Expanding the Scope of Asinger Chemistry towards Enantiomerically Pure Secondary Amines and ?-Aminothiols through Chemoenzymatic Derivatization of 3-Thiazolines



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. General Information
2.2. Construction and Preparation of Whole-Cell Catalyst
2.3. 2,2,4,5,5-Pentamethyl-3-thiazoline (1)
2.4. Achiral Gas Chromatography Analysis
2.5. (S)-2,2,4,5,5-Pentamethyl-3-thiazolidine (2)
2.6. N-Isopropyl-3-methylbutan-2-amine (rac-3)
2.7. General Procedure 1 for Derivatization Using Phenylisocyanate
rac-1-Isopropyl-1-(3-methylbutan-2-yl)-3-phenylurea (rac-4)
2.8. Attempts for the Synthesis of (S)-3 According to Ni/Al Alloy Routes a–c
2.8.1. Attempt a
2.8.2. Attempt b
2.8.3. Attempt c
2.9. Attempts for the Synthesis of (S)-3 by Using Raney Ni (W4)
2.9.1. Attempt a
2.9.2. Attempt b
2.10. (S)-N-Isopropyl-3-methylbutan-2-amine (S-3)
2.11. (S)-1-Isopropyl-1-(3-methylbutan-2-yl)-3-phenylurea (S-4)
2.12. General Procedure 2 for Synthesis of β-aminothiols
2.12.1. S-(3-(1-Isopropyl-3-phenylureido)-2-methylbutan-2-yl)phenylcarbamothioate (rac-6)
2.12.2. (S)-S-(3-(1-Isopropyl-3-phenylureido)-2-methylbutan-2-yl)phenylcarbamothioate ((S)-6)
2.13. Chiral Supercritical Fluid Chromatography–High Performance Liquid Chromatography Analysis
3. Results and Discussion
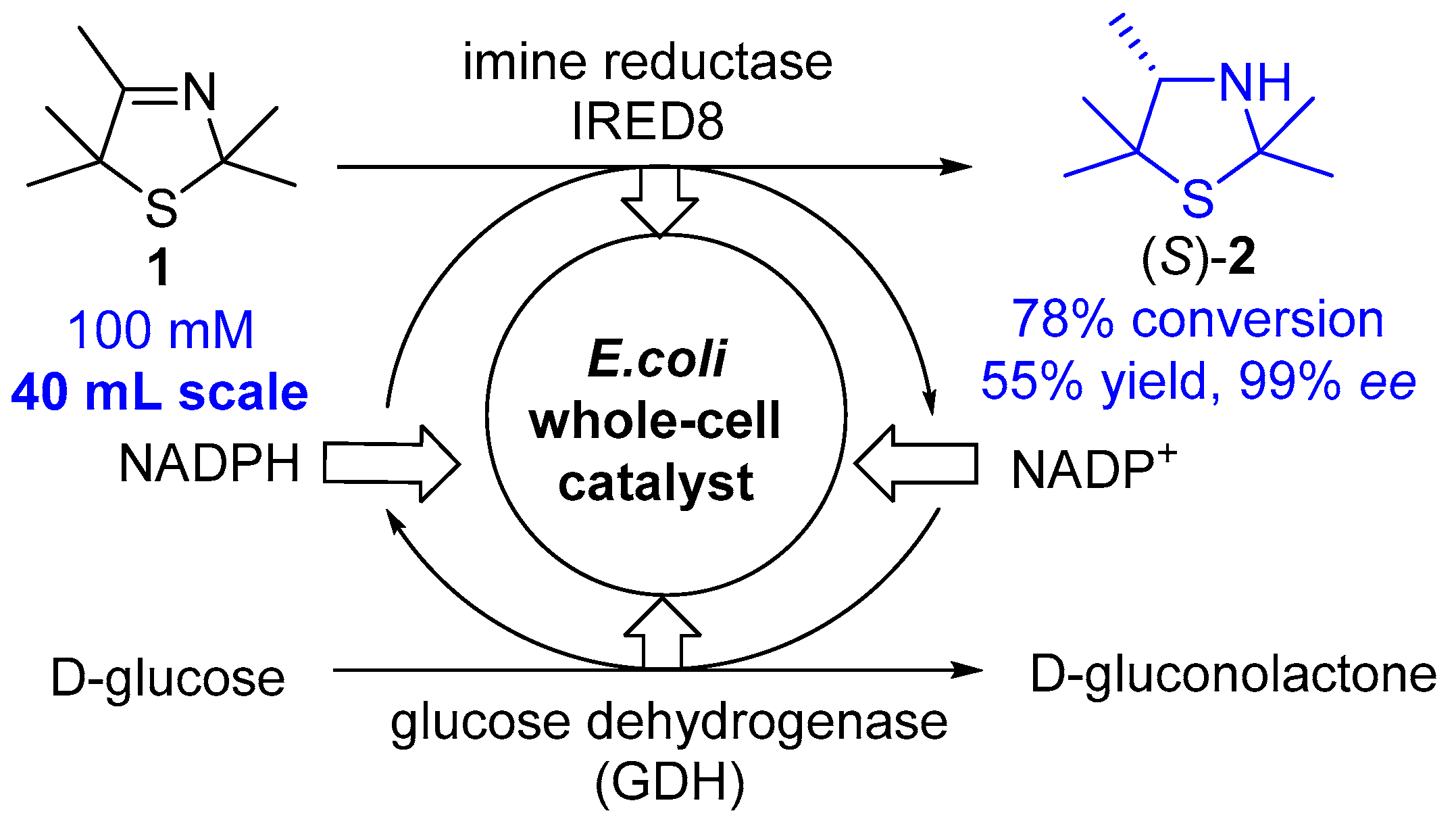
3.1. Biocatalytic Synthesis of an Enantiomerically Pure Thiazolidine
3.2. Reductive Desulfurization of Thiazolidine (S)-2
3.3. Reductive Ring Opening Toward Synthesis of β-aminothiols
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keim, W.; Offermanns, H. Friedrich Asinger (1907–1999): A mediator between basic and applied research. Angew. Chem. Int. Ed. 2007, 46, 6010–6013. [Google Scholar] [CrossRef] [PubMed]
- Asinger, F.; Offermanns, H. Syntheses with ketones, sulfur, and ammonia or amines at room temperature. Angew. Chem. Int. Ed. 1967, 6, 907–919. [Google Scholar] [CrossRef]
- Asinger, F. Über die gemeinsame Einwirkung von Schwefel und Ammoniak auf Ketone. Angew. Chem. 1956, 68, 413. [Google Scholar] [CrossRef]
- Weigert, W.M.; Offermanns, H.; Scherberich, P. d-Penicillamine--production and properties. Angew. Chem. Int. Ed. 1975, 14, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.; Kintscher, J.; Arnold, W. Synthese von 4-Thiazolidinessigsäuren und β-Homopenicillamin. Synthesis 1991, 1991, 497–498. [Google Scholar] [CrossRef]
- Kintscher, J.; Martens, J. Vereinfachte Peptidsynthese mit schutzgruppenfreien Aminosäure-Hydrochloriden nach dem Prinzip der Vierkomponenten-Kondensation. Synthesis 1992, 1992, 837–838. [Google Scholar] [CrossRef]
- Gröger, H.; Hatam, M.; Kintscher, J.; Martens, J. Synthesis of Glutathione Analogues, Peptide Nucleic Acids and Phosphonooligopeptides from Heterocyclic Imines. Synth. Commun. 1996, 26, 3383–3394. [Google Scholar] [CrossRef]
- Drauz, K.; Koban, H.G.; Martens, J.; Schwarze, W. Phosphonic and Phosphinic Acid Analogs of Penicillamine. Liebigs Ann. Chem. 1985, 1985, 448–452. [Google Scholar] [CrossRef]
- Gröger, H.; Saida, Y.; Arai, S.; Martens, J.; Sasai, H.; Shibasaki, M. First catalytic asymmetric hydrophosphonylation of cyclic imines: Highly efficient enantioselective approach to a 4-thiazolidinylphosphonate via chiral titanium and lanthanoid catalysts. Tetrahedron Lett. 1996, 37, 9291–9292. [Google Scholar] [CrossRef]
- Gröger, H.; Saida, Y.; Sasai, H.; Yamaguchi, K.; Martens, J.; Shibasaki, M. A New and Highly Efficient Asymmetric Route to Cyclic α-Amino Phosphonates: The First Catalytic Enantioselective Hydrophosphonylation of Cyclic Imines Catalyzed by Chiral Heterobimetallic Lanthanoid Complexes. J. Am. Chem. Soc. 1998, 120, 3089–3103. [Google Scholar] [CrossRef]
- Schlemminger, I.; Saida, Y.; Gröger, H.; Maison, W.; Durot, N.; Sasai, H.; Shibasaki, M.; Martens, J. Concept of improved rigidity: How to make enantioselective hydrophosphonylation of cyclic imines catalyzed by chiral heterobimetallic lanthanoid complexes almost perfect. J. Org. Chem. 2000, 65, 4818–4825. [Google Scholar] [CrossRef] [PubMed]
- Zumbrägel, N.; Merten, C.; Huber, S.M.; Gröger, H. Enantioselective reduction of sulfur-containing cyclic imines through biocatalysis. Nat. Commun. 2018, 9, 1949. [Google Scholar] [CrossRef] [PubMed]
- Zumbrägel, N.; Gröger, H. Merging Heterocyclic Chemistry and Biocatalysis in One-Pot Processes through Compartmentalization of the Reaction Steps. Bioengineering 2018, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Zumbrägel, N.; Gröger, H. One-pot synthesis of a 3-thiazolidine through combination of an Asinger-type multi-component-condensation reaction with an enzymatic imine reduction. J. Biotechnol. 2019, 291, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Meinzer, A.; Breckel, A.; Thaher, B.A.; Manicone, N.; Otto, H.-H. Properties and Reactions of Substituted 1,2-Thiazetidine 1,1-Dioxides: Chiral Mono- and Bicyclic 1,2-Thiazetidine 1,1-Dioxides from α-Amino Acids. Helv. Chim. Acta 2004, 87, 90–105. [Google Scholar] [CrossRef]
- Reiners, I.; Gröger, H.; Martens, J. A New Enantioselective Synthetic Approach to β-Aminothio-Compoundsvia enantioselective reduction of N,S-heterocyclic imines. J. Prakt. Chem. 1997, 339, 541–546. [Google Scholar] [CrossRef]
- Papa, D.; Schwenk, E.; Whitman, B. Reductions with nickel-aluminium alloy and aqueous alkali. J. Org. Chem. 1942, 07, 587–590. [Google Scholar] [CrossRef]
- Tsukinoki, T.; Kanda, T.; Liu, G.-B.; Tsuzuki, H.; Tashiro, M. Organic reaction in water. Part 3: A facile method for reduction of aromatic rings using a raney Ni–Al alloy in dilute aqueous alkaline solution under mild conditions. Tetrahedron Lett. 2000, 41, 5865–5868. [Google Scholar] [CrossRef]
- Mozingo, R.; Wolf, D.E.; Harris, S.A.; Folkers, K. Hydrogenolysis of Sulfur Compounds by Raney Nickel Catalyst. J. Am. Chem. Soc. 1943, 65, 1013–1016. [Google Scholar] [CrossRef]
- Mozingo, R. Catalyst, Raney Nickel, W-2. Org. Synth. 1941, 21, 15. [Google Scholar] [CrossRef]
- Rentner, J.; Kljajic, M.; Offner, L.; Breinbauer, R. Recent advances and applications of reductive desulfurization in organic synthesis. Tetrahedron 2014, 70, 8983–9027. [Google Scholar] [CrossRef]
- Keefer, L.K.; Lunn, G. Nickel-aluminum alloy as a reducing agent. Chem. Rev. 1989, 89, 459–502. [Google Scholar] [CrossRef]
- Meng, X.; Li, L.; Li, K.; Zhou, P.; Zhang, H.; Jia, J.; Sun, T. Desulfurization of fuels with sodium borohydride under the catalysis of nickel salt in polyethylene glycol. J. Clean Prod. 2018, 176, 391–398. [Google Scholar] [CrossRef]
- Ohshima, T.; Xu, Y.; Takita, R.; Shimizu, S.; Zhong, D.; Shibasaki, M. Enantioselective total synthesis of (-)-strychnine using the catalytic asymmetric Michael reaction and tandem cyclization. J. Am. Chem. Soc. 2002, 124, 14546–14547. [Google Scholar] [CrossRef] [PubMed]
- Wetzl, D.; Berrera, M.; Sandon, N.; Fishlock, D.; Ebeling, M.; Müller, M.; Hanlon, S.; Wirz, B.; Iding, H. Expanding the Imine Reductase Toolbox by Exploring the Bacterial Protein-Sequence Space. ChemBioChem 2015, 16, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Figueroa, E.; Chaparro-Riggers, J.; Bommarius, A.S. Development of a thermostable glucose dehydrogenase by a structure-guided consensus concept. ChemBioChem 2007, 8, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Hutchby, M.; Houlden, C.E.; Ford, J.G.; Tyler, S.N.G.; Gangné, M.R.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Hindered Ureas as Masked Isocyanates: Facile Carbamolyation of Nucleophiles under Neutral Conditions. Angew. Chem. Int. Ed. 2009, 48, 8721–8724. [Google Scholar] [CrossRef]
- Thiel, M.; Asinger, F.; Häussler, K.; Körner, T. Über die gemeinsame Einwirkung von elementarem Schwefel und gasförmigem Ammoniak auf Ketone, XXII: Mercaptoamine durch Reduktion von Thiazolinen-Δ3 oder Dihydro-metathiazinen-Δ3 mit Lithiumalanat. Liebigs Ann. Chem. 1959, 622, 107–116. [Google Scholar] [CrossRef]
- Jeon, H.B.; Jang, Y. Reversible inactivation of bovine plasma amine oxidase by cysteamine and related analogs. Biochem. Biophys. Res. Commun. 2010, 403, 442–446. [Google Scholar] [CrossRef]
- Reisser, M.; Schmidt, B.F.; Brown, W. Synthesis, Characterization, and Solvolysis of Mono- and Bis-S-(glutathionyl) Adducts of Methylene-bis-(phenylisocyate) (MDI). Chem. Res. Toxicol. 2002, 15, 1235–1241. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyseni, M.; Zumbrägel, N.; Offermanns, H.; Gröger, H. Expanding the Scope of Asinger Chemistry towards Enantiomerically Pure Secondary Amines and ?-Aminothiols through Chemoenzymatic Derivatization of 3-Thiazolines. Chemistry 2019, 1, 180-191. https://doi.org/10.3390/chemistry1010012
Hyseni M, Zumbrägel N, Offermanns H, Gröger H. Expanding the Scope of Asinger Chemistry towards Enantiomerically Pure Secondary Amines and ?-Aminothiols through Chemoenzymatic Derivatization of 3-Thiazolines. Chemistry. 2019; 1(1):180-191. https://doi.org/10.3390/chemistry1010012
Chicago/Turabian StyleHyseni, Mentor, Nadine Zumbrägel, Heribert Offermanns, and Harald Gröger. 2019. "Expanding the Scope of Asinger Chemistry towards Enantiomerically Pure Secondary Amines and ?-Aminothiols through Chemoenzymatic Derivatization of 3-Thiazolines" Chemistry 1, no. 1: 180-191. https://doi.org/10.3390/chemistry1010012
APA StyleHyseni, M., Zumbrägel, N., Offermanns, H., & Gröger, H. (2019). Expanding the Scope of Asinger Chemistry towards Enantiomerically Pure Secondary Amines and ?-Aminothiols through Chemoenzymatic Derivatization of 3-Thiazolines. Chemistry, 1(1), 180-191. https://doi.org/10.3390/chemistry1010012