
Molecular Docking and Molecular Dynamics Simulation Studies of Quinoline-3-Carboxamide Derivatives with DDR Kinases–Selectivity Studies towards ATM Kinase


Abstract
:1. Introduction
2. Materials and Methods
Tools and Software
3. General Procedure
3.1. Molecular Docking
3.2. Molecular Dynamics Simulation
3.3. Multiple Sequence Alignment Analysis
4. Results
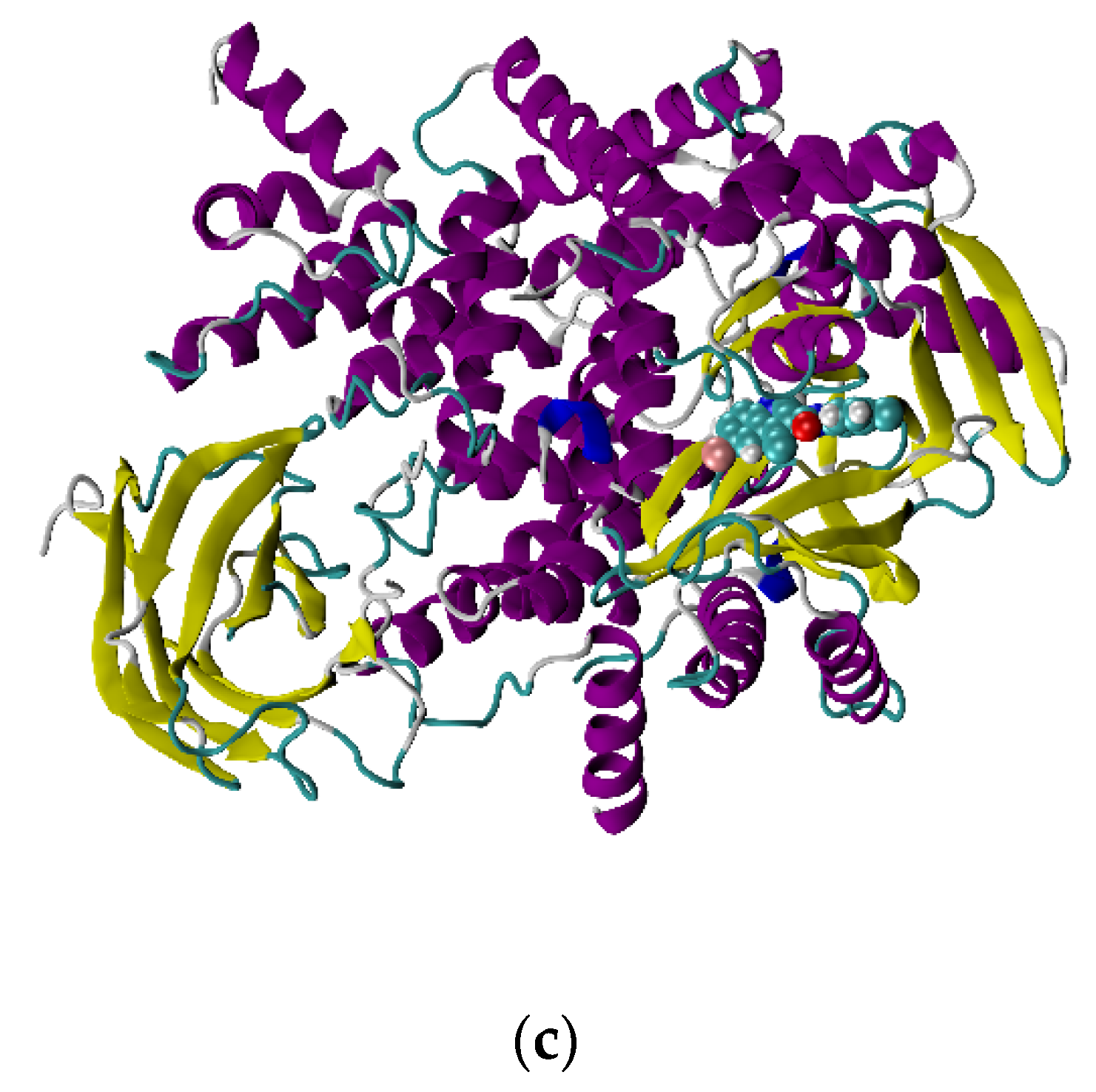
4.1. Molecular Docking Studies
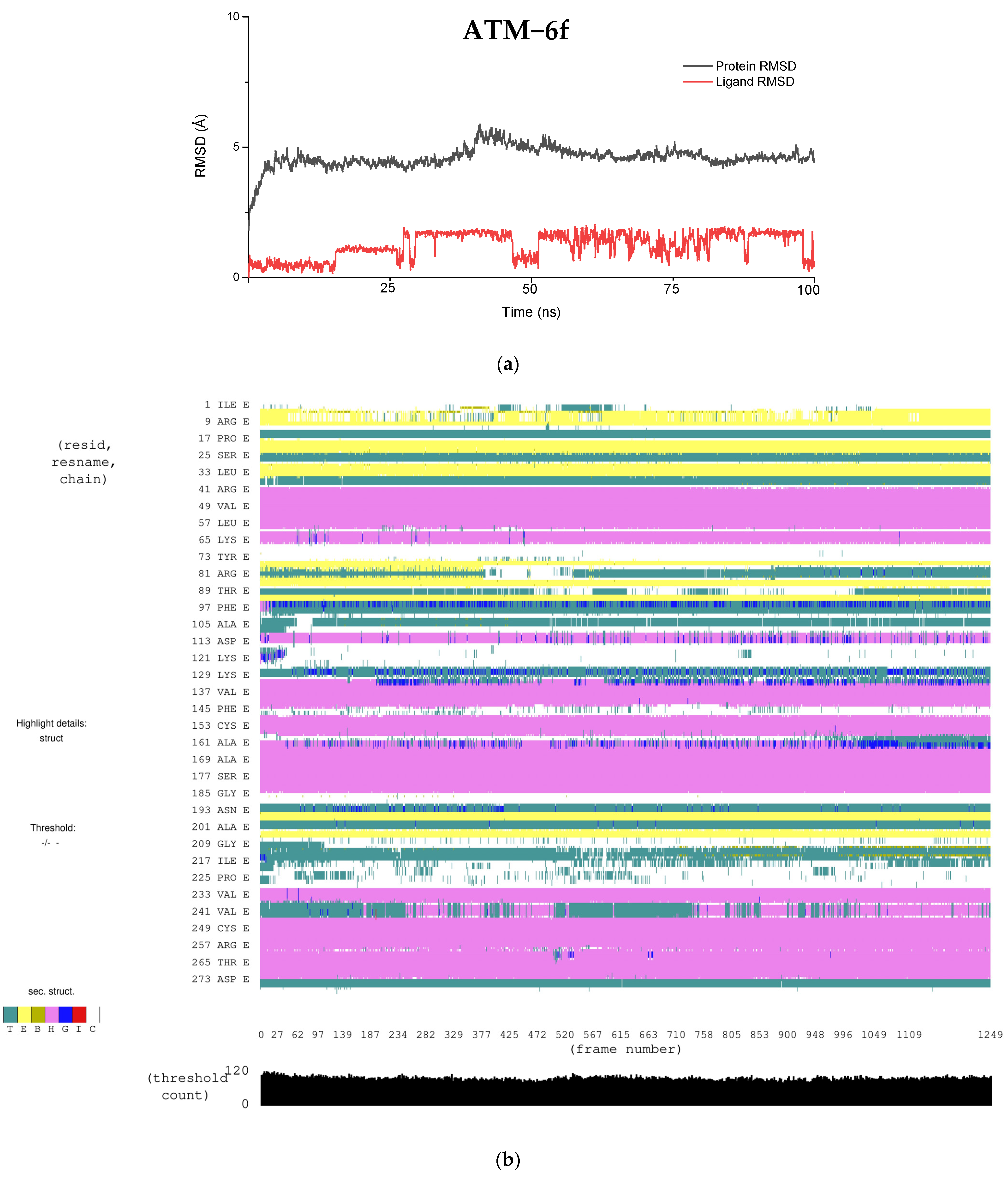
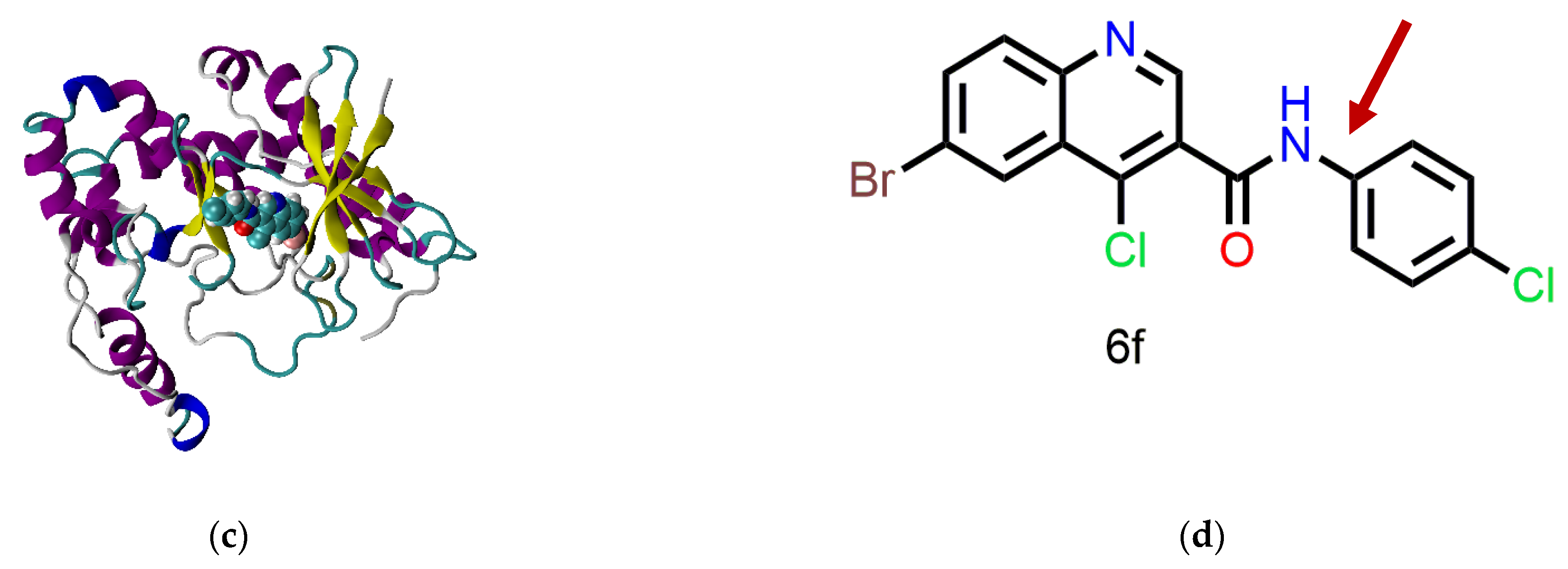
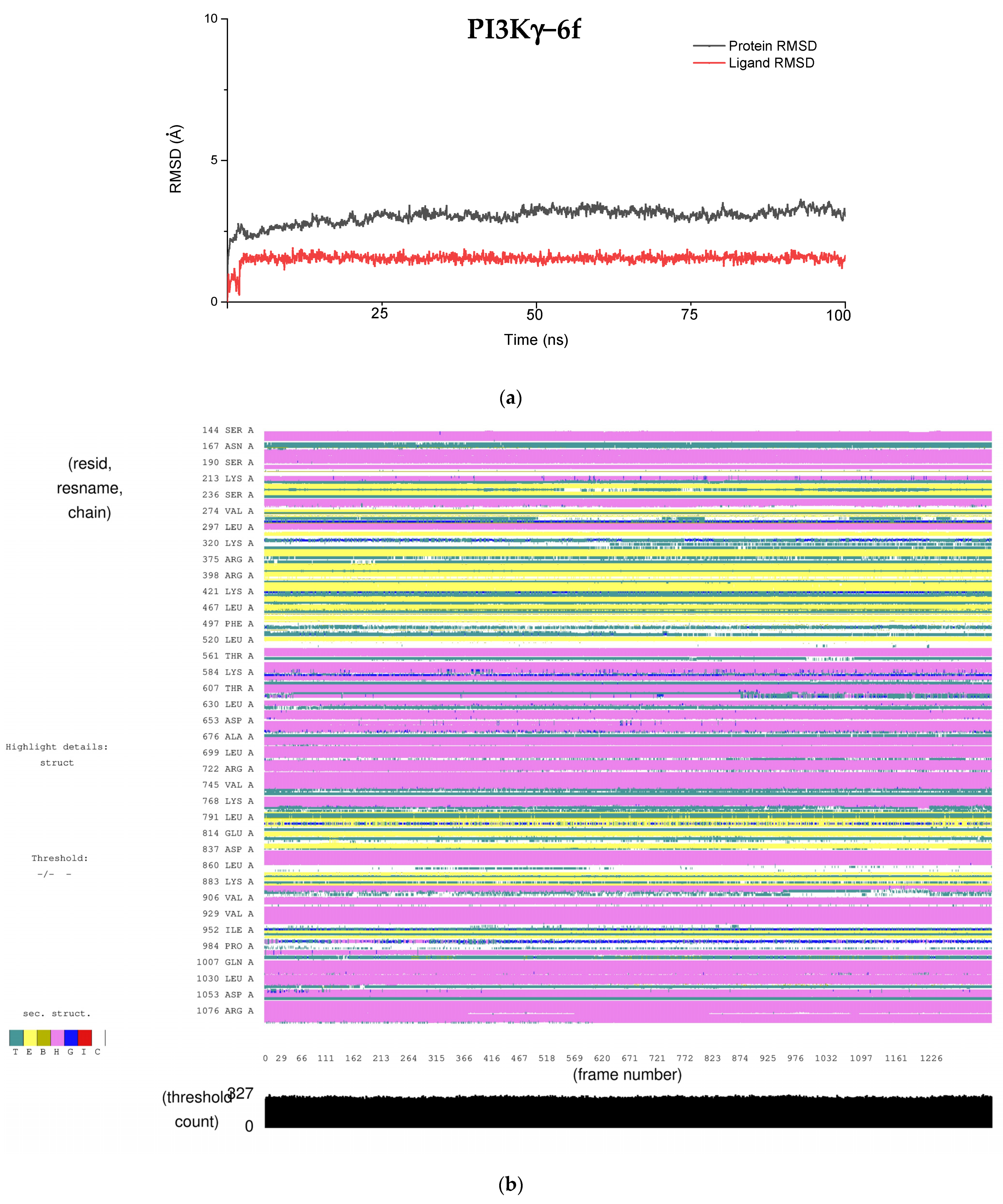
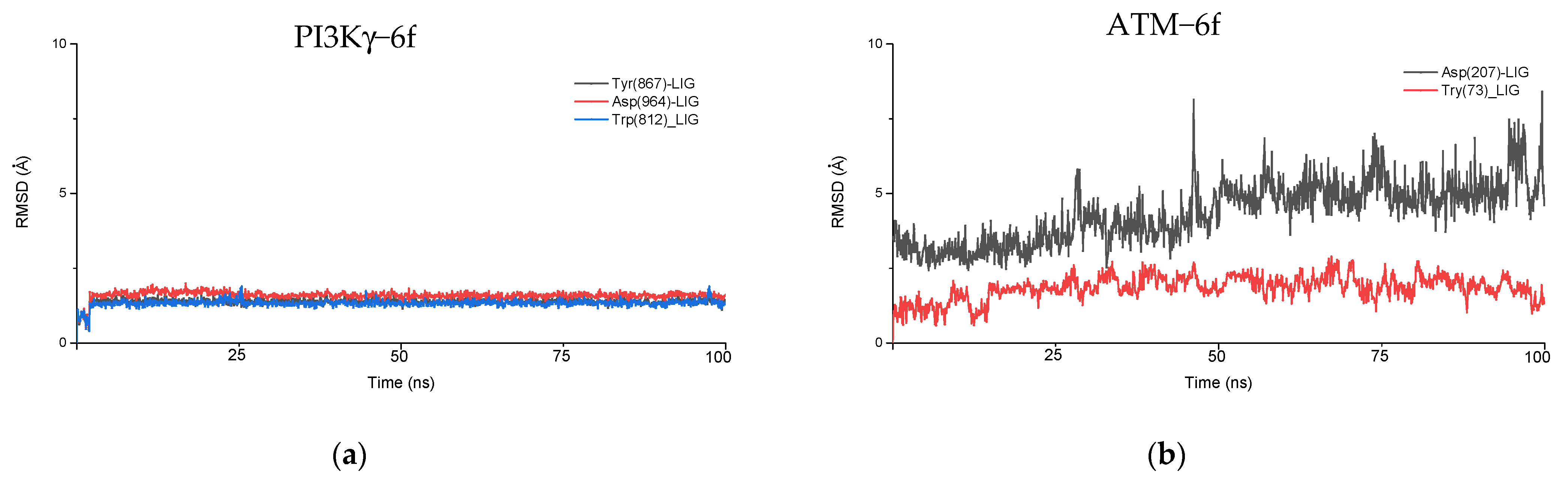
4.2. Molecular Dynamics Simulation
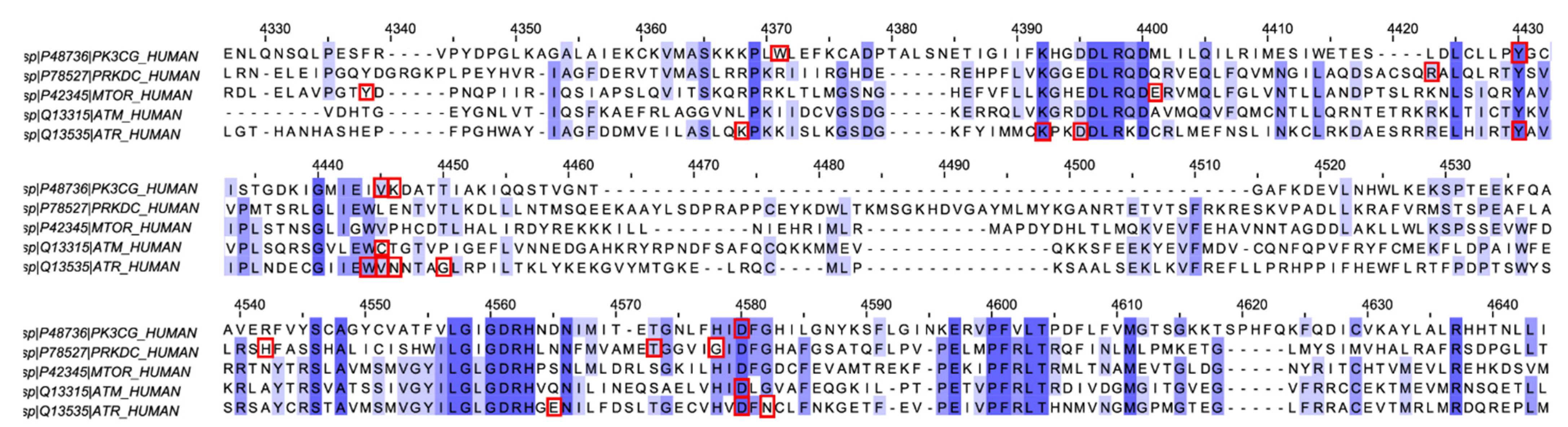
4.3. Multiple Sequence Alignment (MSA) Analysis
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PIKKs | Phosphatidylinositol 3−kinase−related kinases |
| ATP | Adenosine triphosphate |
| ATM | Ataxia−Telangiesctasia Mutated |
| DNA | Deoxyribonucleic acid |
| DDR | DNA Damage and Response |
| ATR | Ataxia Telangiectasia and Rad3−related |
| DNA-PKcs | DNA dependent Protein Kinase catalytic subunit |
| mTOR | mammalian Target of Rapamycin |
| PI3Kγ | Phosphoinositide 3−kinaseγ |
| ADME | Adsorption, Distribution, Metabolism, Excretion |
| MD | Molecular Dynamics |
| SMG1 | Suppressor of Morphogenesis in Genitalia |
| TRRAP | Transformation/transcription domain−associated protein |
| DSB | Double−stranded break |
| SAR | Structure Activity Relationship |
| MSA | Multiple Sequence Alignment |
| CryoEM | Cryo−Electron Microscopy |
| NAMD | Nanoscale Molecular Dynamics |
| CHARRM36 | Chemistry at Harvard Macromolecular Mechanics 36 |
| CHARMM | GUI−Chemistry at Harvard Macromolecular Mechanics–Graphic User Interface |
| LJ cutoff | Lennard Jones |
| PME | Particle Mesh Ewald |
| NPT | Isothermal−Isobaric ensemble |
| VMD | Visual Molecular Dynamics |
| RMSD | Root Mean Square Deviation |
References
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, Y.; Lu, X. Regulators in the DNA damage response. Arch. Biochem. Biophys. 2016, 594, 18–25. [Google Scholar] [CrossRef]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Yang, J.; Yu, Y.; Hamrick, H.E.; Duerksen-Hughes, P.J. ATM, ATR and DNA-PK: Initiators of the cellular genotoxic stress responses. Carcinogenesis 2003, 24, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [Green Version]
- Velic, D.; Couturier, A.M.; Ferreira, M.T.; Rodrigue, A.; Poirier, G.G.; Fleury, F.; Masson, J.Y. DNA damage signalling and repair inhibitors: The long-sought-after achilles’ heel of cancer. Biomolecules 2015, 5, 3204–3259. [Google Scholar] [CrossRef] [Green Version]
- Davidson, D.; Amrein, L.; Panasci, L.; Aloyz, R.; Batist, G.; Wu, J.H. Small molecules, inhibitors of DNA-PK, targeting DNA repair, and beyond. Front. Pharmacol. 2013, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Finlay, M.R.V.; Griffin, R.J. Modulation of DNA repair by pharmacological inhibitors of the PIKK protein kinase family. Bioorg. Med. Chem. Lett. 2012, 22, 5352–5359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Calzada, A.; López-Perrote, A.; Melero, R.; Boskovic, J.; Muñoz-Hernandez, H.; Martino, F.; Llorca, O. Structure and Assembly of the PI 3 K-like Protein Kinases (PIKKs) Revealed by Electron Microscopy. AIMS Biophys. 2015, 2, 36–57. [Google Scholar] [CrossRef]
- Stracker, T.H.; Roig, I.; Knobel, P.A.; Marjanović, M. The ATM signaling network in development and disease. Front. Genet. 2013, 4, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ca, C.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Yan, H.; Guo, W.; Tang, M.; Zhao, X.; Tong, A.; Peng, Y.; Li, Q.; Yuan, Z. ATM inhibition induces synthetic lethality and enhances sensitivity of PTEN-deficient breast cancer cells to cisplatin. Exp. Cell Res. 2018, 366, 24–33. [Google Scholar] [CrossRef]
- Khalil, H.; Tummala, H.; Chakarov, S.; Zhelev, N.; Lane, D. Targeting ATM pathway for therapeutic intervention in cancer. Biodiscovery 2012, 1, e8920. [Google Scholar] [CrossRef] [Green Version]
- Ravi, S.; Barui, S.; Kirubakaran, S.; Duhan, P.; Bhowmik, K. Synthesis and Characterization of Quinoline-3-Carboxamide Derivatives as Inhibitors of the ATM Kinase. Curr. Top. Med. Chem. 2020, 20, 2070–2079. [Google Scholar] [CrossRef]
- Degorce, S.L.; Barlaam, B.; Cadogan, E.; Dishington, A.; Ducray, R.; Glossop, S.C.; Hassall, L.A.; Lach, F.; Lau, A.; McGuire, T.M.; et al. Discovery of Novel 3-Quinoline Carboxamides as Potent, Selective, and Orally Bioavailable Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase. J. Med. Chem. 2016, 59, 6281–6292. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, X.; Chen, Y.; Cheung, J.C.; Wang, H.; Jiang, J.; de Val, N.; Fox, T.; Gellert, M.; Yang, W. Structure of an activated DNA-PK and its implications for NHEJ. Mol. Cell 2021, 81, 801–810.e3. [Google Scholar] [CrossRef]
- Shaik, A.; Bhakuni, R.; Kirubakaran, S. Design, synthesis, and docking studies of New Torin2 analogs as potential ATR/mTOR kinase inhibitors. Molecules 2018, 23, 992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys 2020, 153, 44130. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
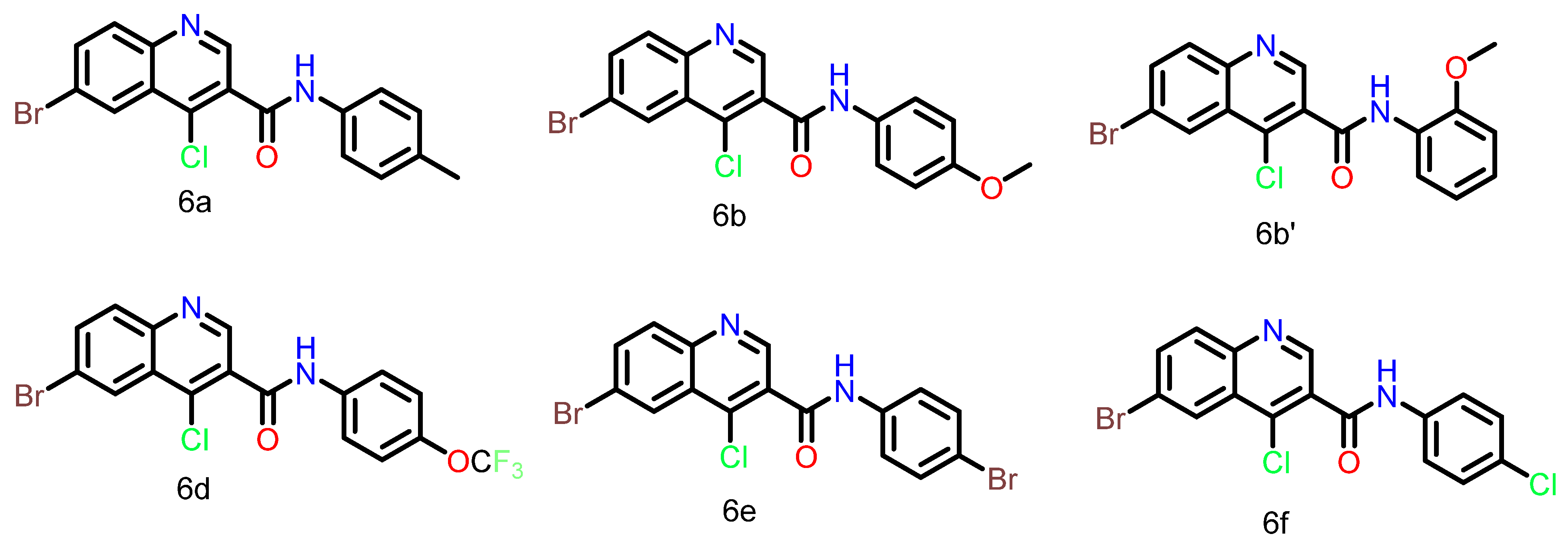
| Compound | HCT116 | MDA-MB-468 | MDA-MB-231 |
|---|---|---|---|
| 6a | 14.31 | 20.16 | 27.14 |
| 6b | 11.80 | 17.51 | 23.74 |
| 6b’ | 21.03 | Not available | Not available |
| 6d | 33.24 | 35.60 | 31.81 |
| 6e | 57.49 | 85.84 | 192.5 |
| 6f | 53.51 | 43.47 | 39.20 |
| KU60019 | 9.80 | 8.04 | 8.21 |
| ATM | ATR | DNA-PKcs | mTOR | PI3Kγ | |
|---|---|---|---|---|---|
| PDB ID | Modeled using 5G55 and 5NP0 | Modeled using 4JSP | Modeled using 7k0y.1 | 5WBY | 5G55 |
| Residues used for docking | 2683–2962 | 2293–2567 | 3747–4015 | Full length | Full length |
| Organism | Human | Human | Human | Human | Human |
| Ligand | QPlogS | QPPCaCo | % Human Oral Absorption | QPlogKhsa | QLogBB | Mol. Wt. | HBD | HBA | QPlogP (o/w) |
|---|---|---|---|---|---|---|---|---|---|
| 6a | −5.992 | 2599.950 | 100 | 0.570 | 0.143 | 375.652 | 1 | 3.50 | 4.498 |
| 6b | −5.739 | 2601.497 | 100 | 0.428 | 0.080 | 391.651 | 1 | 4.25 | 4.308 |
| 6b’ | −5.681 | 2513.798 | 100 | 0.443 | 0.063 | 391.651 | 1 | 4.25 | 4.315 |
| 6d | −6.937 | 2612.762 | 100 | 0.687 | 0.368 | 445.623 | 1 | 3.50 | 5.337 |
| 6e | −6.262 | 2594.035 | 100 | 0.550 | 0.332 | 440.521 | 1 | 3.50 | 4.754 |
| 6f | −6.143 | 2590.807 | 100 | 0.526 | 0.321 | 396.070 | 1 | 3.50 | 4.675 |
| Ligands | ATM | ATR | DNA-PKcs | mTOR | PI3Kγ |
|---|---|---|---|---|---|
| 6a | −9.8 | −3.9 | −3.7 | −3.6 | −4.6 |
| 6b | −10.1 | −2.5 | −4.0 | −2.7 | −5.0 |
| 6b’ | −10.2 | −3.0 | −1.7 | −2.7 | −6.1 |
| 6d | −10.3 | −3.3 | −3.0 | −3.2 | −4.8 |
| 6e | −10.1 | −3.0 | −3.8 | −2.6 | −4.5 |
| 6f | −10.4 | −3.5 | −3.8 | −2.1 | −4.7 |
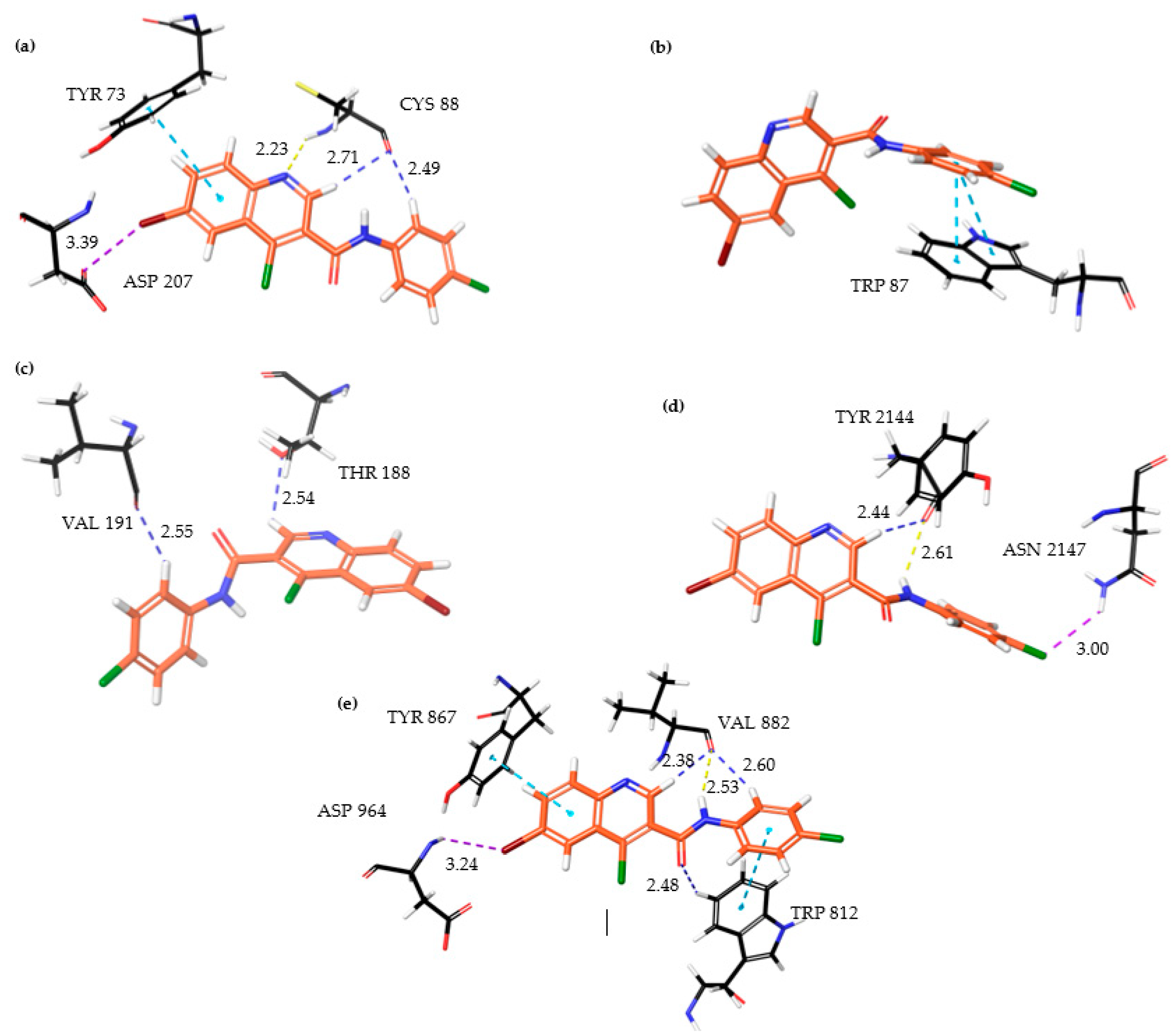
| Protein | Interacting Residues | Type of Interactions |
|---|---|---|
| ATM | ASP 207, TYR 73, CYS 88 | Halogen bonding, π−π stacking interaction, hydrogen bonding, aromatic hydrogen bonding |
| ATR | TRP 87 | π−π stacking interaction |
| DNA−PKcs | THR 188, VAL 191 | Aromatic hydrogen bonding |
| mTOR | TYR 2144, ASN 2147 | Aromatic hydrogen bonding, hydrogen bonding, halogen bonding |
| PI3Kγ | ASP 964, TYR 867, VAL 882, TRP 812 | Halogen bonding, π−π stacking interaction, aromatic hydrogen bonding, hydrogen bonding |
| Property | ATM (6f) | PI3Kγ (6f) |
|---|---|---|
| LipophilicEvdW | −4.87 | −3.76 |
| PhobEnHB | −1.5 | −1.5 |
| HBond | −1.03 | −0.7 |
| Electro | −0.31 | −0.49 |
| Penalties | 0.17 | 1.81 |
| ExposPenal | 0 | 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravi, S.; Priya, B.; Dubey, P.; Thiruvenkatam, V.; Kirubakaran, S. Molecular Docking and Molecular Dynamics Simulation Studies of Quinoline-3-Carboxamide Derivatives with DDR Kinases–Selectivity Studies towards ATM Kinase. Chemistry 2021, 3, 511-524. https://doi.org/10.3390/chemistry3020036
Ravi S, Priya B, Dubey P, Thiruvenkatam V, Kirubakaran S. Molecular Docking and Molecular Dynamics Simulation Studies of Quinoline-3-Carboxamide Derivatives with DDR Kinases–Selectivity Studies towards ATM Kinase. Chemistry. 2021; 3(2):511-524. https://doi.org/10.3390/chemistry3020036
Chicago/Turabian StyleRavi, Srimadhavi, Bhanu Priya, Pankaj Dubey, Vijay Thiruvenkatam, and Sivapriya Kirubakaran. 2021. "Molecular Docking and Molecular Dynamics Simulation Studies of Quinoline-3-Carboxamide Derivatives with DDR Kinases–Selectivity Studies towards ATM Kinase" Chemistry 3, no. 2: 511-524. https://doi.org/10.3390/chemistry3020036
APA StyleRavi, S., Priya, B., Dubey, P., Thiruvenkatam, V., & Kirubakaran, S. (2021). Molecular Docking and Molecular Dynamics Simulation Studies of Quinoline-3-Carboxamide Derivatives with DDR Kinases–Selectivity Studies towards ATM Kinase. Chemistry, 3(2), 511-524. https://doi.org/10.3390/chemistry3020036