Abstract
This review summarizes the results on the aromaticity of a series of synthesized and hypothetical neutral heterocirculene molecules and their double charged ions. The aromaticity of heterocirculenes is a direct reflection of their electronic structure responsible for the specific optoelectronic and photophysical properties. We show how the presence of a heteroatom in the outer macrocycle affects the aromaticity of hetero[8]circulenes. In addition, we also describe the change in aromaticity and strain energy for a series of the “lower” (n < 8) and “higher” (n > 8) hetero[n]circulenes. It was demonstrated that the loss of planarity with increased strain leads to an increased antiaromaticity of the lower hetero[n]circulenes, whereas higher hetero[n]circulenes demonstrate a more pronounced aromatic nature because of the small departure from planarity of each heteroarene ring in hetero[n]circulene molecule. Finally, we discuss the aromatic nature of the first examples of π-extended hetero[8]circulenes.
1. Introduction
Aromaticity is one of the central concepts in chemistry [1], which implies a set of special features (criteria) inherent to a number of cyclic conjugated molecules to one degree or another. In organic chemistry, aromaticity is associated with a stable (often symmetric) structure, such as benzene, with delocalized π-electrons satisfying the 4n + 2 rule, which exhibits a diamagnetic ring current in the external magnetic fields.
Aromaticity has a quantum nature, and it is impossible to explain aromaticity from the standpoint of classical structural or resonance theory. Thus, a number of approaches were proposed to understand aromaticity in a more profound meaning, to evaluate the aromaticity measure qualitatively as well as quantitatively. Among them, structural [2,3], energetic [4], magnetic [5,6,7,8,9,10], electronic [11,12], and reactivity-based [13] criteria were developed. The problem of aromaticity definition is associated with its multidimensional nature, which is not completely determined [14].
The study of the aromaticity of polyheterocyclic systems is a complex task and requires the application of several simultaneous criteria. For such interesting compounds as heterocirculenes, aromaticity is a key feature that explains their specific electronic properties [15,16,17,18]. During the last decade, heterocirculenes have attracted much attention [19,20,21,22] because of their applicability in modern optoelectronic devices, such as organic light emitting diodes (OLEDs) [23,24,25,26] and organic field-effect transistors (OFETs) [27,28,29,30,31]. From the point of view of aromaticity, heterocirculenes can be considered as a specific class of compounds exhibiting a particular ‘‘bifacial’’ aromatic/antiaromatic nature. In particular, all the heterocirculene molecules contain the inner antiaromatic hub-system due to the presence of magnetically induced paratropic ring currents, and the aromatic outer part of the macrocycle (the rim-system), which possesses the diatropic (“aromatic”) ring current [32,33,34]. Thus, the heterocirculene molecules can be considered as nonaromatic compounds, because the sum of the diatropic and paratropic ring currents of the hub and the rim almost vanishes.
The most simple and widespread criterion for assessing the aromaticity of heterocirculenes is considered to be magnetism, which is implemented within the framework of the nucleus independent chemical shift (NICS) [7,35], the gauge including magnetically induced current (GIMIC) [9,10], and anisotropy of the current-induced density (ACID) plot [36] approaches. NICS is a probe (local measure) of the induced magnetic field (derived from the shielding tensor), which is the consequence of the induced currents. NICS cannot be used to correctly describe “global” aromaticity in complex systems [37,38], such as polycyclic hydrocarbons [39,40], porphyrins [41], and all-metal aromatic compounds [42,43,44], as well as some dianionic [8]circulenes [34]. The NICS criterion is useful to evaluate local aromaticity of an individual ring included in the polyheterocyclic system [45,46,47,48]. Therefore, it is appropriate to apply the GIMIC method, which allows for evaluating the aromaticity of molecules as a whole, or ACID plot calculations for visualizing ring currents and electron delocalization in polyheterocyclic systems. Aromatic molecules are characterized by a reduced molecular energy due to the specific cyclic conjugation of the π-electron systems. In this regard, the topological graph-theoretical energetic indices including topological resonance energy (TRE), bond resonance energy (BRE) and circuit resonance energy (CRE) were developed, which made it possible to unify the graph theories of aromatic stabilization and ring current diamagnetism [49]. Both TRE and ring current diamagnetism have the same nature of origin, arising from the same set of circuits in the π-system. An energetic quantity equivalent to TRE can be theoretically derived from the diamagnetic ring current’s susceptibility, although the diamagnetic ring current’s susceptibility itself is not proportional to the TRE [49].
This review presents the current state of aromaticity research regarding a series of partially and fully heteroannelated circulenes with different heteroatoms in the outer macrocycle.
2. Aromaticity of Partially Heteroannelated[8]Circulenes
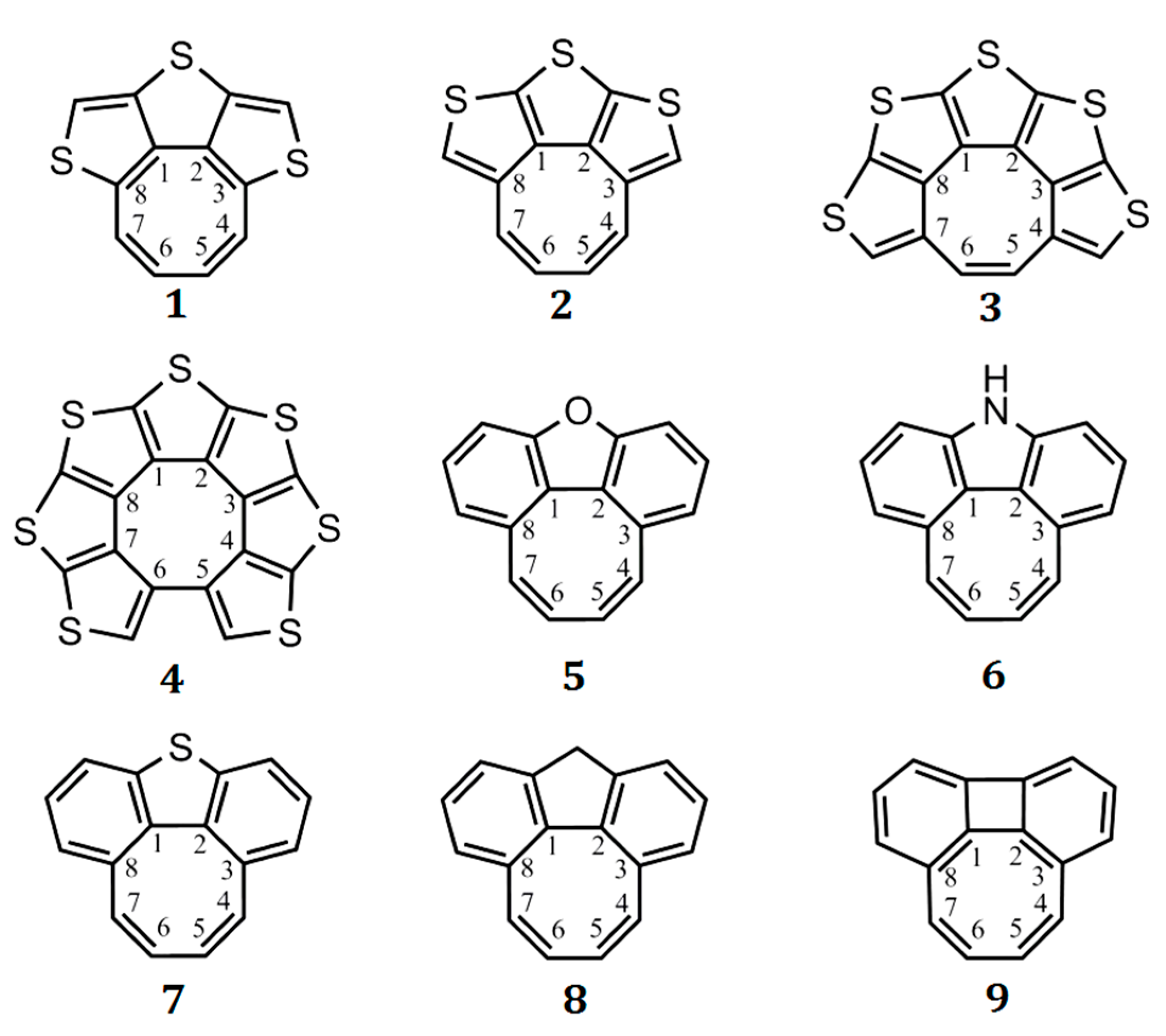
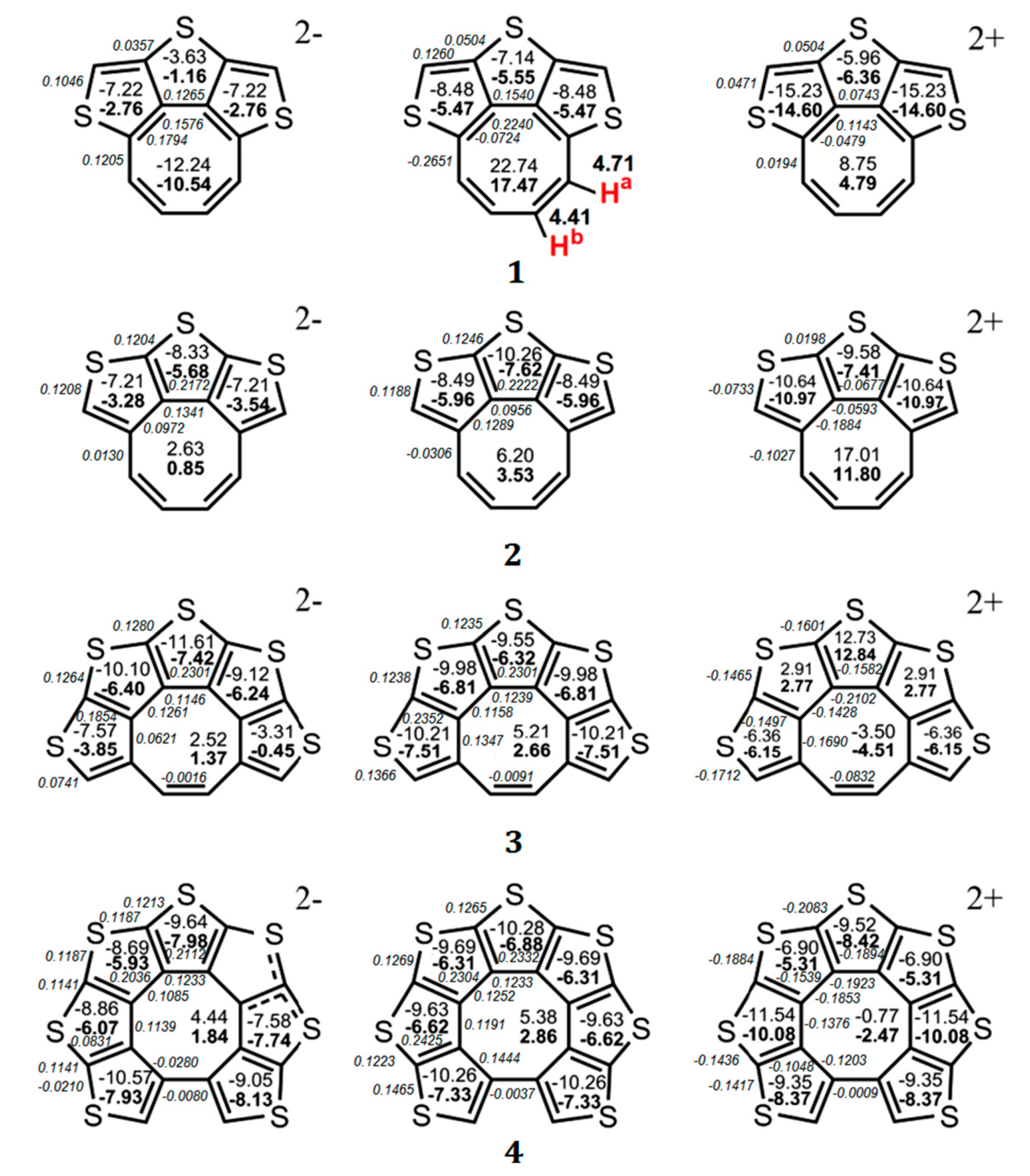
Partially heteroannelated circulenes (quasicirculenes) belong to a family of planar (or quasi-planar) cyclooctatetraenes (Figure 1), and their electronic and spectral properties were clearly interpreted in Refs. [50,51]. The studied molecules of quasicirculenes (except for 4) contain olefinic protons directly bound to the eight-membered ring [52,53,54], which allow us to study their aromaticity using 1H NMR spectroscopy. It was shown that in the 1H NMR spectra, the proton signals of the eight-membered ring appear in the region of 4.4–4.7 ppm for molecule 1 [55], 5.7–5.9 ppm for molecule 8 [53] and 4.2–4.8 ppm for molecule 9 [54], which provides evidence for the antiaromatic nature of the octatetraene ring.
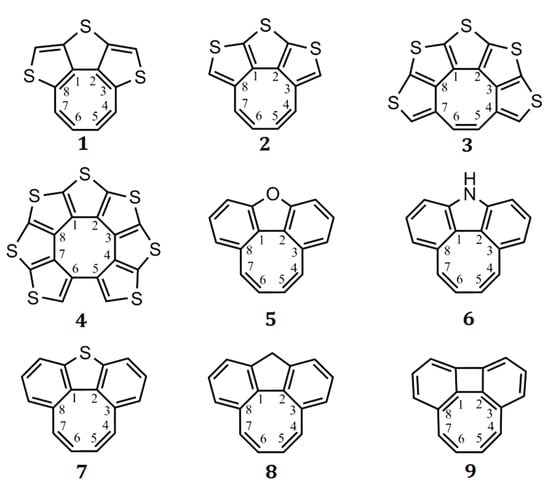
Figure 1.
Structure of the quasicirculenes 1–9.
Quasicirculenes are of practical interest, being promising organic semiconducting materials, as well as initial reagents for the synthesis of new circulenes and n-dimensional materials based on them. This chapter presents the results of the quantum chemical study of the structure and aromaticity for a series of the synthesized 1, 4, 8, 9 (Figure 1) and the simulated model 2, 3, 5–7 quasicirculenes (Figure 1).
2.1. Structural and NICS Criterion
The partially annelated heterocirculene molecules (quasicirculenes) in the ground singlet state are planar and belong to the C2v symmetry point group (except the compounds 7 and 8). Quasicirculenes 7 and 8 undergo a weak out-of-plane deformation of their molecular skeleton, which leads to symmetry reduction in the C2 point group.
An important parameter for the interpretation of the structural features of quasicirculene molecules is the parameter of the bonds length alternation (ΔR) in the octatetraene ring, which can be calculated as the average difference in the adjacent C–C bond lengths:
where a and a* are the lengths of two adjacent C–C bonds (a total of eight pairs in the octatetraene ring).
The ΔR parameter can be interpreted as a structural aromaticity criterion, i.e., the pronounced alternation of the single and double bonds within a ring determines a large ΔR value and indicates the antiaromatic nature of the ring. For example, for a planar hypothetical cyclooctatetraene of the D4h symmetry, this value is equal to 0.121 Å [56,57,58], for cyclobutadiene, ∆R = 0.227 Å [59]), while a ΔR value close to zero (or equal to zero, as in the benzene molecule) indicates the cyclic conjugation, stabilization and aromatic nature of such a ring system.
On the basis of this criterion, it was shown [50] that the ΔR values for the octatetraene ring vary in the range from 0.11 to 0.02 Å, indicating its antiaromatic or weakly antiaromatic nature in the quasicirculene molecules 1–9 [50]. The nuclear independent chemical shift (NICS) indices were calculated at the middle point of the ring, NICS(0), and 1 Å above it, NICS(1). The positive NICS(0) and NICS(1) values calculated at the GIAO/B3LYP/6-311++G(d,p) level of the magnetic chemical-shift theory confirm the antiaromatic nature of the eight-membered ring of quasicirculenes 1–9 (Figure 2 and Figure 3) [50]. As one can see from Figure 2, the octatetraene ring of quasicirculene 1 is strictly antiaromatic (NICS(0) = +22.7 ppm, NICS(1) = +17.5 ppm, ∆R = 0.091 Å) and shows similar properties to the free cyclooctatetraene molecule of the D4h symmetry (NICS(0) = +41.7, ∆R = 0.121 Å). For a modeled quasicirculene 2, the NICS(0), NICS(1) and ∆R parameters have much lower values (+6.2 ppm, +3.5 ppm and 0.053 Å, respectively) which corresponds to the weaker antiaromatic nature of the octatetraene ring [50]. Such a difference in the NICS indices and ∆R values for two isomers 1 and 2 is due to violation of the C–C bond alternation in the octatetraene ring for the quasicirculene molecule 2. Thus, the C1–C8, C1–C2 and C2–C3 bond lengths underlying the three thiophene rings (Figure 1) have almost identical values, with a ∆R variation of 0.01 Å [50]. As a result, the structural aromaticity criterion of the octatetraene ring is violated, as it requires a strict alternation of single and double bonds.
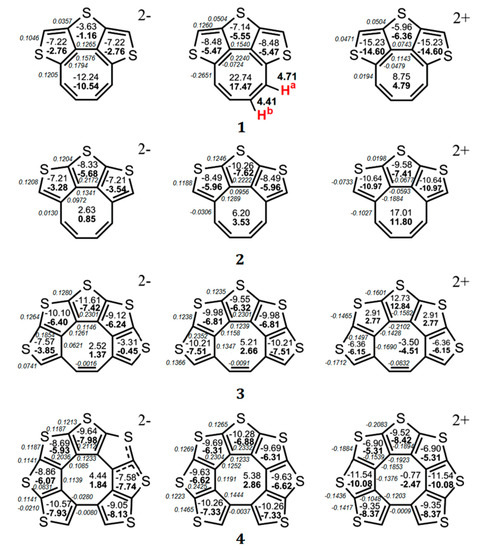
Figure 2.
The values of NICS(0) (top number) and NICS(1) (bottom number indicated in bold) indices calculated at the GIAO/B3LYP/6-311++G(d,p) level of the theory for the neutral molecules of quasicirculenes 1–4 and their corresponding dianions and dications [50] and experimental values of the 1H NMR chemical shifts [52]. The BREs in units of |β| for the neutral and doubly charged ions of 1–4 were taken from Ref. [51] and are presented in italics.
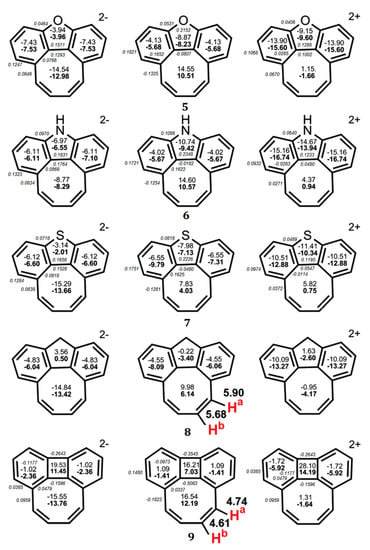
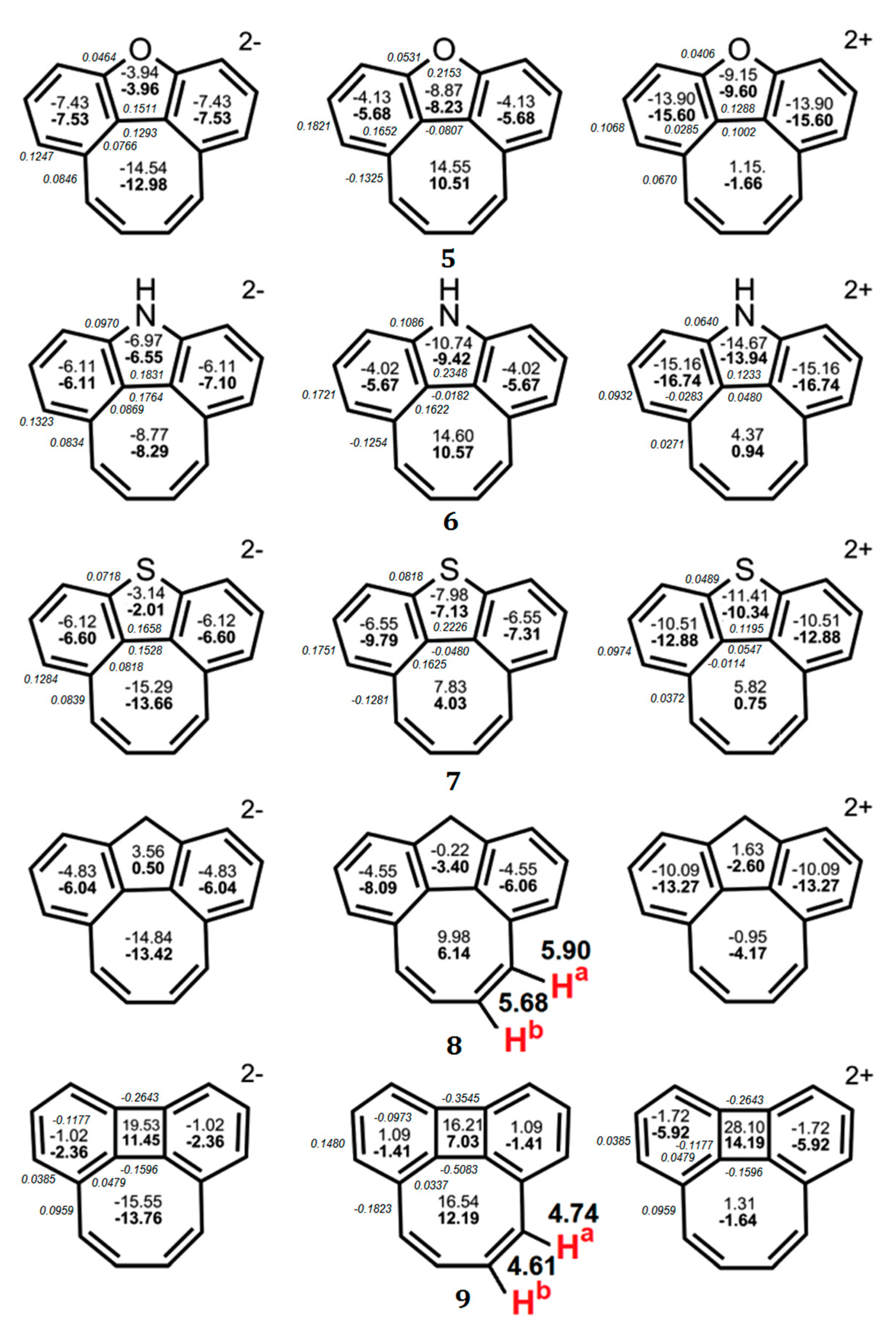
Figure 3.
The values of NICS(0) (top number) and NICS(1) (bottom number indicated in bold) indices calculated at the GIAO/B3LYP/6-311++G(d,p) level of the theory for the neutral molecules of quasicirculenes 5–9 and their corresponding dianions and dications [50] and experimental values of the 1H NMR chemical shifts [53,54]. The BREs in units of |β| for the neutral and doubly charged ions of 1–4 were taken from Ref. [51] and are presented in italics.
In the case of quasicirculene dianions 1, 5–9 the octatetraene ring acquires an aromatic nature, as evidenced by the negative values of the NICS(0) and NICS(1) indices, and therefore the whole system becomes aromatic (Figure 2 and Figure 3). The doubly reduced (2–) ions of hypothetical quasicirculenes 2–4 have a weakly antiaromatic (almost non-aromatic) octatetraene ring because of the small positive NICS(0) and NICS(1) values [50]. Upon double oxidation of quasicirculenes 7–9, the octatetraene ring undergoes out-of-plane deformation and acquires a non-aromatic nature in all dications of molecules 5–9. In this case, the annelating fragments retain the presence of diatropic ring currents [50].
The dications of molecules 1 and 2 have a planar structure, and the eight-membered ring retains an antiaromatic character similar to neutral molecules (Figure 2).
2.2. Graph-Theoretical Analysis
In 2018, Kerim et al. [51] systematically examined the aromaticity for a series of thiophene, furan, pyrrole, and benzene annelated cyclooctatetraene derivatives using the graph theory of aromatic stabilization and topology [49] for the study of aromaticity. They compared the results of the calculated bond resonance energy (BRE) and circuit resonance energy (CRE) indices with those of the NICS(0) and NICS(1) indices.
The topological resonance energy (TRE) method is considered as one of the most reliable energetic measures of global aromaticity [60,61,62]. Positive TRE values suggest aromaticity and negative TRE values indicate antiaromaticity. The resonance energy per π-electron (REPE) was calculated by dividing the TRE by the number of π-electrons in the corresponding quasicirculene in order to compare molecules with different numbers of π-electrons. The BRE and the CRE methods provide the local aromatic pattern for any ring in the polycyclic aromatic compounds [63,64,65,66,67,68]. The larger the TRE, BRE, CRE, and REPE values, the greater the aromaticity.
According to REPEs, cyclooctatetraene shows a highly antiaromatic nature (REPE is −0.074 β), but its dianion and dication possess an aromatic nature, with REPE values of 0.019 and 0.031 β, respectively [60,61,69].
The positive TRE values indicate aromaticity of the neutral species, with the exception of 1 and 9 (Table 1). The dianions of the compounds 1, 2, 5−7 are more aromatic than neutral molecules or the dicationic species. It is noteworthy that the dications 2, 3 and 4 exhibit relatively smaller aromaticity than their corresponding neutral molecules and dianions. The negative TRE values for molecule 9 both in the neutral and the doubly charged states (−0.146 for the neutral and −0.084 for dication and dianion) predict their antiaromatic nature.
The BRE and CRE indices were used to estimate the local aromaticity of the quasicirculenes. As one can see in Figure 2 and Figure 3, the BRE values of the bonds are positive, indicating that all of the bonds in five- and six-membered rings for the compounds 1−7 show local aromaticity. This is due to the fact that all these rings correspond to Hückel’s 4n + 2 rule of aromaticity. In contrast, the BRE values of the C−C bonds in the octatetraene ring are significantly large and negative, pointing to local antiaromaticity of the octatetraene ring in the composition of quasicirculenes. This fact can be explained by the fact that the octatetraene ring does not satisfy Hückel’s rule of aromaticity. Figure 2 and Figure 3 show that with an increase in the number of benzene or heterocyclic rings annelating the octatetraene ring, the BREs have negative values only for the unfused C−C bonds of the octatetraene ring, while all the fused C−C bonds display relatively large and positive BREs.

Table 1.
TREs and REPEs for quasicirculenes 1−7, 9 and their double charged ions.
Table 1.
TREs and REPEs for quasicirculenes 1−7, 9 and their double charged ions.
| Species | TRE | REPE | ||||
|---|---|---|---|---|---|---|
| Dianion | Neutral | Dication | Dianion | Neutral | Dication | |
| 1 | 0.355 | −0.006 | 0.137 | 0.018 | 0.000 | 0.009 |
| 2 | 0.352 | 0.365 | −0.145 | 0.018 | 0.020 | −0.009 |
| 3 | 0.527 | 0.655 | −0.063 | 0.020 | 0.027 | −0.003 |
| 4 | 0.591 | 0.933 | 0.166 | 1.743 | 2.597 | 0.576 |
| 5 | 0.320 | 0.302 | 0.247 | 0.016 | 0.017 | 0.015 |
| 6 | 0.378 | 0.330 | 0.177 | 0.019 | 0.018 | 0.011 |
| 7 | 0.349 | 0.314 | 0.193 | 0.017 | 0.017 | 0.012 |
| 9 | −0.084 | −0.146 | −0.084 | −0.005 | −0.009 | −0.006 |
The TRE and REPE values are given in units of |β|, where β represents the standard resonance integral in Hückel theory. Reprinted with permission from Ref. [51]. Copyright 2018 Springer.
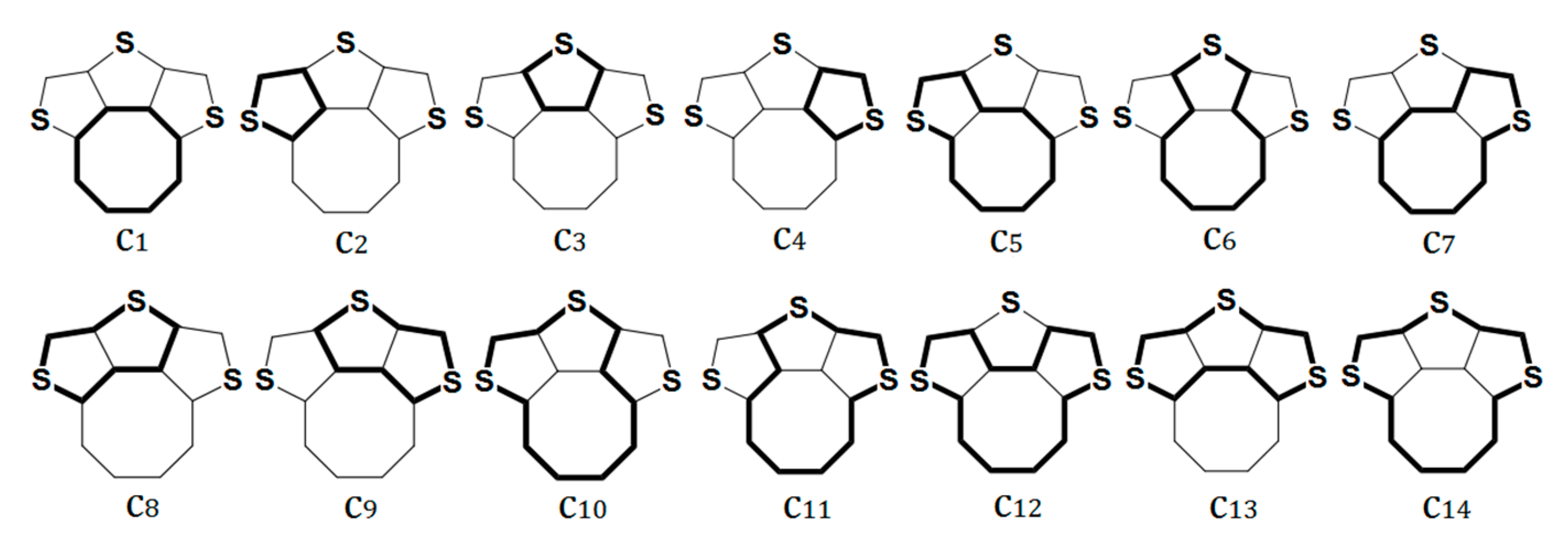
The CRE index indicates the individual contributions of different circuits to global aromaticity. The π-electronic systems of quasicirculenes 1, 2, 5−7, 9 contain 14 circuits numerated as c1−c14 (Figure 4). Compounds 3 and 4 contain 47 and 156 circuits, respectively, which are considered in detail in Ref. [51]. For molecules of the quasicirculenes 1, 2, 5−7, the benzene and heterocyclic rings exhibit an aromatic nature, as evidenced by the positive CRE values (Table 2). It was found that the c2 and c4 circuits possess the largest positive CRE values and make the main aromatic contribution, while the c1 circuit has the largest negative CRE value and makes the largest antiaromatic contribution to the global aromaticity of quasicirculenes 1, 2, 5−7.
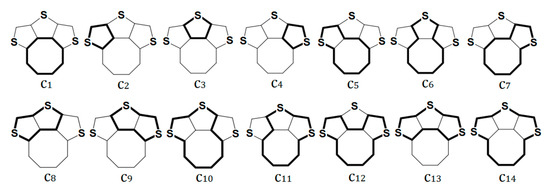
Figure 4.
The geometrically non-identical π-electron circuits in compound 1. The specific circuits studied are shown in bold. Reprinted with permission from Ref. [51]. Copyright 2018 Springer.
For quasicirculene 9, a large antiaromatic contribution arises from the c2 and c4 circuits, while the c1 and c3 circuits make a large antiaromatic contribution. Interestingly, the CRE value for the cyclobutadiene ring (c3) circuit is more negative than the same CRE value for the octatetraene ring (c1) circuit (Table 2). Thus, the results obtained from the BRE and CRE calculations are in good agreement with the NICS(0) and NICS(1) indices for the local aromaticity of the octatetraene and thiophene rings in compounds 1 and 2 for both neutral molecules and their double charged ions. However, the magnitude of the NICS(0) and NICS(1) indices for 52−, 5, 62−, 6, 7 and 72− species do not correspond to the BRE and CRE results. For instance, the octatetraene ring in dianions 52−, 62− and 72− show more negative NICS(0) and NICS(1) values compared with the same values for benzene rings.
Table 2.
CREs in units of |β| for quasicirculenes 1, 2, 5−7, 9 and their double charged ions.
Table 2.
CREs in units of |β| for quasicirculenes 1, 2, 5−7, 9 and their double charged ions.
| c1 | c2 | c3 | c4 | c5 | c6 | c7 | c8 | c9 | c10 | c11 | c12 | c13 | c14 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12− | 0.0629 | 0.0681 | 0.0233 | 0.0681 | 0.0173 | −0.0018 | 0.0173 | 0.0048 | 0.0048 | 0.0014 | 0.0014 | 0.0049 | 0.0006 | 0.0006 |
| 1 | −0.2423 | 0.2250 | 0.0246 | 0.2250 | −0.0910 | −0.0006 | −0.0910 | 0.0103 | 0.0103 | 0.0024 | 0.0024 | −0.0335 | 0.0117 | −0.0121 |
| 12+ | −0.0360 | −0.0093 | 0.0503 | −0.0093 | 0.0232 | −0.0264 | 0.0232 | 0.0103 | 0.0103 | 0.0024 | 0.0024 | 0.0266 | −0.0141 | 0.0137 |
| 22− | −0.0027 | 0.0821 | 0.0850 | 0.0821 | 0.0022 | −0.0052 | 0.0022 | 0.0113 | 0.0113 | 0.0030 | 0.0030 | 0.0100 | 0.0012 | 0.0012 |
| 2 | −0.0049 | 0.1099 | 0.0846 | 0.1099 | −0.0055 | −0.0029 | −0.0055 | 0.0150 | 0.0150 | 0.0012 | 0.0012 | −0.0110 | 0.0034 | −0.0037 |
| 22+ | 0.1661 | 0.0097 | 0.2429 | 0.0097 | −0.1880 | −0.1533 | −0.1880 | −0.0228 | −0.0228 | 0.0266 | 0.0266 | 0.1202 | −0.0560 | 0.0642 |
| 52− | −0.0009 | 0.0556 | 0.0298 | 0.0556 | 0.0196 | −0.0046 | 0.0196 | 0.0066 | 0.0066 | 0.0023 | 0.0023 | 0.0324 | −0.0021 | 0.0043 |
| 5 | −0.0506 | 0.2112 | 0.0284 | 0.2112 | −0.0365 | −0.0017 | −0.0365 | 0.0112 | 0.0112 | 0.0008 | 0.0008 | −0.0303 | 0.0073 | −0.0065 |
| 52+ | −0.0166 | 0.0424 | 0.0297 | 0.0424 | 0.0136 | −0.0058 | 0.0136 | 0.0040 | 0.0040 | 0.0028 | 0.0028 | 0.0415 | −0.0066 | 0.0094 |
| 62− | 0.0030 | 0.0547 | 0.0574 | 0.0547 | 0.0197 | −0.0097 | 0.0197 | 0.0149 | 0.0149 | 0.0048 | 0.0048 | 0.0243 | −0.0029 | 0.0077 |
| 6 | −0.0470 | 0.1796 | 0.0532 | 0.1796 | −0.0314 | −0.0034 | −0.0314 | 0.0234 | 0.0234 | 0.0015 | 0.0015 | −0.0248 | 0.0159 | −0.0144 |
| 62+ | −0.0405 | 0.0389 | 0.0567 | 0.0389 | −0.0003 | −0.0146 | −0.0003 | 0.0034 | 0.0034 | 0.0073 | 0.0073 | 0.0418 | −0.0226 | 0.0299 |
| 72− | 0.0013 | 0.0545 | 0.0427 | 0.0545 | 0.0197 | −0.0075 | 0.0197 | 0.0112 | 0.0112 | 0.0037 | 0.0037 | 0.0278 | −0.0025 | 0.0062 |
| 7 | −0.0486 | 0.1915 | 0.0398 | 0.1915 | −0.0333 | −0.0026 | −0.0333 | 0.0183 | 0.0183 | 0.0012 | 0.0012 | −0.0268 | 0.0125 | −0.0114 |
| 72+ | −0.0335 | 0.0421 | 0.0426 | 0.0421 | 0.0037 | −0.0116 | 0.0037 | 0.0022 | 0.0022 | 0.0058 | 0.0058 | 0.0415 | −0.0183 | 0.0241 |
| 92− | −0.0212 | −0.0274 | −0.2167 | −0.0274 | 0.0487 | 0.0371 | 0.0487 | −0.0212 | −0.0212 | −0.0180 | −0.0180 | 0.0487 | 0.0756 | −0.0667 |
| 9 | −0.1041 | 0.3809 | −0.2461 | 0.3809 | −0.0736 | 0.0136 | −0.0736 | −0.1041 | −0.1041 | −0.0060 | −0.0060 | −0.0736 | −0.0648 | 0.0675 |
| 92+ | −0.0212 | −0.0274 | −0.2167 | −0.0274 | 0.0487 | 0.0371 | 0.0487 | −0.0212 | −0.0212 | −0.0180 | −0.0180 | 0.0487 | 0.0756 | −0.0667 |
Reprinted with permission from Ref. [51]. Copyright 2018 Springer.
2.3. ACID Calculations
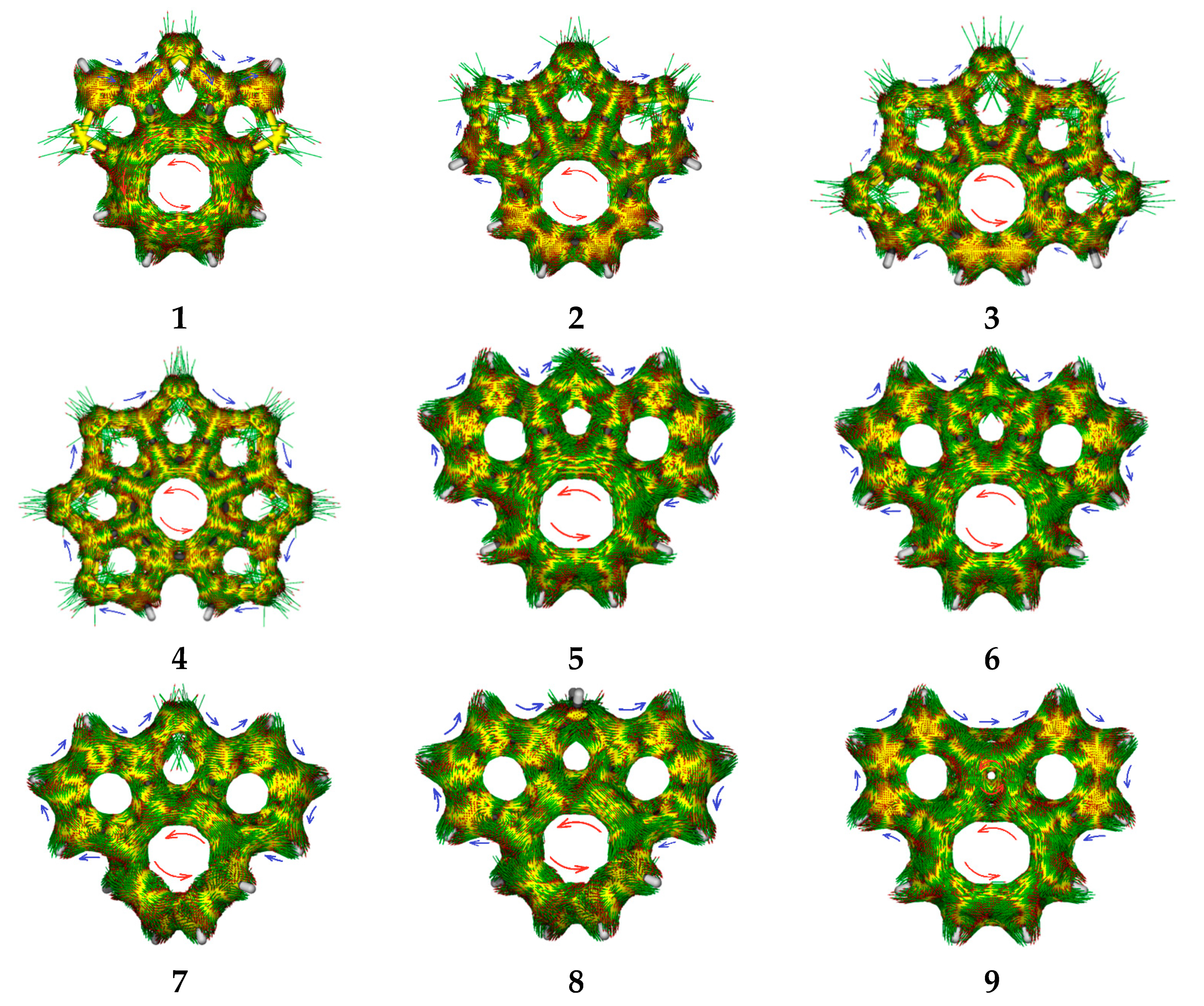
To summarize the complete picture of aromaticity of quasicirculene molecules, we calculated the ACID plots for visualizing ring-currents and electron delocalization. The ACID plots for 1–9 are presented in Figure 5, and clearly show a counterclockwise 8π ring current flowing along the central octatetraene ring, indicating its antiaromaticity, while a clockwise (“aromatic”) ring current circulates along the outer fused rings.
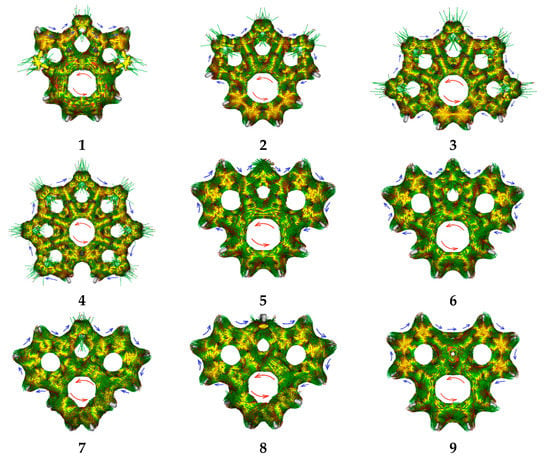
Figure 5.
ACID plots. Blue arrows—local diatropic currents, red arrows—paratropic currents.
Thus, the ACID calculations suggest the presence of two major electronic sub-systems in quasicirculene molecules—one includes the antiaromatic octatetraene ring, and the other outer system includes the aromatic benzene and heteroarene rings.
3. Aromaticity of Completely Annelated Heterocirculenes
3.1. NICS Aromaticity Criterion
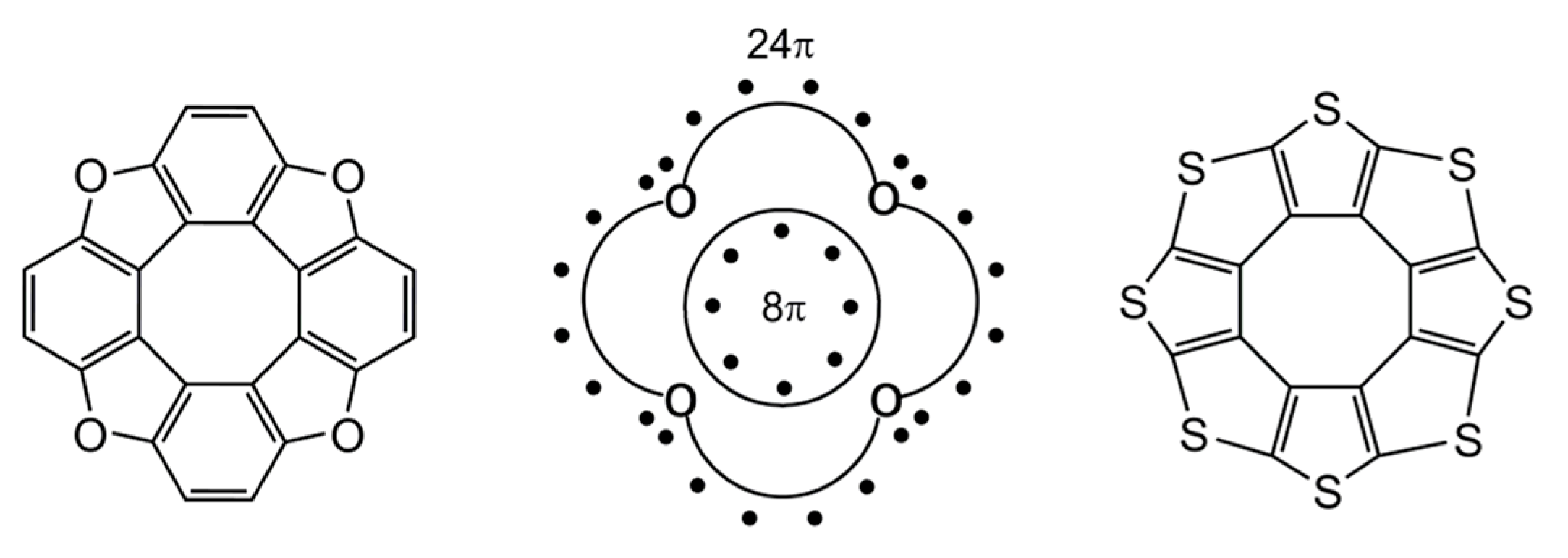
The molecules of hetero[8]circulenes contain 32 π-electrons in a polycondensed system (Figure 6). At first glance, heterocirculenes do not satisfy the well-known Hückel 4n + 2 rule, as one of the aromaticity criteria; on the contrary, they belong to anti-Hückel 4n π-electronic systems (n = 8). In this case, the “annulene in annulene” concept is not applicable: the 32 π-electrons in the polycondensed system of hetero[8]circulenes comprises an outer perimeter containing 24 π-electrons and an inner perimeter with eight π-electrons (Figure 6). However, it is known that the Hückel rule is strictly valid for monocyclic conjugated compounds (in general case for annulenes) and is often not performed for polycyclic systems such as pyrene, benzo[e]pyrene, and [8]circulene [70]). Therefore, the aromaticity of heterocirculenes was studied using the NICS, GIMIC and ACID plot criterions.
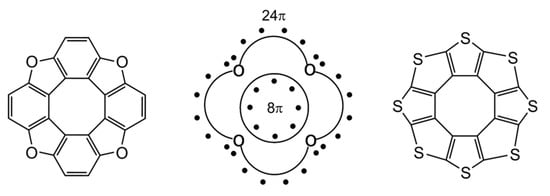
Figure 6.
π-electronic structure of hetero[8]circulenes [46].
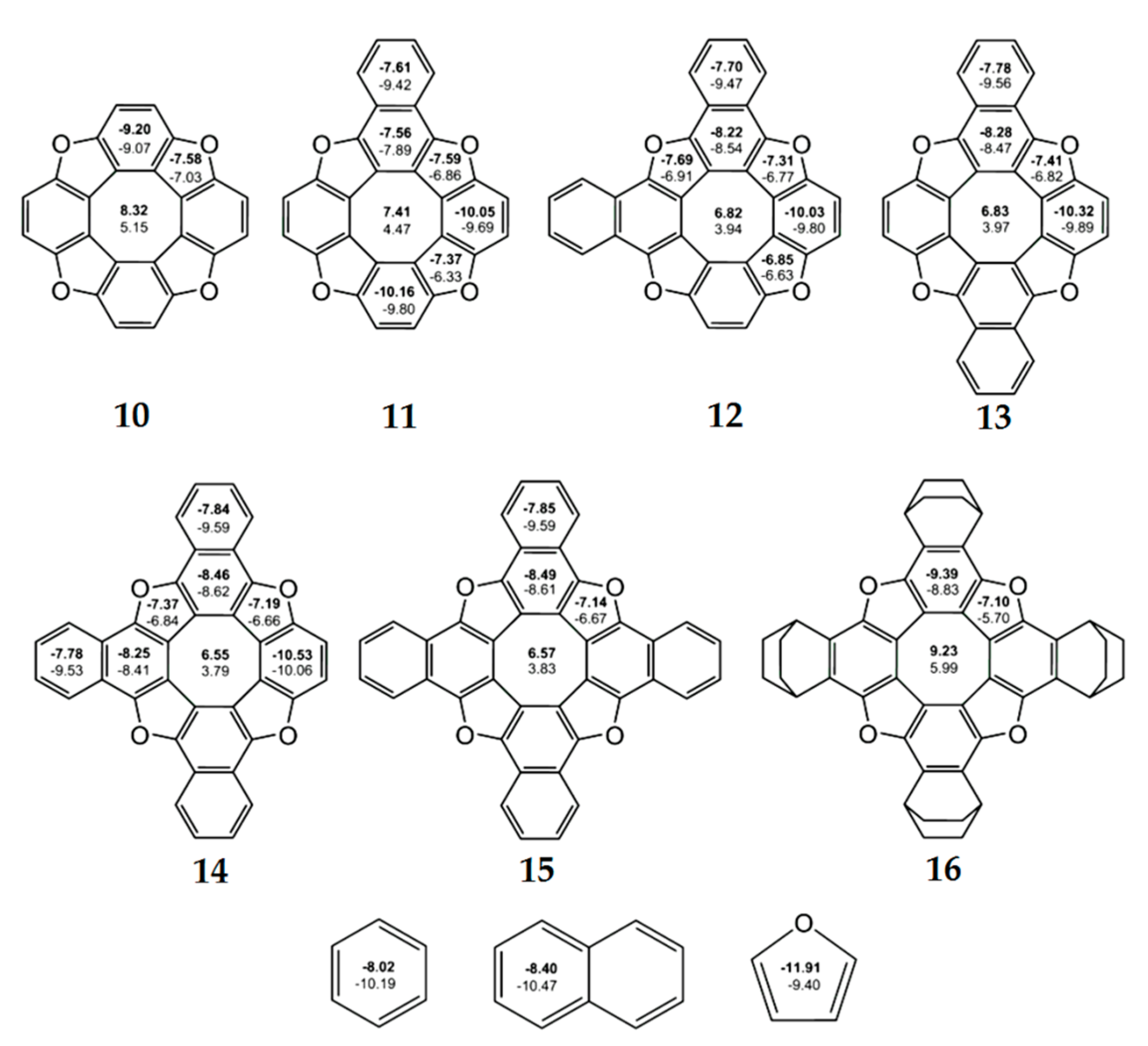
According to the NICS criterion, the inner eight-membered ring for a series of synthesized tetraoxa[8]circulenes 10–16(R = H) [46], azaoxa[8]circulenes 17 and 18 [71,72], ketocirculene 19 [33,73], thia[8]circulenes 20 and 21 [33], tetrasilatetrathia[8]circulenes 22 [74], tetrathia[8]circulene 23 and tetraselena[8]circulene 24 [75,76,77], diazadithia[8]circulene 25 and diazadiselena[8]circulenes 27 [78], tetragermatetrathia[8]circulene 26 [79] and hypothetical circulenes 28–38 [33,34,80,81,82,83] is characterized by positive NICS(0) and NICS(1) values (Figure 7, Figure 8 and Figure 9, Table 3), indicating its antiaromatic nature. In particular, for tetraoxa[8]circulene molecules, the NICS(0) and NICS(1) indices calculated at the center of the eight-membered ring are positive and vary in the range of 6.55–9.23 ppm and 3.79–5.99 ppm, respectively (Figure 7). At the same time, all benzene rings in the tetraoxa[8]circulene molecules are characterized by significantly negative NICS(0) and NICS(1) values, which vary in the range from −10.53 to −7.56 ppm and from −10.06 to −7.89 ppm, respectively. These values are close to the NICS(0) and NICS(1) values for the free benzene molecule (Figure 7). Similar results were obtained for the naphthalene fragments of the tetraoxa[8]circulenes, which yield NICS(0) and NICS(1) values close to those for the free naphthalene (Figure 7). However, it should be noted that the NICS indices differ significantly for the free furan molecule and conjugated furan moieties. This is due to the significant delocalization of the furan π-system into adjacent benzene rings. Thus, a common π-extended ribbon-like system of benzene and furan rings is formed in the tetraoxa[8]circulene molecules, which is closed around the antiaromatic octatetraene ring. In fact, the 1H NMR spectra of tetraoxa[8]circulenes show the characteristic signals of the “aromatic” protons at 7.7 ppm [23].
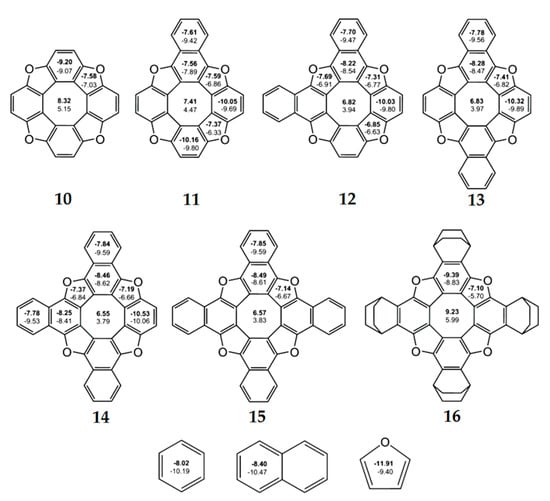
Figure 7.
The values of NICS(0) (top number indicated in bold) and NICS(1) (bottom number) indices calculated at the GIAO/B3LYP/6-311+G(d,p) level of the theory for tetraoxa[8]circulene, benzene, naphthalene and furan molecules [46].
Table 3.
The total ring current strength (Itot, nA∙T−1) and the balance between the diatropic (d) and paratropic (p) currents in the rim- and hub-subsystems for the neutrally and doubly charged hetero[8]circulene molecules.
S. Radenković et al. [45] came to a similar conclusion when calculating the ring current densities for a series of tetraoxa[8]circulenes. They found the presence of two types of ring currents in tetraoxa[8]circulene molecules: the paratropic ring current induced by the internal octatetraene ring, and the diatropic ones induced by the outer system of the furan and benzene rings [45].
As a general trend, the dianionic tetraoxa[8]circulenes formed under conditions of electrochemical experiments are predicted to be completely aromatic, because the inner octatetraene ring and all surrounding benzene and furan moieties are strongly aromatic (the NICS indices are significantly negative for these rings) (Figure 8). However, for the dicationic tetraoxa[8]circulenes identified in the mass spectrum [84], the inner eight-membered ring gains an antiaromatic character (for the tetraoxa[8]circulene 10 NICS(0) = 2.6 ppm, Figure 8). Sequential replacement of furan rings by the pyrrole rings does not significantly affect the aromaticity of the tetraoxa[8]circulenes [71,72]. In the case of neutral molecules and dications, the central octatetraene core of azaoxa[8]circulenes 17 and 18 is antiaromatic and becomes aromatic in the azaoxa[8]circulenes dianions (Figure 8). Similarly to the tetraoxa[8]circulenes, the pyrolle, furan and benzene rings maintain aromaticity upon double ionization.
As one can see from Figure 9, for most of the hetero[8]circulenes, the internal eight-membered ring is antiaromatic and is characterized by positive NICS(0) and NICS(1) values, which are approximately equal (with the opposite sign) to the corresponding NICS values for the five- and six-membered aromatic rings (we can except aromatic circulene molecules 30 and 31, which contain the electron-deficient boron atoms in the external macrocycle). Thus, the NICS indices indicate qualitatively the nonaromatic character of most of the hetero[8]circulenes, with the exception of the antiaromatic circulenes 35 and 36 (Figure 9).
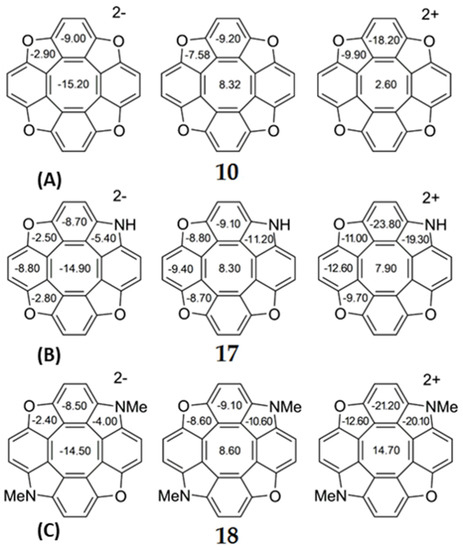
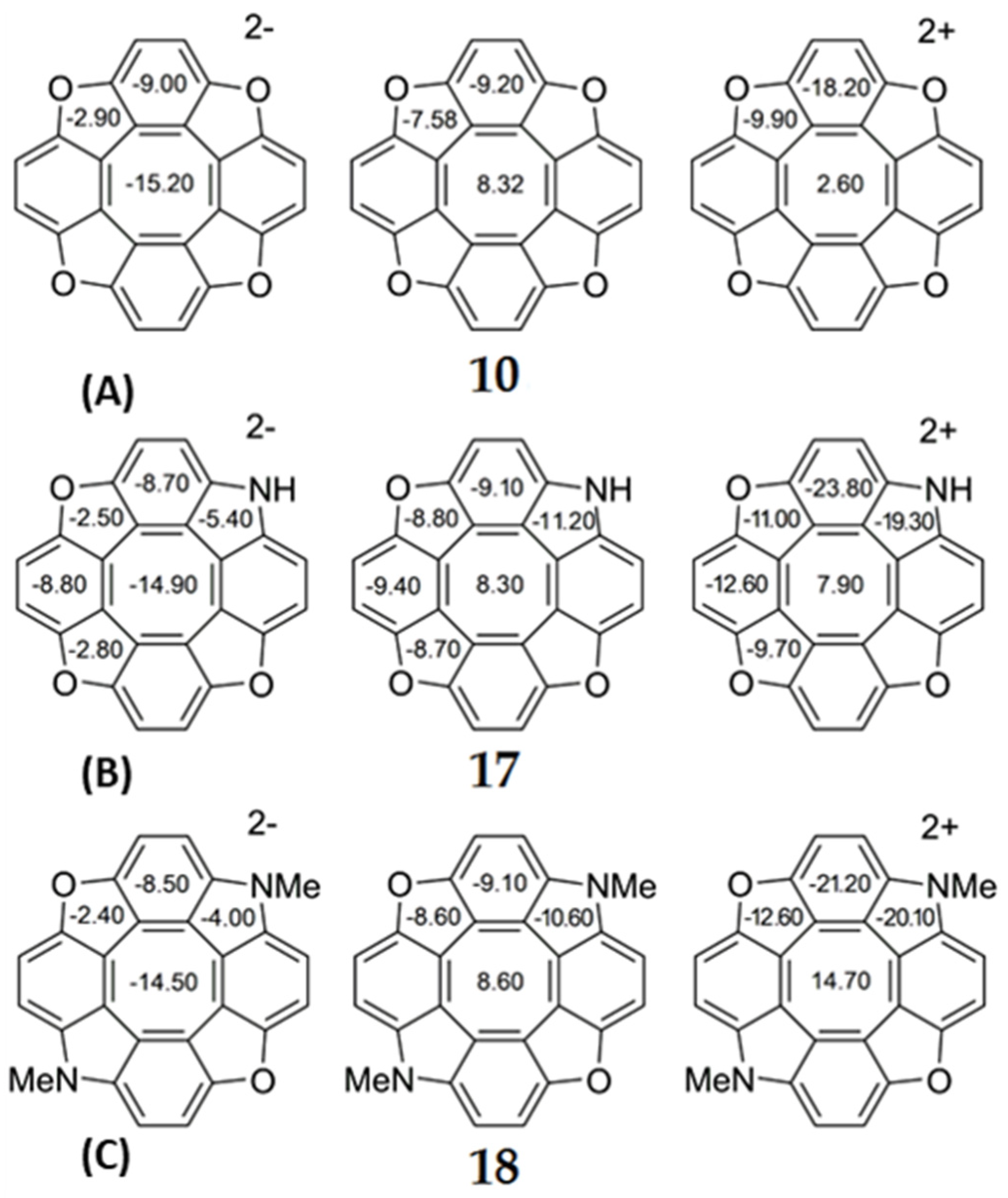
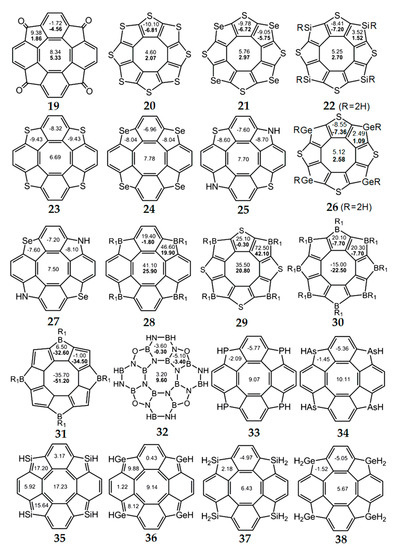
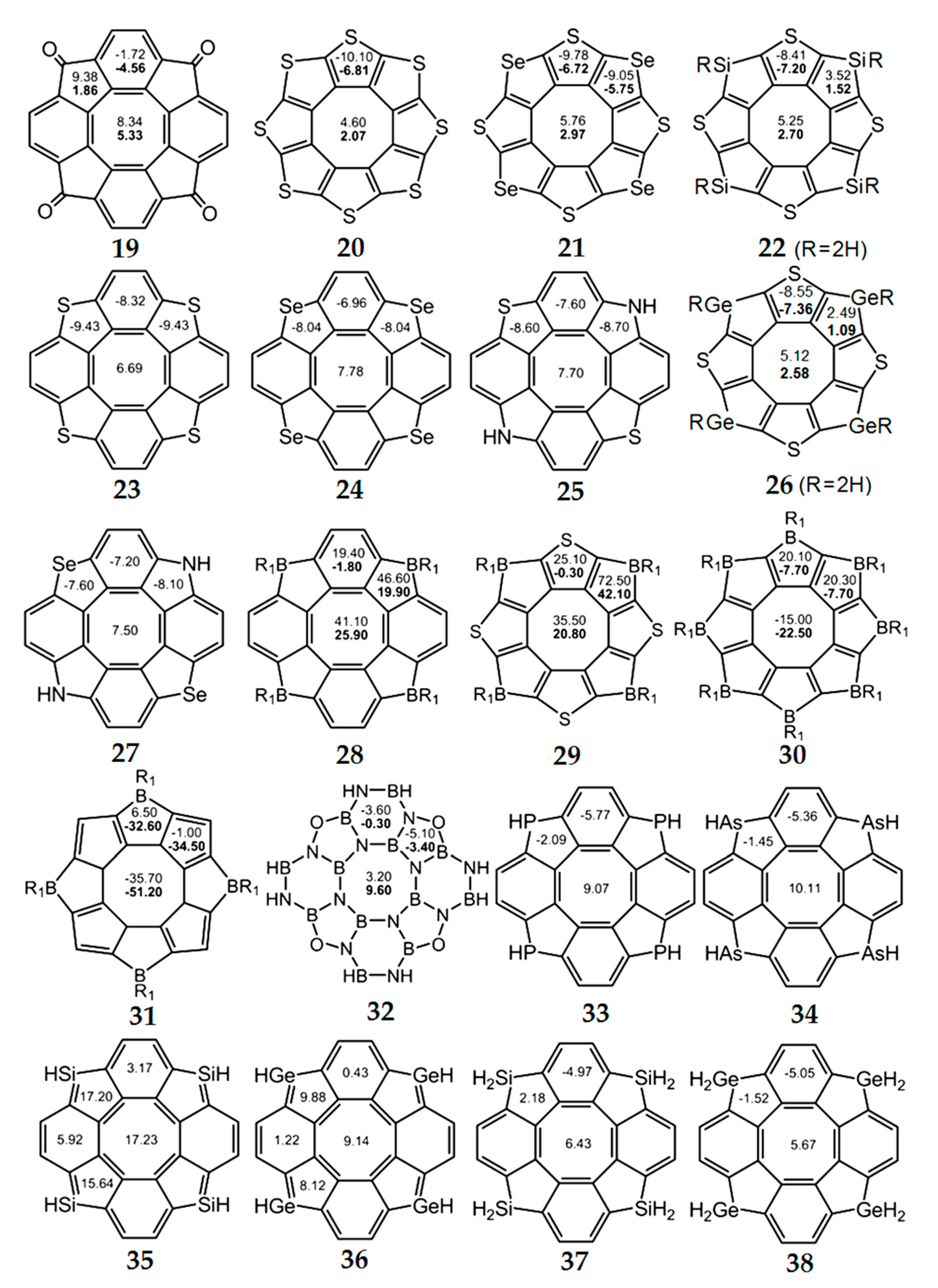
Figure 8.
The values of NICS(0) indices for neutral and doubly ionized tetraoxa[8]circulene (A), azaoxa[8]circulene (B) and N,N-dimethyldiazadioxa[8]circulene (C) molecules.
Figure 8.
The values of NICS(0) indices for neutral and doubly ionized tetraoxa[8]circulene (A), azaoxa[8]circulene (B) and N,N-dimethyldiazadioxa[8]circulene (C) molecules.
Figure 9.
The values of NICS(0) (top number) and NICS(1) (bottom number in bold) indices for hetero[8]circulene molecules [33,34,74,75,76,77,78,79,80,81]. For planar molecules, NICS(0) and NICS(1) values are given, and for non-planar molecules, only NICS(0) values are shown.
Figure 9.
The values of NICS(0) (top number) and NICS(1) (bottom number in bold) indices for hetero[8]circulene molecules [33,34,74,75,76,77,78,79,80,81]. For planar molecules, NICS(0) and NICS(1) values are given, and for non-planar molecules, only NICS(0) values are shown.
3.2. GIMIC Aromaticity Approach
A more informative criterion for assessing aromaticity of polyheterocyclic compounds is the gauge including magnetically induced currents (GIMIC) approach. As was shown in studies conducted by Fliegl et al. [9] and Jusélius et al. [10], the GIMIC method, similarly to the NICS concept, is based on the approximation of gauge-invariant atomic orbitals (GIAOs), and provides detailed information about electron delocalization properties, aromatic character, and magnetically induced current pathways in molecules.
It is known that for aromatic compounds such as benzene, the diatropic ring current induced from the outside of the molecule (16.7 nA∙T−1) dominates over the paratropic ring current inside the ring (−4.9 nA∙T−1), yielding a total diatropic current (Itot) equal to 11.8 nA∙T−1 [10]. For antiaromatic cyclobutadiene molecules, the paratropic ring current (−23.4 nA∙T−1) dominates over the diatropic ring current (3.5 nA∙T−1), yielding a total paratropic ring current equal to −19.9 nA∙T−1 [10]. For nonaromatic molecules, such as cyclohexane, diatropic and paratropic components cancel one another out, yielding an approximately zero total current (Itot = 0.2 nA∙T−1) [10]. The above statements are true for many molecules, but there are some exceptions in which the “canonical” aromaticity (energetic criteria) does not coincide with the “magnetic” aromaticity [85]. From the well-known phenalenyl example with various substituents [85], chemists realize that electron counting alone cannot be fully informative regarding the magnetic current response [86]. Just the allied considerations of both orbital energy and orbital symmetry provide proper prediction of current-density GIMIC patterns and the attendant respective aromaticity. Thus, the direction of the rotation of the current density depends strictly on the orbital symmetry of the magnetically induced transitions, as has been explained in detail by E. Steiner and P. W. Fowler [86].
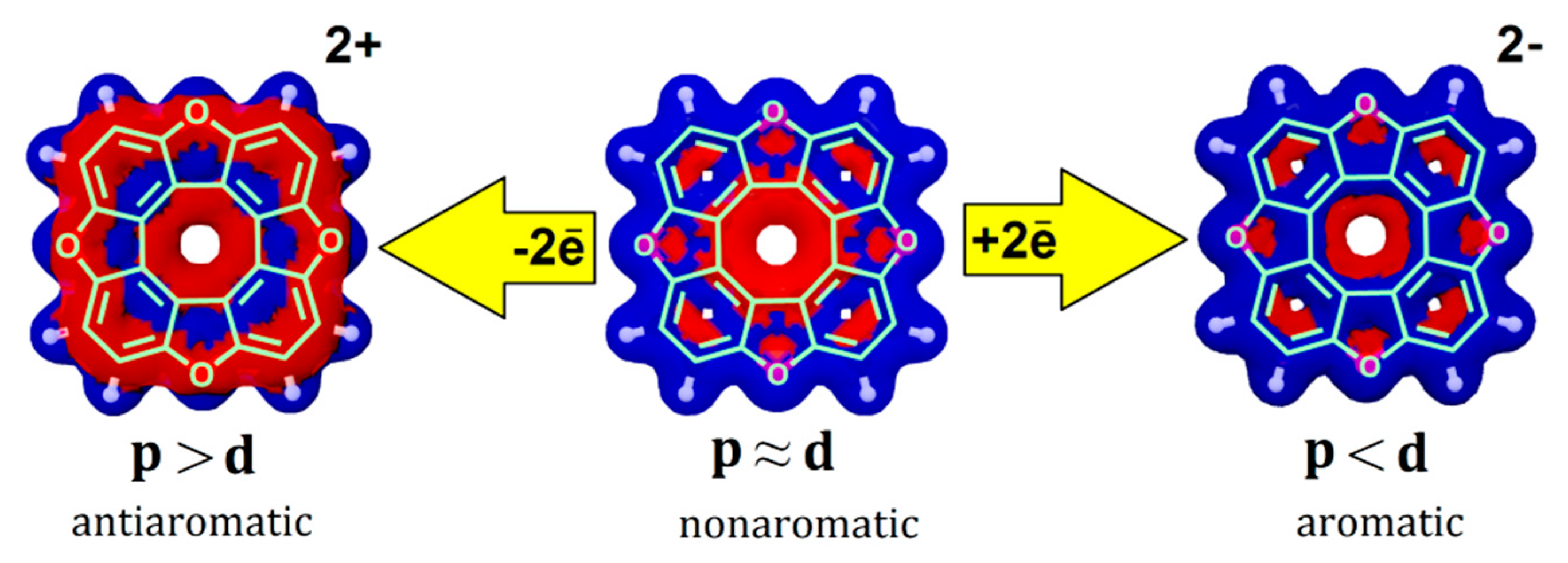
According to the above classification, most neutral hetero[8]circulene molecules represent nonaromatic species [32,33,34] because the paratropic internal currents substantially cancel out the diatropic current component in the outside part of the molecules (Figure 10, Table 3), similar to the well-known non-aromatic cyclic hydrocarbons, fullerene C60 and its multi-charged ion C6010+ [87,88]. A similar pattern is observed for the other molecules of hetero[8]circulenes (Table 3).
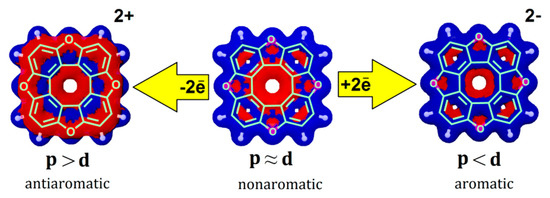
Figure 10.
Balance of magnetically induced ring currents in the tetraoxa[8]circulene molecule 10 and its doubly charged ions. Paratropic current densities are shown in red and the diatropic ones are shown in blue.
In the case of the doubly charged heterocirculene species, the balance of magnetically induced ring currents is significantly disturbed. As a result, the dianions and the OC-, SiH-, GeH-, SiH2-, GeH2-containing dications of circulene molecules become completely aromatic (Table 3) due to the dominant role of the diatropic ring current component, as it is shown for tetraoxa[8]circulene dication in Figure 10. Upon double oxidation of the O-, S-, Se-, NH-, PH- and AsH-containing circulenes, the paratropic ring current component dominates significantly, which leads to the formation of antiaromatic dications (Figure 10, Table 3).
Circulene 19 differs from others by the presence of a specific system of two adjacent antiaromatic five- and eight-membered rings, while the benzene rings retain an aromatic character (Figure 9). Therefore, circulene 19 is a weakly antiaromatic compound (Itot = −4 nA∙T−1, Table 3), i.e., the paratropic current component inside the octatetraene ring of the molecule dominates over the diatropic current component of the outside benzene rings [33].
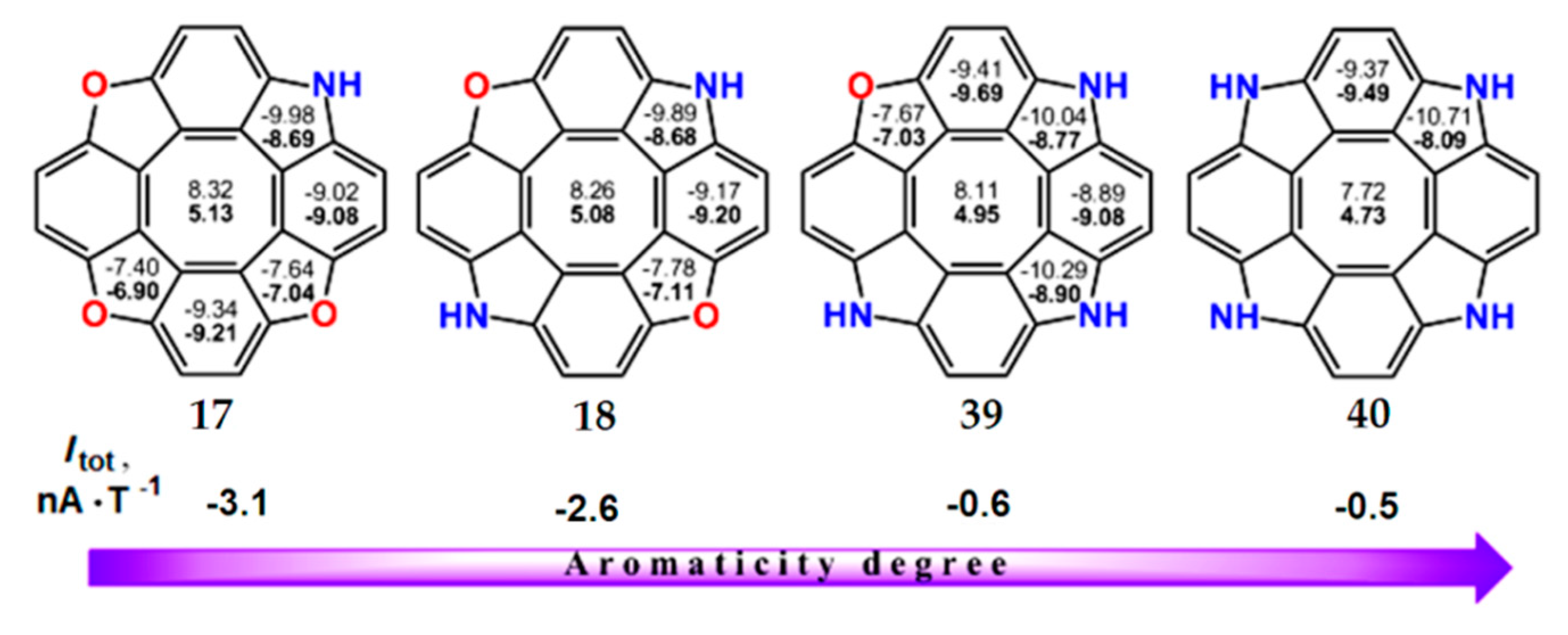
An interesting feature of azacirculenes 17, 18, 39 and 40 is an increase in the total ring-current strength with an increase in the number of nitrogen atoms in the molecule. As shown in Figure 11, the NICS(0) and NICS(1) indices for the central octatetraene ring of azacirculenes are monotonously decreased, i.e., paratropic contributions to the total ring current also decrease in the series 17, 18, 39, 40. However, the diatropic component of the net ring current slightly increases for circulenes 17, 18, 39, 40 (the NICS indices for pyrrole rings demonstrare more negative values). Thus, the total ring current of azacirculenes 17, 18, 39 and 40 decreases to −3.1 nA∙T−1 for 17 and to −0.5 nA∙T−1 for 40. Therefore, circulene 17 can be considered as a slightly antiaromatic molecule, whereas circulene 40 represents an almost nonaromatic species (Figure 11).
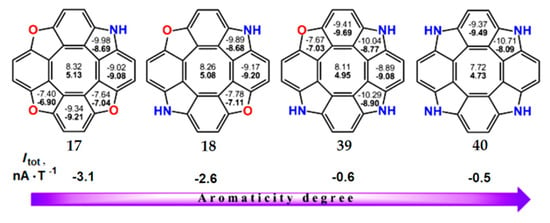
Figure 11.
The NICS(0) (top number) and NICS(1) (bottom number in bold) indices in the aza[8]circulene series and the total ring current strength (Itot), calculated by the GIAO/B3LYP/6-311++G(d,p) method.
Baryshnikov et al. [33,34] have shown that the aromaticity of hetero[8]circulenes depends on the type of heteroatom inserted into the outer framework of the molecules. In particular, an insertion of heavy heteroatoms into the outer macrocycle of heterocirculenes leads to the twist-type distortion of molecules with subsequent formation of the saddle-shaped conformation. This phenomenon is clearly observed in a series of circulenes with different heteroatoms of oxygen, sulfur, selenium, phosphorus, arsenic: circulene 10 is strictly planar, circulene 23 is almost planar, circulene 24 is characterized by a non-planar structure, circulene 33 is non-planar, circulene 34 has a more distorted non-planar structure of the molecule in comparison with circulene 33. Such structural changes directly affect the aromaticity of heterocirculenes.
It is known that the NICS(0) and NICS(1) values are consistently decreasing in a series of furan, selenophene, and thiophene species. Thus, the furan molecule is slightly less aromatic than the selenophene and thiophene molecules [7]. A similar conclusion can be made for heterocirculenes with oxygen (10), sulfur (23) and selenium (24) heteroatoms according to the total ring current strength (Itot). The less aromatic furan fragment in molecule 10 leads to the weakly antiaromatic nature of a whole tetraoxa[8]circulene 10. Circulenes 23 and 24 are nonaromatic species, because the aromaticity of selenophene and thiophene fragments is stronger than that of the furan molecule. The total ring current strength in the other part of the molecule stays almost the same in this series. Thus, the type of heteroatom inserted into the outer framework of heterocirculene molecules 10, 23 and 24 plays a crucial role.
The effect of heteroatom on the aromaticity of heterocirculenes is more clearly pronounced for the series of circulene molecules with nitrogen (40), phosphorus (33) and arsenic (34) heteroatoms. It is known [89] that the aromaticity degree of free arsole molecule is two times less than that of pyrrole; the phosphole molecule is less aromatic than pyrrole and more aromatic than arsole. As a result, the more aromatic pyrrole fragment causes almost zero total ring current of molecule 40, while the presence of a less aromatic arsole ring provides antiaromaticity of compound 34. In this case, the P-containing circulene 33 is located between compounds 34 and 40 in a series of circulenes 33, 34 and 40. Therefore, the paratropic ring-current component induced by the tetraphenylene fragments dominates over the diatropic ring-current component induced by the heterocyclic fragments in a series of compounds 33, 34 and 40.
It should be noted that NICS can be affected by the currents of adjacent rings in polyheterocyclic systems, i.e., when there are several circuits in a molecule, all of them will contribute to the NICS calculated at some selected point [90]. Consequently, despite the NICS local nature, it cannot be used to assess the degree of benzenoid character for a particular ring in polyheterocyclic systems, since it contains not only the ring current of an individual benzenoid circuits, but also has a contribution from neighboring benzene and heteroarene circuits. Moreover, it has also been shown that NICS could wrongly indicate an increase in aromaticity due to out-of-plane distortion in polycyclic molecules [91,92].
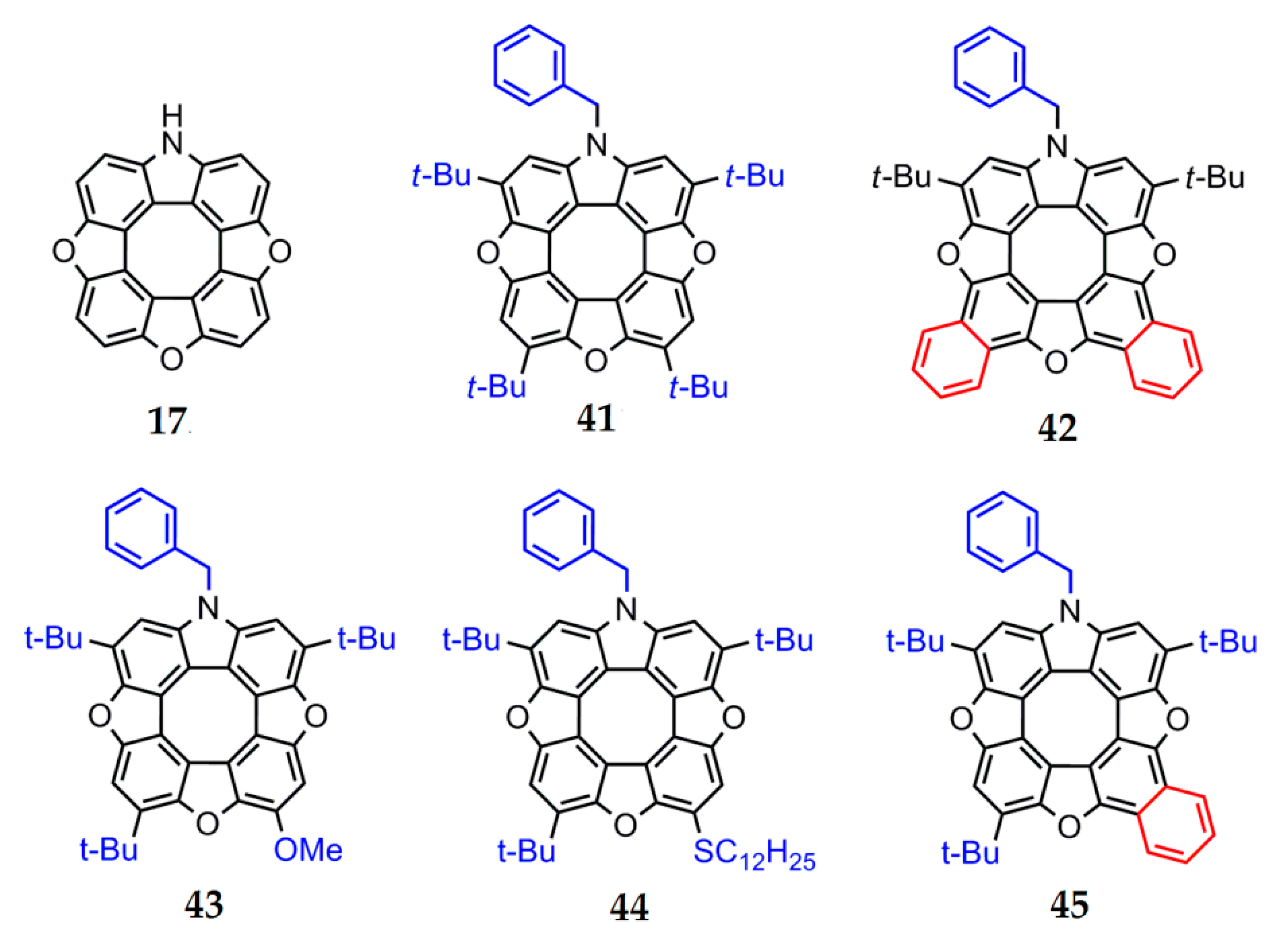
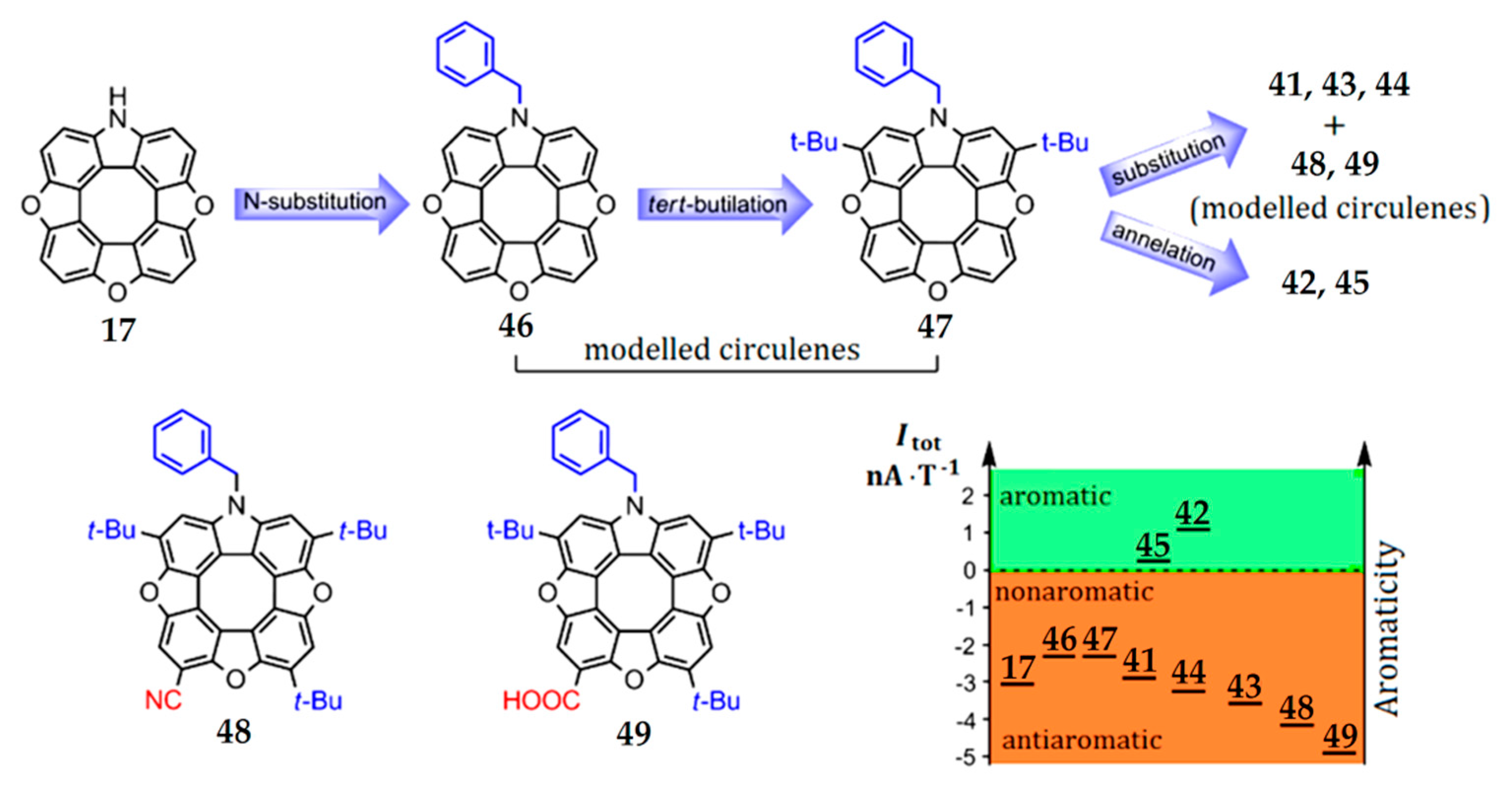
The presence of electron-donor and electron-acceptor substituents in the outer macrocycle of the molecule has a significant influence on the aromaticity of heterocirculenes. This effect is seen in the example of azatrioxa[8]circulenes [93]. The total ring current (Itot) for azatrioxa[8]circulene 17 (Figure 12) is equal to 3.1 nA∙T−1, which corresponds to the sum of the paratropic ring-current component, induced inside the internal octatetraene ring (−12.4 nA∙T−1), and the diatropic ring-current component, induced by the outer macrocycle of the circulene molecule (9.3 nA∙T−1) (we can note that the calculation of circulene 17 was carried out without the side substituents for simplicity). The introduction of one N-benzyl (Bn) substituent (circulene 46, Figure 13) into the initial structure of the molecule 17 leads to an increase in the total ring current strength to −2.2 nA∙T−1. This fact can be explained by an increase in the diatropic ring-current component contribution on the outside the molecule (10.2 nA∙T−1) in comparison with the paratropic ring-current component inside the octatetraene ring, which have the same value as the molecule of the initial circulene 17.
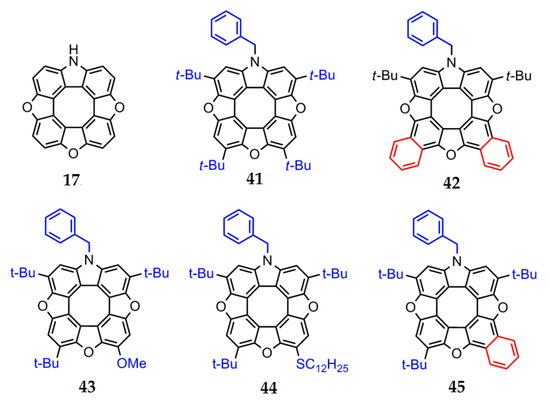
Figure 12.
The structure of the parent ‘‘hypothetical’’ azatrioxa[8]circulene frame 17 (without the side substituents for simplicity) and related synthesized unsymmetrical compounds 41–45 [93].
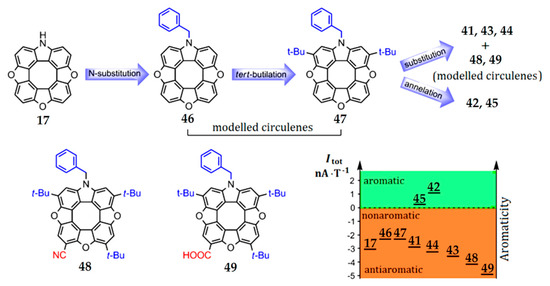
Figure 13.
Logical scheme for subsequent functionalization of the simplest hypothetical azatrioxa[8]circulene 17 vs. changing diagram for the net magnetically induced ring current strengths [93].
The introduction of two tert-butyl groups into the structure of the molecule 46 (circulene 47, Figure 13) does not affect the overall balance between the paratropic and diatropic ring-current components. In a series of substituted heterocirculenes 41, 43 and 44, there is a tendency towards a decrease in the total ring current strength 41 (4t-Bu, –3.1 nA∙T–1), 44 (3t-Bu+SMe, −3.4 nA∙T−1), 43 (3t-Bu+OMe, −3.7 nA∙T−1) in comparison with the circulene molecule 46 (Table 4).
Table 4.
The total ring current strength (Itot) and aromaticity of the azatrioxa[8]circulene molecules.
The introduction of electron-acceptor groups instead of one t-Bu substituent (modeled circulenes 48 and 49, Figure 13) provides a strong reduction in the paratropic ring-current component inside the octatetraene ring (−12.8 and −13.0 nA∙T−1 for –CN and –COOH substituted molecules 48 and 49, respectively, Figure 13). At the same time, the diatropic component in the outer macrocycle of the molecules remains the same value in the circulene molecules 41, 43 and 44. As a result, the total ring-current strength for the modeled circulenes 48 and 49 is equal to −4.1 and −5.0 nA∙T−1, respectively, indicating their antiaromatic nature (Table 4). Therefore, one can conclude that the fivefold substitution of the initial circulene 17 does not impose a clear effect on the aromaticity degree. However, compared with the modeled Bn-substituted circulene 46, one can see that the fourfold substitution in the outer macrocycle of azatrioxacirculene molecule provides an increase in the antiaromaticity degree for the synthesized molecules 41, 43 and 44, as well as for the modeled compounds 48 and 49. It should be noted that the effect of the electron-acceptor groups –CN and –COOH is more pronounced in comparison with that of the t-Bu, –OMe and –SMe substituents (Figure 13).
The mono- and double benzoannelation leads to an increase in the aromaticity degree in molecules 42 and 45 in comparison with the modeled molecule 47, i.e., the annelation effect is opposite to the substitution effect. This fact can be explained by the additional expansion of the π-conjugation system of azatrioxa[8]circulene 17, which causes an increase in the diatropic component’s contribution to the total ring current strength. Formally, we can talk about the aromaticity inversion during benzoannelation, since the total ring current strength changes the sign from negative “–” (in the case of all nonbenzoannelated circulenes) to positive “+” (1.0 nA∙T−1 for circulene 42 and 0.3 nA∙T−1 for circulene 45). It should be noted that a similar benzoannelation effect is observed for the π-conjugated tetraoxa[8]circulenes [45,46].
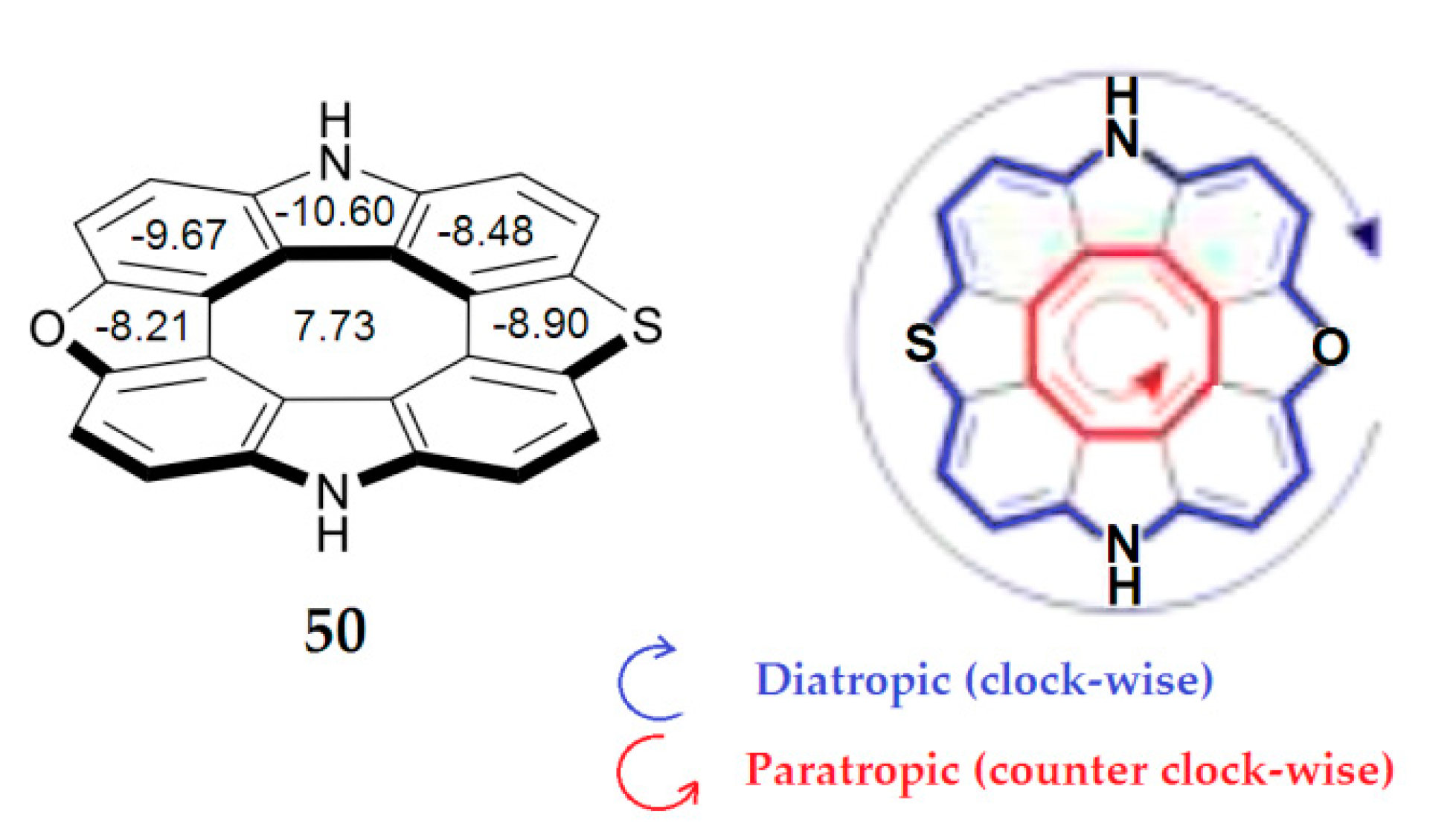
In 2020, Pittelkow’s group reported the first antiaromatic heterocyclic [8]circulene with three different heterocyclic rings—diazaoxathia[8]circulene 50 (Figure 14)—using a novel synthetic sequential annulation strategy, where heterocyclic [7]helicenes was planarized [94].
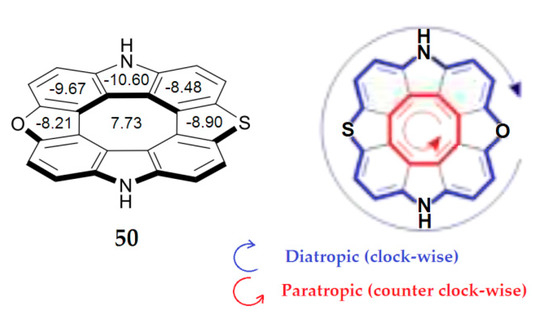
Figure 14.
The structure of diazaoxathia[8]circulene. The NICS indices for 50 calculated in the center of each ring by the B3LYP/6-311++G(d,p) method in GIAO approximation [94].
As one can see from Figure 14, the central eight-membered planar ring shows a positive NICS(0)-value of 7.73 ppm [94], suggesting that the structure shares the paratropic and antiaromatic nature of the inner octatetraene ring. The surrounding benzene, furan and pyrrole demonstrate considerably negative NICS indices, indicating a diatropic ring current and aromatic nature.
Thus, a combination of NICS and GIMIC approaches allows us to estimate the measure of competition between the diatropic and paratropic ring currents in the heterocirculene molecules and to make a general conclusion about their aromatic nature.
4. Aromaticity of the “Lower” (n < 8) and “Higher” (n > 8) Hetero[n]circulenes
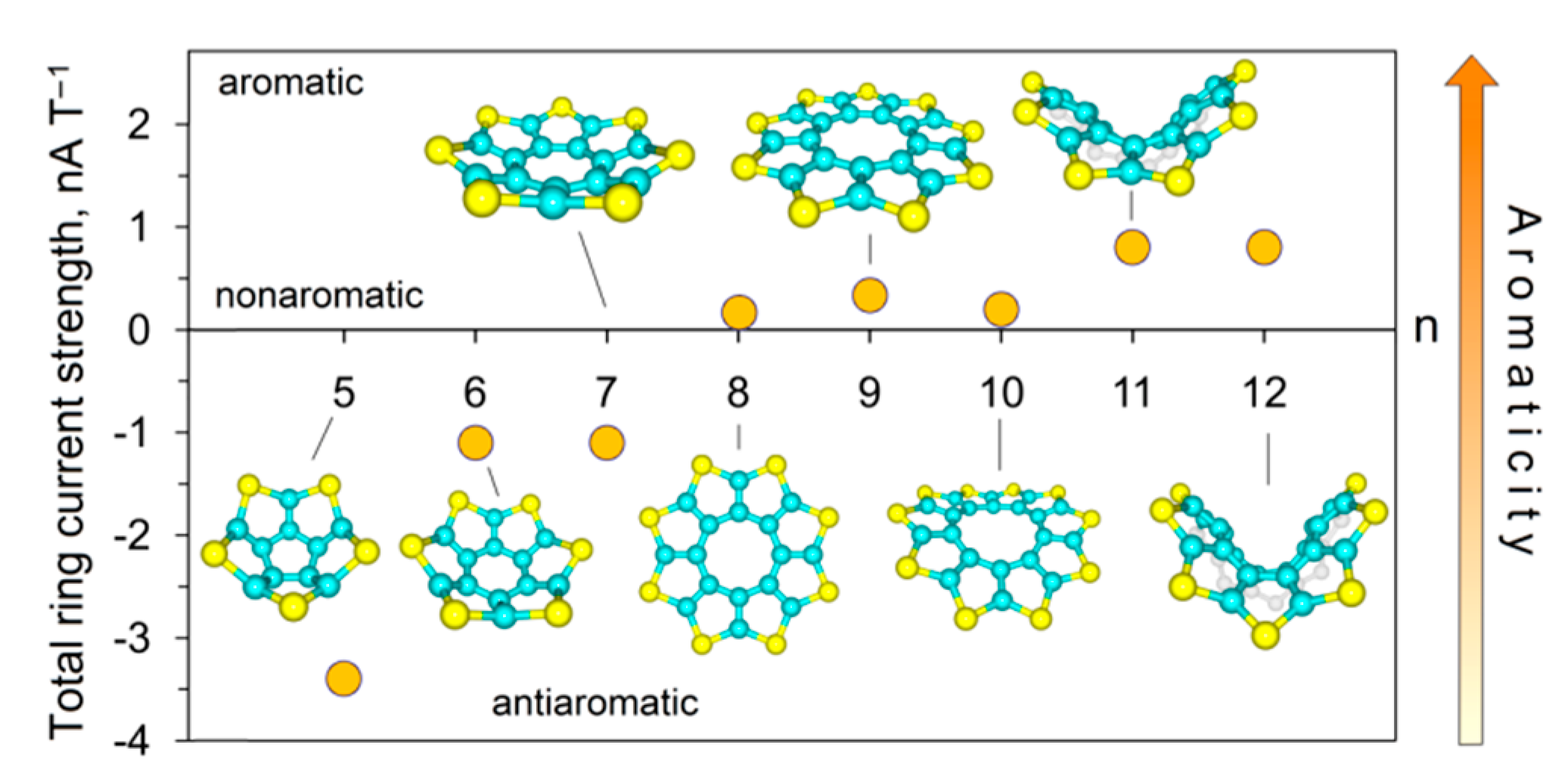
4.1. Thia[n]circulenes (n = 5–12)
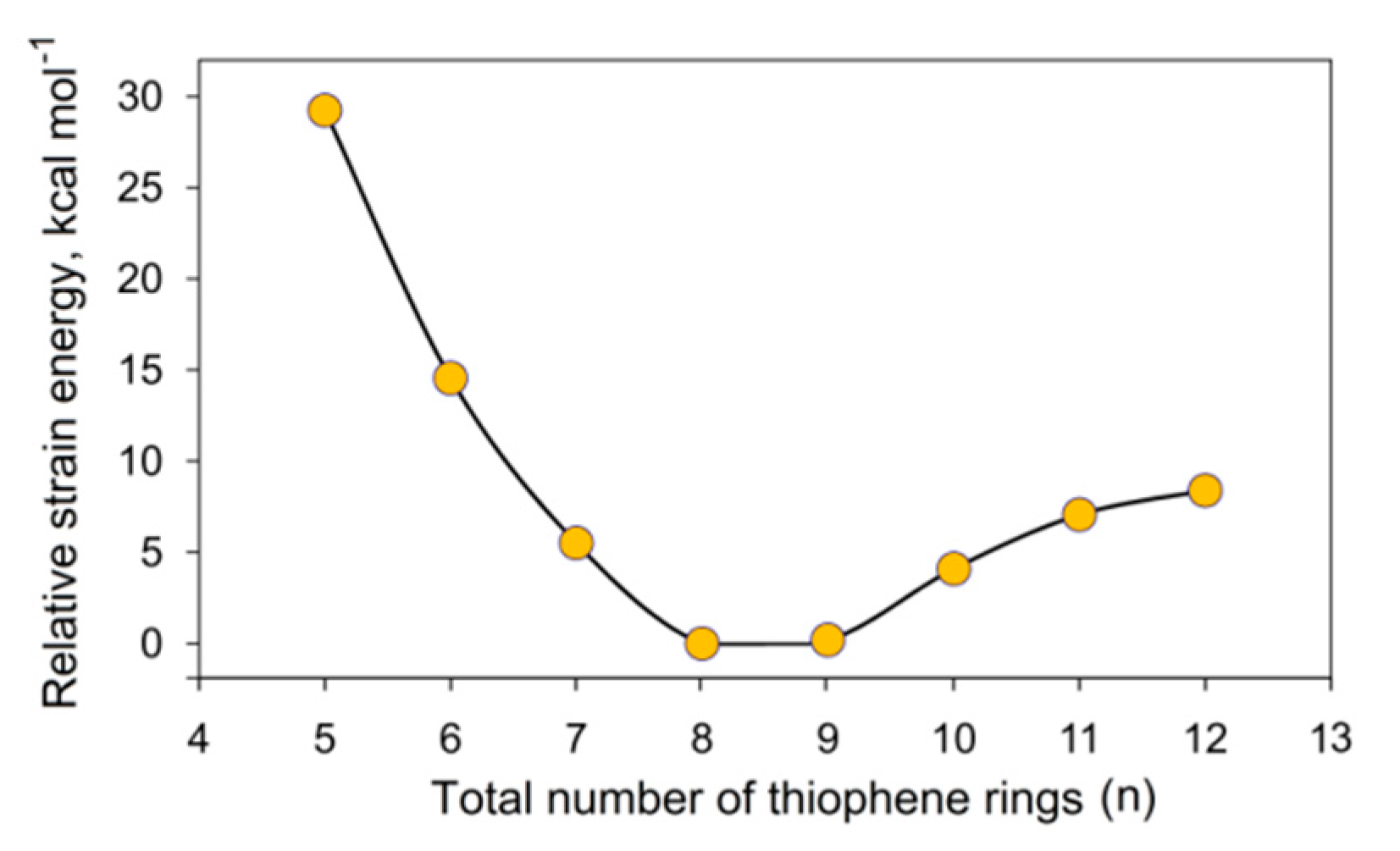
Karaush-Karmazin et al. [95] examined the change in aromaticity for a series of thia[n]circulenes with a different number of fused thiophene rings (n = 5–12) by means of the methodology proposed in Ref. [96]. The calculated strain energies for thia[n]circulenes (n = 5–12) relative to planar octathia[8]circulene 20 are presented in Figure 15. It was shown that the strain energy for the nonathia[9]circulene is only 0.2 kcal∙mol−1; therefore, its molecule has an almost unstrained macrocycle with planar structure [95]. With further extension of the circulene macrocycle from n = 10 up to n = 12, the strain slightly increases up to ≈ 9 kcal mol−1 for thia[12]circulene (Figure 15) because of a gradual bending of the molecular skeleton. One can expect that the loss of planarity in thia[n]circulenes (n = 10–12) should lead to a reduction in aromaticity. However, GIMIC and NICS calculations indicate that moving from planar to corrugated thia[n]circulenes, the aromaticity of their molecules is only weakly changed, and even slightly increased up to 0.8 nA T−1 for thia[n]circulenes with n = 11, 12 (Figure 15). This behavior is similar to the higher carbo[n]helicenes (n > 5) [48] and helical [n]benzofurans (n > 2) [97]. This fact was explained by a gradual increase in the diatropic current component on the outside of the thia[n]circulene molecules (n = 9–12), with macrocycle extension and its dominance over the paratropic current inside the ring yielding a total diatropic current [95].
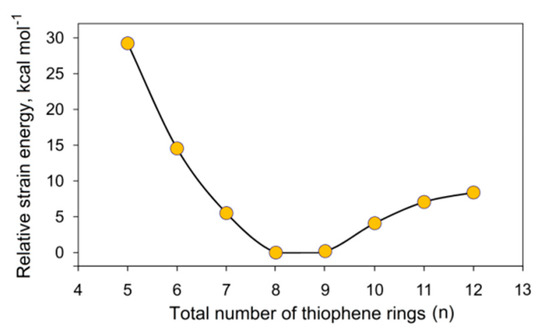
Figure 15.
Calculated strain energies for thia[n]circulene species relative to planar octathia[8]circulene 20 at the B3LYP/6–311++G(d,p) level of theory [95].
The molecules of smaller thia[n]circulenes with n = 5–7 demonstrate a more pronounced increase in the strain energy up to 30 kcal mol−1 for pentathia[5]circulene when macrocycle is reduced. Such strain energy of the macrocycle leads to its distortion into a bowl-shaped structure. Therefore, it is obvious that the molecules of smaller thia[n]circulenes (n = 5–7) should be antiaromatic or nonaromatic species. According to GIMIC calculations, the contribution of the paratropic current component is increased, yielding a total paratropic ring current for smaller thia[n]circulenes with n = 5–7. It was reported in Ref. [95] that the total ring current reduces from −3.4 nA T−1 for pentathia[5]circulene to −1.1 nA T−1 for hexathia[6]circulene and heptathia[7]circulene (Figure 16), i.e., the first circulene was found to be a slightly antiaromatic compound, whereas the latter two compounds are almost nonaromatic species.
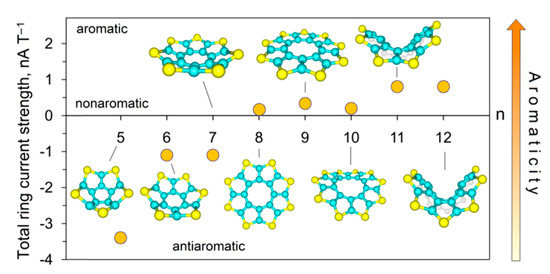
Figure 16.
The dependence of the magnetically induced current strength on the increase in the number of conjugated thiophene rings (n) in a molecule [95].
Summarizing the above, it should be noted that GIMIC also has problems with molecules that are far from being planar, because it is not clear in which direction the magnetic field should point [98]. For example, in the case of [12]circulene in Figure 16, the magnetic field is almost perpendicular to the central twelve-membered ring, but is far from being perpendicular to all the other five-membered rings of the periphery.
4.2. Heterocyclic[9]- and [10]Circulenes
Until recently, the synthetic efforts of heterocyclic [n]circulenes covered up to eight aromatic rings (n = 8), while the higher hetero[n]circulenes (n > 8) have been predicted only by quantum chemical modeling [95,97]. In 2020, the first synthesis of a fully aromatic [9]circulene, formally a diazatrioxa[9]circulene 51, along with a tetrahydrodiazatetraoxa[10]circulene 52, was reported by Pittelkow’s group (Figure 17) [99]. The structure of [9]circulene’s macrocyclic core is perfectly planar, while tetrahydro[10]circulene adopts a V-shaped cis form with unilaterally directed hydrogen atoms on four bridging sp3 carbon atoms. The angle between the mean plane of the two carbazoles is 73.7° [99].
Figure 17.
Molecular structures of the studied heterocyclic [9]- and [10]circulenes.
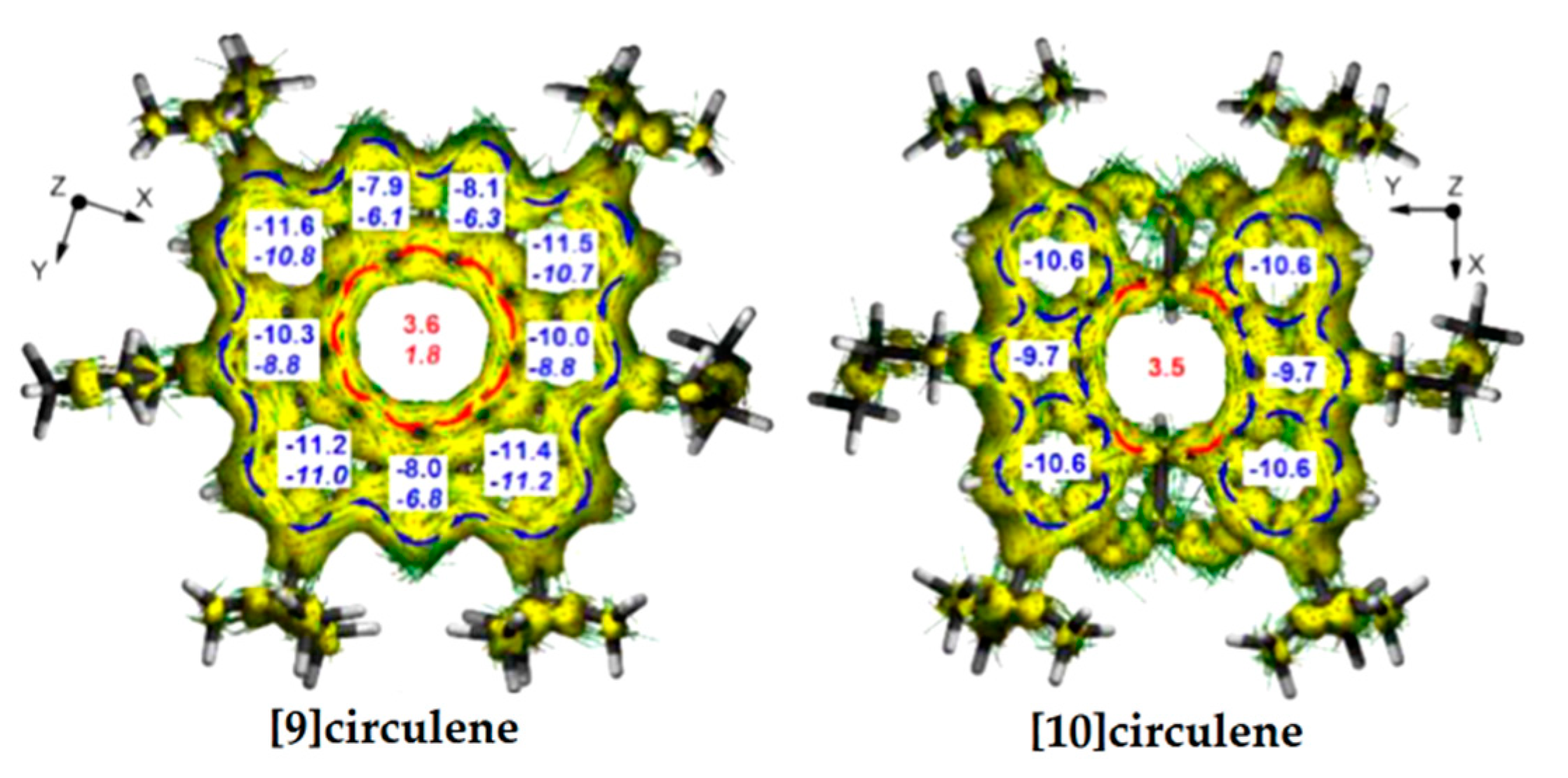
According to the ACID plots of [9]circulene 51, the diatropic ring currents appear in the outer rim (blue arrows in Figure 18), while the 9-membered hub ring sustains paratropic currents (red arrows in Figure 18) similar to those of the hetero[8]circulenes presented above.
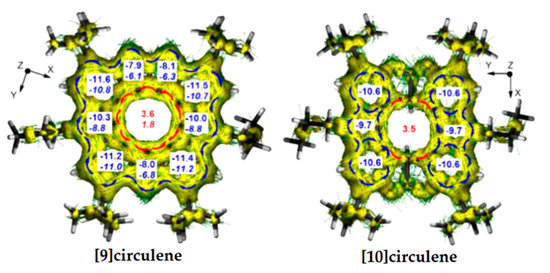
Figure 18.
ACID plots. Blue arrows—local diatropic currents, red arrows—paratropic currents. The NICS(0) (top number) and NICS(1) (bottom number in italics) indices in the [9]circulene and [10]circulene calculated by the GIAO/B3LYP/6-311++G(d,p) method [99].
In the case of tetrahydrotetraoxadiaza[10]circulene 52, the π-conjugation in the rim and hub subsystems is broken. The strong local diatropic currents flow in the two carbazole fragments (blue arrows in Figure 18) and the radial C−C bonds are also involved; only weak paratropic currents circulate inside the 10-membered ring through the σ-conjugation (red arrows in Figure 18). NICS calculations of [9]- and [10]circulenes are in agreement with the results obtained from the ACID plots. NICS indices are negative for all individual aromatic rings constructing these circulenes, due to the diatropic ring current, and positive for the central ring due to the paratropic ring current. The presence of paratropic ring currents in the central nonaromatic rings of [9]- and [10]circulenes can be explained by the concept of topologically induced paratropicity [100].
It should be noted that the NICS(1) and NICS(0) (1.8, 3.6, respectively) of the central nonaromatic ring of [9]circulene are significantly lower than those values of the related diazadioxa[8]circulene, indicating a contribution from the antiaromatic cyclooctatetraene in the diazadioxa[8]circulene molecule.
5. Aromaticity of the π-Extended Hetero[8]circulenes
5.1. Diaza[n]circulenes
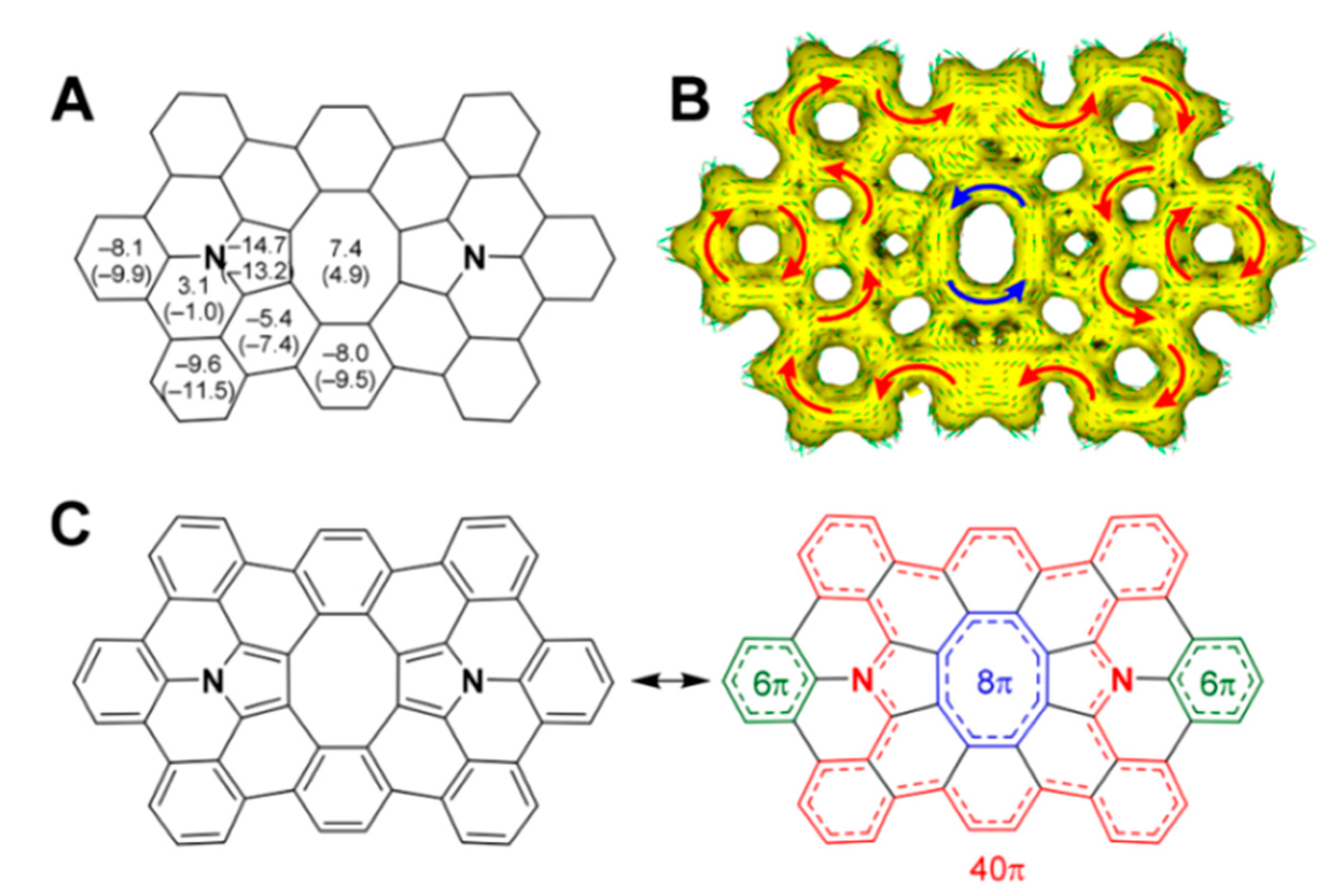
The authors of Ref. [101] described on-surface synthesis of the first example of a π-extended diaza[8]circulene 53 surrounded by and fused with six hexagons and two pentagons (Figure 19). It was found that some molecules are planar on Au(111) with highly distorted octagon structure. This is due to the large sum of the wedge angles of the surrounding rings (six hexagons and two pentagons), thus creating a strained conformation of 53. According to DFT calculations for 53 in the gas phase, the twisted structure is the most stable in comparison with the saddle (2.7 kcal/mol) and planar (12.7 kcal/mol) conformations [101].
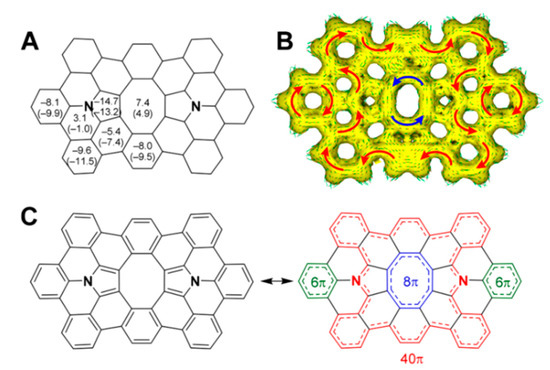
Figure 19.
(A) NICS(0) values (in ppm) for planar 53 calculated in the gas phase. NICS(1) values are shown in parentheses. (B) ACID plot for 53 calculated in the gas phase. (C) Bond alternation patterns of 53. Reprinted with permission from Ref. [101]. Copyright 2020 American Chemical Society.
In 53, the central antiaromatic octatetraene ring shows an NICS(0) value of 7.4 ppm, which is comparable to those of the hetero[8]circulenes discussed above. The ACID plot clearly shows a counterclockwise 8π ring current flowing along the central octatetraene ring as well as a clockwise 40π ring current flowing along the outer fused rings, including the two pyrrole rings. The observation of a diatropic 40π ring current is consistent with those of other hetero[8]circulenes, which typically show internal paratropic 8π ring currents surrounded by outer diatropic 24π ring currents.
Therefore, the NICS and ACID calculations suggest the presence of two major contributing electronic systems, one having two 6π pyrrole rings and eight 6π benzene rings (left in Figure 19C) and the other having inner 8π, outer 40π, and two 6π conjugations (right in Figure 19C).
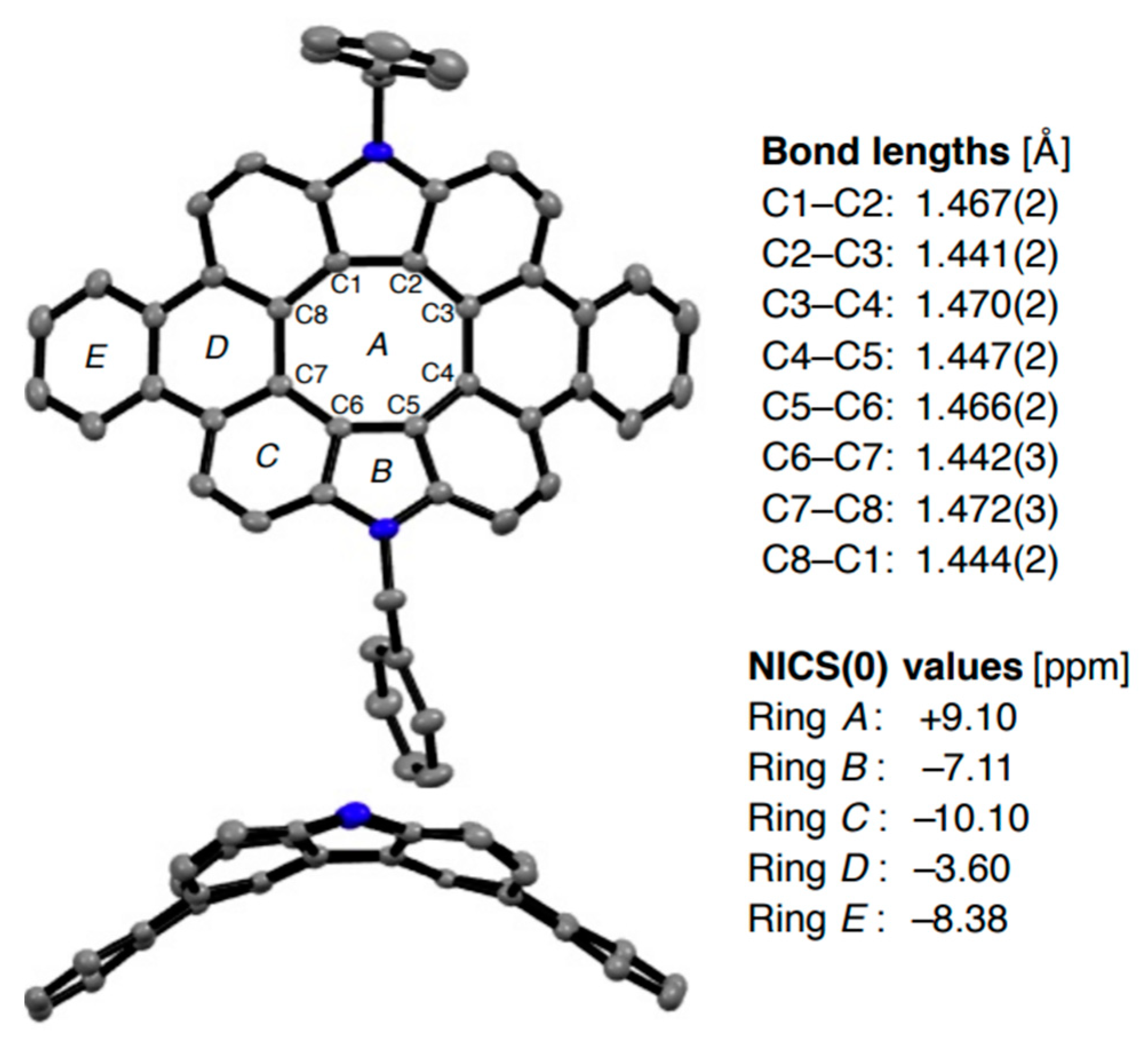
In 2021, Maeda et al. reported on the solution-based synthesis of the deeply saddle-distorted dibenzodiaza[8]circulene 54 [102], which made it possible to unambiguously determine the structure of π-extended diaza[8]circulenes. The mean plane deviation of the dibenzodiaza[8]circulene moiety is 0.90 Å indicating the distorted π -framework. Aromaticity nature of 54 is similar to the previously discussed hetero[8]circulenes, i.e., the central antiaromatic core surrounded by an aromatic system of benzene and pyrrole rings (Figure 20).
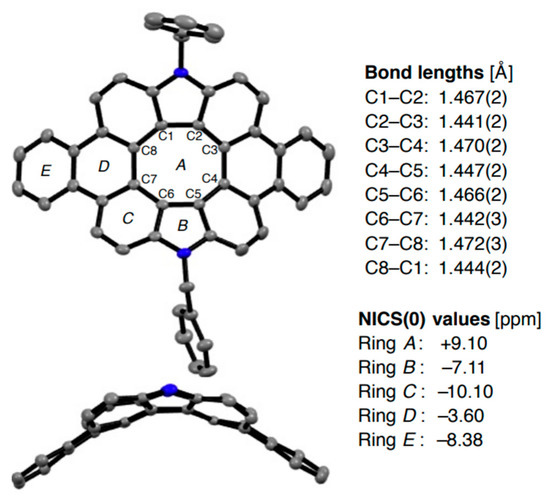
Figure 20.
X-ray crystal structure, bond lengths and NICS(0) values of saddle-distorted dibenzodiaza[8]circulene 54. Hydrogen atoms, solvent molecules, and peripheral substituents in the side views are omitted for clarity. Reprinted with permission from Ref. [102]. Copyright 2021 John Wiley & Sons.
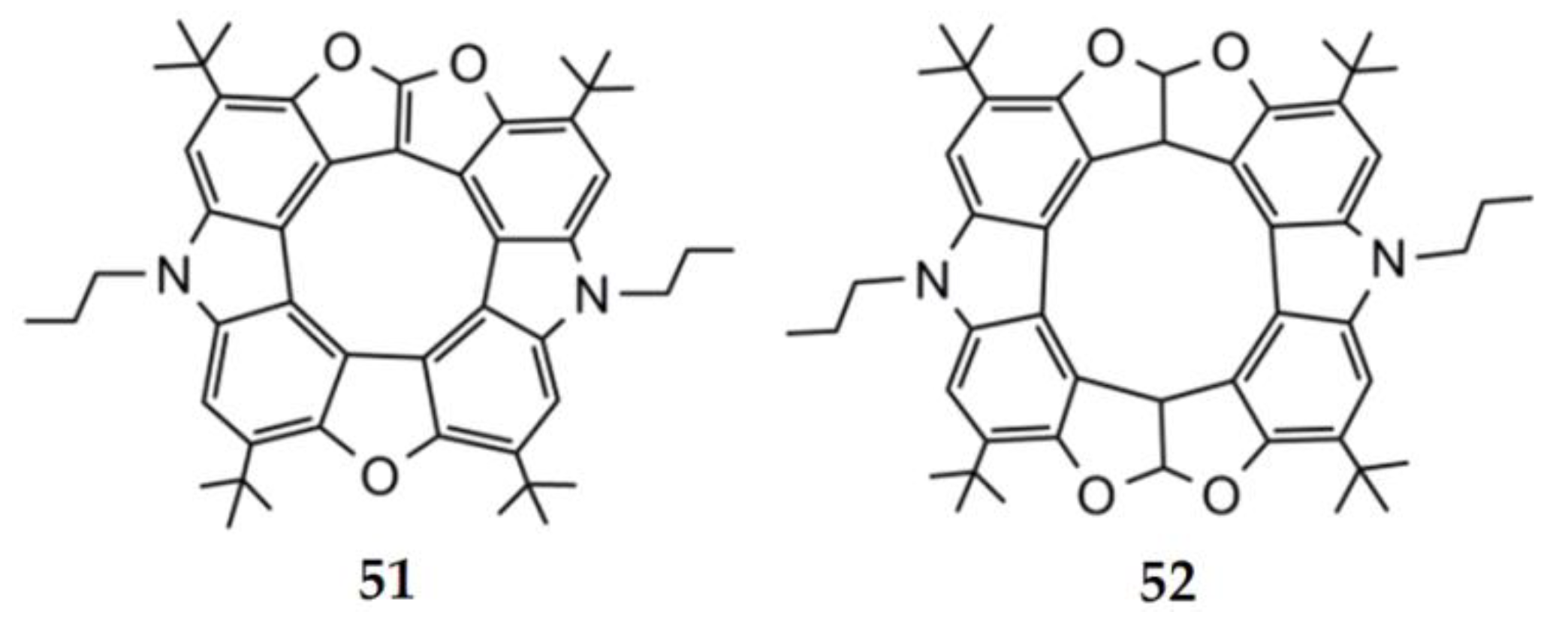
5.2. Dianthracenylazatrioxa[8]circulene
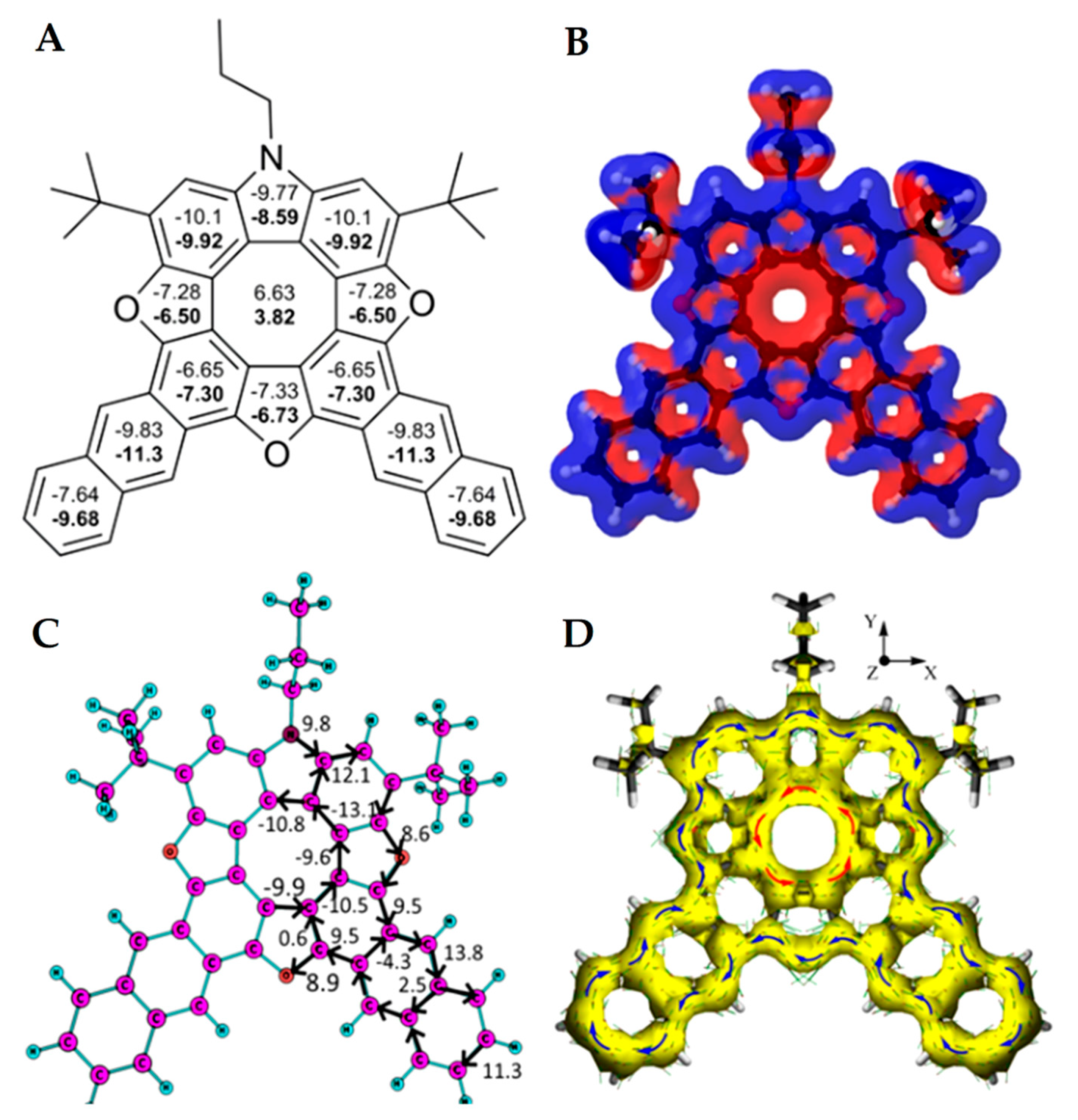
In 2021, Pittelkow’s group presented the synthesis of a π-extended azatrioxa[8]circulene—dianthracenylazatrioxa[8]circulene 55 (Figure 21) [26]. The dianthracenylazatrioxa[8]circulene possesses cyclic 8π electron conjugation of the central octatetraene core, and the structure is almost completely planar according to X-ray data [26]. This circulene demonstrates a similar aromaticity behavior compared to other hetero[8]circulenes. The positive NICS(0) and NICS(1) values confirm the presence of strong paratropicity (i.e., antiaromaticity) inside the inner eight-membered core, while all of the remaining five- and six-membered rings show negative values for the NICS(0) and NICS(1) indices, implying the presence of a predominant diatropicity (i.e., aromaticity).
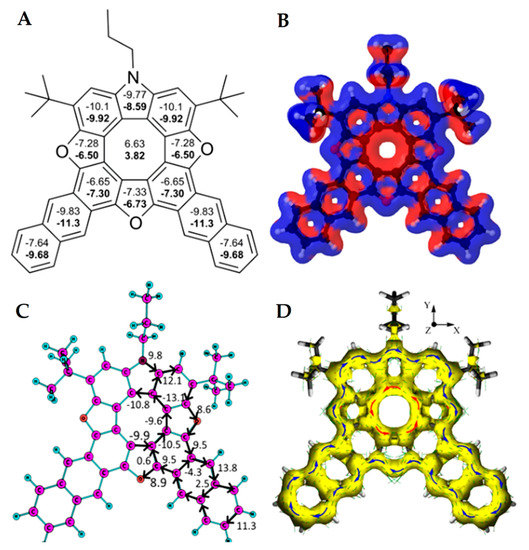
Figure 21.
NICS(0) and NICS(1) (in bold) indices (A), the signed modulus of the MIC densities (B), MIC strengths vs. MIC pathway (C) and anisotropy of the induced current density plot (D) for the dianthracenylazatrioxa[8]circulene molecule. Paratropic current densities are shown in red and the diatropic ones are shown in blue in (B) [26].
The GIMIC results are in complete agreement with the NICS indices. As can be seen from the magnetically induced current (MIC) density plots (Figure 21B), the inner eight-membered core possesses a strong paratropic (anticlockwise) MIC, while the outer perimeter of 55 sustains a diatropic (clockwise) MIC circulation. The MIC strength for paratropic and diatropic components was found to be similar in absolute values (−13.1 nA T−1 vs. 12.1 nA T−1, respectively). The resulting MIC strength is only −1 nA T−1, meaning that the studied circulene molecule is globally non-aromatic. This feature implies the existence of two self-cancelling MIC systems, paratropic in the inner eight-membered core, and diatropic along the outer perimeter. The MIC strength along the radial CC bonds is very small (0.3–2.3 nA T−1), in excellent agreement with the ACID plot in Figure 21D.
6. Conclusions
The considered hetero[8]circulenes and their partially heteroannelated analogues, the [9]- and [10]circulenes, show common complicated aromatic features. Their analysis sheds new light on the differences in physico-chemical properties of various series of heterocirculenes. All types of classifications analyzed in this review are used in practical implementation of heterocirculenes in molecular electronics and in the IR, UV, and NMR spectral interpretations [103,104,105,106,107,108,109]. During the preparation of this paper, a new hybrid aromatic molecule with alternating thiophene and furan rings, called an oxisulflower (with 10-fused rings), and “pure” oxiflower (with 14-furan fused) rings was modeled as a potential structure for synthesizing sulflower (octathia[8]circulene) [110].
Author Contributions
Conceptualization, N.N.K.-K., G.V.B. and B.F.M.; writing—original draft preparation, N.N.K.-K.; writing—review and editing, N.N.K.-K., G.V.B. and B.F.M.; visualization, N.N.K.-K.; supervision, B.F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grunenberg, J. Ill-defined chemical concepts: The problem of quantification. Int. J. Quantum Chem. 2017, 117, e25359. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Cyrański, M.K. Structural Aspects of Aromaticity. Chem. Rev. 2001, 101, 1385–1420. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Szatylowicz, H.; Stasyuk, O.A.; Dominikowska, J.; Palusiak, M. Aromaticity from the Viewpoint of Molecular Geometry: Application to Planar Systems. Chem. Rev. 2014, 114, 6383–6422. [Google Scholar] [CrossRef]
- Cyrański, M.K. Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. Chem. Rev. 2005, 105, 3773–3811. [Google Scholar] [CrossRef] [PubMed]
- Zanasi, R.; Lazzeretti, P.; Malagoli, M.; Piccinini, F. Molecular magnetic properties within continuous transformations of origin of the current density. J. Chem. Phys. 1995, 102, 7150–7157. [Google Scholar] [CrossRef]
- Mitchell, R.H. Measuring aromaticity by NMR. Chem. Rev. 2001, 101, 1301–1315. [Google Scholar] [CrossRef]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Gershoni-Poranne, R.; Stanger, A. Magnetic criteria of aromaticity. Chem. Soc. Rev. 2015, 44, 6597–6615. [Google Scholar] [CrossRef] [PubMed]
- Fliegl, H.; Taubert, S.; Lehtonen, O.; Sundholm, D. The gauge including magnetically induced current method. Phys. Chem. Chem. Phys. 2011, 13, 20500–20518. [Google Scholar] [CrossRef] [PubMed]
- Jusélius, J.; Sundholm, D.; Gauss, J. Calculation of current densities using gauge-including atomic orbitals. J. Chem. Phys. 2004, 121, 3952–3963. [Google Scholar] [CrossRef]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef]
- Feixas, F.; Matito, E.; Poater, J.; Solà, M. Quantifying aromaticity with electron delocalisation measures. Chem. Soc. Rev. 2015, 44, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Mucsi, Z.; Viskolcz, B.; Csizmadia, I.G. A quantitative scale for the degree of aromaticity and antiaromaticity: A comparison of theoretical and experimental enthalpies of hydrogenation. J. Phys. Chem. A 2007, 111, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Solà, M. Aromaticity. In Encyclopedia of Physical Organic Chemistry; Wang, Z., Ed.; John Wiley and Sons: Weinheim, Germany; London, UK, 2017; Volume 1, pp. 511–542. [Google Scholar]
- Minaev, B.F.; Baryshnikov, G.V.; Minaeva, V.A. Density functional theory study of electronic structure and spectra of tetraoxa[8]circulenes. Comput. Theor. Chem. 2011, 972, 68–74. [Google Scholar] [CrossRef]
- Valiev, R.R.; Baryshnikov, G.V.; Sundholm, D. Relations between the aromaticity and magnetic dipole transitions in the electronic spectra of hetero[8]circulenes. Phys. Chem. Chem. Phys. 2018, 20, 30239–30246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiev, R.R.; Gadirov, R.M.; Degtyarenko, K.M.; Grigoryev, D.; Nasubullin, R.T.; Baryshnikov, G.; Minaev, B.F.; Pedersen, S.K.; Pittelkow, M. The blue vibronically resolved electroluminescence of azatrioxa[8]circulene. Chem. Phys. Lett. 2019, 732, 136667–136671. [Google Scholar] [CrossRef]
- Karaush-Karmazin, N.N.; Baryshnikov, G.V.; Kuklin, A.V.; Saykova, D.I.; Ågren, H.; Minaev, B.F. Impact of molecular and packing structure on the charge-transport properties of hetero[8]circulenes. J. Mat. Chem. C 2021, 9, 1451–1466. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Minaev, B.F.; Minaeva, V.A. Electronic structure, aromaticity and spectra of hetero[8]circulenes. Russ. Chem. Rev. 2015, 84, 455–484. [Google Scholar] [CrossRef]
- Hensel, T.; Andersen, N.N.; Plesner, M.; Pittelkow, M. Synthesis of Heterocyclic [8]Circulenes and Related Structures. Synlett. 2016, 27, 498–525. [Google Scholar] [CrossRef] [Green Version]
- Karaush, N.N.; Baryshnikov, G.V.; Minaeva, V.A.; Ågren, H.; Minaev, B.F. Recent progress in quantum chemistry of hetero[8]circulenes. Mol. Phys. 2017, 115, 2218–2230. [Google Scholar] [CrossRef]
- Miyake, Y.; Shinokubo, H. Hetero[8]circulenes: Synthetic progress and intrinsic properties. Chem. Commun. 2020, 56, 15605–15614. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Brock-Nannestad, T.; Reenberg, T.K.; Hammershøj, P.; Christensen, J.B.; Stouwdam, J.W.; Pittelkow, M. Organic Light-Emitting Diodes from Symmetrical and Unsymmetrical π-Extended Tetraoxa[8]circulenes. Chem. Eur. J. 2010, 16, 13030–13034. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Valiev, R.R.; Karaush, N.N.; Minaeva, V.A.; Sinelnikov, A.N.; Pedersen, S.K.; Pittelkow, M.; Minaev, B.F.; Ågren, H. Benzoannelated aza-, oxa- and azaoxa[8]circulenes as promising blue organic emitters. Phys. Chem. Chem. Phys. 2016, 18, 28040–28051. [Google Scholar] [CrossRef]
- Ivaniuk, K.B.; Baryshnikov, G.V.; Stakhira, P.Y.; Pedersen, S.K.; Pittelkow, M.; Lazauskas, A.; Volyniuk, D.; Grazulevicius, J.V.; Minaev, B.F.; Ågren, H. New WOLEDs based on p-extended azatrioxa[8]circulenes. J. Mater. Chem. C 2017, 5, 4123–4128. [Google Scholar] [CrossRef]
- Pedersen, S.K.; Pedersen, V.B.; Kamounah, F.S.; Broløs, L.M.; Baryshnikov, G.V.; Valiev, R.R.; Ivaniuk, K.; Stakhira, P.; Karaush-Karmazin, N.N.; Minaev, B.; et al. Dianthracenylazatrioxa[8]circulene: Synthesis, Characterization and Application in OLEDs. Chem.-A Eur. J. 2021, 27, 11609–11617. [Google Scholar] [CrossRef]
- Fujimoto, T.; Matsushita, M.M.; Awaga, K. Ionic-Liquid Component Dependence of Carrier Injection and Mobility for Electric-Double-Layer Organic Thin-Film Transistors. J. Phys. Chem. C 2012, 116, 5240–5245. [Google Scholar] [CrossRef]
- Fujimoto, T.; Matsushita, M.M.; Awaga, K. Dual-gate field-effect transistors of octathio[8]circulene thin-films with ionic liquid and SiO2 gate dielectrics. Appl. Phys. Lett. 2010, 97, 123303–123305. [Google Scholar] [CrossRef]
- Dadvand, A.; Cicoira, F.; Chernichenko, K.Y.; Balenkova, E.S.; Osuna, R.M.; Rosei, F.; Nenajdenko, V.G.; Perepichka, D.F. Heterocirculenes as a new class of organic semiconductors. Chem. Commun. 2008, 5354–5356. [Google Scholar] [CrossRef] [PubMed]
- Kuklin, A.V.; Baryshnikov, G.V.; Minaev, B.F.; Ignatova, N.; Ågren, H. Strong Topological States and High Charge Carrier Mobility in Tetraoxa[8]circulene Nanosheets. J. Phys. Chem. C 2018, 122, 22216–22222. [Google Scholar] [CrossRef]
- Begunovich, L.V.; Kuklin, A.V.; Baryshnikov, G.V.; Valiev, R.R.; Ågren, H. Single-layer Polymeric Tetraoxa [8] circulene Modified by s-Metals: Toward Stable Spin Qubits and Novel Superconductors. Nanoscale 2021, 13, 4799–4811. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Valiev, R.R.; Karaush, N.N.; Minaev, B.F. Aromaticity of the planar hetero[8]circulenes and their doubly charged ions: NICS and GIMIC characterization. Phys. Chem. Chem. Phys. 2014, 16, 15367–15374. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikov, G.V.; Karaush, N.N.; Valiev, R.R.; Minaev, B.F. Aromaticity of the completely annelated tetraphenylenes: NICS and GIMIC characterization. J. Mol. Model. 2015, 21, 136. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikov, G.V.; Valiev, R.R.; Karaush, N.N.; Sundholm, D.; Minaev, B.F. Aromaticity of the doubly charged [8]circulenes. Phys. Chem. Chem. Phys. 2016, 18, 8980–8992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, H.C.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Anisotropy of the Induced Current Density (ACID), a General Method to Quantify and Visualize Electronic Delocalization. Chem. Rev. 2005, 105, 3758–3772. [Google Scholar] [CrossRef]
- Badri, Z.; Pathak, S.; Fliegl, H.; Rashidi-Ranjbar, P.; Bast, R.; Marek, R.; Foroutan-Nejad, C.; Ruud, K. All-Metal Aromaticity: Revisiting the Ring Current Model among Transition Metal Clusters. J. Chem. Theory Comput. 2013, 9, 4789–4796. [Google Scholar] [CrossRef]
- Foroutan-Nejad, C. Is NICS a reliable aromaticity index for transition metal clusters? Theor. Chem. Acc. 2015, 134, 8–16. [Google Scholar] [CrossRef]
- Jusélius, J.; Sundholm, D. Abinitio determination of the induced ring current in aromatic molecules. Phys. Chem. Chem. Phys. 1999, 1, 3429–3435. [Google Scholar] [CrossRef]
- Steiner, E.; Fowler, P.; Soncini, A.; Jenneskens, L.W. Current-density maps as probes of aromaticity: Global and Clar π ring currents in totally resonant polycyclic aromatic hydrocarbons. Faraday Discuss. 2007, 135, 309–323. [Google Scholar] [CrossRef]
- Jusélius, J.; Sundholm, D. The Aromatic Character of Magnesium Porphyrins. J. Org. Chem. 2000, 65, 5233–5237. [Google Scholar] [CrossRef] [PubMed]
- Fowler, P.W.; Havenith, R.W.A.; Steiner, E. Ring current and electron delocalisation in an all-metal cluster, Al42−. Chem. Phys. Lett. 2002, 359, 530–536. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Jusélius, J.; Sundholm, D.; Gauss, J. Magnetically induced current densities in Al4 (2-) and Al4 (4-) species studied at the coupled-cluster level. J. Chem. Phys. 2005, 122, 214308. [Google Scholar] [CrossRef]
- Tsipis, C.A. DFT study of “all-metal” aromatic compounds. Coord. Chem. Rev. 2005, 249, 2740–2762. [Google Scholar] [CrossRef]
- Radenković, S.; Gutman, I.; Bultinck, P. Comparative Study of Aromaticity in Tetraoxa[8]circulenes. J. Phys. Chem. A 2012, 116, 9421–9430. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikov, G.V.; Minaev, B.F.; Pittelkow, M.; Nielsen, C.B.; Salcedo, R. Nucleus-independent chemical shift criterion for aromaticity in π-extended tetraoxa[8]circulenes. J. Mol. Model. 2013, 19, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikov, G.V.; Minaev, B.F.; Karaush, N.N.; Minaeva, V.A. The art of the possible: Computational design of the 1D and 2D materials based on the tetraoxa[8]circulene monomer. RSC Adv. 2014, 4, 25843–25851. [Google Scholar] [CrossRef]
- Portella, G.; Poater, J.; Bofill, J.M.; Alemany, P.; Solà, M. Local Aromaticity of [n]Acenes, [n]Phenacenes, and [n]Helicenes (n = 1−9). J. Org. Chem. 2005, 70, 2509–2521. [Google Scholar] [CrossRef]
- Aihara, J. Graph Theory of Aromatic Stabilization. Chem. Soc. Jpn. 2016, 89, 1425–1454. [Google Scholar] [CrossRef] [Green Version]
- Baryshnikov, G.V.; Karaush, N.N.; Minaev, B.F. The electronic structure of heteroannelated cyclooctatetraenes and their UV-Vis absorption spectra. Chem. Heterocycl. Comp. 2014, 50, 349–363. [Google Scholar] [CrossRef]
- Abdukadir, A.; Mattursun, A.; Kerim, A.; Omar, K.; Hushur, L. A study of the aromaticity of heteroannelated cyclooctatetraene derivatives. J. Mol. Mod. 2018, 24, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Aita, K.; Ohmae, T.; Takase, M.; Nomura, K.; Kimura, H.; Nishinaga, T. Dithieno[3,4-b:3′,4′-d]thiophene-annelated antiaromatic planar cyclooctatetraene with olefinic protons. Org. Lett. 2013, 15, 3522–3525. [Google Scholar] [CrossRef]
- Willner, I.; Rabinovitz, M. Cycloocta[def]fluorene: A planar cyclooctatetraene derivative. Paratropicity of hydrocarbon and anion. J. Org. Chem. 1980, 45, 1628–1633. [Google Scholar] [CrossRef]
- Wilcox, C.F., Jr.; Uetrecht, J.P.; Grohman, K.K. Preparation of cycloocta[def]biphenylene, a novel benzenoid antiaromatic hydrocarbon. J. Am. Chem. Soc. 1972, 94, 2532–2533. [Google Scholar] [CrossRef]
- Wilcox, C.F. Empirically corrected ab initio calculation of proton NMR chemical shifts of antiaromatic hydrocarbons. J. Mol. Struct. Theochem 2006, 759, 125–132. [Google Scholar] [CrossRef]
- Karadakov, P.B. Aromaticity and antiaromaticity in the low-lying electronic states of cyclooctatetraene. J. Phys. Chem. A 2008, 112, 12707–12713. [Google Scholar] [CrossRef]
- Nishinaga, T.; Uto, T.; Inoue, R.; Matsuura, A.; Treitel, N.; Rabinovitz, M.; Komatsu, K. Antiaromaticity and reactivity of a planar cyclooctatetraene fully annelated with bicyclo[2.1.1]hexane units. Chem. Eur. J. 2010, 14, 2067–2074. [Google Scholar] [CrossRef]
- Nishinaga, T.; Ohmae, T.; Iyoda, M. Recent studies on the aromaticity and antiaromaticity of planar cyclooctatetraene. Symmetry 2010, 2, 76–97. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.J.; Emmert, F.L., III; Slipchenko, L.V. Effects of ethynyl substituents on the electronic structure of cyclobutadiene. J. Phys. Chem. A 2012, 116, 3194–3201. [Google Scholar] [CrossRef]
- Aihara, J. A new definition of Dewar-type resonance energies. J. Am. Chem. Soc. 1976, 98, 2750–2758. [Google Scholar] [CrossRef]
- Gutman, I.; Milun, M.; Trinajstic, N. Graph theory and molecular orbitals. 19. Nonparametric resonance energies of arbitrary conjugated systems. J. Am. Chem. Soc. 1977, 99, 1692–1704. [Google Scholar] [CrossRef]
- Sekine, R.; Nakagami, Y.; Aihara, J. Aromatic character of polycyclic π systems formed by fusion of two or more rings of the same size. J. Phys. Chem. A 2011, 115, 6724–6731. [Google Scholar] [CrossRef]
- Aihara, J. Bond resonance energy and verification of the isolated pentagon rule. J. Am. Chem. Soc. 1995, 117, 4130–4136. [Google Scholar] [CrossRef]
- Aihara, J. Bond resonance energies of polycyclic benzenoid and non-benzenoid hydrocarbons. J. Chem. Soc. Perkin Trans. 1996, 2, 2185–2195. [Google Scholar] [CrossRef]
- Aihara, J.; Ishida, T.; Kanno, H. Bond resonance energy as an indicator of local aromaticity. Bull. Chem. Soc. Jpn. 2007, 80, 1518–1521. [Google Scholar] [CrossRef]
- Aihara, J. Circuit resonance energy: A key quantity that links energetic and magnetic criteria of aromaticity. J. Am. Chem. Soc. 2006, 128, 2873–2879. [Google Scholar] [CrossRef]
- Arkin, R.; Kerim, A. A study on the aromaticity and magnetotropicity of 10π-electron azapentalene derivatives. Chem. Phys. Lett. 2012, 546, 144–149. [Google Scholar] [CrossRef]
- Kerim, A. A study on the aromaticity and magnetic properties of triazolephorphyrazines. New J. Chem. 2014, 383, 3783–3790. [Google Scholar] [CrossRef]
- Ilić, P.; Trinajstić, N. Topological resonance energies of conjugated ions, radicals, and ion radicals. J. Org. Chem. 1980, 45, 1738–1748. [Google Scholar] [CrossRef]
- Salcedo, R.; Sansores, L.E.; Picazo, A.; Sanson, L. [8] Circulene. Theoretical approach. J. Mol. Struct. Theochem 2004, 678, 211–215. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Brock-Nannestad, T.; Hammershøj, P.; Reenberg, T.K.; Schau-Magnussen, M.; Trpcevski, D.; Hensel, T.; Salcedo, R.; Baryshnikov, G.V.; Minaev, B.F.; et al. Azatrioxa[8]circulenes: Planar antiaromatic cyclooctatetraenes. Chem. Eur. J. 2013, 19, 3898–3904. [Google Scholar] [CrossRef] [PubMed]
- Hensel, T.; Trpcevski, D.; Lind, C.; Grosjean, R.; Hammershoj, P.; Nielsen, C.B.; Brock-Nannestad, T.; Nielsen, B.E.; Schau-Magnussen, M.; Minaev, B.; et al. Diazadioxa[8]circulenes: Planar antiaromatic cyclooctatetraenes. Chem. Eur. J. 2013, 19, 17097–17102. [Google Scholar] [CrossRef] [PubMed]
- Hellwinkel, D.; Reiff, G. Cyclooctatetraene systems flattened by steric constraints. Angew. Chem. Int. Ed. Engl. 1970, 9, 527–528. [Google Scholar] [CrossRef]
- Serizawa, Y.; Akahori, S.; Kato, S.; Sakai, H.; Hasobe, T.; Miyake, Y.; Shinokubo, H. Synthesis of tetrasilatetrathia[8]circulenes by a fourfold intramolecular dehydrogenative silylation of C-H bonds. Chem. Eur. J. 2017, 23, 6948–6952. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.-D.; Deng, C.-L.; Peng, X.-S.; Miao, Q.; Wong, H.N.C. Heteroatom-bridged tetraphenylenes: Synthesis, structures, and properties. Org. Lett. 2014, 16, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Deng, C.-L.; Minaev, B.F.; Baryshnikov, G.V.; Peng, X.-S.; Wong, H.N.C. Tetrathio and tetraseleno[8]circulenes: Synthesis, structures, and properties. Chem. Asian J. 2014, 9, 969–977. [Google Scholar] [CrossRef]
- Minaeva, V.A.; Baryshnikov, G.V.; Minaev, B.F.; Karaush, N.N.; Xiong, X.-D.; Lee, D.P.; Wong, H.N.C. Structure and spectroscopic characterization of tetrathia- and tetraselena[8]circulenes as a new class of polyaromatic heterocycles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 151, 247–261. [Google Scholar] [CrossRef]
- Xiong, X.; Deng, C.-L.; Li, Z.; Penga, X.-S.; Wong, H.N.C. Quasi-planar diazadithio and diazodiseleno[8]circulenes: Synthesis, structures and properties. Org. Chem. Front. 2017, 4, 682–687. [Google Scholar] [CrossRef]
- Akahori, S.; Sakai, H.; Hasobe, T.; Shinokubo, H.; Miyake, Y. Synthesis and photodynamics of tetragermatetrathia[8]circulene. Org. Lett. 2018, 20, 304−307. [Google Scholar] [CrossRef]
- Tai, T.B.; Huong, V.T.T.; Nguyen, M.T. Design of aromatic heteropolycyclics containing borole frameworks. Chem. Commun. 2013, 49, 11548–11550. [Google Scholar] [CrossRef]
- Kar, T.; Scheiner, S.; Roy, A.K. Structure and properties of [8]BN-circulenes—Inorganic analogues of [8]circulenes. J. Phys. Chem. C 2015, 119, 15541–15546. [Google Scholar] [CrossRef]
- Gribanova, T.N.; Zefirov, N.S.; Minkin, V.I. Structure and stability of the heteroannulated [8–10]circulenes: A quantum-chemical study. Pure Appl. Chem. 2010, 82, 1011–1024. [Google Scholar] [CrossRef]
- Mandal, T.K.; Jose, D.; Nijamudheen, A.; Datta, A. Structures and electronic properties of heavier congeners of disk-like molecules: (Si, Ge) sulflower and (Si, Ge) olympicene. J. Phys. Chem. C 2014, 118, 12115–12120. [Google Scholar] [CrossRef]
- Erdtman, H.; Högberg, H.-E. Cyclooligomerisation of quinones. Tetrahedron Lett. 1970, 11, 3389–3392. [Google Scholar] [CrossRef]
- Cyrański, M.K.; Havenith, R.W.A.; Dobrowolski, M.A.; Gray, B.R.; Krygowski, T.M.; Fowler, P.W.; Jenneskens, L.W. The phenalenyl motf: A magnetic chameleon. Chem.—Eur. J. 2007, 13, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Steiner, E.; Fowler, P.W. Patterns of Ring Currents in Conjugated Molecules: A Few-Electron Model Based on Orbital Contributions. J. Phys. Chem. A 2001, 105, 9553–9562. [Google Scholar] [CrossRef]
- Johansson, M.P.; Jusélius, J.; Sundholm, D. Sphere Currents of Buckminsterfullerene. Chemie Int. Ed. 2005, 44, 1843–1846. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, J.I.; Corminboeuf, C.; Bohmann, J.; Lu, X.; Hirschf, A.; Schleyer, P.R. Is C60 buckminsterfullerene aromatic? Phys. Chem. Chem. Phys. 2012, 14, 14886–14891. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.P.; Juselius, J. Arsole aromaticity revisited. Let. Org. Chem. 2005, 2, 469–474. [Google Scholar] [CrossRef]
- Fias, S.; Van Damme, S.; Bultinck, P. Multidimensionality of Delocalization Indices and Nucleus Independent Chemical Shifts in Polycyclic Aromatic Hydrocarbons. J. Comput. Chem. 2008, 29, 358–366. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Viglione, R.G.; Zanasi, R. Local Aromaticity of the Six-Membered Rings in Pyracylene. A Difficult Case for the NICS Indicator of Aromaticity. J. Org. Chem. 2004, 69, 7537–7542. [Google Scholar] [CrossRef]
- Solà, M.; Feixas, F.; Jiménez-Halla, J.O.C.; Matito, E.; Poater, J. A Critical Assessment of the Performance of Magnetic and Electronic Indices of Aromaticity. Symmetry 2010, 2, 1156–1179. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Valiev, R.R.; Minaev, B.F.; Ågren, H. A computational study of aromaticity and photophysical properties of unsymmetrical azatrioxa[8]circulenes. New J. Chem. 2017, 41, 2717–2723. [Google Scholar] [CrossRef]
- Lousen, B.; Pedersen, S.K.; Bols, P.; Hansen, K.H.; Pedersen, M.R.; Hammerich, O.; Bondarchuk, S.; Minaev, B.; Baryshnikov, G.V.; Ågren, H.; et al. Compressing a Non-Planar Aromatic Heterocyclic [7]Helicene to a Planar Hetero[8]Circulene. Chem. Eur. J. 2020, 26, 4935–4940. [Google Scholar] [CrossRef]
- Karaush-Karmazin, N.N.; Baryshnikov, G.V.; Valiulina, L.I.; Valiev, R.; Ågren, H.; Minaev, B.F. Impact of heteroatoms (S, Se, and Te) on the aromaticity of heterocirculenes. New J. Chem. 2019, 43, 12178–12190. [Google Scholar] [CrossRef]
- Chernichenko, K.Y.; Sumerin, V.V.; Shpanchenko, R.V.; Balenkova, E.S.; Nenajdenko, V.G. “Sulflower”: A New Form of Carbon Sulfide. Angew. Chem. 2006, 118, 7527–7530. [Google Scholar] [CrossRef]
- Karaush, N.N.; Baryshnikov, G.V.; Ågren, H.; Minaev, B.F. A theoretical study of new representatives of closed- and open-circle benzofuran and benzocyclopentadienone oligomers. New J. Chem. 2018, 42, 11493–11505. [Google Scholar] [CrossRef]
- Barquera-Lozada, J.E. The vorticity of the current density tensor and 3D-aromaticity. Int. J. Quantum Chem. 2019, 119, e25848. [Google Scholar] [CrossRef]
- Pedersen, S.K.; Eriksen, K.; Ågren, H.; Minaev, B.F.; Karaush-Karmazin, N.N.; Hammerich, O.; Baryshnikov, G.V.; Pittelkow, M. A Fully Conjugated Planar Heterocyclic [9] Circulene. J. Am. Chem. Soc. 2020, 142, 14058–14063. [Google Scholar] [CrossRef]
- Pedersen, S.K.; Eriksen, K.; Karaush-Karmazin, N.N.; Minaev, B.; Ågren, H.; Baryshnikov, G.V.; Pittelkow, M. Anti-Aromatic versus Induced Paratropicity: Synthesis and Interrogation of a Dihydro- diazatrioxa[9]circulene with a Proton Placed Directly above the Central Ring. Angew. Chem. Int. Ed. 2020, 59, 5144–5150. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Li, Q.-Q.; Krejčí, O.; Foster, A.S.; Sun, K.; Kawai, S.; Ito, S. On-Surface Synthesis of a π-Extended Diaza[8]circulene. J. Am. Chem. Soc. 2020, 142, 11363–11369. [Google Scholar] [CrossRef]
- Maeda, C.; Nomoto, S.; Akiyama, K.; Tanaka, T.; Ema, T. Facile Synthesis of Azahelicenes and Diaza[8]circulenes through the Intramolecular Scholl Reaction. Chem. Eur. J. 2021, 27, 15699–15705. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Valiev, R.R.; Cherepanov, V.N.; Karaush-Karmazin, N.N.; Minaeva, V.A.; Minaev, B.F.; Ågren, H. Aromaticity and photophysics of tetrasila-and tetragerma-annelated tetrathienylenes as new representatives of the hetero[8]circulene family. Phys. Chem. Chem. Phys. 2019, 21, 9246–9254. [Google Scholar] [CrossRef] [Green Version]
- Karaush, N.N.; Minaev, B.F.; Baryshnikov, G.V.; Minaeva, V.A. A Comparative Study of the Electronic Structure and Spectra of Tetraoxa [8] circulene and Octathio[8]circulene. Opt. Spectrosc. 2014, 116, 33–46. [Google Scholar] [CrossRef]
- Karaush, N.N.; Valiev, R.R.; Baryshnikov, G.V.; Minaev, B.F.; Ågren, H. DFT simulation of the heteroannelated octatetraenes vibronic spectra with the Franck–Condon and Herzberg–Teller approaches including Duschinsky effect. Chem. Phys. 2015, 459, 65–71. [Google Scholar] [CrossRef]
- Karaush, N.N.; Baryshnikov, G.V.; Minaeva, V.A.; Minaev, B.F. A DFT and QTAIM study of the novel d-block metal complexes with tetraoxa[8]circulene-based ligands. New J. Chem. 2015, 39, 7815–7821. [Google Scholar] [CrossRef]
- Karaush, N.N.; Baryshnikov, G.V.; Minaev, B.F. Alkali and alkaline-earth metal complexes with tetraoxa[8]circulene sheet: A computational study by DFT and QTAIM methods. RSC Adv. 2015, 5, 24299–24305. [Google Scholar] [CrossRef]
- Minaeva, V.; Karaush-Karmazin, N.; Baryshnikov, G.; Minaev, B. A complete characterization of vibrational IR and Raman spectra of the highly-symmetrical octathia[8]circulene. Vib. Spectrosc. 2019, 100, 107–116. [Google Scholar] [CrossRef]
- Karaush-Karmazin, N.N.; Kuklin, A.V.; Baryshnikov, G.V.; Begunovich, L.V.; Ågren, H.; Minaev, B.F. Structure, stability and electronic properties of one-dimensional tetrathia-and tetraselena[8]circulene-based materials: A comparative DFT study. New J. Chem. 2020, 44, 6872–6882. [Google Scholar] [CrossRef]
- Nguyen, L.H. A Computational Study of the Electronic Properties of Heterocirculenes: Oxiflowers and Sulflowers. ACS Omega 2021, 6, 30085–30092. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).