; Adam, F.; Wasmiah, M.D.; Al-Lohedan, H.A. Review on Carbon Dioxide Utilization for Cycloaddition of Epoxides by Ionic Liquid-Modified Hybrid Catalysts: Effect of Influential Parameters and Mechanisms Insight. Catalysts 2020, 11, 4. [Google Scholar] [CrossRef]
- Yue, S.; Qu, H.; Song, X.; Zang, S.; Deng, G. Hydroxy Acid-Functionalized Ionic Liquids as Green Alternatives for the Efficient Catalytic Conversion of Epoxides to Cyclic Carbonates under Solvent and Co-Catalyst-Free Conditions. Catal. Sci. Technol. 2021, 11, 6999–7008. [Google Scholar] [CrossRef]
- Li, Z.; Sun, J.; Xu, Q.; Yin, J. Homogeneous and Heterogeneous Ionic Liquid System: Promising “Ideal Catalysts” for the Fixation of CO2 into Cyclic Carbonates. ChemCatChem 2021, 13, 1848–1866. [Google Scholar] [CrossRef]
- Comin, E.; Aquino, A.S.; Favero, C.; Mignoni, M.L.; de Souza, R.F.; de Souza, M.O.; Pergher, S.B.C.; da Silva Campos, C.X.; Bernardo-Gusmão, K. Cyclic Carbonate Synthesis via Cycloaddition of CO2 and Epoxides Catalysed by Beta Zeolites Containing Alkyl Imidazolium Ionic Liquids Used as Structure-Directing Agents. Mol. Catal. 2022, 530, 112624. [Google Scholar] [CrossRef]
- Qu, Y.; Chen, Y.; Sun, J. Conversion of CO2 with Epoxides to Cyclic Carbonates Catalyzed by Amino Acid Ionic Liquids at Room Temperature. J. CO2 Util. 2022, 56, 101840. [Google Scholar] [CrossRef]
- de la Cruz-Martínez, F.; Martínez, J.; Gaona, M.A.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Rodríguez, A.M.; Castro-Osma, J.A.; Otero, A.; Lara-Sánchez, A. Bifunctional Aluminum Catalysts for the Chemical Fixation of Carbon Dioxide into Cyclic Carbonates. ACS Sustain. Chem. Eng. 2018, 6, 5322–5332. [Google Scholar] [CrossRef]
- Mesías-Salazar, Á.; Rojas, R.S.; Carrillo-Hermosilla, F.; Martínez, J.; Antiñolo, A.; Trofymchuk, O.S.; Nachtigall, F.M.; Santos, L.S.; Daniliuc, C.G. Guanidinium Iodide Salts as Single Component Catalysts for CO2 to Epoxide Fixation. New J. Chem. 2024, 48, 105–111. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A Generally Applicable Atomic-Charge Dependent London Dispersion Correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef]
- Ásgeirsson, V.; Birgisson, B.O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector Following. J. Chem. Theory Comput. 2021, 17, 4929–4945. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. J. Chem. Phys. 2008, 128, 84106. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural Triple Excitations in Local Coupled Cluster Calculations with Pair Natural Orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse Maps—A Systematic Infrastructure for Reduced-Scaling Electronic Structure Methods. II. Linear Scaling Domain Based Pair Natural Orbital Coupled Cluster Theory. J. Chem. Phys. 2016, 144, 24109. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A. Efficient and Accurate Local Approximations to Coupled-Electron Pair Approaches: An Attempt to Revive the Pair Natural Orbital Method. J. Chem. Phys. 2009, 130, 114108. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Martínez, J.; de la Cruz-Martínez, F.; Gaona, M.A.; Pinilla-Peñalver, E.; Fernández-Baeza, J.; Rodríguez, A.M.; Castro-Osma, J.A.; Otero, A.; Lara-Sánchez, A. Influence of the Counterion on the Synthesis of Cyclic Carbonates Catalyzed by Bifunctional Aluminum Complexes. Inorg. Chem. 2019, 58, 3396–3408. [Google Scholar] [CrossRef] [PubMed]
- Castro-Osma, J.A.; Martínez, J.; de la Cruz-Martínez, F.; Caballero, M.P.; Fernández-Baeza, J.; Rodríguez-López, J.; Otero, A.; Lara-Sánchez, A.; Tejeda, J. Development of Hydroxy-Containing Imidazole Organocatalysts for CO2 Fixation into Cyclic Carbonates. Catal. Sci. Technol. 2018, 8, 1981–1987. [Google Scholar] [CrossRef]
- Martínez de Sarasa Buchaca, M.; de la Cruz-Martínez, F.; Francés-Poveda, E.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Garcés, A.; Castro-Osma, J.A.; Lara-Sánchez, A. Synthesis of Nonisocyanate Poly(Hydroxy)Urethanes from Bis(Cyclic Carbonates) and Polyamines. Polymers 2022, 14, 2719. [Google Scholar] [CrossRef]
- Pronoitis, C.; Hakkarainen, M.; Odelius, K. Structurally Diverse and Recyclable Isocyanate-Free Polyurethane Networks from CO2-Derived Cyclic Carbonates. ACS Sustain. Chem. Eng. 2022, 10, 2522–2531. [Google Scholar] [CrossRef]
- Schmitt, M.; Strehmel, V. Chemical Reaction of Carbon Dioxide with Bisepoxides for Synthesis of Organic Cyclic Dicarbonates at Ambient Pressure for Polyhydroxy Urethane Synthesis. Org. Process Res. Dev. 2020, 24, 2521–2528. [Google Scholar] [CrossRef]
- Grosjean, M.; Berne, D.; Caillol, S.; Ladmiral, V.; Nottelet, B. Dynamic PEG–PLA/Hydroxyurethane Networks Based on Imine Bonds as Reprocessable Elastomeric Biomaterials. Biomacromolecules 2023, 24, 3472–3483. [Google Scholar] [CrossRef]
- de la Cruz-Martínez, F.; Martínez de Sarasa Buchaca, M.; Martínez, J.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Rodríguez-Diéguez, A.; Castro-Osma, J.A.; Lara-Sánchez, A. Synthesis of Bio-Derived Cyclic Carbonates from Renewable Resources. ACS Sustain. Chem. Eng. 2019, 7, 20126–20138. [Google Scholar] [CrossRef]
- de la Cruz-Martínez, F.; Francés-Poveda, E.; North, M.; Castro-Osma, J.A.; Lara-Sánchez, A. Base-Catalyzed Highly Regioselective Synthesis of Bio-Based Hydroxyurethanes. Eur. J. Org. Chem. 2024, 27, e202400275. [Google Scholar] [CrossRef]
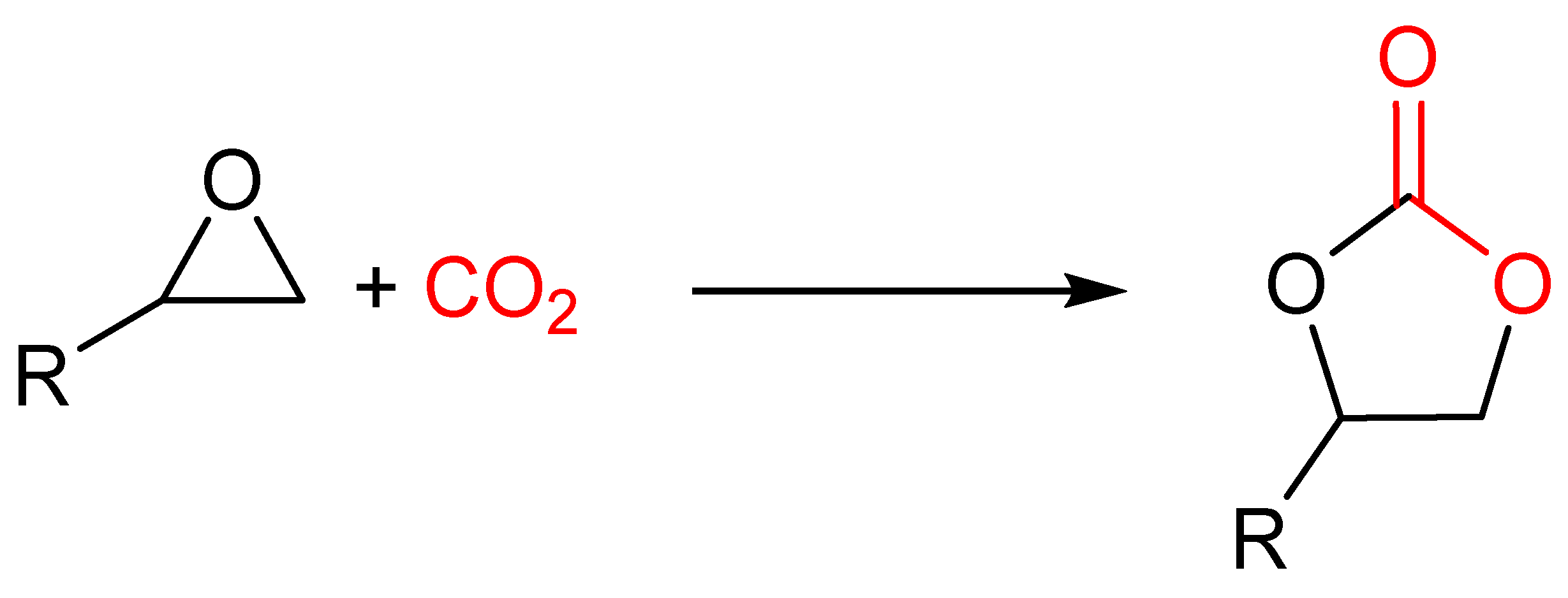



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Conversion 2 (%) | TOF 3 (h−1) |
|---|---|---|---|
| 1 | 1 | 100 | 10 |
| 2 | 2 | 100 | 10 |
| 3 | 3 | 100 | 10 |
| 4 | 4 | 100 | 10 |
| 5 | 5 | 100 | 10 |
| 6 | 1 | 100 | 50 |
| 7 | 2 | 26 | 13 |
| 8 | 3 | 12 | 6 |
| 9 | 4 | 82 | 41 |
| 10 | 5 | 100 | 50 |
| 11 4 | 1 | 97 (95) 5 | 97 |
| 12 4 | 5 | 83 | 83 |
| 13 6 | - | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francés-Poveda, E.; Navarro, M.; Beroíza-Duhart, M.; Mahecha, G.L.; Urzúa, J.I.; Valenzuela, M.L.; de la Cruz-Martínez, F.; Douglas-Gallardo, O.A.; Werlinger, F.; Lara-Sánchez, A.; et al. Effective One-Component Organocatalysts for Eco-Friendly Production of Cyclic Carbonates. Reactions 2025, 6, 8. https://doi.org/10.3390/reactions6010008
Francés-Poveda E, Navarro M, Beroíza-Duhart M, Mahecha GL, Urzúa JI, Valenzuela ML, de la Cruz-Martínez F, Douglas-Gallardo OA, Werlinger F, Lara-Sánchez A, et al. Effective One-Component Organocatalysts for Eco-Friendly Production of Cyclic Carbonates. Reactions. 2025; 6(1):8. https://doi.org/10.3390/reactions6010008
Chicago/Turabian StyleFrancés-Poveda, Enrique, Marta Navarro, Monserrat Beroíza-Duhart, Genesys L. Mahecha, Julio I. Urzúa, María Luisa Valenzuela, Felipe de la Cruz-Martínez, Oscar A. Douglas-Gallardo, Francisca Werlinger, Agustín Lara-Sánchez, and et al. 2025. "Effective One-Component Organocatalysts for Eco-Friendly Production of Cyclic Carbonates" Reactions 6, no. 1: 8. https://doi.org/10.3390/reactions6010008
APA StyleFrancés-Poveda, E., Navarro, M., Beroíza-Duhart, M., Mahecha, G. L., Urzúa, J. I., Valenzuela, M. L., de la Cruz-Martínez, F., Douglas-Gallardo, O. A., Werlinger, F., Lara-Sánchez, A., & Martínez, J. (2025). Effective One-Component Organocatalysts for Eco-Friendly Production of Cyclic Carbonates. Reactions, 6(1), 8. https://doi.org/10.3390/reactions6010008