
Photoelectron Circular Dichroism as a Probe of Chiral Hydrocarbons


Abstract
:1. Introduction
2. The Theoretical Approach
3. Results and Discussion
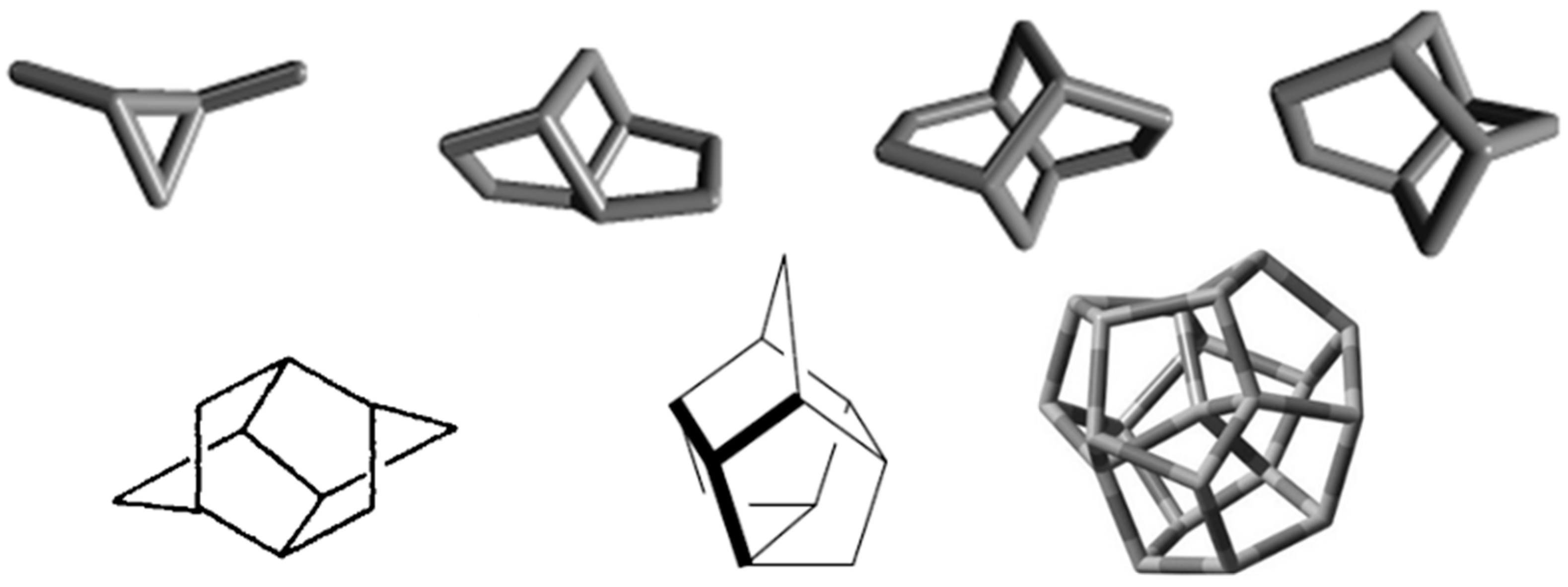
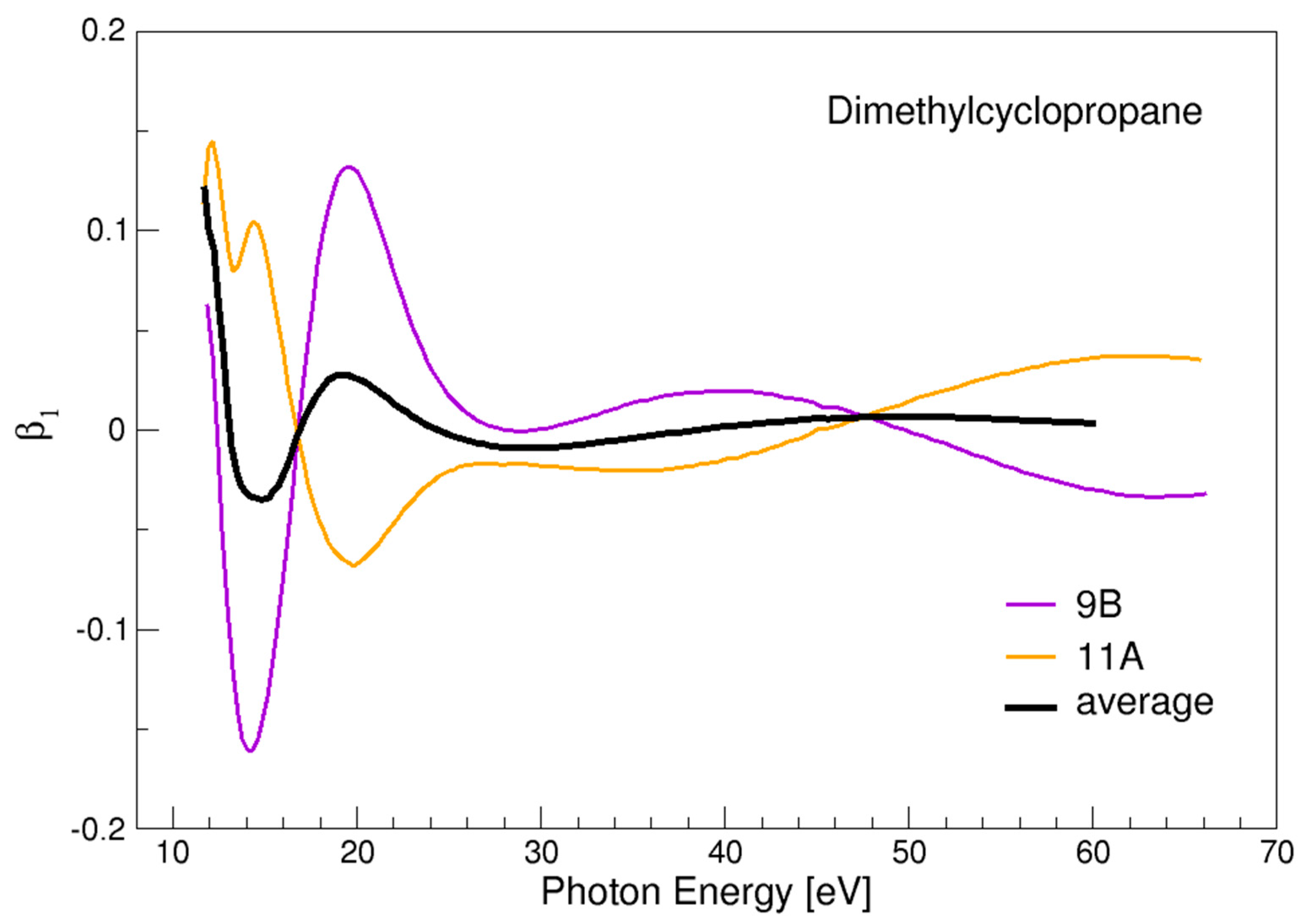
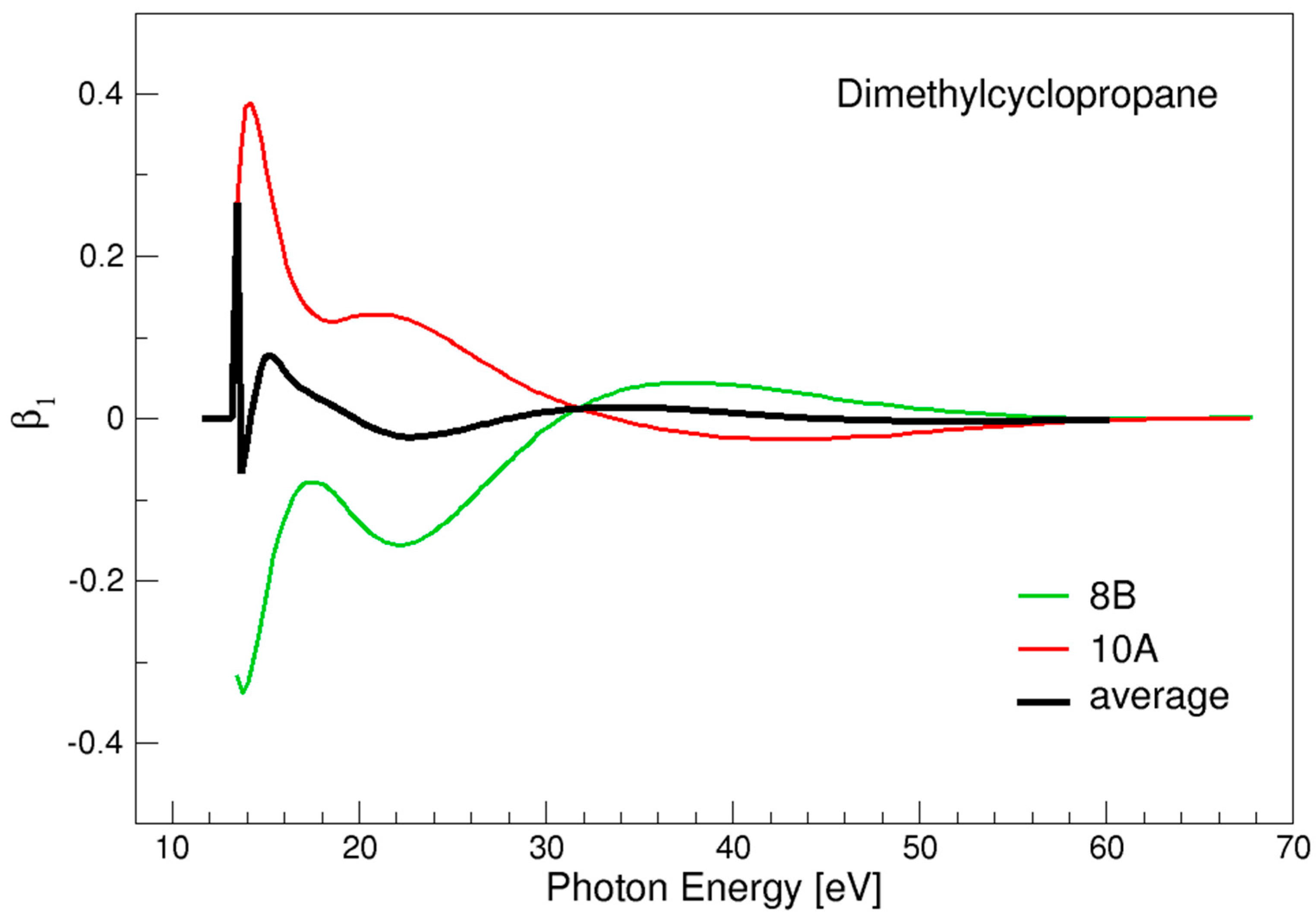
3.1. Dimethylcyclopropane
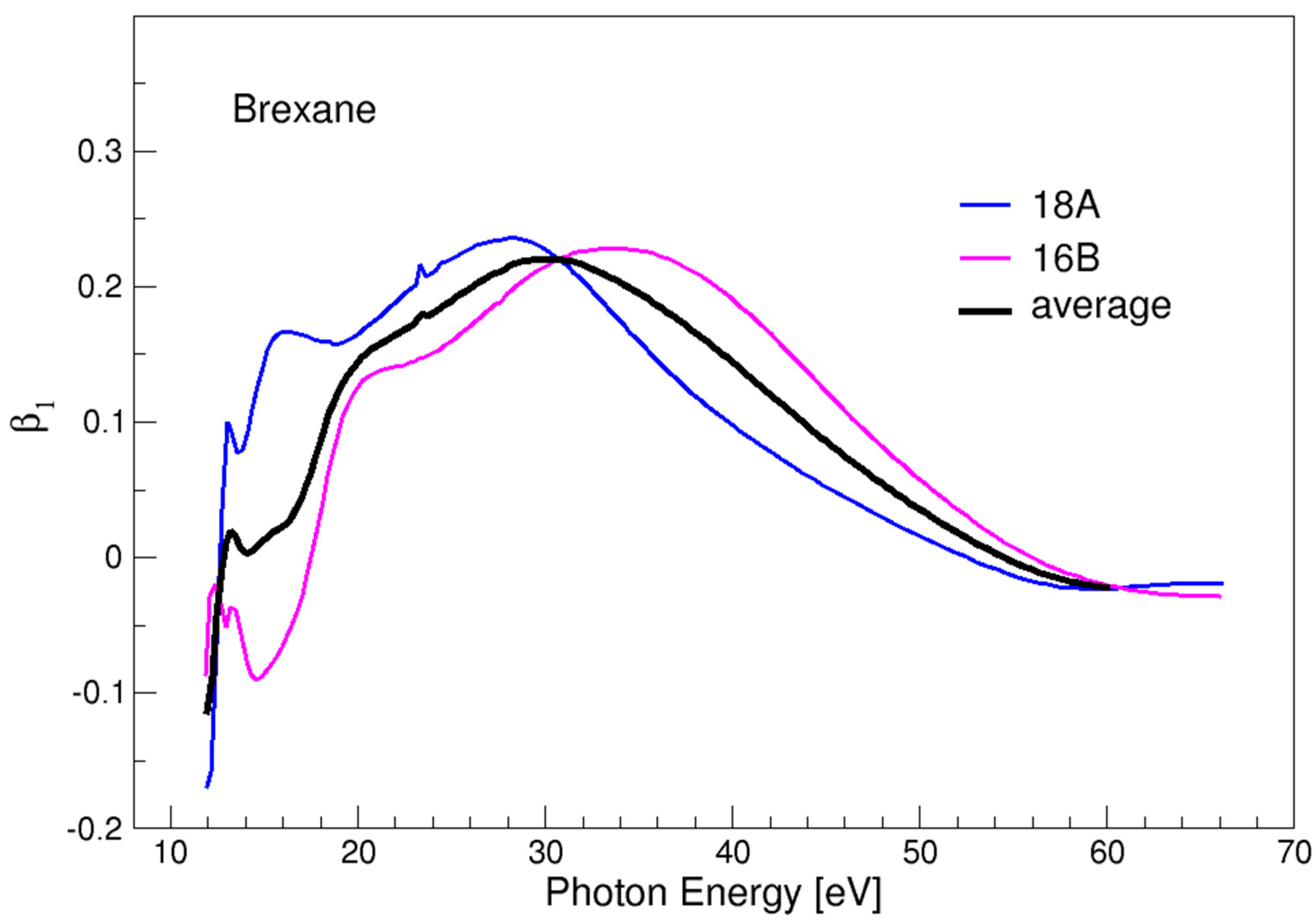
3.2. Brexane
3.3. Twistane
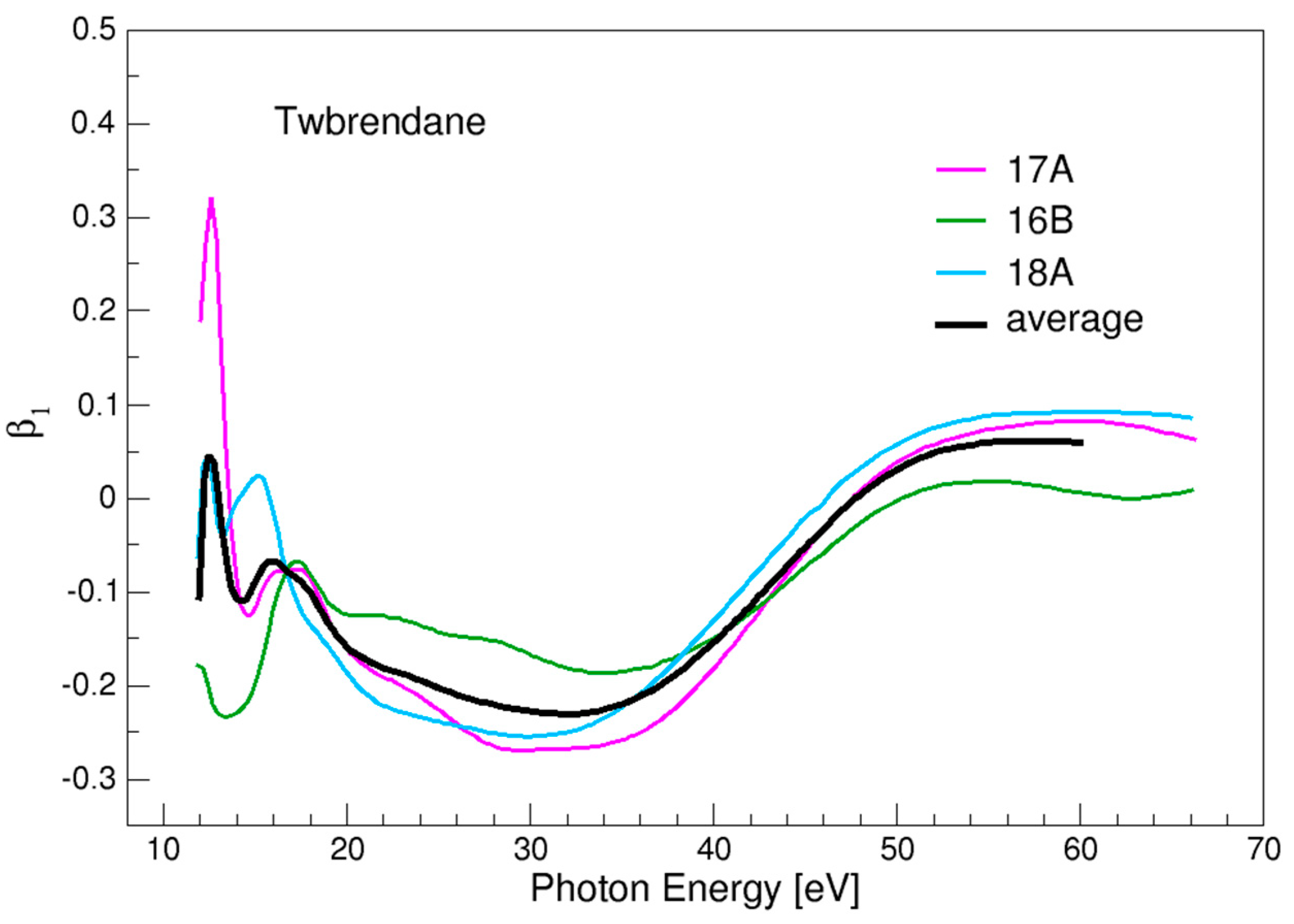
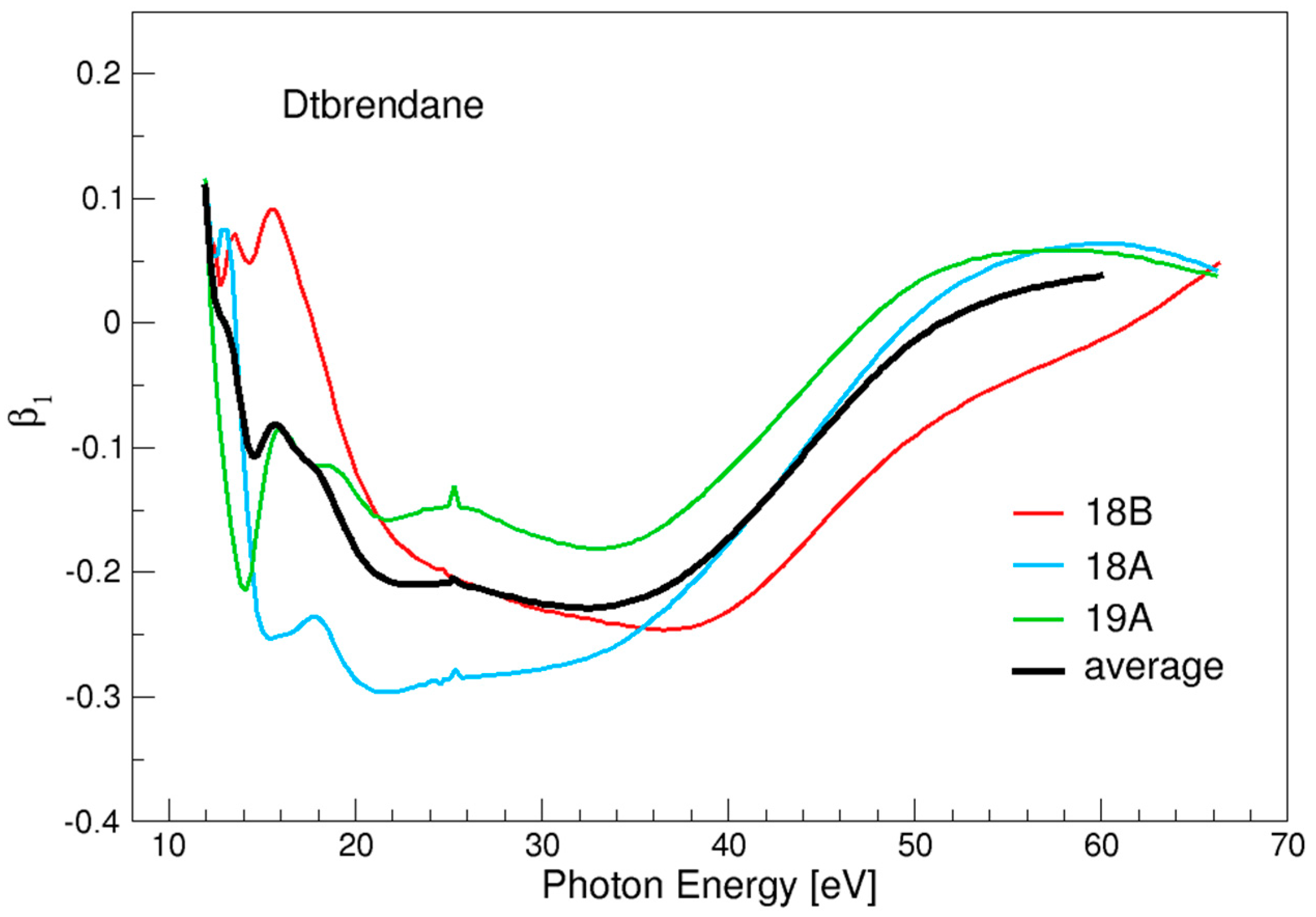
3.4. Twistbrendane and Ditwistbrendane
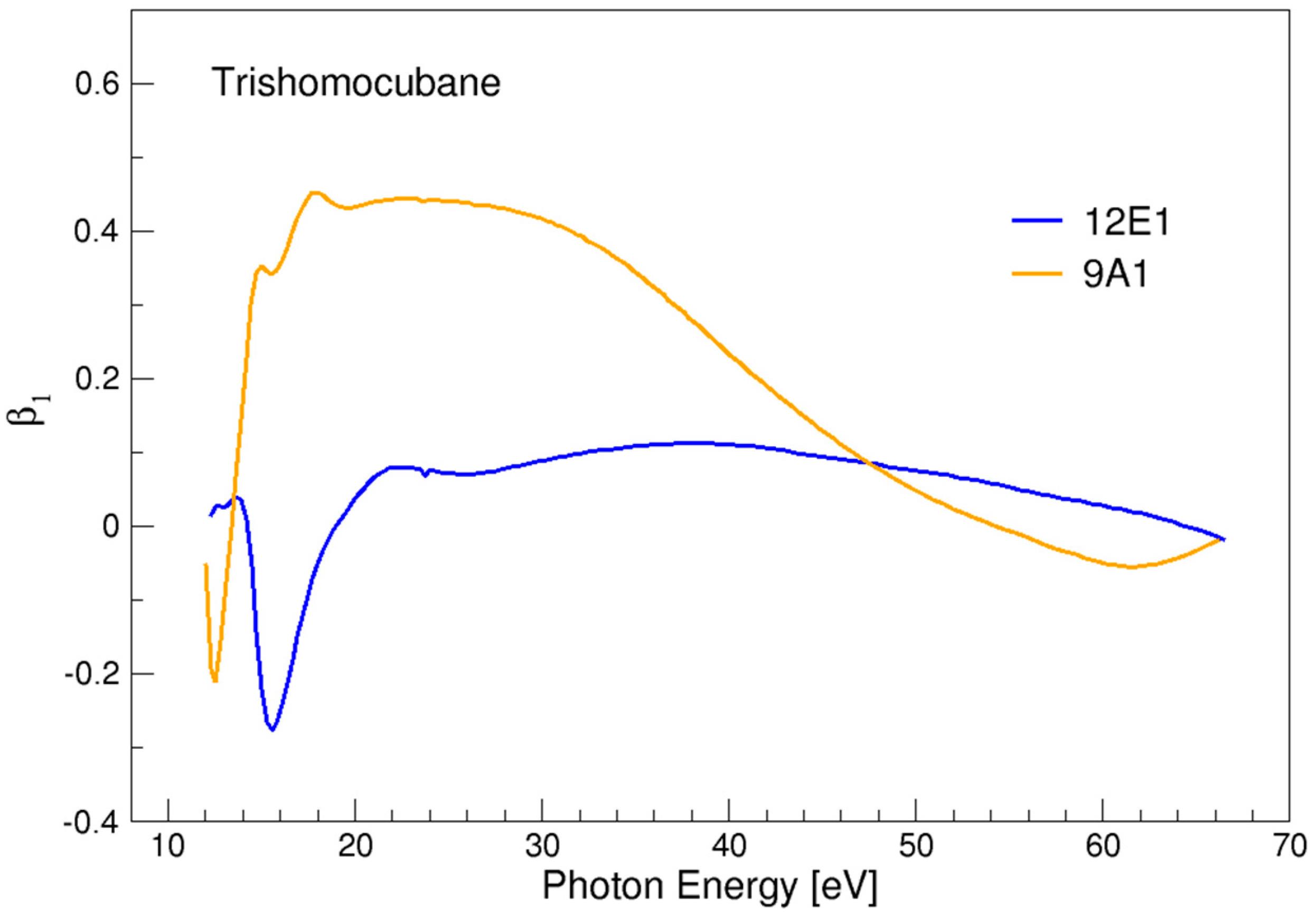
3.5. Trishomocubane
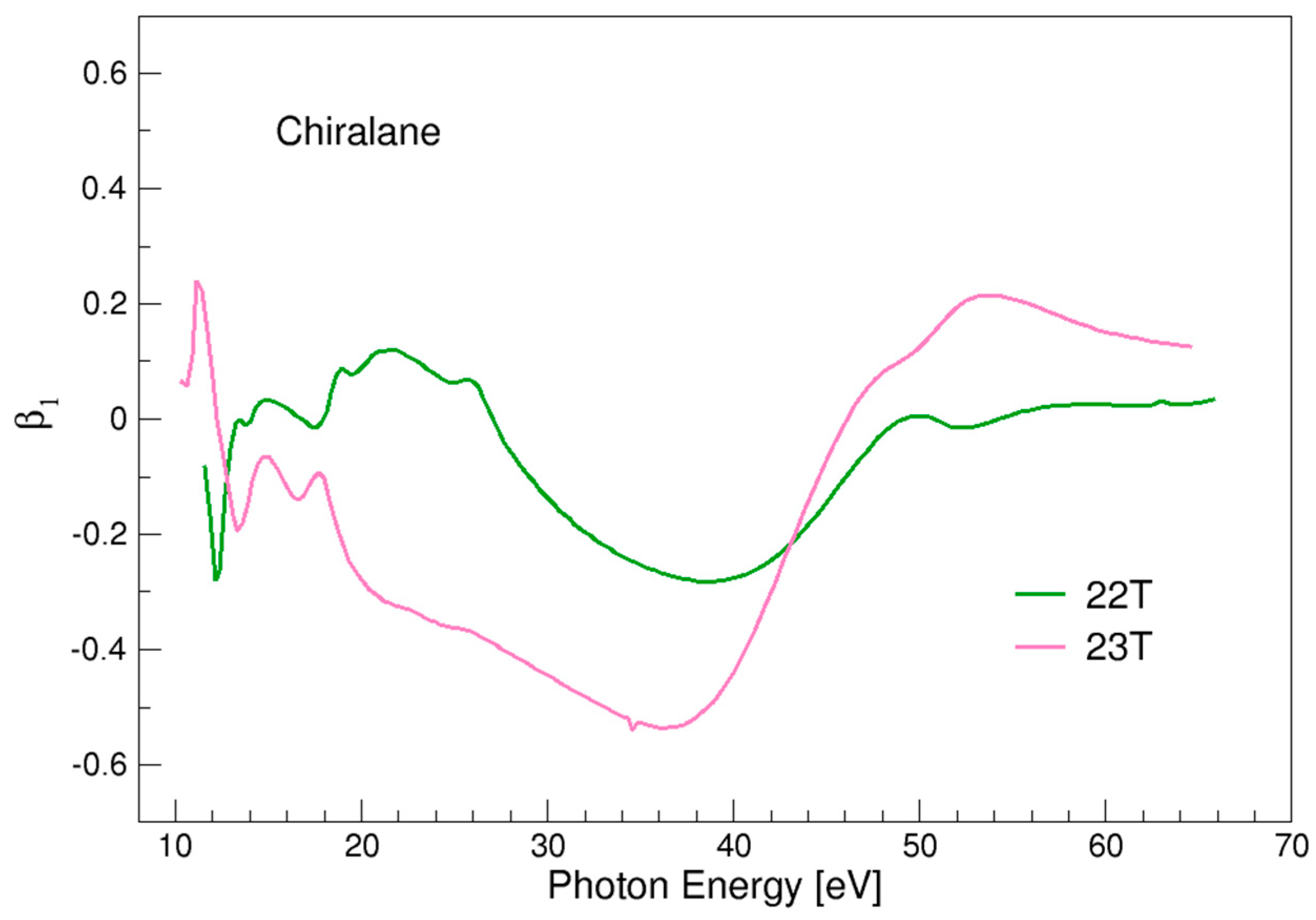
3.6. Chiralane
- -
- PECD is sensitive to chirality in saturated hydrocarbons, and gives generally a rather large signal, apart from the special case of dimethylcyclopropane, probably associated with a peculiar interference-effect.
- -
- The signature of absolute configuration is therefore very strong and unmistakable in the cases considered.
- -
- For most of the systems considered the magnitude of β1 values is among the strongest recorded, and in trishomocubane and chiralane vastly exceeds the values previously known. For comparison, a record value of β1~0.18 was reported for α-pinene [22] (note that in the literature often chiral asymmetry is reported as 2 β1). The results seem to suggest that the chiral response increases with molecular size in this class of compounds.
- -
- Furthermore, the energy range covered by β1 profiles is remarkably large, extending from threshold to several tens of eV, again significantly exceeding current known cases.
- -
- In the case of relatively similar molecules, with the same type of atoms and bonds, the β1 profiles are not very specific. This similarity has been appreciated only recently [23], also because of the limited number, and quite different nature, of the systems hitherto analyzed, often by the trivial requirement of being readily available.
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berova, N.; Polavarapu, P.L. Comprehensive Chiroptical Spectroscopy: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, and Biomolecules; Nakanishi, K., Woody, R.W., Eds.; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Polavarapu, P.L. Chiroptical Spectroscopy: Fundamentals and Applications; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- McCann, D.M.; Stephens, P.J.; Cheeseman, J.R. Determination of absolute configuration using density functional theory calculation of optical rotation: Chiral alkanes. Org. Chem. 2004, 69, 8709–8717. [Google Scholar] [CrossRef]
- Saito, F.; Schreiner, P.R. Determination of the Absolute Configurations of Chiral Alkanes–An Analysis of the Available Tools. Eur. J. Org. Chem. 2020, 40, 6328–6339. [Google Scholar] [CrossRef]
- Müller, T.; Wiberg, K.B.; Vaccaro, P.H. Cavity ring-down polarimetry (CRDP): A new scheme for probing circular birefringence and circular dichroism in the gas phase. J. Phys. Chem. A 2002, 104, 5959–5968. [Google Scholar] [CrossRef]
- Powis, I. Photoelectron circular dichroism in chiral molecules. Adv. Chem. Phys. 2008, 138, 267–330. [Google Scholar]
- Janssen, M.H.; Powis, I. Detecting chirality in molecules by imaging photoelectron circular dichroism. Phys. Chem. Chem. Phys. 2014, 16, 856–871. [Google Scholar] [CrossRef]
- Nahon, L.; Garcia, G.A.; Powis, I. Valence shell one-photon photoelectron circular dichroism in chiral systems. J. Electron. Spectr. Rel. Phen. 2015, 204, 322. [Google Scholar] [CrossRef]
- Rescigno, T.; McKoy, V.; Schneider, B. Electron-Molecule and Photon-Molecule Collisions; Plenum Press: New York, NY, USA, 1979. [Google Scholar]
- Huo, W.; Gianturco, F. (Eds.) Computational Methods for Electron–Molecule Collisions; Springer: New York, NY, USA, 1995. [Google Scholar]
- Burke, P.G. R-Matrix Theory of Atomic Collisions: Application to Atomic, Molecular and Optical Processes; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Stranges, S.; Turchini, S.; Alagia, M.; Alberti, G.; Contini, G.; Decleva, P.; Fronzoni, G.; Stener, M.; Zema, N.; Prosperi, T. Valence photoionization dynamics in circular dichroism of chiral free molecules: The methyl-oxirane. J. Chem. Phys. 2015, 122, 244303. [Google Scholar] [CrossRef] [PubMed]
- Powis, I. Photoelectron circular dichroism of the randomly oriented chiral molecules glyceraldehyde and lactic acid. J. Chem. Phys. 2000, 112, 301–310. [Google Scholar] [CrossRef]
- Ilchen, M.; Hartmann, G.; Rupprecht, P.; Artemyev, A.N.; Coffee, R.N.; Li, Z.; Ogasawara, H.; Osipov, T.; Ray, D.; Schmidt, P. Emitter-site-selective photoelectron circular dichroism of trifluoromethyloxirane. Phys. Rev. A 2017, 95, 053423. [Google Scholar] [CrossRef] [Green Version]
- Chandra, N. Photoelectron spectroscopic studies of polyatomic molecules. I. Theory. J. Phys. B At. Mol. Phys. 1987, 20, 3405. [Google Scholar] [CrossRef]
- Toffoli, D.; Stener, M.; Fronzoni, G.; Decleva, P. Convergence of the multicenter B-spline DFT approach for the continuum. Chem. Phys. 2002, 276, 25. [Google Scholar] [CrossRef]
- Bachau, H.; Cormier, E.; Decleva, P.; Hansen, J.E.; Martín, F. Applications of B-splines in atomic and molecular physics. Rep. Prog. Phys. 2001, 64, 1815. [Google Scholar] [CrossRef]
- Te Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF 2021.1, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 1 December 2021).
- Van Leeuwen, R.; Baerends, E.J. Exchange-correlation potential with correct asymptotic behavior. Phys. Rev. A 1994, 49, 2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, A.; Petitjean, M. [6.6]Chiralane: A Remarkably Symmetric Chiral Molecule. Symmetry Cult. Sci. 2008, 19, 307–316. [Google Scholar]
- Ganjitabar, H.; Hadidi, R.; Garcia, G.A.; Nahon, L.; Powis, I. Vibrationally-resolved photoelectron spectroscopy and photoelectron circular dichroism of bicyclic monoterpene enantiomers. J. Mol. Spectrosc. 2018, 353, 11–19. [Google Scholar] [CrossRef]
- Catone, D.; Zema, N.; Prosperi, T.; Stener, M.; Decleva, P.; Nitti, P.; Turchini, S. PECD study of a single conformer molecule: A critical comparison of experiment and theory Phys. Chem. Chem. Phys. 2022; submitted. [Google Scholar]
- Ponzi, A.; Angeli, C.; Cimiraglia, R.; Coriani, S.; Decleva, P. Dynamical photoionization observables of the CS molecule: The role of electron correlation. J. Chem. Phys. 2014, 140, 204304. [Google Scholar] [CrossRef] [PubMed]
- Moitra, T.; Ponzi, A.; Koch, H.; Coriani, S.; Decleva, P. Accurate description of photoionization dynamical parameters. J. Phys. Chem. Lett. 2020, 11, 5330–5337. [Google Scholar] [CrossRef]
- Moitra, T.; Coriani, S.; Decleva, P. Capturing correlation effects on photoionization dynamics. J. Chem. Theory Comput. 2021, 17, 5064–5079. [Google Scholar] [CrossRef]
- Ferré, A.; Handschin, C.; Dumergue, M.; Burgy, F.; Comby, A.; Descamps, D.; Fabre, B.; Garcia, G.A.; Géneaux, R.; Merceron, L.; et al. A table-top ultrashort light source in the extreme ultraviolet for circular dichroism experiments. Nat. Photonics 2015, 9, 93–98. [Google Scholar] [CrossRef]
- Lux, C.; Wollenhaupt, M.; Bolze, T.; Liang, Q.; Köhler, J.; Sarpe, C.; Baumert, T. Circular dichroism in the photoelectron angular distributions of camphor and fenchone from multiphoton ionization with femtosecond laser pulses. Angew. Chem. Int. Ed. 2012, 51, 5001–5005. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.S.; Ram, N.B.; Powis, I.; Janssen, M.H. Imaging photoelectron circular dichroism of chiral molecules by femtosecond multiphoton coincidence detection. J. Chem. Phys. 2013, 139, 234307. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dimethylcyclopropane | |
| 8B | 13.34 |
| 10A | 13.22 |
| 9B | 11.64 |
| 11A | 11.41 |
| Brexane | |
| 18A | 11.74 |
| 16B | 11.63 |
| Twistane | |
| 19A | 11.45 |
| 20A | 11.42 |
| 18B | 11.31 |
| Twistbrendane | |
| 17A | 11.85 |
| 16B | 11.67 |
| 18A | 11.65 |
| Ditwistbrendane | |
| 18B | 11.94 |
| 18A | 11.84 |
| 19A | 11.74 |
| Trishomocubane | |
| 11E1 | 12.31 |
| 12E1 | 12.04 |
| 9A1 | 11.76 |
| Chiralane | |
| 22T | 11.46 |
| 23T | 10.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Decleva, P. Photoelectron Circular Dichroism as a Probe of Chiral Hydrocarbons. Chemistry 2022, 4, 31-41. https://doi.org/10.3390/chemistry4010003
Decleva P. Photoelectron Circular Dichroism as a Probe of Chiral Hydrocarbons. Chemistry. 2022; 4(1):31-41. https://doi.org/10.3390/chemistry4010003
Chicago/Turabian StyleDecleva, Piero. 2022. "Photoelectron Circular Dichroism as a Probe of Chiral Hydrocarbons" Chemistry 4, no. 1: 31-41. https://doi.org/10.3390/chemistry4010003
APA StyleDecleva, P. (2022). Photoelectron Circular Dichroism as a Probe of Chiral Hydrocarbons. Chemistry, 4(1), 31-41. https://doi.org/10.3390/chemistry4010003