Abstract
Nowadays, global climate change is likely the most compelling problem mankind is facing. In this scenario, decarbonisation of the chemical industry is one of the global challenges that the scientific community needs to address in the immediate future. Catalysis and catalytic processes are called to play a decisive role in the transition to a more sustainable and low-carbon future. This critical review analyses the unique advantages of structured reactors (isothermicity, a wide range of residence times availability, complex geometries) with the multifunctional design of efficient catalysts to synthesise chemicals using CO2 and renewable H2 in a Power-to-X (PTX) strategy. Fine-chemistry synthetic methods and advanced in situ/operando techniques are essential to elucidate the changes of the catalysts during the studied reaction, thus gathering fundamental information about the active species and reaction mechanisms. Such information becomes crucial to refine the catalyst’s formulation and boost the reaction’s performance. On the other hand, reactors architecture allows flow pattern and temperature control, the management of strong thermal effects and the incorporation of specifically designed materials as catalytically active phases are expected to significantly contribute to the advance in the valorisation of CO2 in the form of high added-value products. From a general perspective, this paper aims to update the state of the art in Carbon Capture and Utilisation (CCU) and PTX concepts with emphasis on processes involving the transformation of CO2 into targeted fuels and platform chemicals, combining innovation from the point of view of both structured reactor design and multifunctional catalysts development.
1. Introduction
Nowadays, it seems evident that the average surface temperature of our planet has the risk of increasing 6 °C by 2050 if the ongoing trend of CO2 emissions is maintained [1]. The objective of keeping the global temperature increase below 2 °C necessarily implies negative CO2 emissions for the second half of the 21st century [2]. This will produce a radical concept change since carbon dioxide must not longer be considered a by-product but a raw material.
Nowadays, the use of CO2 is restricted to around 200 megatons per year, mainly in urea synthesis, mineral carbonates production, or methanol synthesis, which is negligible in comparison to the global anthropogenic emissions of more than 30,000 megatons per year [3], as depicted in Table 1. Moreover, this CO2 was previously produced along the production of the commodities and, therefore, has no effect on GHG (greenhouse gases) abatement. Considering the massive magnitude of the annual anthropogenic CO2 emitted, a reasonable estimation suggests that the potential use of CO2 may be around two gigatons per year [4]. Currently, carbon dioxide (CO2) transformation into valuable fuels or chemicals is, in most cases, a challenge far from readiness. Most discussion panels on Carbon Capture and Utilisation (CCU) agree on describing CO2 transformation into fuels, construction materials, or polymers as promising technologies [5,6]. The production of fuels and chemicals are among the promising technologies for increasing the use of CO2 in the short and medium terms; however, in most cases, the Technology Readiness Level (TRL) is quite low [5]. It seems obvious that the highest scientific and technological deficit is found in the synthesis of chemical compounds, either intermediates or final products, of high added value. The production of methanol, formic acid, or Fischer–Tropsch products allows access to most products derived from the petrochemical industry; they have, therefore, high added value, but, in some cases, the storage time for CO2 may be short. To increase the storage time, focus on platform chemicals is required. The concept of platform chemical is addressed to potential intermediate molecules derived from biomass that possess a structure able to be transformed into a long variety of products such as biofuels, sugars, organic acids, alcohols, and amino acids. Considering factors as specific as the mass of CO2 as a feedstock, CO2 avoidance potential, relative added value, and independence from fossil fuels reactants, Otto et al. [7] generated a ranking for 23 bulk chemicals indicating the most favorable ones to be produced from CO2. Among them, formaldehyde, dimethyl ether (DME), ethylene oxide, oxalic acid, acetic acid, methanol, salicylic acid, ethylene carbonate or acrylic acid are widely used in the industry. According to this ranking, acetic acid is within the top ten bulk chemicals most dependent on fossil fuels, the main penalty despite its very high added value.

Table 1.
Anthropogenic CO2 emission by sector and conventional uses for producing chemicals. Adapted from references [3,8].
These pathways fit in the circular economy concept, in which materials at the end of their lifecycle and wastes generated in the production of goods are fully recovered and recycled, reducing the environmental impact. However, H2 is required in most cases to run the reactions, either directly or indirectly. This may be considered the main drawback of the utilisation strategy, as energy must be consumed to produce H2; hence, additional CO2 emissions would take place. Renewable routes for producing H2 were investigated as water electrolysers readily accessible for medium to high power requirements, although they are not yet available at a very large scale [9,10]. A summary of possible utilisation routes is shown in the scheme in Figure 1, which emphasises that H2 must be accessible to utilise CO2. The Power-to-X (PTX) strategy allows the synthesis of a wide portfolio of fuels and chemicals by using this electrolytic hydrogen produced from renewable sources (e.g., wind, solar photovoltaic) during periods of low demand (valley periods). It should be emphasised that this strategy has the additional benefit of promoting the penetration rate of renewables in the energy mix in line with the European Union’s long-term objective of reducing greenhouse gas (GHG) emissions by 80–95% by 2050 when compared to 1990 levels [11].
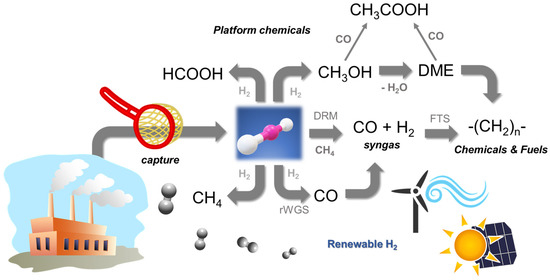
Figure 1.
Schematic representation of CO2 capture and possible utilisation routes.
The CCU and PTX concepts provide the framework for this approach that refers to the transformation of CO2, either pure or diluted (flue gases), into fuels or platform chemicals by using H2 of renewable origin [12]. In this work review, we will explore the most attractive routes presented in Figure 1 that allow the synthesis of fuels, FT hydrocarbons, CH4, and DME (a second-generation fuel), and of platform chemicals such as CH3OH, HCOOH, and CH3COOH. It is worth noting that the successful development of biorefineries or solar refineries depends on the valorisation of the whole set of processes involved that, therefore, must consider the conversion of CO2 to chemicals using renewable H2 when moving to a low-carbon economy.
To successfully achieve CO2 transformation into the proposed set of fuels and chemicals, key catalytic and chemical reaction engineering aspects must be considered: the design of multifunctional catalysts, the knowledge of the reaction from the molecular level to the reactor architecture, and the advanced technologies for the design and synthesis of nanostructured catalysts. Moreover, the control of the selectivity of multiple reactions in series, required for most considered processes (see Figure 1), can be tuned by controlling the reaction temperature, the residence time, a suitable catalyst formulation, and the reactor configuration. Herein, all of these aspects will be discussed and analysed, offering a clear definition of CO2 transformation processes.
2. Carbon Capture and Storage (CCS)
Carbon capture and storage (CCS) is one of the potential solutions to decrease CO2 emissions from stationary sources. Among the different CCS technologies, Ca-looping seems to be the best suited for the decarbonisation of flue gases due to its lower efficiency penalty among the different technologies tested so far [13]. This process, initially proposed by Shimidzu et al. [14], is based on the reversible reaction between calcium and carbon oxides:
Typically, the process is carried out using two connected fluidised beds, one operated in the temperature range of 600–700 °C, which acts as a carbonator, and the other in the range of 750–950 °C, acting as a cracker in which calcination takes place (Figure 2). The mechanical properties of the sorbent particles must be considered since they must present the highest possible attrition resistance [15]. In a recent work, Han et al. [16] critically reviewed all the fundamental aspects of calcium looping CO2 capture technology, focusing on the CaCO3 calcination step. Among the different parameters that must be stressed: reducing the calcination temperature, the injection of steam, the doping of salts, and the reduction of crystallinity.
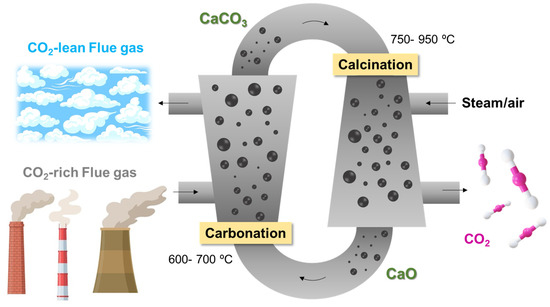
Figure 2.
Schematic representation of Ca-looping CO2 capture process.
Since Shimidzu et al. [14] published their concept, bench-scale plants and, more recently, pilot plant projects have been undertaken, and the Ca-looping process has successfully been demonstrated [13,17,18]. From these experiences, it must be concluded that the presence of 15 wt.% water in the flue gases results in a capture efficiency curve fully adapted to the equilibrium concentrations in the temperature range 700–740 °C, which implies CO2 capture efficiencies as high as ~85% [18,19]. The actual water concentration in flue gases, up to 30%, opens the possibility of further efficiency improvements. Measurements of the rate constant for the carbonation of commercial limestones in the absence and presence of water have demonstrated the positive effect of water in the capture process [20,21], the porosity associated with small macropores and mesopores being an important parameter in the carbonation reaction [22]. The promoting effect of water on CO2 capture is related to the decreasing diffusion resistance and enhanced CO2 adsorption kinetics. Enhancement of the capture efficiency may be attained by different methods, among them doping of natural limestone with different inorganic salts, usually chlorides or carbonates of sodium [23], potassium [24], or a variety of inorganic chlorides, nitrates, or mineral acids [25]. All these studies have been carried out using limestones as absorbents that, once spent, may be reused in cement plants [26]. For instance, Telesca et al. [27] reported a successful production of clinker using the spent lime of a pilot plant designed for the Ca-looping process. Alternatively, the spent limestone may be recovered for the desulphurisation of flue gases since it shows a high reactivity against SO2 [28]. However, the presence of water at high temperatures may favor the sintering behavior and, therefore, reduce the capture capacity.
Flue gases usually contain SO2 and NOx as minor components, which may result in the formation of CaSO4 and nitrates, thus competing with the carbonation process. The amounts of these contaminants depend on the fuel nature and the furnace temperature. The decomposition of CaSO4 in reducing environments allows a procedure for eliminating it from the sorbents. However, the presence of metallic-based catalysts in subsequent downstream steps raises the question of catalyst deactivation. Indeed, Muller et al. [29] demonstrated that the catalytic activity in the Sabatier reaction continuously decreases in the presence of 80 ppm of SO2.
Limestone and dolomite are by far the most studied sorbents for CO2 capture because they are inexpensive and widely available and because of the favorable thermodynamic constraints. However, industrial processes may result in abundant and inexpensive wastes of basic character. Among the various processing routes for sequestering CO2, the direct method seems to be the most adequate when using residues as sorbents [30]. Steel slags, including metallurgical secondary slags and other mineral wastes of the steel industry typically present high Ca content, making them suitable candidates for the capture processes [31]. A panorama for the utilisation of steel slags for this process may be found in the work reported by Yi et al. [32]. Moreover, iron and steel production worldwide result in a considerable amount of basic mineral waste that might be used for the abatement of ca. 40% of the CO2 emissions from electric arc furnaces (EAF) [33]. In general, these residues are advantageous with respect to raw minerals since they require less energy-intensive carbonation conditions. Moreover, mining is not required to have a much lower environmental impact and present low prices being generally available near the CO2 emission location [34]. Considering just the UK industry, the available mineral waste resources sent to landfill may account for 1 Mt of CO2 captured per year [35].
Some authors claim that the kinetics of the carbonation reaction may be too slow for practical purposes [34]. Pan et al. [36] proposed direct carbonation accelerated by the presence of moisture, although the operation conditions must be optimised to make this process economically feasible. Yu and Wang attempted to understand the fundamentals of CO2 capture by steel slags in the so-called direct process [37]. These authors found a dependence of carbonation extent on temperature and CO2 concentration. At low concentrations (<20%) high temperatures are more efficient, while at higher concentrations (>75%) lower temperatures increase the capture efficiency. The main reason for this behavior is that the carbonation rate decreases upon increasing the CO2 concentration. However, these fundamental studies that demonstrate the advantages of steel slag as a potential feedstock for CO2 capture have not resulted in a deeper insight into the process. The use of steel slag for CO2 capture under realistic Ca-looping conditions has been attempted recently in a thermogravimetric device for analysing fundamental parameters [38,39]. After modifying the slag with acetic acid, these authors found that the calcination residence time for regeneration is smaller when using the steel slag instead of limestone and that the furnace temperature is slightly lower than that required when using limestone.
A different approach consists in synthesising absorbents starting from industrial residues [40]. The use of aluminum-containing residues with phosphogypsum, a calcium-containing waste, allowed a highly efficient capture material. Although this procedure seems to be unsuitable from an economic point of view, it provides some evidence of the role of the formed carbonates in preventing the leaching of environmentally hazardous metals contained in the phosphogypsum residue. In a similar approach, the use of the bag house dust residue of the steelmaking process was considered [41], showing that the residue carbonation leaching of heavy elements is drastically reduced, in particular for Pb, Cr, and Zn. Although the capture seems to be very high (0.657 kg of CO2 per kg of dust), the complexity of the mixture may result in complex environmental issues associated with heavy metal volatilisation in the capture/calcination cycles. As for the successful implementation of CO2 capture using steel slags, the first pilot plant showing effective results was developed by Aalto University (Finland) in 2014 [42]. The plant simply makes use of steel slags to produce precipitated calcium carbonate (PCC) via reaction with CO2. According to these studies, if all the calcium in the slag could be used, approximately 13 Mt of PCC per year could be produced, simultaneously sequestering nearly 6 Mt of CO2 per year.
3. Catalytic CO2 Conversion: From Active Sites to Reactor Architecture
The concept of circular economy is addressed to reduce the potential CO2 emissions by transforming the CO2 captured into added-value products so that the carbon dioxide must not longer be considered a by-product but a building block. The production of fuels and chemicals are among the promising technologies for increasing the use of CO2 in the short and medium terms. Indeed, the production of methanol, formic acid, or Fischer–Tropsch hydrocarbon fuels allows access to most products derived from the petrochemical industry; they have, therefore, high added value, but, in some cases, the storage time for CO2 may be short. It seems obvious that the highest scientific and technological deficit is found in the synthesis of chemical compounds, either intermediates or final products, of high added value. To increase the storage time, focus on platform chemicals is required; among them, one of the less explored are C2 compounds, such as acetic acid.
Despite the efforts made, carbon dioxide (CO2) transformation into valuable fuels or chemicals is still, in most cases, a challenge far from being satisfactorily addressed. Motivated by the desire to understand and improve the selectivity of catalytic CO2 transformation processes, numerous efforts have been involved in the development of heterogeneous catalysts that are more active and selective in energy-related reactions, as reflected in diverse publications [43,44,45,46]. The careful design of the nature of the active site has been the focus of multiple activities research over the last years. Furthermore, the use of solid state chemistry and materials science concepts has allowed design synthesis methods for the catalysts that enhance their stability by preventing the sintering of the active phase or tailoring the nature of the active site [47]. The advanced characterisation of the designed catalysts by in situ and operando techniques guides the catalyst design and aids in a better understanding of the phenomena controlling the processes of interest at the microscopic level [48,49,50].
On the other hand, the development of selective catalytic materials for carbon dioxide conversion into desirable products also implies major chemical and chemical engineering challenges, e.g., multifunctional active and selective catalysts and careful design of heat and mass transport tailored reactors that allow a comprehensive valorisation of CO2 and result in zero emissions next-generation industries. In this context, reactors architecture allowing flow pattern and temperature control, the managing of strong thermal effects, and incorporating specifically designed materials as catalytically active phases are expected to significantly contribute to the advancement in the valorisation of CO2 in the form of high added-value products [48]. Herein, microchannel reactor technology combines innovation from the point of view of both structured reactor design and multifunctional catalyst synthesis.
As illustrated in Figure 3, this review is driven by the ambition of discussing the fundamental aspects that involve the control of material surfaces at the nanoscale for optimizing the nature of the active sites to materials and chemical engineering to properly control heat and mass transport mechanism in the catalytic reaction for transforming CO2 into added-value products.
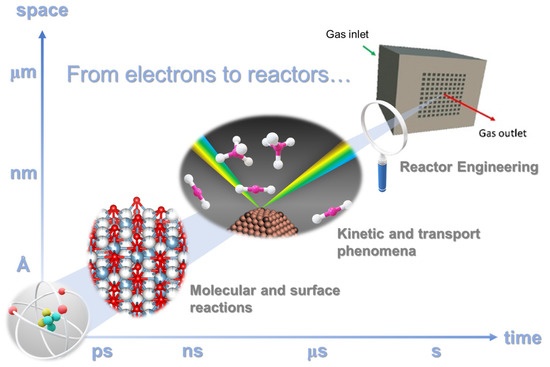
Figure 3.
Time-space multiscale approach: from fundamental level to reactor engineering.
3.1. Design of Nanoscale Catalysts with Atomic-Scale Precision
The control of nanoscale materials with atomic-scale precision, as well as the characterisation and in situ probing of catalytic processes at the atomic level, is the first step in designing processes to obtain high value-added chemicals from CO2. The process consists of three main stages, which include the CO2 activation, the formation of surface adsorbed species, and the final reaction of the adsorbed species. These step processes can be controlled either by designing a multifunctional system able to reduce the number of steps or by designing systems that efficiently couple the three stages. Hence, the development of nanoscale materials with atomic precision is the first step to achieving such a catalytic system using the so-called “LEGO approach” illustrated in Figure 4. The main challenge of this approach is to generate new synthetic protocols for the development of these materials as well as to characterise in depth, at the atomic scale, their active surface and adsorbed species during the catalytic process.
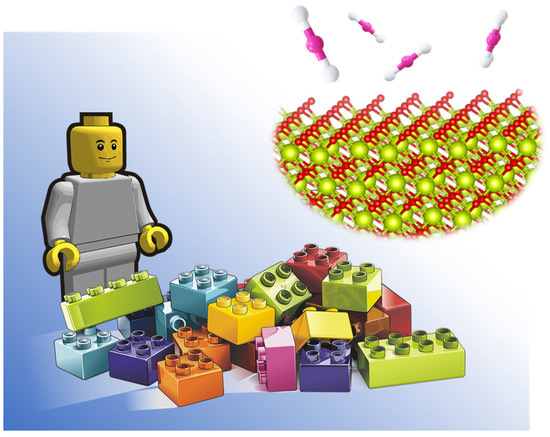
Figure 4.
Rational design of efficient catalysts at atomic level: LEGO approach.
The challenges of converting CO2 in high value-added chemicals are:
- ▪
- To generate multifunctional catalysts formed by nanoscale particles of metals and bimetallic alloys by fine-tuning of the interfaces.
- ▪
- To understand the nature and composition of the active surface, its evolution with time, and the role of the adsorbed species along the reaction time that may modify the catalytic activity, as well as the selectivity of the reactions.
- ▪
- To selectively activate the very strong C=O bonds of the CO2 molecule at moderate temperatures.
- ▪
- To understand the reaction pathways that generate the desired products.
The control of the atomic scale of materials allows the control of the catalyst performances. Metal nanoclusters on the surface of metal oxides create strong metal-support interactions on providing higher metal-support interfacial surface area, thereby, prevent sintering of active metal clusters and maintaining the active surface area of the catalyst and its activity [51,52,53]. On the other hand, defects control the properties of all solid-state materials, their type and distribution, intentionally incorporated in metal oxides, and tailor and optimise their catalytic behavior [54,55,56]. The metal nanocluster/nanoparticle-metal oxide interface is of paramount importance in the catalytic process driving reactant activation and conversions [57]. CO2 can adsorb on stoichiometric and non-stoichiometric metal oxides mostly by the terminal oxygen atoms [58,59]. In the case of non-stoichiometric metal oxides, the surface oxygen vacancies can act as a catalytic center for activation of the C=O bond by interacting with the terminal oxygen atom and may be activated by H-spillover from the metal nanocluster/nanoparticle or by homo- or heterolytically dissociated H2 molecule on stoichiometric and non-stoichiometric metal oxides [60]. The changes in electronic properties of the interfacial atoms and the changes in the chemical state or oxidation state of the active metal atoms of the catalysts will change the interaction and bonding mode of CO2 and the surface adsorbed species with the catalyst surface and with each other, which will influence on the activation of CO2 molecule, its hydrogenation/reduction, and C-C coupling tendency. On interaction with Lewis acid/base centers, the C=O bond of CO2 will weaken, and then the dissociation and hydrogenation of CO2 will be much easier [46]. Therefore, metal oxides can activate H2 and CO2 molecules simultaneously, which makes metal oxides appealing for CO2 reduction/hydrogenation.
3.2. Structured Catalysts and Microreactors
Intensification of catalytic processes requires catalysts with high activity per unit of reactor volume [61]. For this to be practiced, a suitable catalyst arrangement is required in the reactor to ensure sufficient flow of reagents and products and heat transfer in the proper direction. These flows can be controlled by the external diffusion processes (from the bulk fluid phase to the catalyst surface or the inverse), internal diffusion (in the porous network of the catalyst), and by heat transfer within the catalytic bed. Such processes depend on both the properties of the fluid and the operating conditions as well as on the characteristics of the catalyst, the substrate in the case of structured catalysts, and the reactor itself in the case of structured reactors. Furthermore, to enhance the external diffusion rate is convenient to increase the linear velocity of the fluid and the turbulence, but looking for a trade-off with a pressure drop that is favored by the same conditions [62].
Microchannel reactors have unique advantages over conventional catalytic reactors, mostly derived from their high surface area to volume ratio, e.g., reduced diffusion distance between the reactants, excellent mass, and heat transfer. Moreover, in general, microreactors are particularly well suited for fast, highly endo- or exothermic, and explosive reactions [63,64,65,66]. However, despite these advantages, very scarce prototypes have been developed, particularly in the field of high-temperature gas-solid catalytic reactions. Freunds’s group is particularly interested in the heat transfer characteristics of the structured catalysts [67,68,69,70]. They analysed the heat transfer characteristics of different materials and their effect on the catalytic activity and selectivity designing periodic open cellular structures (POCS) made out of materials having different thermal conductivities, e.g., ABS, aluminum alloy, or titanium alloy (Figure 5). They developed models for these materials finding that strut diameter, window opening length, cell length, porosity, and specific surface area substantially affect pressure drop and heat transfer characteristics.

Figure 5.
Periodic open cellular structures (POCS) made of different materials, with varying wall coupling and morphologies. Adapted from reference [68].
Additive manufacturing has the potential to generate a breakthrough in chemical reactor engineering since process intensification drives current advances in chemical engineering. However, the number of catalytic reactors and processes being investigated taking advantage of these technologies is still reduced and mostly reduced to low-temperature processes, as highlighted by Parra-Cabrera et al. [71] in their review. Interestingly, additive manufacturing (AM) allows new approaches as the one initially proposed by Coppens’ nature-inspired microreactors [72]. This approach is based on the recognition of the existence of hierarchical fractal architectures in many trees and mammalian networks. Reactors based on these fractal structures have the advantages of low pressure drop and uniform distribution of temperature, as well as rapid and direct scale-up. The most challenging aspect of this approach is the design of optimised structures of 3D-tailored microstructured reactors that allows for adapting mass and heat transport to the reaction requirements. The combination of modelling, analysis of the material substrate behavior at the reaction conditions, and AM techniques is crucial to provide protocols for the designing tailored prototypes that help in the development of distributed production plants.
At this point, we can conclude that the combination of atomic-scale design of multifunctional catalysts together with the implementation of new tailored to function nature-inspired chemical reactors opens a wholly new scientific avenue, as the only abundant worldwide carbon source, carbon dioxide, can be transformed into long-life products through and for catalysis. The major challenge is the discovery and development of strategies for boosting process intensification by adapting reactors and structured catalysts to the reaction requirements through the knowledge of the relationships between materials, geometry, and transport properties.
3.3. Modelling and Simulation of Structured Catalytic Reactors
Modelling and simulation are essential steps in the analysis and design of structured catalytic reactors. Modelling consists of their physicochemical and mathematical description, which is based on the mass, momentum, energy, and species conservation equations. The first two are known as the Navier–Stokes equations, which are of great importance as they provide the velocity and pressure fields that are needed for describing the residence times distribution and species contact, and hence the reactor performance. As concerns the structured reactors, the main challenge remains the modelling of the whole device [73]. Complexity mainly lies in the multi-scale character of the problem that involves dimensions ranging from a few microns (catalytic layer) to millimeters (characteristic dimension of the channel/microchannel), and finally several centimeters (complete device). As the model resolution is performed using numerical methods based on finite elements (or volumes), modelling the entire device would require an enormous amount of total elements with sizes adapted to each scale, resulting in extremely high computational requirements that are unmanageable in practice [74]. In the case of monolithic converters, the laminar nature of the flow pattern significantly reduces the complexity. For that reason, one- and bi-dimensional models have traditionally been used with either lumped or distributed parameters, but considering a single channel that is assumed to be representative of the ensemble [75,76]. These approaches have allowed us to advance and gain knowledge on the modelling of structured reactors, but they are not always suitable. It should be noted that the channels are isolated regarding mass transport but not as concerns heat transfer. For that reason, in the case of reactions such as most of the ones considered in this review, which are characterised by strong thermal effects, the single-channel models risk incorrect descriptions of the reactor performance due to possible significant temperature profiles. Moreover, flow maldistribution is another issue that may compromise the assumption of equivalent performance among the several channels. As a result, 3D simulations are required in these cases, considering the ensemble of flow ducts.
At present, several computational fluid dynamics (CFD) software are developed and commercially available. This fact, together with the remarkable recent advancement in computer hardware capabilities, has allowed us to take a large step forward in chemical reactor modelling and simulation [74,77,78,79,80,81]. However, even in these conditions, analyzing the whole device remains a challenge. The implementation of CFD models in order to simulate the performance of structured catalytic systems in CO2 transformation processes offers a huge potential for process intensification.
4. Chemical Routes for CO2 Conversion
The production of fuels (methane, DME, liquid fuels, etc.) and chemicals (methanol, syngas, acetic acid, formic acid, etc.) are among the promising chemical applications for increasing the use of CO2 in the short and medium terms. Table 2 depicts a wide range of the valuable products that can be obtained from CO2 conversion available and competitive in the current market [82]. Because CO2 can be transformed into a long variety of valuable chemicals, not every possible product can be mentioned. This section will highlight the most common routes of CO2 conversion for producing valuable products that could have a major impact not only on climate change mitigation but also on diverse sectors such as industry, agriculture, and transportation.
Table 2.
Chemical reactions and usages for some relevant valuable products derived from CO2 as chemical feedstock. Adapted from reference [82].
4.1. Reverse Water—Gas Shift (r-WGS) Reaction
The hydrogenation of CO2 to produce CO is a thermodynamically limited reaction favored at high temperatures because of its endothermicity. This reaction competes with the methanation (Sabatier reaction) of carbon oxides, which are highly exothermic reactions [83]. The equilibrium constant at 400 °C for the transformation into CO is just 0.1 for stoichiometric H2/CO2 mixtures; it is necessary to reach 800 °C to attain an equilibrium constant equal to 1 [84,85].
Overcoming the high temperature required for r-WGS and, at the same time, increasing the selectivity to CO membrane reactors has been proposed [86]. On the other hand, microchannel reactors may be an attractive alternative to membrane reactors as an option to enhance the selectivity of r-WGS against the methanation. An early but unfortunately isolated study by Tonkovich’s group at PNNL (USA) uses Ru catalysts in microchannel reactors at temperatures ranging from 500–540 °C and atmospheric pressure. Under these conditions, the equilibrium conversion is attained (~90% and 100% selectivity to CH4). However, on decreasing the residence time to 30–60 ms, the equilibrium conversion decreases to 30%, as expected, but a selectivity to CO of 70% is attained [87]. More recently, ceramic monoliths washcoated with Ru(Ce)-ZrO2 have been described by Gaudillere et al. [88] for the same reaction at similar space velocities, concluding that by structuring the catalyst the selectivity of the r-WGS is improved. It is important to note that elevated H2/CO2 molar ratios favor methanation and, therefore, mass transport limitations may affect the selectivity due to the higher diffusivity of H2 compared to CO2; therefore, the thickness of the catalytic layer may also play a relevant role. On the other hand, the Sabatier reaction is exothermic while the r-WGS is endothermic, which leads to the need to pay special attention to the management of the thermal effects and the control of the temperature as a powerful strategy for controlling the selectivity of the process.
Typically, the r-WGS reaction is carried out using supported metallic catalysts, with Cu, Pt, and Rh being the most frequently used over a panoply of supports, but reducible supports are able to produce oxygen vacancies upon reduction (TiO2, Fe2O3, ZnO, or CeO2) and are at the forefront of the preferences [83]. The role of oxygen vacancies at the metal-support interface was recently highlighted for Au/TiO2 catalysts [55]. In general, for selecting the active phase, the reaction temperature and the active phase ability for methanation are key issues. For instance, the use of copper reduces the r-WGS temperature to values as low as 165 °C, keeping an elevated selectivity to CO [89,90], but the H2/CO2 molar ratio must be quite high to favor CO2 dissociation on the copper surface [91]. On the other hand, the nature of the support is also a determinant factor in the selectivity of the CO2 hydrogenation. Almost atomically dispersed Pd catalysts supported either on Al2O3 or carbon nanotubes (MWCNT) drive the CO2 hydrogenation in different pathways. The Al2O3 supported catalyst presents a high selectivity to CO, but these atomically dispersed Pd phases show no activity in this reaction. However, by adding La2O3 to the carbon support, the observed activity and selectivity resemble those of the Pd/Al2O3 catalyst [92]. Herein, the support basicity modifies the interaction of CO2 with the support surface. A weak interaction must be expected in interacting with γ-Al2O3 mostly resulting in bicarbonate species, while more stable carbonates are formed upon adsorption on the more basic lanthanide oxides. In any case, surface hydroxyls determine the nature of the species formed. Moreover, all these species may result in the formation of adsorbed formate species by H2-reduction through a well-documented spillover mechanism. Therefore, adding alkaline promoters to the support alters the stability of the formed carbonate species and increases the CO2 adsorption capacity through electrostatic interactions [93]. Additionally, the alkaline promoters affect the sintering behavior of the active phase or in the metal dispersion [93,94,95].
The reaction mechanism is far from being universally accepted. Although the CO2 activation mechanism is not yet well established, there is agreement ast to the effect of the active phase dispersion. The CO2 conversion increases upon increasing the dispersion. This effect has been described for Cu [96,97], Co [98], Ru [99], Rh [100], PdZn [101], Pt [102], or generally for all noble metals, although remarkably Pt and Pd favor the r-WGS while Rh and Ru favor methanation [103]. Panagiotopoulou relates the enhancement of activity and selectivity in the r-WGS on decreasing particle sizes with the activation of CO2 at the perimeter of the metal particles [103]. On the other hand, an increase in the particle size favors the methanation reaction as observed for Ru/TiO2 and Ru/Al2O3 catalysts with 100% selectivities to CH4 for particle sizes above 2.5 nm and 1.5 nm, respectively [103]. In an interesting study, Matsubu et al. [104] found a positive correlation between the r-WGS turnover frequency (TOF) and the number of isolated Rh sites in Rh/TiO2 catalysts. The importance of the metal-support interface in the activity and selectivity has been a recurrent conclusion in the last few years. The presence of Ti3+ at Pt-TiO2 interface determines the catalyst behavior [105]. These authors proposed the H2-reduction of the support generating oxygen vacancies Pt-Ov-Ti3+ (Equation (3)) that will further be reoxidised by CO2, resulting in CO formation. Bobadilla et al. [55] proposed, for Au/TiO2 catalysts, a change in the mechanism as a function of the reaction temperature. At high temperatures, decomposition of the adsorbed formate species on the support formed by the reduction of adsorbed carbonates upon H2 spillover will take place, whereas, at low temperatures, the Ti3+-OH groups at the metal-support interface will play a key role in the formation of hydroxycarbonyl (HOCO) intermediates.
Although the interface has been emphasised by many authors when using reducible supports [55,105,106], the elevated mobility of oxygen vacancies in Cu/CeO2 [97] or Co/CeO2 [107] proposes that the whole support surface is active in the r-WGS. DFT studies point to the formation of HOCO species either at the active phase surface [108] or at the metal-support interface with the participation of both the metal and the support in the transition state [109]. These studies clearly show that a sequential route is the most probable in the CO2 hydrogenation. The hydrogenation of CO species assisted by adsorbed H* and OH* species in the active phase would result in the formation of formyl (HCO) as a first step [110] for CH4 formation. Therefore, this route must be inhibited to increase the production of CO.
4.2. CO2 Methanation: Sabatier Reaction
The catalytic CO2 hydrogenation for producing methane (Equation (4)) was first described by Sabatier and Senderens in 1902 [111]. Despite being thermodynamically favored [112,113], CO2 methanation is still far from being industrially implemented. The main problem is related to the high chemical stability of CO2 molecule, in which the C=O double bond breaking is disfavored, considering both entropy and enthalpy [114]. At 2000 °C, just 2% dissociation into CO and O2 is estimated to occur [115]. Nevertheless, the resulting slow kinetics may be overcome by using catalysts resistant to deactivation and impurities.
There are a number of relevant reviews on the catalytic hydrogenation of carbon dioxide into methane [116,117]. The efficiency of the different active phases tested for CO2 methanation follows the order Ru > Ni > Co > Fe > Mo [118]. Suitable supports have been compared, resulting in the efficiency order CeO2 > Al2O3 > TiO2 > MgO [117]. Although Ru-based catalysts are stable under operating conditions and more active, Ni-based catalysts are preferred for industrial applications due to the metal cost [112,119]. However, Ni-based catalysts suffer rapid deactivation mainly due to the sintering of metallic particles, which occurs through the formation of volatile Ni(CO)4 molecules [120], and the formation of carbon deposits that decrease the number of nickel-active sites for the adsorption of CO2 and H2 dissociation. Among the different strategies to restrain the deactivation, resistance to sintering can be improved by strengthening the metal-support interaction [121], while the addition of water may prevent the carbon deposits [113], although the latter influences the sintering behavior of the catalyst.
In a comprehensive review, Jalama analyses all the aspects that affect the kinetics of CO2 hydrogenation, including active phase, support, promoters, and even pore size and particle shapes [119]. The nature of the support plays a crucial role in the high activity of CO2 methanation. This has resulted in a considerable amount of work that considers the effect of supports and promoters on the catalyst efficiency. The addition of alkaline or alkaline-earth cations promotes dissociation and helps gasifying carbon deposits. The high oxygen storage capacity of reducible oxides as CeO2 or CexZr1−xO2 mixed oxides, the easiness of the reducibility of the active phase, and the metal dispersion or the support basicity are key factors in the catalyst activity [122,123,124]. Concerning the redox properties, perovskite-type materials have been successfully tested as promissory materials for CO2 methanation [125]. Although there are certain discrepancies, it is generally accepted that CO2 methanation is a sensitive reaction to the structure of the catalyst [126], its preparation method, and conditions [127,128], and the composition has a significant influence on its performance [129,130]. Therefore, to improve the catalyst activity, efforts must be devoted to the synthesis conditions, the selection of high surface area supports with tailored pore sizes and optimised basicity and a careful selection of the active phase, including bimetallic or multimetallic active phases. In general, most studies have been carried out in relatively simple systems, and, in consequence, there is a strong need to develop a fundamental understanding of this reaction. In such a way, the application of the LEGO approach to generate multifunctional catalysts could satisfy this challenge. For instance, perovskite-based materials have tunable basic and redox properties that can be adjusted to design optimal catalytic materials for CO2 methanation.
From a mechanistic point of view, two main reaction pathways have been proposed for CO2 methanation. The first involves the formation of CO as an intermediate, which, depending on the reaction conditions, becomes dissociated into C* and O* species that are both hydrogenated to generate CH4 and H2O, respectively. In the second proposed path, CO is formed and hydrogenated without C-O bond scission forming formate or other oxygenated intermediates, such as formyls which are finally hydrogenated, forming methane and water. Recent operando spectroscopic study catalysts have shown that the nature of the support and the density of surface hydroxyls influence the CO2 hydrogenation mechanism [131,132]. In general, adsorbed CO and formate species are nowadays accepted to form on Ni- and Ru-based catalysts, acting as active intermediate species in the hydrogenation of CO2. In some cases, CO may be formed through formate decomposition, and both mechanisms are simultaneously participating in the reaction. The relative proportion of the active intermediate species is determined by the physicochemical properties of the support and, therefore, active supports influence the reaction mechanism.
It is well known that the process efficiency is controlled by the configuration of the catalytic reactor due to the reaction’s high exothermicity, but it is also controlled by the catalyst activity throughout its lifetime [133]. The activity and selectivity of the catalysts for CO2 methanation are drastically influenced by heat and mass transfer in the reactor. To avoid these limitations, several reactor concepts were proposed. Among them, cascades of fixed bed reactors with limited conversion, wall-cooled fixed bed reactors, fluidised bed reactors, or slurry bubble reactors are used or currently investigated [118]. However, very few studies reported process intensification for this reaction using microchannel structured reactors, which is an alternative concept allowing the effective reaction heat removal because of their advantageous surface-to-volume ratio. By structuring the catalyst on proper metallic substrates, the temperature hot spot is reduced to moderate values, and the lifetime of the catalyst is extended by decreasing the activity loss with time. In our recently published critical reviews, the recent advances in structured reactors and microreactors in the Sabatier reaction are analysed in detail [134].
Over the last decade, the rapid evolution of 3D printing and additive manufacturing technologies, as well as the consolidation of knowledge in the structing of catalysts, enabled the development of a wide range of possibilities in the fabrication of microreactors with the optimal properties for Sabatier reaction [135]. For instance, Danaci and coworkers generated a tridimensional structure by 3D printing a metallic paste that, after calcination, is wall-coated with the desired catalyst [136,137,138]. Pressure drop and heat transfer using these structured catalysts for the Sabatier reaction depend on the microreactor architecture (strut stacking and fiber diameter as well as macroporosity), with the macroporosity of the 3D-printed structure being the main parameter influencing the effective thermal conductivity of the structured device.
In a recent collaboration of our research group [139,140], we assessed, for the first time, the impact of the geometric variations of gyroid-based metal substrates on the CO2 methanation catalytic performance using NiRu bimetallic catalysts. In comparison to traditional honeycomb monoliths, the tailored 3D gyroid geometries metal substrates promote the mass and heat diffusion processes, boosting overall process efficiencies, as shown in Figure 6. This promising work reveals that tailored G-3D-printed monoliths are promising for designing advanced microreactors for low-carbon technologies.
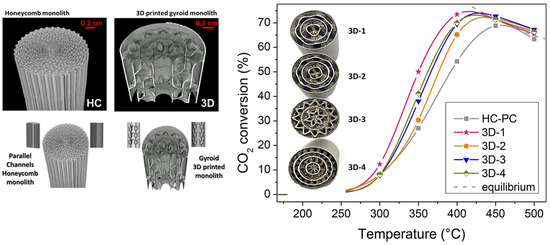
Figure 6.
Comparison of the catalytic performance between conventional honeycomb monolith and different tailored 3D gyroid geometries metallic substrates on CO2 methanation. Adapted from references [139,140]. Reprinted with permission from reference [140]. Copyright 2021 American Chemical Society.
4.3. Synthesis of Formic Acid (FA) as Energy Vector
The utilisation of hydrogen as an alternative energy carrier requires a carefully designed infrastructure satisfying all safety conditions and easing its storage, transport, and handling. Those requirements can be easily satisfied by using chemically bounded hydrogen, allowing its delocalised availability via hydrogenation/dehydrogenation cycles. This scenario conducts the hydrogen economy towards the use of organic compounds able to absorb and release free hydrogen, known as liquid organic hydrogen carriers (LOHCs). The LOHCs present a safe alternative for H2 storage and handling, allowing the use of the existing fuel infrastructure and reassuring society about the negative concerns of directly employing hydrogen gas as a fuel [141]. A successful LOHC should possess sufficiently high gravimetric energy density and should easily allow reversible hydrogenation/dehydrogenation cycles. A large variety of LOHCs were evaluated based on their chemical properties and compared to each other by applying different evaluation criteria based on energy storage, energy transport, and mobility application [142]. In comparison to other LOHCs, such as dibenzyltoluene or N-ethylcarbazole, formic acid (FA) is especially attractive as a hydrogen vector due to its relatively high gravimetric energy density (4.4 wt.%), specific energy (5.3 MJ kg−1), and volumetric energy density (6.4 MJ L−1) [143]. Although lower than what is established by the US Energy Agency, the FAs gravimetric energy density is as high as that of several chemical hybrids and presents additional benefits, such as simplicity, stability, low toxicity, inflammability, and high biodegradability [144].
Formic acid comes with an important advantage as a hydrogen vector: H2 storage can be achieved by CO2 captured hydrogenation into formic acid, and H2 production from FA is considered zero CO2 emission due to the fact that the CO2 liberated during the dehydrogenation cycle can be reused in another hydrogen storage cycle (Figure 7). The complete dehydrogenation/hydrogenation cycle is an environmentally friendly technology within the CCU concept, allowing hydrogen storage and liberation in delocalised applications. Both involved processes in the cycle (hydrogenation of CO2 and dehydrogenation of formic acid) are catalytic reactions. In fact, a recent techno-economic study of the market and the use of CO2 for the production of formic acid reveals that this technology will only be economically viable if decreasing the operating costs is linked to the use of catalysts [145].
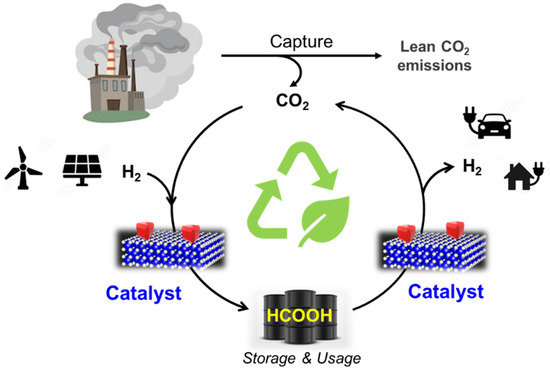
Figure 7.
Hydrogenation/dehydrogenation cycle established by using formic acid as a liquid organic hydrogen carrier (LOHC).
Inoue et al. [146] were pioneers in reporting the direct synthesis of formic acid from CO2 and H2 under mild conditions using homogeneous catalysts based on group VIII transition-metal coordinated with phosphines. Since then, a large variety of homogeneous catalysts based on complexes of Ru, Rh, Pd, Ir, Fe, Co, Ni, Ti, and Mo have been successfully tested [147]. These efforts in the study of homogeneous catalysts for this reaction resulted in the development of very efficient catalytic systems achieving turnover frequency (TOF) values greater than 200,000 h−1 [148]. However, the large-scale production of formic acid from CO2 hydrogenation is still restricted due to the difficulties encountered in separating the homogeneous catalyst from the final product. Moreover, the decomposition of part of the formic acid formed may also occur during the recovery stage, limiting even further the process [149]. Because of such limitations, diverse solid catalysts which can be easily separated from the FA produced have been reported within the last decade. Among the most successful heterogeneous catalytic systems tested, supported catalysts based on dispersed Pd, Au, Rh, and Ru are predominant [149,150,151,152,153,154,155,156,157,158,159], although Ni-based catalysts have also been described [160,161]. Metal oxides [151,158,159,162], layered-double hydroxides [153,154,155], and carbon-based materials [150,163,164,165] are commonly used as supports. It must be emphasised that the role of the support is still controversial. Some authors propose that hydroxyl groups in oxidic supports play a fundamental role in the reaction mechanism [153,154,155], while other researchers suggest that hydrophobic carbonaceous supports with high surface area are beneficial for the FA production [150,151].
The chemically stored H2 in the formic acid platform must be liberated in a controlled manner in the presence of the appropriate catalyst. To increase the efficacy of FA as a hydrogen storage material, one must follow the desired hydrogenation reaction pathway (Equation (5)) preferentially against the unwanted dehydration reaction (Equation (6)). The use of water as a solvent instead of organic solvents, as well as the choice of temperatures equal or lower than 50 °C, favor the dehydrogenation against dehydration reaction [166].
Among the heterogeneous catalysts reported for hydrogen production via FA dehydrogenation, predominate are those based on noble metals (Pd, Au, Rh, and Ru), with Pd being the most frequently used [166,167,168,169]. In a recent work, Santos et al. [168] demonstrated the size-activity dependence of Pd-based catalyst in FA dehydrogenation reaction. As shown in Figure 8, an optimal size of Pd particles exists, and it is controlled by the preparation conditions. The catalytic performance is influenced more strongly by the low coordinated states than by the total number of available atoms. Nevertheless, the use of Pd-based catalysts for FA dehydrogenation presents some drawbacks, such as the high price and important rate of deactivation along time on stream. Therefore, some efforts must be oriented towards noble-metal-free catalytic technologies. The use of Ni, Cu, or bimetallic compounds has been reported [170]. Despite the higher energy barrier found for Ni in comparison to Pd for this reaction, the catalyst’s behaviour can be modulated either by the addition of a second metal (such as Cu or Co) or the right choice of support [171]. Furthermore, the use of carbonaceous supports produced from lignocellulosic biomass with high specific surface area and easiness of modification were recently reported as beneficial for the hydrogenation and dehydrogenation reactions [166,172].
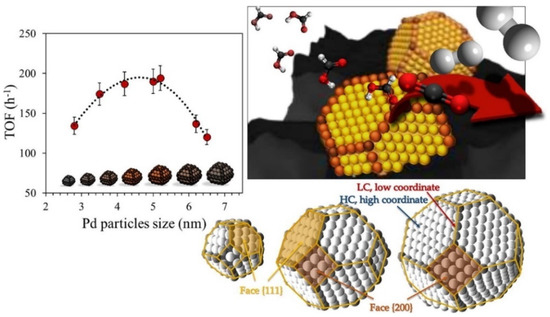
Figure 8.
Illustration of structure-sensitivity character of formic acid dehydrogenation over Pd nanoparticles dispersed on active carbon as support. Adapted from reference [168].
The crucial challenge for implementing such an advanced energy storage approach lies in the development of economically feasible and reliable catalytic systems that do not only store H2 at high energy density but can also operate reversibly under realistic working conditions [165]. A “hydrogen battery” requires a suitable catalyst able to efficiently produce FA from CO2 hydrogenation and release H2 reversibly via dehydrogenation of FA, both under mild conditions. In general, the performance of solid catalysts in both hydrogenation/dehydrogenation reactions depends on three factors: (i) composition, dispersion, and morphology of the metallic particles, (ii) nature of the support, and (iii) the presence of additives. At this point, the LEGO approach plays a key role in optimal catalyst design. Typically, most of the catalysts reported in the literature require extra basic additives and/or elevated temperatures (above ambient temperature) to achieve optimal catalytic performance in both reversible reactions [173,174]. A more sustainable approach involves the use of heterogeneous catalysts in the absence of additives at room temperature [163,165,175,176]. So far, the most successful catalytic systems are constituted by dispersed Pd nanoparticles over graphite oxide or active carbon as supports, and the H2 storage/release cycles are based on the bicarbonate/formate redox equilibrium at near ambient conditions (Equation (7)) [163,165]. The catalytic performance obtained is comparable to that of the homogeneous counterpart system owing to high volumetric energy density and occurs in an aqueous medium avoiding the utilisation of organic solvents and/or additives.
Bath reactors are usually used for the hydrogenation/dehydrogenation of FA. However, the lifetime of the catalysts as a key performance indicator can hardly be evaluated in this type of reactor [177,178]. In order to establish the true potential of this technology, it is mandatory to study the formic acid hydrogenation/dehydrogenation in continuous flow reactors. In an interesting study, Caiti et al. [177] compared the production of hydrogen via FA decomposition over Pd/C catalysts in a continuous stirred tank reactor (CSTR) and a plug-flow reactor (PFR). Detailed kinetics studies and mechanistic insights allowed the establishment of a structure-activity-lifetime relationship. Although the CSTR presented lower activity-specific rates than the analogous PFR, the relevant overall high stability (35-fold more stable) resulted in a more productive and favorable process for the continuous production of hydrogen.
The application of microreactors for continuous process presents important advantages in terms of process control, security, simplified operations, shorter residence time, heat exchange control, and enhanced reaction selectivity [179]. In this context, the multiple advantages of microreactors and process intensification make these systems suitable for FA synthesis and reversible decomposition reactions. Although microreactors have been applied for fuel synthesis and on-line H2 production for polymer electrolyte membrane fuel cells (PEMFCs) [65], the number of papers found in the literature for FA is still very scarce. Thus, it is necessary to explore innovative microreactor configurations for FA decomposition and reversible hydrogenation to obtain optimal yields and high stability in continuous mode. Very recently, Haffez et al. [178,180] published the first studies in which membrane, packed-bed, and coated-wall microreactor configurations are compared in FA decomposition using commercial Pd-based catalysts. By means of computational fluid dynamics (CFD) models, the authors provided innovative insights into the formic acid decomposition reaction, revealing that internal and external mass transfer limitations for all microreactor configurations were insignificant. The coated-wall and membrane microreactors presented superior performance than the packed-bed configuration. This pioneering work offers a practical basis to implement microreactors for the current reaction working in continuous mode.
4.4. Direct Synthesis of Dimethyl Ether (DME)
The transformation of methanol into higher hydrocarbons using the appropriate acid catalysts, such as zeolites, includes an initial dehydration step to produce dimethyl ether (DME). Subsequently, DME is transformed into the corresponding hydrocarbons.
As an oxygenated compound, dimethyl ether (DME) is an excellent fuel that does not generate particles during combustion [181,182]. Its toxicity is very low, and it is not known to contribute to global warming or the stratospheric ozone layer depletion [183]. The conventional manufacturing process starts from the synthesis of gas and consists of two stages: the first produces methanol, while the second dehydrates the methanol into DME. The synthesis of methanol has two clear limitations in temperature. On the one hand, the catalyst using Cu as an active phase is very sensitive to deactivation by sintering, and, above all, it is a reaction limited by thermodynamic equilibrium due to its reversible exothermic character. Therefore, low temperatures favor the stability of the catalyst and increase the maximum conversion obtainable but penalize the process kinetics [184].
4.4.1. One-Step Process
Throughout the last few decades, the interest in carrying out the process of synthesis of DME in a single stage from synthesis gas has become evident. The idea is to arrange in a single reactor the two catalytic functions of interest and to dehydrate the methanol as it is produced. This achieves a simpler system, a single reactor versus two, but above all, the thermodynamic limitation of methanol synthesis is overcome. Numerous industrial groups have shown their interest and have begun their development. Among them, we can mention the JFE Group (Japan) [185,186,187], Haldor Topsøe (Denmark) [188,189], Air Products (USA) [190,191], Snamprogetti S.p.a. (Italy) [192], Shell Oil Company (Holland) [192], and Mobil Oil Corporation (USA) [193]. Both the stage of methanol synthesis and dehydration are exothermic, as is the WGS reaction, which is also fundamental in this process. Most of the proposed processes are performed in a slurry reactor to enhance the heat of reaction elimination and thus achieve good temperature control.
4.4.2. Catalysts for the One-Step Process
The one-step process uses a bifunctional catalytic system with activity for the synthesis of methanol from synthesis gas and with dehydrating properties of the methanol produced [194]. The catalysts cited in the literature and patents are all inspired by the methanol synthesis catalyst developed by ICI in the 1960s. It is a copper/zinc oxide/alumina catalyst that is prepared by precipitating the Cu, Zn, and Al nitrates with sodium carbonate [195]. The most commonly used dehydration catalyst in this process was alumina, although recently the use of zeolites has arisen. The role of the dehydrating phase of the catalyst is really very important and complex because it controls the final selectivity of the process since the DME can also be dehydrated to produce hydrocarbons. Therefore, if there is little dehydrating capacity, the selectivity to DME will be low because a lot of methanol remains. However, if the dehydrating activity is excessive, the selectivity to DME will also be low because it dehydrates to hydrocarbons. This low or high dehydrating capacity can be related, in a simplistic way, to low or high acidity [196,197], but a more detailed analysis must also take into account the type of acidic center (Lewis or Brønsted) that is involved in the reaction and its influence or lack thereof in the methanol synthesis reaction when using hybrid catalysts with both functionalities. A. Martínez et al. [198,199,200,201,202] have made an excellent systematic study of this problem, reflecting the confusing starting situation existing in the previous bibliography (see for example reference [201]) due largely to the complexity of the problem but also to the fact that many of the studies were probably conducted under conditions in which dehydration was not the process controlling step. The main conclusions of A. Martínez’s group, based on their experimental results and the bibliographic review carried out, can be summarised as follows: (i) zeolite ZSM-5 is considered to be more suitable than γ-alumina as the dehydrating phase of the hybrid catalyst; (ii) in the case of zeolites, both strong Brønsted and Lewis centers play important roles [201]; (iii) the stability of the hybrid catalyst with zeolites depends on the preparation mode or contact between the active phases. A final important aspect of the hybrid catalyst is how the two phases are contacted to manufacture the final catalyst. Catalysts were proposed in which the Cu phase is precipitated on an alumina core [187,203], and a Cu–Zn nanoalloy is formed, generating cooperative sites [204] or mixtures with some binders to carry out pelletisation [193,197]. Finally, as already mentioned, the works by Martínez’s group show that excessively intimate contact between both phases can be negative if it promotes the mechanism of deactivation of Cu by migration of the extra-framework Al of the zeolite.
4.4.3. Dimethyl Ether from CO2
The direct hydrogenation of CO2 to DME is a possible reaction that adds to the global decarbonisation strategy and the reinforcement of renewable energies via the production of hydrogen. Saravanan et al. [205] recently reviewed this reaction confirming its viability but highlighting its critical points, such as the significant production of H2O by the r-WGS reaction that negatively affects the process introducing thermodynamic limitations and causing catalyst deactivation. Therefore, these authors conclude that new catalytic systems and new contact strategies for the two involved phases must be explored: the phase active in the synthesis of methanol and the dehydrating phase.
4.4.4. Production of DME in Compact Systems
The structured catalytic systems allow substantial improvements in the one-step DME production process. On the one hand, they allow taking advantage of the thermal conductivity improvements of a substrate using catalysts structured on metals [206,207]. Moreover, the use of microchannel reactors provides additional improvements associated with the intensification of processes [208,209]. These reactors offer high surface/volume ratios, which increase heat and mass transfer rates while increasing volumetric productivity (process intensification). In addition, the microchannel reactors are organised in a hierarchical way by means of blocks that are replicated until reaching the desired production capacity, avoiding problems associated with the scale-up (rapid development) and offering the flexibility to assume changes in production capacity using more or fewer blocks connected in parallel. Finally, the dimension of the channels means that the pressure drop is very low and the processes are intrinsically safe. There are few works in the literature that explore the potential of microchannel technology in the direct synthesis of DME. These studies show that this technology is highly promising because it allows excellent temperature control (a key point of this process), good catalyst stability, and a much higher volumetric productivity than conventional technology [210,211,212,213,214]. For instance, Hu et al. [210] demonstrated that DME space-time yield is three times superior for microchannel reactors over conventional ones.
4.5. The Synthesis of Acetic Acid Using CO2-Rich Feedstocks
Acetic acid is one of the most important fine chemicals whose production of 6.5 Mt per year worldwide creates thousands of jobs, and it is used to produce added-value products that we use in our daily life, such as plastics, pesticides, polymers, textiles, and pharmaceuticals.
The hydrogenation of CO2 to acetic acid is an exothermic process that is thermodynamically favoured [215] (Equation (8)), although the kinetic constraints associated with the formation of C-C bonds have avoided obtaining high acetic acid yields.
In an early work, Noriyuki et al. [216] observed the production of acetic acid on Ag-promoted Rh/SiO2 catalysts at 190 °C. At such low temperatures, these authors suggested that previous reduction to CO is not required for the formation of acetic acid. Direct carboxylation reactions are of great importance since they allow the synthesis of highly demanded products such as acetic acid. However, the catalytic carboxylation of C-H bonds is still challenging and deserves more effort to design simpler and more C-efficient routes than the ones currently set up in industry [217].
In a patent issued in June 2014, BP researchers disclosed a multistep gas phase procedure for producing acetic acid using CO2-enriched syngas for the synthesis of methanol that uses CuZn/Al2O3 catalysts (Katalco, Johnson, Matthey) that is further dehydrated to DME (dimethyl ether) in a second unit and this is, in a third unit, carbonylated to acetic acid using the previously obtained syngas [218]. Different zeolitic materials are used in these processes, mordenite being (MOR) the preferred catalyst for DME carbonylation. Among the advantages announced by BP: the separation problem of the homogeneous precious metal complexes is eliminated as well as the expensive and corrosive liquid halides. However, most of the examples reported in the patent refer to product distribution estimated using ASPEN Plus software, and just the methanol production is experimentally exemplified.
Recently, Somiari and Manousiouthakis conceptually designed a process for producing acetic acid from CH4 using standard technologies that are techno-economically feasible [219]. They propose a reaction cluster that leads to a conceptual process organised into six subsystems that realises the overall reaction. The subsystems are a partial methane oxidation unit that produces CO and H2O; a methane steam reforming unit resulting in CO, CO2, and H2O; a subsystem for the r-WGS that uses pure H2; a gas separation system for H2 and CO2; the methanol synthesis unit; and finally, a methanol carbonylation subsystem. This scheme allows the production of acetic acid and H2 at zero CO2 emissions.
Reports on the direct synthesis of oxygenates from CO2 are quite scarce. Dagle et al. [220] report a multifunctional catalytic system formed by mixing several catalysts accounting for different functions. By mixing an Fe-based FTS catalyst, and CuZn-based and PdZn-based methanol synthesis catalysts in a 1:1:1 weight ratio around 500 g Lcat−1 h−1 total STY to oxygenates (methanol, ethanol, and acetic acid) was obtained at 350 °C, 80 bar, and 50,000 h−1. Factors such as temperature, nature of the methanol synthesis catalysts, or relative amount of each catalyst were investigated, being especially relevant to the results concerning the method of mixing the different catalysts that significantly alter the total STY to oxygenates as well as their product distribution. By pelletising each catalyst (70–100 mesh) separately, the total STY increases, and C2 oxygenates are favoured over C1 ones. These results indicate that ethanol and acetic acid formation occur by carbonylation of initially formed methanol precursors; therefore, their formation can be facilitated by the proximity of methanol synthesis sites and chain growth sites. This synthesis requires either a series of catalysts or the efficient design of a multifunctional catalytic system including Fischer–Tropsch, methanol, r-WGS, and hydrogenation functions. The interest in developing catalysts for the direct synthesis of C2+ products is an area of current interest challenged by a precise control of different functionalities that must work together. Active sites for CO2 reduction, either in the r-WGSR or methanol synthesis, must be present. A detailed analysis of the reaction mechanisms allows for identifying adsorbed formate species at the beginning of the transformation pathway from CO2 to CO or CH3OH [221]. The pathway to any of these products mainly depends on pressure and H2/CO2 molar ratio. Once activated, these C1 primary products need a second active site for chain propagation. CO insertion into noble metal adsorbed methyl species [222] at high pressures or into methoxy groups at low/moderate pressures [109] may drive the reaction to adsorbed acyl intermediates that further evolve to acetic acid.
Early work demonstrated the feasibility of the acetic acid one pot synthesis starting from CO using Rh catalysts [222]. Two main factors favour acetic acid production on Rh-based catalysts, the presence of cationic Rh+ species and small metal nanoparticles [223,224]. Alternatively, acetic acid may be produced by gas-phase carbonylation of methanol or DME using acidic zeolites [225] or polyoxometalate [226] catalysts at low temperatures and pressures, the acylation of adsorbed methoxy species being the key step in the proposed reaction mechanism. This mechanism is similar to the one proposed for DME formation, the methylation of the adsorbed methoxy groups. On the other hand, methanol is formed at low temperatures and high pressures by hydrogenation of CO and/or CO2. The reaction mechanism implies, in the first stage, the formation of adsorbed HCO or HCOO species that are further hydrogenated up to adsorbed methoxy species [109]. Therefore, it may be possible to shift the reaction to the desired products by modifying the reaction conditions (temperature, pressure, and reactant concentrations) once the catalyst active sites have been carefully designed. In the conventional Cu/ZnO/Al2O3 catalyst for the synthesis of methanol, the active site has been postulated to be partially oxidised Zn cations decorating step edges of Cu (211) facets pointing to the Cu and ZnO synergy and the presence of defects in the Cu surface for designing active catalysts for methanol synthesis [227]. The presence of Zn strengthens the binding energy of intermediate species to the copper surface while decreasing the activation barriers. The active sites for methanol carbonylation are Brønsted sites inside the zeolitic cavity; when placing Cu+ close to these sites, the catalyst activity increases, and the catalyst is less prone to deactivate by water [228]. Brønsted sites are located in both eight-membered rings (8MRs), connected to 12- or 10-membered ring channels, in mordenite and ferrierite, respectively. However, the carbonylation of methanol to acetic acid occurs selectively in the 8MR pockets, while hydrocarbon formation resulting in deactivation is easier in 12MR pockets [229,230,231]. Considering DME carbonylation, the existence of a synergic effect between Cu and Zn in the carbonylation reaction has been shown. Zn cations are preferentially located in T3 sites of the 8MR pockets, but exchanging H-MOR with Zn and Cu results in the presence of Cu in these sites while Zn2+ is now located in the T4 sites of the 12MR pockets [232]. Finally, it should be mentioned that a similar mechanism has been proposed for the polyoxometalate-based catalysts, where methoxy groups adsorbed con acid sites react with cationic species that hold CO molecules [233].
A complete understanding of the reaction mechanism and catalyst dynamics during the reaction provides extremely useful information for refining the catalyst formulation and boosting the catalytic performance. However, along with high catalytic activity, the catalyst’s stability is of paramount importance for commercial applications. In this regard, the main culprit of zeolites is that they suffer from rapid deactivation because their channels are easily blocked by the formation of coke deposits. The stability of the catalysts is compromised by the formation of coke in the zeolitic cavities since zeolite is also active in the transformation of methanol into hydrocarbons. By controlling the acidity, the dehydration rate may be enhanced with respect to coke formation. The careful design of the nature of the active sites and the application of solid-state chemistry concepts have allowed design synthesis methods for the catalysts that enhance their stability by preventing coke deposition [234]. Moreover, the temperature control of the reaction also plays a key role since the hydrocarbon formation occurs at higher temperatures than the dehydration process; therefore, the control of hot spots should improve the selectivity. It is well-known that the presence of hot spots at the reactor compromises the activity as well as the selectivity of the catalytic process; moreover, mass transport effects play a substantial role in the selectivity. In this regard, the utilisation of microstructured catalytic reactors instead of conventional reactors can not only improve the quality of obtained products it but also reduces energy consumption and minimises the production of wastes. Temperature control and flow pattern regulation are crucial to run the process safely and maximise the selectivity towards the desired products [235]. Indeed, safety is another added value of microchannel reactor technology since they can cope with high pressures, hot spots are ruled out, and explosive reactions cannot be propagated within the microchannels. Microchannel reactors represent a step ahead in fine chemicals manufacturing, and, to the best of our knowledge, they have never been applied for acetic acid production. Boosting the selectivity of methanol carbonylation to acetic acid by implementing microchannel reactors could certainly have a remarkable impact on the current acetic acid synthesis technologies.
5. Concluding Remarks and Future Trends
CO2 emissions resulting from fossil fuel use and their contribution to global warming have raised serious environmental concerns, and they have oriented the current EU policy towards a circular economy where products, materials, and resources are maintained as long as possible. The circular economy is expected to promote economic growth (job creation and business opportunities) by implying policies of material cost savings, waste control, greenhouse gas emission reduction, and material loops (use, conversion, and reuse).
The production of fuels and chemicals are among the promising technologies for increasing the use of CO2 in the short and medium terms. The production of methane, DME, synthesis gas, or acetic acid allows access to most products derived from the conventional industry; they have, therefore, high added value, but in some cases, the storage time for CO2 may be short. However, in most cases, the technology readiness level (TRL) is quite low. Therefore, it is mandatory to advance basic and applied sciences to cope with the challenges of the utilisation and valorisation of CO2 emissions. It seems obvious that the highest scientific and technological deficit is found in the synthesis of chemical compounds, either intermediates or final products, of high added value. This review has shortly analysed the current state of the art on the carbon capture and utilisation (CCU) and Power-to-X (PTX) concepts with an emphasis on processes involving the valorisation of CO2 in the form of fuels and platform chemicals.
As for future development, advances in heterogeneous catalysis are required to reveal fundamental phenomena (such as strong metal-support interactions, spillover, and structure-sensitivity relations) that are very useful for understanding the microscopic performance of supported metallic catalysts and as a guide for optimal catalyst design. The reductive C-O bond breaking to activate the stable CO2 molecule requires the design of catalysts at the nanoscale with as close as possible atomic position precision, taking into consideration the careful design of the metal particle size and their interaction with the supports where the number and acidity of surface hydroxyls determine the reaction path and the presence of promoters determine the resistance to sintering and the adsorption capacity of CO2. This is the essence of the LEGO approach concept. Furthermore, the requirements for sustainable development also include the use of waste and economic precursor materials for the synthesis of added-value chemicals.
On the other hand, microchannel reactor technology opens a new scientific avenue, as reaction selectivity can be tailored by controlling residence time and heat and mass transport properties. 3D printing is particularly useful in reactions where diffusion, mass and/or heat transport are key limitations for the desired performances. The combination of the unique advantages of structured reactors (isothermicity, a wide range of residence times availability, and complex geometries) with a multifunctional design of the catalyst is fundamental to efficiently synthesise chemicals using CO2 and renewable H2 in a Power-to-X strategy. The possibility of obtaining a kinetic model and its eventual use in the modelling of the device by CFD must be explored.
Currently, the application of microchannel reactors is still an immature technology for CO2 scale-up valorisation at the industrial scale due to its high manufacture cost. Nevertheless, the important advantages shown by these devices can compete with their price in the near future.
Author Contributions
Conceptualisation, J.A.O., M.A.C. and T.R.-R.; Resources, L.F.B., S.I., J.A.O. and T.R.-R.; Writing—original draft preparation, L.A., M.G.-C., L.F.B., L.A.L.-Á., G.T.-S. and L.P.-P.; Writing—review and editing, T.R.-R., M.A.C., S.I. and J.A.O.; Visualisation, J.Y.; Supervision, J.A.O., M.A.C. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support for this work has been obtained from the Spanish Ministerio de Ciencia, Innovación y Universidades (Grant: RTI2018-096294-B-C33) and Junta de Andalucia projects with references US-1263288, P18-RT-3405, and P20-00594, all of them co-funded by the European Union FEDER. This work is also sponsored by the Spanish Ministry of Science and Innovation through the projects PID2019-108502RJ-I00 and grant IJC2019-040560-I.
Data Availability Statement
Not applicable.
Acknowledgments
Ligia A. Luque Álvarez thanks VI-PPITUS (University of Sevilla) for her current predoctoral contract.
Conflicts of Interest
The authors declare no conflict of interest.
References
- IEA. Energy Technology Perspectives 2016. Towards Sustainable Urban Energy Systems; OECD/IEA: Paris, France, 2016. [Google Scholar]
- Smith, P.; Davis, S.J.; Creutzig, F.; Fuss, S.; Minx, J.; Gabrielle, B.; Kato, E.; Jackson, R.B.; Cowie, A.; Kriegler, E.; et al. Biophysical and economic limits to negative CO2 emissions. Nat. Clim. Chang. 2016, 6, 42–50. [Google Scholar] [CrossRef]
- Rafiee, A.; Rajab Khalilpour, K.; Milani, D.; Panahi, M. Trends in CO2 conversion and utilization: A review from process systems perspective. J. Environ. Chem. Eng. 2018, 6, 5771–5794. [Google Scholar] [CrossRef]
- Dairanieh, I.; Bernard, D.; Mason, F.; Stokes, G.; de Brun, S. Carbon Dioxide Utilization—ICEF Roadmap 1.0; Innovation for Cool Earth Forum (ICEF): Tokyo, Japan, 2016. [Google Scholar]
- Onarheim, K.; Hannula, A.A.I.; Karki, J.; Lehtonen, J.; Vainikka, P. Carbon Capture and Utilization; A VTT Discussion Paper; VTT: Espoo, Finland, 2017. [Google Scholar]
- Arasto, A.; Onarheim, K.; Tsupari, E.; Kärki, J. Bio-CCS: Feasibility comparison of large scale carbon-negative solutions. Energy Procedia 2014, 63, 6756–6769. [Google Scholar] [CrossRef]
- Otto, A.; Grube, T.; Schiebahn, S.; Stolten, D. Closing the loop: Captured CO2 as a feedstock in the chemical industry. Energy Environ. Sci. 2015, 8, 3283–3297. [Google Scholar] [CrossRef]
- Ritchie, H.; Roser, M.; Rosado, P. CO2 and Greenhouse Gas Emissions. Published Online at OurWorldInData.org. 2020. Available online: https://ourworldindata.org/co2-and-other-greenhouse-gas-emissions (accessed on 7 July 2022).
- Ursua, A.; Gandia, L.M.; Sanchis, P. Hydrogen production from water electrolysis: Current status and future trends. Proc. IEEE Inst. Electr. Electron. Eng. 2012, 100, 410–426. [Google Scholar] [CrossRef]
- Gandía, L.M.; Oroz, R.; Ursúa, A.; Sanchis, P.; Diéguez, P.M. Renewable hydrogen production: Performance of an alkaline water electrolyzer working under emulated wind conditions. Energy Fuels 2007, 21, 1699–1706. [Google Scholar] [CrossRef]
- Vázquez, F.V.; Koponen, J.; Ruuskanen, V.; Bajamundi, C.; Kosonen, A.; Simell, P.; Ahola, J.; Frilund, C.; Elfving, J.; Reinikainen, M.; et al. Power-to-X technology using renewable electricity and carbon dioxide from ambient air: SOLETAIR proof-of-concept and improved process concept. J. CO2 Util. 2018, 28, 235–246. [Google Scholar] [CrossRef]
- Aicher, T. Renewable Hydrogen Technologies. Production, Purification, Storage, Applications, and Safety. Edited by Luis M. Gandía, Gurutze Arzamendi, and Pedro M. Diéguez. Energy Technol. 2015, 3, 90–91. [Google Scholar] [CrossRef]
- Fennell, P.; Anthony, B. (Eds.) Calcium and Chemical Looping Technology for Power Generation and Carbon Dioxide (CO2) Capture; Woodhead Publishing: Boca Raton, FL, USA, 2015; pp. 439–446. [Google Scholar]
- Shimizu, T.; Hirama, T.; Hosoda, H.; Kitano, K.; Inagaki, M.; Tejima, K. A twin fluid-bed reactor for removal of CO2 from combustion processes. Chem. Eng. Res. Des. 1999, 77, 62–68. [Google Scholar] [CrossRef]
- Stanmore, B.R.; Gilot, P. Review—Calcination and carbonation of limestone during thermal cycling for CO2 sequestration. Fuel Process. Technol. 2005, 86, 1707–1743. [Google Scholar] [CrossRef]
- Han, R.; Wang, Y.; Xing, S.; Pang, C.; Hao, Y.; Song, C.; Liu, Q. Progress in reducing calcination reaction temperature of Calcium-Looping CO2 capture technology: A critical review. Chem. Eng. J. 2022, 450, 137952. [Google Scholar] [CrossRef]
- Hornberger, M.; Spörl, R.; Scheffknecht, G. Calcium looping for CO2 capture in cement plants—Pilot scale test. Energy Procedia 2017, 114, 6171–6174. [Google Scholar] [CrossRef]
- Dieter, H.; Bidwe, A.R.; Varela-Duelli, G.; Charitos, A.; Hawthorne, C.; Scheffknecht, G. Development of the calcium looping CO2 capture technology from lab to pilot scale at IFK, University of Stuttgart. Fuel 2014, 127, 23–37. [Google Scholar] [CrossRef]
- Sanchez-Biezma, A.; Paniagua, J.G.; Díaz, L.Á.V.; Lorenzo, M.; Álvarez, J.F.G.; Martinez, D.; Arias, B.; Diego, M.E.; Abanades, J.C. Testing postcombustion CO2 capture with CaO in a 1.7 MWt pilot facility. Energy Procedia 2013, 37, 1–8. [Google Scholar] [CrossRef]
- Adánez, J.; Gayán, P.; García-Labiano, F. Comparison of mechanistic models for the sulfation reaction in a broad range of particle sizes of sorbents. Ind. Eng. Chem. Res. 1996, 35, 2190–2197. [Google Scholar] [CrossRef]
- Agnew, J.; Hampartsoumian, E.; Jones, J.M.; Nimmo, W. The simultaneous calcination and sintering of calcium based sorbents under a combustion atmosphere. Fuel 2000, 79, 1515–1523. [Google Scholar] [CrossRef]
- Abanades, J.C.; Álvarez, D. Conversion limits in the reaction of CO2 with lime. Energy Fuels 2003, 17, 308–315. [Google Scholar] [CrossRef]
- Salvador, C.; Lu, D.; Anthony, E.J.; Abanades, J.C. Enhancement of CaO for CO2 capture in an FBC environment. Chem. Eng. J. 2003, 96, 187–195. [Google Scholar] [CrossRef]
- Fennell, P.S.; Pacciani, R.; Dennis, J.S.; Davidson, J.F.; Hayhurst, A.N. The effects of repeated cycles of calcination and carbonation on a variety of different limestones, as measured in a hot fluidized bed of sand. Energy Fuels 2007, 21, 2072–2081. [Google Scholar] [CrossRef]
- Al-Jeboori, M.J.; Nguyen, M.; Dean, C.; Fennell, P.S. Improvement of limestone-based CO2 sorbents for Ca-Looping by HBr and other mineral acids. Ind. Eng. Chem. Res. 2013, 52, 1426–1433. [Google Scholar] [CrossRef]
- Dean, C.C.; Dugwell, D.; Fennell, P.S. Investigation into potential synergy between power generation, cement manufacture and CO2 abatement using the calcium looping cycle. Energy Environ. Sci. 2011, 4, 2050–2053. [Google Scholar] [CrossRef]
- Telesca, A.; Calabrese, D.; Marroccoli, M.; Tomasulo, M.; Valenti, G.L.; Duelli, G.; Montagnaro, F. Spent limestone sorbent from calcium looping cycle as a raw material for the cement industry. Fuel 2014, 118, 202–205. [Google Scholar] [CrossRef]
- Hanak, D.P.; Anthony, E.J.; Manovic, V. A review of developments in pilot-plant testing and modelling of calcium looping process for CO2 capture from power generation systems. Energy Environ. Sci. 2015, 8, 2199–2249. [Google Scholar] [CrossRef]
- Müller, K.; Fleige, M.; Rachow, F.; Schmeißer, D. Sabatier based CO2-methanation of flue gas emitted by conventional power plants. Energy Procedia 2013, 40, 240–248. [Google Scholar] [CrossRef]
- Huijgen, W.J.J.; Comans, R.N.J.; Witkamp, G.-J. Cost evaluation of CO2 sequestration by aqueous mineral carbonation. Energy Conv. Manag. 2007, 48, 1923–1935. [Google Scholar] [CrossRef]
- Domínguez, M.I.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. Physicochemical characterization and use of wastes from stainless steel mill. Environ. Prog. Sustain. Energy 2010, 29, 471–480. [Google Scholar] [CrossRef]
- Yi, H.; Xu, G.; Cheng, H.; Wang, J.; Wan, Y.; Chen, H. An Overview of Utilization of Steel Slag. Procedia Environ Sci. 2012, 16, 791–801. [Google Scholar] [CrossRef]
- Doucet, F.J. Effective CO2-specific sequestration capacity of steel slags and variability in their leaching behaviour in view of industrial mineral carbonation. Miner. Eng. 2010, 23, 262–269. [Google Scholar] [CrossRef]
- Bobicki, E.R.; Liu, Q.; Xu, Z.; Zeng, H. Carbon capture and storage using alkaline industrial wastes. Prog. Energy Combust. Sci. 2012, 38, 302–320. [Google Scholar] [CrossRef]
- Sanna, A.; Dri, M.; Hall, M.R.; Maroto-Valer, M. Waste materials for carbon capture and storage by mineralisation (CCSM)—A UK perspective. Appl. Energy 2012, 99, 545–554. [Google Scholar] [CrossRef]
- Pan, S.-Y.; Chang, E.E.; Chiang, P.-C. CO2 capture by accelerated carbonation of alkaline wastes: A review on its principles and applications. Aerosol Air Qual. Res. 2012, 12, 770–791. [Google Scholar] [CrossRef]
- Yu, J.; Wang, K. Study on characteristics of steel slag for CO2 capture. Energy Fuels 2011, 25, 5483–5492. [Google Scholar] [CrossRef]
- Miranda-Pizarro, J.; Perejón, A.; Valverde, J.M.; Sánchez-Jiménez, P.E.; Pérez-Maqueda, L.A. Use of steel slag for CO2 capture under realistic calcium-looping conditions. RSC Adv. 2016, 6, 37656–37663. [Google Scholar] [CrossRef]
- Valverde, J.M.; Miranda-Pizarro, J.; Perejón, A.; Sánchez-Jiménez, P.E.; Pérez-Maqueda, L.A. Calcium-Looping performance of steel and blast furnace slags for thermochemical energy storage in concentrated solar power plants. J. CO2 Utili. 2017, 22, 143–154. [Google Scholar] [CrossRef]
- Romero-Hermida, I.; Santos, A.; Pérez-López, R.; García-Tenorio, R.; Esquivias, L.; Morales-Flórez, V. New method for carbon dioxide mineralization based on phosphogypsum and aluminium-rich industrial wastes resulting in valuable carbonated by-products. J. CO2 Util. 2017, 18, 15–22. [Google Scholar] [CrossRef]
- El-Naas, M.H.; El Gamal, M.; Hameedi, S.; Mohamed, A.-M.O. CO2 sequestration using accelerated gas-solid carbonation of pre-treated EAF steel-making bag house dust. J. Environ. Manag. 2015, 156, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Aalto University. Available online: http://eng.aalto.fi/en/current/current_archive/news/2014-09-16/ (accessed on 12 July 2022).
- Torres-Sempere, G.; Pastor-Pérez, L.; Odriozola, J.A.; Yu, J.; Durán-Olivencia, F.J.; Bobadilla, L.F.; Reina, T.R. Recent advances on gas-phase CO2 conversion: Catalysis design and chemical processes to close the carbon cycle. Curr. Opin. Green Sustain. Chem. 2022, 36, 100647. [Google Scholar] [CrossRef]
- Zhao, G.; Huang, X.; Wang, X.; Wang, X. Progress in catalyst exploration for heterogeneous CO2 reduction and utilization: A critical review. J. Mater. Chem. A 2017, 5, 21625–21649. [Google Scholar] [CrossRef]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef]
- Álvarez, A.; Borges, M.; Corral-Pérez, J.J.; Olcina, J.G.; Hu, L.; Cornu, D.; Huang, R.; Stoian, D.; Urakawa, A. CO2 Activation over catalytic surfaces. ChemPhysChem 2017, 18, 3135–3141. [Google Scholar] [CrossRef]
- Sharma, D.; Sharma, R.; Chand, D.; Chaudhary, A. Nanocatalysts as potential candidates in transforming CO2 into valuable fuels and chemicals: A review. Environ. Nanotechnol. Monit. Manag. 2022, 18, 100671. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, C.; Sun, K.; Jia, X.; Ye, J.; Liu, C.-J. Advances in studies of the structural effects of supported Ni catalysts for CO2 hydrogenation: From nanoparticle to single atom catalyst. J. Mater. Chem. A 2022, 10, 5792–5812. [Google Scholar] [CrossRef]
- Heidary, N.; Ly, K.H.; Kornienko, N. Probing CO2 conversion chemistry on nanostructured surfaces with operando vibrational spectroscopy. Nano Lett. 2019, 19, 4817–4826. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Wang, Y.; Guo, M.; Zhang, J.; Li, Z.; Deng, T.; Zhang, Z.; Yan, B. In-situ/operando techniques to identify active sites for thermochemical conversion of CO2 over heterogeneous catalysts. J. Energy Chem. 2021, 62, 153–171. [Google Scholar] [CrossRef]
- van Deelen, T.W.; Hernández Mejía, C.; de Jong, K.P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Lou, Y.; Xu, J.; Zhang, Y.; Pan, C.; Dong, Y.; Zhu, Y. Metal-support interaction for heterogeneous catalysis: From nanoparticles to single atoms. Mater. Today Nano 2020, 12, 100093. [Google Scholar] [CrossRef]
- Fujiwara, K.; Okuyama, K.; Pratsinis, S.E. Metal-support interactions in catalysts for environmental remediation. Environ. Sci. Nano 2017, 4, 2076–2092. [Google Scholar] [CrossRef]
- Hernández, W.Y.; Centeno, M.A.; Romero-Sarria, F.; Odriozola, J.A. Synthesis and characterization of Ce1−xEuxO2−x/2 mixed oxides and their catalytic activities for CO oxidation. J. Phys. Chem. C 2009, 113, 5629–5635. [Google Scholar] [CrossRef]
- Bobadilla, L.F.; Santos, J.L.; Ivanova, S.; Odriozola, J.A.; Urakawa, A. Unravelling the role of oxygen vacancies in the mechanism of the reverse water–gas Shift reaction by operando DRIFTS and Ultraviolet–Visible spectroscopy. ACS Catal. 2018, 8, 7455–7467. [Google Scholar] [CrossRef]
- Laguna, O.H.; Romero Sarria, F.; Centeno, M.A.; Odriozola, J.A. Gold supported on metal-doped ceria catalysts (M = Zr, Zn and Fe) for the preferential oxidation of CO (PROX). J. Catal. 2010, 276, 360–370. [Google Scholar] [CrossRef]
- Cargnello, M.; Doan-Nguyen, V.V.T.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science 2013, 341, 771–773. [Google Scholar] [CrossRef]
- Azancot, L.; Bobadilla, L.F.; Centeno, M.A.; Odriozola, J.A. Effect of potassium loading on basic properties of Ni/MgAl2O4 catalyst for CO2 reforming of methane. J. CO2 Util. 2021, 52, 101681. [Google Scholar] [CrossRef]
- Azancot, L.; Bobadilla, L.F.; Centeno, M.A.; Odriozola, J.A. IR spectroscopic insights into the coking-resistance effect of potassium on nickel-based catalyst during dry reforming of methane. Appl. Catal. B Environ. 2021, 285, 119822. [Google Scholar] [CrossRef]
- Jia, J.; Qian, C.; Dong, Y.; Li, Y.F.; Wang, H.; Ghoussoub, M.; Butler, K.T.; Walsh, A.; Ozin, G.A. Heterogeneous catalytic hydrogenation of CO2 by metal oxides: Defect engineering—perfecting imperfection. Chem. Soc. Rev. 2017, 46, 4631–4644. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.; Moulijn, J.A. Process Intensification. Ind. Eng. Chem. Res. 2002, 41, 1920–1924. [Google Scholar] [CrossRef]
- Giani, L.; Groppi, G.; Tronconi, E. Heat transfer characterization of metallic foams. Ind. Eng. Chem. Res. 2005, 44, 9078–9085. [Google Scholar] [CrossRef]
- Tonkovich, A.Y.; Zilka, J.L.; Lamont, M.; Wang, Y.; Wegeng, R.S. Microchannel reactors for fuel processing applications: Water gas shift reactor. Chem. Eng. Sci. 1999, 54, 2947–2951. [Google Scholar] [CrossRef]
- Domínguez, M.I.; Centeno, M.A.; Martínez, T.M.; Bobadilla, L.F.; Laguna, Ó.H.; Odriozola, J.A. Current scenario and prospects in manufacture strategies for glass, quartz, polymers and metallic microreactors: A comprehensive review. Chem. Eng. Res. Des. 2021, 171, 13–35. [Google Scholar] [CrossRef]
- Laguna, O.H.; Domínguez, M.I.; Centeno, M.A.; Odriozola, J.A. Chapter 4—Catalysts on Metallic Surfaces: Monoliths and Microreactors. In New Materials for Catalytic Applications; Parvulescu, V.I., Kemnitz, E., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 81–120. [Google Scholar]
- Ivanova, S.; Laguna, O.H.; Centeno, M.Á.; Eleta, A.; Montes, M.; Odriozola, J.A. Chapter 10—Microprocess Technology for Hydrogen Purification. In Renewable Hydrogen Technologies; Gandía, L.M., Arzamendi, G., Diéguez, P.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 225–243. [Google Scholar]
- Lämmermann, M.; Schwieger, W.; Freund, H. Experimental investigation of gas-liquid distribution in periodic open cellular structures as potential catalyst supports. Catal. Today 2016, 273, 161–171. [Google Scholar] [CrossRef]
- Busse, C.; Freund, H.; Schwieger, W. Intensification of heat transfer in catalytic reactors by additively manufactured periodic open cellular structures (POCS). Chem. Eng. Proc. 2018, 124, 199–214. [Google Scholar] [CrossRef]
- Lämmermann, M.; Horak, G.; Schwieger, W.; Freund, H. Periodic open cellular structures (POCS) for intensification of multiphase reactors: Liquid holdup and two-phase pressure drop. Chem. Eng. Process. 2018, 126, 178–189. [Google Scholar] [CrossRef]
- Klumpp, M.; Inayat, A.; Schwerdtfeger, J.; Körner, C.; Singer, R.F.; Freund, H.; Schwieger, W. Periodic open cellular structures with ideal cubic cell geometry: Effect of porosity and cell orientation on pressure drop behavior. Chem. Eng. J. 2014, 242, 364–378. [Google Scholar] [CrossRef]
- Parra-Cabrera, C.; Achille, C.; Kuhn, S.; Ameloot, R. 3D printing in chemical engineering and catalytic technology: Structured catalysts, mixers and reactors. Chem. Soc. Rev. 2018, 47, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Coppens, M.-O. A nature-inspired approach to reactor and catalysis engineering. Curr. Opin. Chem. Eng. 2012, 1, 281–289. [Google Scholar] [CrossRef]
- Kumar, A.; Mazumder, S. Toward simulation of full-scale monolithic catalytic converters with complex heterogeneous chemistry. Comput. Chem. Eng. 2010, 34, 135–145. [Google Scholar] [CrossRef]
- Mazumder, S. Modeling full-scale monolithic catalytic converters: Challenges and possible solutions. J. Heat Transfer 2006, 129, 526–535. [Google Scholar] [CrossRef]
- Groppi, G.; Belloli, A.; Tronconi, E.; Forzatti, P. A comparison of lumped and distributed models of monolith catalytic combustors. Chem. Eng. Sci. 1995, 50, 2705–2715. [Google Scholar] [CrossRef]
- Hayes, R.E.; Kolaczkowski, S.T. A study of Nusselt and Sherwood numbers in a monolith reactor. Catal. Today 1999, 47, 295–303. [Google Scholar] [CrossRef]
- Fox, R.O. CFD models for analysis and design of chemical reactors. In Advances in Chemical Engineering; Marin, G.B., Ed.; Academic Press: Salt Lake City, UT, USA, 2006; Volume 31, pp. 231–305. [Google Scholar]
- Nien, T.; Mmbaga, J.P.; Hayes, R.E.; Votsmeier, M. Hierarchical multi-scale model reduction in the simulation of catalytic converters. Chem. Eng. Sci. 2013, 93, 362–375. [Google Scholar] [CrossRef]
- Heidebrecht, P.; Pfafferodt, M.; Sundmacher, K. Multiscale modelling strategy for structured catalytic reactors. Chem. Eng. Sci. 2011, 66, 4389–4402. [Google Scholar] [CrossRef]
- Bertrand, F.; Devals, C.; Vidal, D.; Préval, C.S.d.; Hayes, R.E. Towards the simulation of the catalytic monolith converter using discrete channel-scale models. Catal. Today 2012, 188, 80–86. [Google Scholar] [CrossRef]
- Štěpánek, J.; Kočí, P.; Marek, M.; Kubíček, M. Catalyst simulations based on coupling of 3D CFD tool with effective 1D channel models. Catal. Today 2012, 188, 87–93. [Google Scholar] [CrossRef]
- Kamkeng, A.D.N.; Wang, M.; Hu, J.; Du, W.; Qian, F. Transformation technologies for CO2 utilisation: Current status, challenges and future prospects. Chem. Eng. J. 2021, 409, 128138. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- le Saché, E.; Pastor-Pérez, L.; Haycock, B.J.; Villora-Picó, J.J.; Sepúlveda-Escribano, A.; Reina, T.R. Switchable catalysts for chemical CO2 recycling: A step forward in the methanation and reverse water–gas shift reactions. ACS Sustain. Chem. Eng. 2020, 8, 4614–4622. [Google Scholar] [CrossRef]
- Whitlow, J.E.; Parrish, C.F. Operation, modeling and analysis of the reverse water gas shift process. AIP Conf. Proc. 2003, 654, 1116–1123. [Google Scholar]
- van der Wiel, D.; Zilka, J.; Wang, Y.; Tonkovich, A.; Wegeng, R. Carbon Dioxide Conversions in Microreactors. In Proceedings of the 4th International Conference on Microreaction Technology, Atlanta, GA, USA, 23–25 June 2014; AIChE Spring National Meeting: Atlanta, GA, USA, 2000; p. 187. [Google Scholar]
- Gaudillere, C.; Navarrete, L.; Serra, J.M. CO2 hydrogenation on Ru/Ce based catalysts dispersed on highly ordered micro-channelled 3YSZ monoliths fabricated by freeze-casting. Int. J. Hydrogen Energy 2017, 42, 895–905. [Google Scholar] [CrossRef]
- Fujita, S.-i.; Usui, M.; Takezawa, N. Mechanism of the reverse water gas shift reaction over Cu/ZnO catalyst. J. Catal. 1992, 134, 220–225. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Evans, J.; Feria, L.; Vidal, A.B.; Liu, P.; Nakamura, K.; Illas, F. CO2 hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC catalysts: Production of CO, methanol, and methane. J. Catal. 2013, 307, 162–169. [Google Scholar] [CrossRef]
- Liu, C.; Cundari, T.R.; Wilson, A.K. CO2 reduction on transition metal (Fe, Co, Ni, and Cu) surfaces: In comparison with homogeneous catalysis. J. Phys. Chem. C 2012, 116, 5681–5688. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. Heterogeneous catalysis on atomically dispersed supported metals: CO2 reduction on multifunctional Pd catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Baibars, F.; Le Sache, E.; Arellano-García, H.; Gu, S.; Reina, T.R. CO2 valorisation via Reverse Water-Gas Shift reaction using advanced Cs doped Fe-Cu/Al2O3 catalysts. J. CO2 Util. 2017, 21, 423–428. [Google Scholar] [CrossRef]
- Chen, C.-S.; Cheng, W.-H.; Lin, S.-S. Study of reverse water gas shift reaction by TPD, TPR and CO2 hydrogenation over potassium-promoted Cu/SiO2 catalyst. Appl. Catal. A Gen. 2003, 238, 55–67. [Google Scholar] [CrossRef]
- Chen, C.-S.; Cheng, W.-H.; Lin, S.-S. Study of iron-promoted Cu/SiO2 catalyst on high temperature reverse water gas shift reaction. Appl. Catal. A Gen. 2004, 257, 97–106. [Google Scholar] [CrossRef]
- Chen, C.S.; Lin, J.H.; You, J.H.; Chen, C.R. Properties of Cu(thd)2 as a precursor to prepare Cu/SiO2 catalyst using the atomic layer epitaxy technique. J. Am. Chem. Soc. 2006, 128, 15950–15951. [Google Scholar] [CrossRef]
- Zhou, G.; Dai, B.; Xie, H.; Zhang, G.; Xiong, K.; Zheng, X. CeCu composite catalyst for CO synthesis by reverse water–gas shift reaction: Effect of Ce/Cu mol ratio. J. CO2 Util. 2017, 21, 292–301. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H.; Chen, Y.; Yang, S. Reverse water–gas shift reaction over co-precipitated Co–CeO2 catalysts: Effect of Co content on selectivity and carbon formation. Int. J. Hydrogen Energy 2017, 42, 3682–3689. [Google Scholar] [CrossRef]
- Tang, R.; Zhu, Z.; Li, C.; Xiao, M.; Wu, Z.; Zhang, D.; Zhang, C.; Xiao, Y.; Chu, M.; Genest, A.; et al. Ru-catalyzed reverse water gas shift reaction with near-unity selectivity and superior stability. ACS Mater. Lett. 2021, 3, 1652–1659. [Google Scholar] [CrossRef]
- Tang, Y.; Asokan, C.; Xu, M.; Graham, G.W.; Pan, X.; Christopher, P.; Li, J.; Sautet, P. Rh single atoms on TiO2 dynamically respond to reaction conditions by adapting their site. Nat. Commun. 2019, 10, 4488. [Google Scholar] [CrossRef]
- Lebarbier, V.; Dagle, R.; Datye, A.; Wang, Y. The effect of PdZn particle size on reverse-water–gas-shift reaction. Appl. Catal. A Gen. 2010, 379, 3–6. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, M.; Ma, P.; Zheng, Y.; Chen, J.; Li, H.; Zhang, X.; Zheng, K.; Kuang, Q.; Xie, Z.-X. Atomically dispersed Pt/CeO2 catalyst with superior CO selectivity in reverse water gas shift reaction. Appl. Catal. B Environ. 2021, 291, 120101. [Google Scholar] [CrossRef]
- Panagiotopoulou, P. Hydrogenation of CO2 over supported noble metal catalysts. Appl. Catal. A Gen. 2017, 542, 63–70. [Google Scholar] [CrossRef]
- Matsubu, J.C.; Yang, V.N.; Christopher, P. Isolated metal active site concentration and stability control catalytic CO2 reduction selectivity. J. Am. Chem. Soc. 2015, 137, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Lee, H.H.; Hong, S.C. The effect of the morphological characteristics of TiO2 supports on the reverse water–gas shift reaction over Pt/TiO2 catalysts. Appl. Catal. B Environ. 2012, 119-120, 100–108. [Google Scholar] [CrossRef]
- Vourros, A.; Garagounis, I.; Kyriakou, V.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Marnellos, G.E.; Konsolakis, M. Carbon dioxide hydrogenation over supported Au nanoparticles: Effect of the support. J. CO2 Util. 2017, 19, 247–256. [Google Scholar] [CrossRef]
- Wang, L.C.; Tahvildar Khazaneh, M.; Widmann, D.; Behm, R.J. TAP reactor studies of the oxidizing capability of CO2 on a Au/CeO2 catalyst—A first step toward identifying a redox mechanism in the Reverse Water–Gas Shift reaction. J. Catal. 2013, 302, 20–30. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef]
- Ma, S.; Song, W.; Liu, B.; Zheng, H.; Deng, J.; Zhong, W.; Liu, J.; Gong, X.-Q.; Zhao, Z. Elucidation of the high CO2 reduction selectivity of isolated Rh supported on TiO2: A DFT study. Catal. Sci. Technol. 2016, 6, 6128–6136. [Google Scholar] [CrossRef]
- Avanesian, T.; Gusmão, G.S.; Christopher, P. Mechanism of CO2 reduction by H2 on Ru(0001) and general selectivity descriptors for late-transition metal catalysts. J. Catal. 2016, 343, 86–96. [Google Scholar] [CrossRef]
- Sabatier, P.; Senderens, J. Hydrogénation directe des oxydes du carbone en présence de divers métaux divisés. Comptes Rendus 1902, 134, 689–691. [Google Scholar]
- Xiaoding, X.; Moulijn, J.A. Mitigation of CO2 by chemical conversion: Plausible chemical reactions and promising products. Energy Fuels 1996, 10, 305–325. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Jacquemin, M.; Beuls, A.; Ruiz, P. Catalytic production of methane from CO2 and H2 at low temperature: Insight on the reaction mechanism. Catal. Today 2010, 157, 462–466. [Google Scholar] [CrossRef]
- Dubois, L.H.; Somorjai, G.A. Comments on “why CO2 does not dissociate on Rh at low temperature” by W.H. Weinberg. Surf. Sci. 1983, 128, L231–L235. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Türks, D.; Mena, H.; Armbruster, U.; Martin, A. Methanation of CO2 on Ni/Al2O3 in a Structured Fixed-Bed Reactor—A Scale-Up Study. Catalysts 2017, 7, 152. [Google Scholar] [CrossRef]
- Jalama, K. Carbon dioxide hydrogenation over nickel-, ruthenium-, and copper-based catalysts: Review of kinetics and mechanism. Catal. Rev. 2017, 59, 95–164. [Google Scholar] [CrossRef]
- Agnelli, M.; Swaan, H.M.; Márquez-Alvarez, C.; Martín, G.A.; Mirodatos, C. CO hydrogenation on a nickel catalyst: II. A mechanistic study by transient kinetics and infrared spectroscopy. J. Catal. 1998, 175, 117–128. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Wissing, L.; Skjøth-Rasmussen, M.S. High temperature methanation: Catalyst considerations. Catal. Today 2013, 215, 233–238. [Google Scholar] [CrossRef]
- Ghaib, K.; Nitz, K.; Ben-Fares, F.-Z. Chemical methanation of CO2: A Review. ChemBioEng Rev. 2016, 3, 266–275. [Google Scholar] [CrossRef]
- Xu, Y.; Wan, H.; Du, X.; Yao, B.; Wei, S.; Chen, Y.; Zhuang, W.; Yang, H.; Sun, L.; Tao, X.; et al. Highly active Ni/CeO2/SiO2 catalyst for low-temperature CO2 methanation: Synergistic effect of small Ni particles and optimal amount of CeO2. Fuel Process. Technol. 2022, 236, 107418. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Roger, A.-C. Methanation of carbon dioxide over nickel-based Ce0.72Zr0.28O2 mixed oxide catalysts prepared by sol–gel method. Appl. Catal. A Gen. 2009, 369, 90–96. [Google Scholar] [CrossRef]
- Safdar, M.; González-Castaño, M.; Penkova, A.; Centeno, M.A.; Odriozola, J.A.; Arellano-García, H. CO2 methanation on Ni/YMn1−xAlxO3 perovskite catalysts. Appl. Mater. Today 2022, 29, 101577. [Google Scholar] [CrossRef]
- Vogt, C.; Groeneveld, E.; Kamsma, G.; Nachtegaal, M.; Lu, L.; Kiely, C.J.; Berben, P.H.; Meirer, F.; Weckhuysen, B.M. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 2018, 1, 127–134. [Google Scholar] [CrossRef]
- Gil, A.; Díaz, A.; Gandía, L.M.; Montes, M. Influence of the preparation method and the nature of the support on the stability of nickel catalysts. Appl. Catal. A Gen. 1994, 109, 167–179. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Chemistry of nickel-alumina catalysts. J. Catal. 1976, 45, 41–53. [Google Scholar] [CrossRef]
- Jia, C.; Gao, J.; Li, J.; Gu, F.; Xu, G.; Zhong, Z.; Su, F. Nickel catalysts supported on calcium titanate for enhanced CO methanation. Catal. Sci. Technol. 2013, 3, 490–499. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.; Li, H.; Yu, Z. CO2 Methanation: The effect of catalysts and reaction conditions. Energy Procedia 2017, 105, 2022–2027. [Google Scholar] [CrossRef]
- Aldana, P.A.U.; Ocampo, F.; Kobl, K.; Louis, B.; Thibault-Starzyk, F.; Daturi, M.; Bazin, P.; Thomas, S.; Roger, A.C. Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy. Catal. Today 2013, 215, 201–207. [Google Scholar]
- Navarro-Jaén, S.; Szego, A.; Bobadilla, L.F.; Laguna, Ó.H.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. Operando spectroscopic evidence of the induced effect of residual species in the reaction intermediates during CO2 hydrogenation over ruthenium nanoparticles. ChemCatChem 2019, 11, 2063–2068. [Google Scholar] [CrossRef]
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S. Production of synthetic natural gas (SNG) from coal and dry biomass—A technology review from 1950 to 2009. Fuel 2010, 89, 1763–1783. [Google Scholar] [CrossRef]
- Navarro, J.C.; Centeno, M.A.; Laguna, O.H.; Odriozola, J.A. Policies and motivations for the CO2 Valorization through the Sabatier reaction using structured catalysts. A review of the most recent advances. Catalysts 2018, 8, 578. [Google Scholar]
- Lawson, S.; Li, X.Z.; Thakkar, H.V.; Rownaghi, A.A.; Rezaei, F. Recent advances in 3D printing of structured materials for adsorption and catalysis applications. Chem. Rev. 2021, 121, 6246–6291. [Google Scholar] [CrossRef] [PubMed]
- Danaci, S.; Protasova, L.; Snijkers, F.; Bouwen, W.L.; Bengaouer, A.; Marty, P. Innovative 3D-manufacture of structured copper supports post-coated with catalytic material for CO2 methanation. Chem. Eng. Process. 2018, 127, 168–177. [Google Scholar] [CrossRef]
- Danaci, S.; Protasova, L.; Try, R.; Bengaouer, A.; Marty, P. Experimental and numerical investigation of heat transport and hydrodynamic properties of 3D-structured catalytic supports. Appl. Therm. Eng. 2017, 126, 167–178. [Google Scholar] [CrossRef]
- Danaci, S.; Protasova, L.; Lefevere, J.; Bedel, L.; Guilet, R.; Marty, P. Efficient CO2 methanation over Ni/Al2O3 coated structured catalysts. Catal. Today 2016, 273, 234–243. [Google Scholar] [CrossRef]
- González-Castaño, M.; Baena-Moreno, F.; Carlos Navarro de Miguel, J.; Miah, K.U.M.; Arroyo-Torralvo, F.; Ossenbrink, R.; Odriozola, J.A.; Benzinger, W.; Hensel, A.; Wenka, A.; et al. 3D-printed structured catalysts for CO2 methanation reaction: Advancing of gyroid-based geometries. Energy Conv. Manag. 2022, 258, 115464. [Google Scholar] [CrossRef]
- Baena-Moreno, F.M.; González-Castaño, M.; Navarro de Miguel, J.C.; Miah, K.U.M.; Ossenbrink, R.; Odriozola, J.A.; Arellano-García, H. Stepping toward efficient microreactors for CO2 methanation: 3D-printed gyroid geometry. ACS Sustain. Chem. Eng. 2021, 9, 8198–8206. [Google Scholar] [CrossRef]
- Preuster, P.; Papp, C.; Wasserscheid, P. Liquid Organic Hydrogen Carriers (LOHCs): Toward a hydrogen-free hydrogen economy. Acc. Chem. Res. 2017, 50, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC)—Assessment based on chemical and economic properties. Int. J. Hydrogen Energy 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Eppinger, J.; Huang, K.-W. Formic acid as a hydrogen energy carrier. ACS Energy Lett. 2017, 2, 188–195. [Google Scholar] [CrossRef]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source—recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Pérez-Fortes, M.; Schöneberger, J.C.; Boulamanti, A.K.; Harrison, G.; Tzimas, E. Formic acid synthesis using CO2 as raw material: Techno-economic and environmental evaluation and market potential. Int. J. Hydrogen Energy 2016, 41, 16444–16462. [Google Scholar] [CrossRef]
- Inoue, Y.; Izumida, H.; Sasaki, Y.; Hashimoto, H. Catalytic fixation of carbon dioxide to formic acid by transition-metal complexes under mild conditions. Chem. Lett. 1976, 5, 863–864. [Google Scholar] [CrossRef]
- Behr, A.; Nowakowski, K. Book Series: Advances in Inorganic Chemistry; CO2 Chemistry; Elsevier: Amsterdam, The Netherlands, 2014; Volume 66. [Google Scholar]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Gunasekar, G.H.; Park, K.; Jung, K.-D.; Yoon, S. Recent developments in the catalytic hydrogenation of CO2 to formic acid/formate using heterogeneous catalysts. Inorg. Chem. Front. 2016, 3, 882–895. [Google Scholar] [CrossRef]
- Song, H.; Zhang, N.; Zhong, C.; Liu, Z.; Xiao, M.; Gai, H. Hydrogenation of CO2 into formic acid using a palladium catalyst on chitin. New J. Chem. 2017, 41, 9170–9177. [Google Scholar] [CrossRef]
- Upadhyay, P.R.; Srivastava, V. Selective hydrogenation of CO2 gas to formic acid over nanostructured Ru-TiO2 catalysts. RSC Adv. 2016, 6, 42297–42306. [Google Scholar] [CrossRef]
- Nguyen, L.T.M.; Park, H.; Banu, M.; Kim, J.Y.; Youn, D.H.; Magesh, G.; Kim, W.Y.; Lee, J.S. Catalytic CO2 hydrogenation to formic acid over carbon nanotube-graphene supported PdNi alloy catalysts. RSC Adv. 2015, 5, 105560–105566. [Google Scholar] [CrossRef]
- Maru, M.S.; Ram, S.; Shukla, R.S.; Khan, N.-u.H. Ruthenium-hydrotalcite (Ru-HT) as an effective heterogeneous catalyst for the selective hydrogenation of CO2 to formic acid. Mol. Catal. 2018, 446, 23–30. [Google Scholar] [CrossRef]
- Maru, M.S.; Ram, S.; Adwani, J.H.; Shukla, R.S. Selective and Direct Hydrogenation of CO2 for the Synthesis of Formic Acid over a Rhodium Hydrotalcite (Rh-HT) Catalyst. ChemistrySelect 2017, 2, 3823–3830. [Google Scholar] [CrossRef]
- Mori, K.; Taga, T.; Yamashita, H. Isolated single-atomic Ru catalyst bound on a layered double hydroxide for hydrogenation of CO2 to formic acid. ACS Catal. 2017, 7, 3147–3151. [Google Scholar] [CrossRef]
- Umegaki, T.; Satomi, Y.; Kojima, Y. Catalytic properties of palladium nanoparticles for hydrogenation of carbon dioxide into formic acid. J. Japan Inst. Energy 2017, 96, 487–492. [Google Scholar] [CrossRef]
- Preti, D.; Resta, C.; Squarcialupi, S.; Fachinetti, G. Carbon dioxide hydrogenation to formic acid by using a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2011, 50, 12551–12554. [Google Scholar] [CrossRef]
- Hao, C.; Wang, S.; Li, M.; Kang, L.; Ma, X. Hydrogenation of CO2 to formic acid on supported ruthenium catalysts. Catal. Today 2011, 160, 184–190. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.; Zhao, Y.; Ma, X. Hydrogenation of scCO2 to formic acid catalyzed by heterogeneous ruthenium(III)/Al2O3 catalysts. Chem. Lett. 2016, 45, 555–557. [Google Scholar] [CrossRef]
- He, C.-S.; Gong, L.; Zhang, J.; He, P.-P.; Mu, Y. Highly selective hydrogenation of CO2 into formic acid on a nano-Ni catalyst at ambient temperature: Process, mechanisms and catalyst stability. J. CO2 Util. 2017, 19, 157–164. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, T.; Wang, X.; Hao, R.; Wang, H. CO2 hydrogenation to formate over nano-scale zero-valent nickel catalyst under atmospheric pressure. Chem. Eng. J. 2018, 347, 860–869. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Vrijburg, W.L.; Hensen, E.J.M.; Pidko, E.A. On the activity of supported Au catalysts in the liquid phase hydrogenation of CO2 to formates. J. Catal. 2016, 343, 97–105. [Google Scholar] [CrossRef]
- Su, J.; Yang, L.; Lu, M.; Lin, H. Highly efficient hydrogen storage system based on ammonium bicarbonate/formate redox equilibrium over palladium nanocatalysts. ChemSusChem 2015, 8, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Lu, M.; Lin, H. High yield production of formate by hydrogenating CO2 derived ammonium carbamate/carbonate at room temperature. Green Chem. 2015, 17, 2769–2773. [Google Scholar] [CrossRef]
- Bi, Q.-Y.; Lin, J.-D.; Liu, Y.-M.; Du, X.-L.; Wang, J.-Q.; He, H.-Y.; Cao, Y. An aqueous rechargeable formate-based hydrogen battery driven by heterogeneous Pd catalysis. Angew. Chem. Int. Ed. 2014, 53, 13583–13587. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Megías-Sayago, C.; Ivanova, S.; Centeno, M.Á.; Odriozola, J.A. Functionalized biochars as supports for Pd/C catalysts for efficient hydrogen production from formic acid. Appl. Catal. B Environ. 2021, 282, 119615. [Google Scholar] [CrossRef]
- Santos, J.L.; León, C.; Monnier, G.; Ivanova, S.; Centeno, M.Á.; Odriozola, J.A. Bimetallic PdAu catalysts for formic acid dehydrogenation. Int. J. Hydrogen Energy 2020, 45, 23056–23068. [Google Scholar] [CrossRef]
- Santos, J.L.; Megías-Sayago, C.; Ivanova, S.; Centeno, M.Á.; Odriozola, J.A. Structure-sensitivity of formic acid dehydrogenation reaction over additive-free Pd NPs supported on activated carbon. Chem. Eng. J. 2021, 420, 127641. [Google Scholar] [CrossRef]
- Qin, Y.-l.; Wang, J.; Meng, F.-z.; Wang, L.-m.; Zhang, X.-B. Efficient PdNi and PdNi@Pd-catalyzed hydrogen generation via formic acid decomposition at room temperature. Chem. Commun. 2013, 49, 10028–10030. [Google Scholar] [CrossRef]
- Iglesia, E.; Boudart, M. Decomposition of formic acid on copper, nickel, and copper-nickel alloys: III. Catalytic decomposition on nickel and copper-nickel alloys. J. Catal. 1983, 81, 224–238. [Google Scholar] [CrossRef]
- Navlani-García, M.; Mori, K.; Salinas-Torres, D.; Kuwahara, Y.; Yamashita, H. New approaches toward the hydrogen production from formic acid dehydrogenation over Pd-based heterogeneous catalysts. Front. Mater. 2019, 6, 44. [Google Scholar] [CrossRef]
- Cazaña, F.; Galetti, A.; Meyer, C.; Sebastián, V.; Centeno, M.A.; Romeo, E.; Monzón, A. Synthesis of Pd-Al/biomorphic carbon catalysts using cellulose as carbon precursor. Catal. Today 2018, 301, 226–238. [Google Scholar] [CrossRef]
- Loges, B.; Boddien, A.; Gärtner, F.; Junge, H.; Beller, M. Catalytic generation of hydrogen from formic acid and its derivatives: Useful hydrogen storage materials. Top. Catal. 2010, 53, 902–914. [Google Scholar] [CrossRef]
- Beloqui Redondo, A.; Morel, F.L.; Ranocchiari, M.; van Bokhoven, J.A. Functionalized ruthenium–phosphine Metal–Organic Framework for continuous vapor-phase dehydrogenation of formic acid. ACS Catal. 2015, 5, 7099–7103. [Google Scholar] [CrossRef]
- Lee, J.H.; Ryu, J.; Kim, J.Y.; Nam, S.-W.; Han, J.H.; Lim, T.-H.; Gautam, S.; Chae, K.H.; Yoon, C.W. Carbon dioxide mediated, reversible chemical hydrogen storage using a Pd nanocatalyst supported on mesoporous graphitic carbon nitride. J. Mater. Chem. A 2014, 2, 9490–9495. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Yan, J.-M.; Wang, H.-L.; Ping, Y.; Jiang, Q. Au@Pd core–shell nanoclusters growing on nitrogen-doped mildly reduced graphene oxide with enhanced catalytic performance for hydrogen generation from formic acid. J. Mater. Chem. A 2013, 1, 12721–12725. [Google Scholar] [CrossRef]
- Caiti, M.; Padovan, D.; Hammond, C. Continuous production of hydrogen from formic acid decomposition over heterogeneous nanoparticle catalysts: From batch to continuous flow. ACS Catal. 2019, 9, 9188–9198. [Google Scholar] [CrossRef]
- Hafeez, S.; Sanchez, F.; Al-Salem, S.M.; Villa, A.; Manos, G.; Dimitratos, N.; Constantinou, A. Decomposition of additive-free formic acid using a Pd/C catalyst in flow: Experimental and CFD modelling studies. Catalysts 2021, 11, 341. [Google Scholar] [CrossRef]
- Katuri, P.; Maralla, Y.S.S.; Tumma, B.N. Production of performic acid through a capillary microreactor by heterogeneous catalyst. Int. J. Chem. Reactor Eng. 2020, 20. In press. [Google Scholar] [CrossRef]
- Hafeez, S.; Al-Salem, S.M.; Bansode, A.; Villa, A.; Dimitratos, N.; Manos, G.; Constantinou, A. Computational investigation of microreactor configurations for hydrogen production fromformic acid decomposition using a Pd/C catalyst. Ind. Eng. Chem. Res. 2022, 61, 1655–1665. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl ether (DME) as an alternative fuel. J. Power Sources 2006, 156, 497–511. [Google Scholar] [CrossRef]
- Arcoumanis, C.; Bae, C.; Crookes, R.J.; Kinoshita, E. The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: A review. Fuel 2008, 87, 1014–1030. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the development of novel cobalt Fischer−Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Rahimpour, M.R. Dimethyl ether: A review of technologies and production challenges. Chem. Eng. Process. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Shikada, T.; Ohno, Y.; Ogawa, T.; Ono, M.; Mizuguchi, M.; Tomura, K.; Fujimoto, K. Direct synthesis of dimethyl ether form synthesis gas. In Studies in Surface Science and Catalysis; Parmaliana, A., Sanfilippo, D., Frusteri, F., Vaccari, A., Arena, F., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; Volume 119, pp. 515–520. [Google Scholar]
- Ohno, Y.; Yoshida, M.; Shikada, T.; Inokoshi, O.; Ogawa, T.; Inoue, N. New Direct Synthesis Technology for DME (Dimethyl Ether) and Its Application Technology; JFE Technical Report, nº 8; JFE Holdings Inc.: Tokyo, Japan, October 2006. [Google Scholar]
- Shikada, T.; Ohno, Y.; Ogawa, T.; Mizuguchi, M.; Ono, M.; Fujimoto, K. Catalyst for Producing Dimethyl Ether, Method for Producing Catalyst and Method for Producing Dimethyl Ether. U.S. Patent 7,033,972 B2, 25 April 2006. [Google Scholar]
- Rostrup-Nielsen, T.; Madsen, J. Process for the Preparation of Pure Dimethyl Ether. U.S. Patent 7,910,630 B2, 22 March 2011. [Google Scholar]
- Joensen, F.; Madsen, J.; Erik, P. Process for the Preparation of Dimethyl Ether. WO 2014032973 A1, 6 March 2014. [Google Scholar]
- Lewnard, J.J.; Hsiung, T.H.; White, J.F.; Bhatt, B.L. Liquid Phase Process for Dimethyl Ether Synthesis. U.S. Patent 5,218,003, 8 June 1993. [Google Scholar]
- Peng, X. Use of Aluminum Phosphate as the Dehydration Catalyst in Single Step Dimethyl Ether Process. U.S. Patent 5,753,716, 19 May 1998. [Google Scholar]
- Pagani, G. Process for the Production of Dimethyl Ether. U.S. Patent 4,098,809, 4 July 1978. [Google Scholar]
- Bell, W.K.; Chang, C.D. Dimethyl Ether Synthesis Catalysts. U.S. Patent 4,423,155, 27 December 1983. [Google Scholar]
- Khandan, N.; Kazemeini, M.; Aghaziarati, M. Direct production of dimethyl ether from synthesis gas utilizing bifunctional catalysts. Appl. Petrochem. Res. 2012, 1, 21–27. [Google Scholar] [CrossRef]
- Bridger, G.W.; Spencer, M.S. Catalyst Handbook, 2nd ed.; Twigg, M.V., Ed.; Wofle Press: London, UK, 1989. [Google Scholar]
- Stiefel, M.; Ahmad, R.; Arnold, U.; Döring, M. Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas: Influence of dehydration catalysts and operating conditions. Fuel Process. Technol. 2011, 92, 1466–1474. [Google Scholar] [CrossRef]
- Cheiky, M. Low Pressure Dimethyl Ether Synthesis Catalyst. U.S. Patent 2013/0211147 A1, 15 August 2013. [Google Scholar]
- García-Trenco, A.; Martínez, A. Direct synthesis of DME from syngas on hybrid CuZnAl/ZSM-5 catalysts: New insights into the role of zeolite acidity. Appl. Catal. A Gen. 2012, 411–412, 170–179. [Google Scholar] [CrossRef]
- García-Trenco, A.; Martínez, A. The influence of zeolite surface-aluminum species on the deactivation of CuZnAl/zeolite hybrid catalysts for the direct DME synthesis. Catal. Today 2014, 227, 144–153. [Google Scholar] [CrossRef]
- García-Trenco, A.; Martínez, A. A rational strategy for preparing Cu–ZnO/H-ZSM-5 hybrid catalysts with enhanced stability during the one-step conversion of syngas to dimethyl ether (DME). Appl. Catal. A Gen. 2015, 493, 40–49. [Google Scholar] [CrossRef]
- García-Trenco, A.; Valencia, S.; Martínez, A. The impact of zeolite pore structure on the catalytic behavior of CuZnAl/zeolite hybrid catalysts for the direct DME synthesis. Appl. Catal. A Gen. 2013, 468, 102–111. [Google Scholar] [CrossRef]
- García-Trenco, A.; Vidal-Moya, A.; Martínez, A. Study of the interaction between components in hybrid CuZnAl/HZSM-5 catalysts and its impact in the syngas-to-DME reaction. Catal. Today 2012, 179, 43–51. [Google Scholar] [CrossRef]
- Xiao, K.; Wang, Q.; Qi, X.; Zhong, L. For better industrial Cu/ZnO/Al2O3 methanol synthesis catalyst: A Compositional Study. Catal. Lett. 2017, 147, 1581–1591. [Google Scholar] [CrossRef]
- Sun, J.; Yang, G.; Ma, Q.; Ooki, I.; Taguchi, A.; Abe, T.; Xie, Q.; Yoneyama, Y.; Tsubaki, N. Fabrication of active Cu–Zn nanoalloys on H-ZSM5 zeolite for enhanced dimethyl ether synthesis via syngas. J. Mater. Chem. A 2014, 2, 8637–8643. [Google Scholar] [CrossRef]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts. Appl. Catal. B Environ. 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Sanz, O.; Banús, E.D.; Goya, A.; Larumbe, H.; Delgado, J.J.; Monzón, A.; Montes, M. Stacked wire-mesh monoliths for VOCs combustion: Effect of the mesh-opening in the catalytic performance. Catal. Today 2017, 296, 76–83. [Google Scholar] [CrossRef]
- Merino, D.; Sanz, O.; Montes, M. Effect of catalyst layer macroporosity in high-thermal-conductivity monolithic Fischer-Tropsch catalysts. Fuel 2017, 210, 49–57. [Google Scholar] [CrossRef]
- Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors: New Technology for Modern Chemistry; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2000. [Google Scholar]
- Hessel, V.; Renken, A.; Schouten, J.C.; Yoshida, J. (Eds.) Micro Process Engineering. A Comprehensive Handbook; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2009. [Google Scholar]
- Hu, J.; Wang, Y.; Cao, C.; Elliott, D.C.; Stevens, D.J.; White, J.F. Conversion of biomass syngas to DME using a microchannel reactor. Ind. Eng. Chem. Res. 2005, 44, 1722–1727. [Google Scholar] [CrossRef]
- Hayer, F.; Bakhtiary-Davijany, H.; Myrstad, R.; Holmen, A.; Pfeifer, P.; Venvik, H.J. Synthesis of dimethyl ether from syngas in a microchannel reactor—Simulation and experimental study. Chem. Eng. J. 2011, 167, 610–615. [Google Scholar] [CrossRef]
- Dagle, R.A.; Wang, Y.; Baker, E.G.; Hu, J. Dimethyl Ether Production from Methanol and/or Syngas. U.S. Patent 8,957,259 B2, 17 February 2015. [Google Scholar]
- Venvik, H.J.; Yang, J. Catalysis in microstructured reactors: Short review on small-scale syngas production and further conversion into methanol, DME and Fischer-Tropsch products. Catal. Today 2017, 285, 135–146. [Google Scholar] [CrossRef]
- Allahyari, S.; Haghighi, M.; Ebadi, A. Direct synthesis of DME over nanostructured CuO–ZnO–Al2O3/HZSM-5 catalyst washcoated on high pressure microreactor: Effect of catalyst loading and process condition on reactor performance. Chem. Eng. J. 2015, 262, 1175–1186. [Google Scholar] [CrossRef]
- Jia, C.; Gao, J.; Dai, Y.; Zhang, J.; Yang, Y. The thermodynamics analysis and experimental validation for complicated systems in CO2 hydrogenation process. J. Energy Chem. 2016, 25, 1027–1037. [Google Scholar] [CrossRef]
- Noriyuki, I.; Kouji, H.; Atsushi, S.; Tadashi, H.; Yuichi, M. Unique temperature dependence of acetic acid formation in CO2 hydrogenation on Ag-promoted Rh/SiO2 catalyst. Chem. Lett. 1994, 23, 263–264. [Google Scholar]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. State of the art and perspectives in catalytic processes for CO2 conversion into chemicals and fuels: The distinctive contribution of chemical catalysis and biotechnology. J. Catal. 2016, 343, 2–45. [Google Scholar] [CrossRef]
- Bristow, T.G. Integrated Process for Making Acetic Acid. WO2014096254, 26 June 2014. [Google Scholar]
- Somiari, I.; Manousiouthakis, V.I. Coproduction of acetic acid and hydrogen/power from natural gas with zero carbon dioxide emissions. AIChE J. 2018, 64, 860–876. [Google Scholar] [CrossRef]
- Dagle, R.A.; Hu, J.; Jones, S.B.; Wilcox, W.; Frye, J.G.; White, J.F.; Jiang, J.; Wang, Y. Carbon dioxide conversion to valuable chemical products over composite catalytic systems. J. Energy Chem. 2013, 22, 368–374. [Google Scholar]
- Cheung, P.; Bhan, A.; Sunley, G.J.; Law, D.J.; Iglesia, E. Site requirements and elementary steps in dimethyl ether carbonylation catalyzed by acidic zeolites. J. Catal. 2007, 245, 110–123. [Google Scholar] [CrossRef]
- Xu, B.-Q.; Sachtler, W.M.H. Rh/NaY: A selective catalyst for direct synthesis of acetic acid from syngas. J. Catal. 1998, 180, 194–206. [Google Scholar] [CrossRef]
- Han, L.; Mao, D.; Yu, J.; Guo, Q.; Lu, G. C2-oxygenates synthesis through CO hydrogenation on SiO2-ZrO2 supported Rh-based catalyst: The effect of support. Appl. Catal. A Gen. 2013, 454, 81–87. [Google Scholar] [CrossRef]
- Yu, J.; Mao, D.; Lu, G.; Guo, Q.; Han, L. Enhanced C2 oxygenate synthesis by CO hydrogenation over Rh-based catalyst supported on a novel SiO2. Catal. Commun. 2012, 24, 25–29. [Google Scholar]
- Cheung, P.; Bhan, A.; Sunley, G.J.; Iglesia, E. Selective carbonylation of dimethyl ether to methyl acetate catalyzed by acidic zeolites. Angew. Chem. Int. Ed. 2006, 45, 1617–1620. [Google Scholar] [CrossRef] [PubMed]
- Volkova, G.G.; Plyasova, L.M.; Shkuratova, L.N.; Budneva, A.A.; Paukshtis, E.A.; Timofeeva, M.N.; Likholobov, V.A. Solid superacids for halide-free carbonylation of dimethyl ether to methyl acetate. Stud. Surf. Sci. Catal. 2004, 147, 403–408. [Google Scholar]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Blasco, T.; Boronat, M.; Concepción, P.; Corma, A.; Law, D.; Vidal-Moya, J.A. Carbonylation of methanol on metal–acid zeolites: Evidence for a mechanism involving a multisite active center. Angew. Chem. Int. Ed. 2007, 46, 3938–3941. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Allian, A.D.; Sunley, G.J.; Law, D.J.; Iglesia, E. Specificity of sites within eight-membered ring zeolite channels for carbonylation of methyls to acetyls. J. Am. Chem. Soc. 2007, 129, 4919–4924. [Google Scholar] [CrossRef]
- Boronat, M.; Martínez-Sánchez, C.; Law, D.; Corma, A. Enzyme-like specificity in zeolites: A unique site position in mordenite for selective carbonylation of methanol and dimethyl ether with CO. J. Am. Chem. Soc. 2008, 130, 16316–16323. [Google Scholar] [CrossRef] [PubMed]
- Boronat, M.; Martínez, C.; Corma, A. Mechanistic differences between methanol and dimethyl ether carbonylation in side pockets and large channels of mordenite. Phys. Chem. Chem. Phys. 2011, 13, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Reule, A.A.C.; Shen, J.; Semagina, N. Copper affects the location of zinc in bimetallic ion-exchanged mordenite. ChemPhysChem 2018, 19, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Luzgin, M.V.; Kazantsev, M.S.; Volkova, G.G.; Stepanov, A.G. Solid-state NMR study of the kinetics and mechanism of dimethyl ether carbonylation on cesium salt of 12-tungstophosphoric acid modified with Ag, Pt, and Rh. J. Catal. 2013, 308, 250–257. [Google Scholar] [CrossRef]
- Xue, H.; Huang, X.; Ditzel, E.; Zhan, E.; Ma, M.; Shen, W. Coking on micrometer- and nanometer-sized mordenite during dimethyl ether carbonylation to methyl acetate. Chin. J. Catal. 2013, 34, 1496–1503. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, Y.; Du, L.; Liu, J.; Yao, J. Review of the applications of microreactors. Renew. Sustain. Energy Rev. 2015, 47, 519–539. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).