
Expanding Heteroaromatic and 2-Aminosugar Chemical Space Accessible from the Biopolymer Chitin



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. General Information
2.1.1. 2-Hydroxyimino-3-acetamido-5-acetylfuran (4)
2.1.2. 2-Cyano-3-acetamido-5-acetylfuran (5)
2.1.3. 5-Acetyl-3-amino-2-carboxamidofuran (6)
2.1.4. 6-Acetylfuro[3,2-d]pyrimidin-4-one (7)
2.1.5. Methyl 4-acetamido-5-formylfuran-2-carboxylate (8)
2.1.6. Methyl 4-Acetamido-5-[(hydroxyimino)methyl]furan-2-carboxylate (9)
2.1.7. Methyl 4-Acetamido-5-cyanofuran-2-carboxylate (10)
2.1.8. Methyl 4-Amino-5-cyanofuran-2-carboxylate (11)
2.1.9. Methyl 4-Aminofuro[3,2-d]pyrimidine-6-carboxylate (12)
2.1.10. (R)-N-(5-(2-((tert-Butyldimethylsilyl)oxy)-1-hydroxyethyl)furan-3-yl)acetamide (15)
2.1.11. N-((6R)-6-(((tert-Butyldimethylsilyl)oxy)methyl)-2-hydroxy-5-oxo-5,6-dihydro-2H-pyran-3-yl)acetamide (16)
2.1.12. (R)-N-(6-(((tert-Butyldimethylsilyl)oxy)methyl)-2,5-dioxo-5,6-dihydro-2H-pyran-3-yl)acetamide (17)
2.1.13. N-((5R,6R)-6-(((tert-Butyldimethylsilyl)oxy)methyl)-5-hydroxy-2-oxo-5,6-dihydro-2H-pyran-3-yl)acetamide (18)
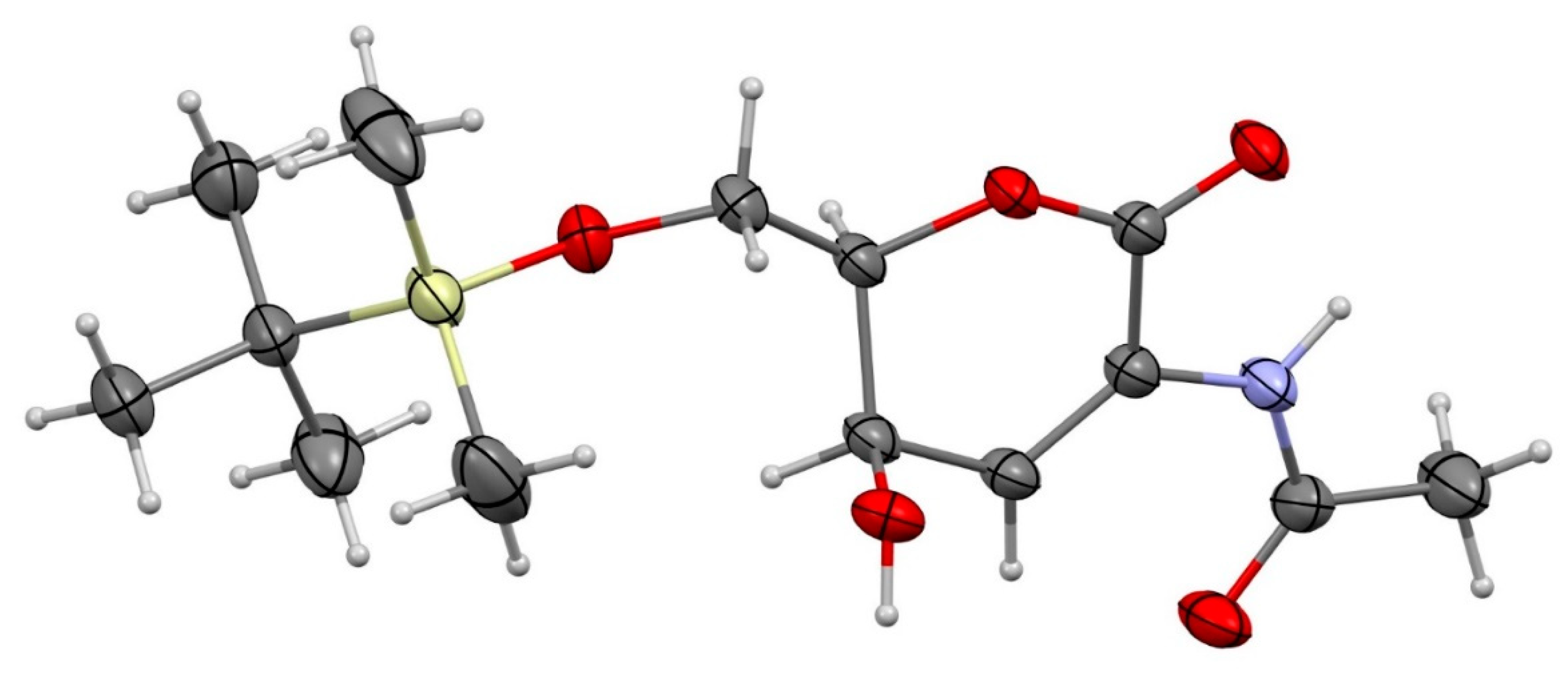
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyriakou, V.; Garagounis, I.; Vourros, A.; Vasileiou, E.; Stoukides, M. An Electrochemical Haber-Bosch Process. Joule 2020, 4, 142–158. [Google Scholar] [CrossRef]
- Song, X.; Basheer, C.; Zare, R.N. Making Ammonia from Nitrogen and Water Microdroplets. Proc. Natl. Acad. Sci. USA 2023, 120, e2301206120. [Google Scholar] [CrossRef] [PubMed]
- Soloveichik, G. Electrochemical Synthesis of Ammonia as a Potential Alternative to the Haber–Bosch Process. Nat. Catal. 2019, 2, 377–380. [Google Scholar] [CrossRef]
- Wang, M.; Khan, M.A.; Mohsin, I.; Wicks, J.; Ip, A.H.; Sumon, K.Z.; Dinh, C.-T.; Sargent, E.H.; Gates, I.D.; Golam Kibria, M. Can Sustainable Ammonia Synthesis Pathways Compete with Fossil-Fuel Based Haber–Bosch Processes? Energy Environ. Sci. 2021, 14, 2535–2548. [Google Scholar] [CrossRef]
- Yan, N.; Chen, X. Sustainability: Don’t Waste Seafood Waste. Nature 2015, 524, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Hülsey, M.J.; Yang, H.; Yan, N. Sustainable Routes for the Synthesis of Renewable Heteroatom-Containing Chemicals. ACS Sustain. Chem. Eng. 2018, 6, 5694–5707. [Google Scholar] [CrossRef]
- Dai, J.; Li, F.; Fu, X. Towards Shell Biorefinery: Advances in Chemical-Catalytic Conversion of Chitin Biomass to Organonitrogen Chemicals. ChemSusChem 2020, 13, 6498–6508. [Google Scholar] [CrossRef]
- Osada, M.; Kikuta, K.; Yoshida, K.; Totani, K.; Ogata, M.; Usui, T. Non-Catalytic Synthesis of Chromogen I and III from N-Acetyl-D-Glucosamine in High-Temperature Water. Green Chem. 2013, 15, 2960–2966. [Google Scholar] [CrossRef]
- Techikawara, K.; Kobayashi, H.; Fukuoka, A. Conversion of N-Acetylglucosamine to Protected Amino Acid over Ru/C Catalyst. ACS Sustain. Chem. Eng. 2018, 6, 12411–12418. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Zhang, J.; Pierson, Y.; Chen, X.; Yan, N. Conversion of Chitin Derived N-Acetyl-D-Glucosamine (NAG) into Polyols over Transition Metal Catalysts and Hydrogen in Water. Green Chem. 2015, 17, 1024–1031. [Google Scholar] [CrossRef]
- Nikahd, M.; Mikusek, J.; Yu, L.-J.; Coote, M.L.; Banwell, M.G.; Ma, C.; Gardiner, M.G. Exploiting Chitin as a Source of Biologically Fixed Nitrogen: Formation and Full Characterization of Small-Molecule Hetero- and Carbocyclic Pyrolysis Products. J. Org. Chem. 2020, 85, 4583–4593. [Google Scholar] [CrossRef] [PubMed]
- Banwell, M.G.; Pollard, B.; Liu, X.; Connal, L.A. Exploiting Nature’s Most Abundant Polymers: Developing New Pathways for the Conversion of Cellulose, Hemicellulose, Lignin and Chitin into Platform Molecules (and Beyond). Chem. Asian J. 2021, 16, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.F.A.; Gonçalves, B.M.F.; Andrade, K.H.S.; Sousa, B.B.; Maulide, N.; Bernardes, G.J.L.; Afonso, C.A.M. Unlocking the Potential of Bio-Based Nitrogen-Rich Furanic Platforms as Biomass Synthons. Angew. Chem. Int. Ed. 2023, 62, e202304449. [Google Scholar] [CrossRef]
- Padovan, D.; Kobayashi, H.; Fukuoka, A. Facile Preparation of 3-Acetamido-5-Acetylfuran from N-Acetyl-d-Glucosamine by Using Commercially Available Aluminum Salts. ChemSusChem 2020, 13, 3594–3598. [Google Scholar] [CrossRef] [PubMed]
- van der Loo, C.H.M.; Borst, M.L.G.; Pouwer, K.; Minnaard, A.J. The Dehydration of N -Acetylglucosamine (GlcNAc) to Enantiopure Dihydroxyethyl Acetamidofuran (Di-HAF). Org. Biomol. Chem. 2021, 19, 10105–10111. [Google Scholar] [CrossRef]
- Sadiq, A.D.; Chen, X.; Yan, N.; Sperry, J. Towards the Shell Biorefinery: Sustainable Synthesis of the Anticancer Alkaloid Proximicin A from Chitin. ChemSusChem 2018, 11, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.T.; Lindsay, A.C.; Chen, X.; Gözaydin, G.; Yan, N.; Sperry, J. Transferring the Biorenewable Nitrogen Present in Chitin to Several N-Functional Groups. Sustain. Chem. Pharm. 2019, 13, 100143. [Google Scholar] [CrossRef]
- Pham, T.T.; Lindsay, A.C.; Kim, S.-W.; Persello, L.; Chen, X.; Yan, N.; Sperry, J. Two-Step Preparation of Diverse 3-Amidofurans from Chitin. ChemistrySelect 2019, 4, 10097–10099. [Google Scholar] [CrossRef]
- Pham, T.T.; Chen, X.; Yan, N.; Sperry, J. A Novel Dihydrodifuropyridine Scaffold Derived from Ketones and the Chitin-Derived Heterocycle 3-Acetamido-5-Acetylfuran. Monatsh. Chem. 2018, 149, 857–861. [Google Scholar] [CrossRef]
- Pham, T.T.; Gözaydın, G.; Söhnel, T.; Yan, N.; Sperry, J. Oxidative Ring-Expansion of a Chitin-Derived Platform Enables Access to Unexplored 2-Amino Sugar Chemical Space. Eur. J. Org. Chem. 2019, 2019, 1355–1360. [Google Scholar] [CrossRef]
- Pham, T.T.; Chen, X.; Söhnel, T.; Yan, N.; Sperry, J. Haber-Independent, Diversity-Oriented Synthesis of Nitrogen Compounds from Biorenewable Chitin. Green Chem. 2020, 22, 1978–1984. [Google Scholar] [CrossRef]
- Neville, J.C.; Lau, M.Y.; Söhnel, T.; Sperry, J. Haber-Independent, Asymmetric Synthesis of the Marine Alkaloid Epi-Leptosphaerin from a Chitin-Derived Chiral Pool Synthon. Org. Biomol. Chem. 2022, 20, 6562–6565. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.G.; Ravasco, J.M.J.M.; Vale, J.R.; Queda, F.; Gomes, R.F.A. A Direct Diels–Alder Reaction of Chitin Derived 3-Acetamido-5-Acetylfuran. Green Chem. 2022, 24, 7131–7136. [Google Scholar] [CrossRef]
- Santos, C.S.; Rodini Mattioli, R.; Soares Baptista, J.; Menezes da Silva, V.H.; Browne, D.L.; Pastre, J.C. Nitrogenated Aromatics from Chitin. Green Chem. 2023, 25, 5059–5067. [Google Scholar] [CrossRef]
- van der Loo, C.H.M.; Schim van der Loeff, R.; Martín, A.; Gomez-Sal, P.; Borst, M.L.G.; Pouwer, K.; Minnaard, A.J. π-Facial Selectivity in the Diels–Alder Reaction of Glucosamine-Based Chiral Furans and Maleimides. Org. Biomol. Chem. 2023, 21, 1888–1894. [Google Scholar] [CrossRef]
- Ikegami, S.; Isomura, H.; Tsuchimori, N.; Hamada, K.; KOBAYASHI, H.; Kojima, Y.; Osano, Y.T.; Kumazawa, S.; Matsuzaki, T. Crystal Structure of an Inhibitor of Starfish Embryonic Development, 4-Oxo-7-(β-D-Ribofuranosyl)-3H-Furo[3, 2-d]Pyrimidine: Revision of Pyrrolosine Structure. Anal. Sci. 1992, 8, 897–898. [Google Scholar] [CrossRef]
- Bhattacharya, B.K.; Lim, M.-I.; Otter, B.A.; Klein, R.S. Synthesis of Furo[3,2-d]Pyrimidine Nucleosides: A Novel c-Nucleoside Isostere of Adenosine. Tetrahedron Lett. 1986, 27, 815–818. [Google Scholar] [CrossRef]
- Bhattacharya, B.K.; Otter, B.A.; Berens, R.L.; Klein, R.S. Studies on the Synthesis of Furo[3,2-d]Pyrimidine C-Nucleosides: New Inosine Analogues with Antiprotozoan Activity. Nucleosides Nucleotides 1990, 9, 1021–1043. [Google Scholar] [CrossRef]
- Hoemann, M.; Wilson, N.; Argiriadi, M.; Banach, D.; Burchat, A.; Calderwood, D.; Clapham, B.; Cox, P.; Duignan, D.B.; Konopacki, D.; et al. Synthesis and Optimization of Furano[3,2-d]Pyrimidines as Selective Spleen Tyrosine Kinase (Syk) Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5562–5567. [Google Scholar] [CrossRef]
- Koshizawa, T.; Morimoto, T.; Watanabe, G.; Watanabe, T.; Yamasaki, N.; Sawada, Y.; Fukuda, T.; Okuda, A.; Shibuya, K.; Ohgiya, T. Optimization of a Novel Series of Potent and Orally Bioavailable GPR119 Agonists. Bioorg. Med. Chem. Lett. 2017, 27, 3249–3253. [Google Scholar] [CrossRef]
- Hancox, T.C.; Pegg, N.A.; Nadin, A.J.; Price, S. Pharmaceutical Compounds. Patent WO2008152394, 12 June 2008. [Google Scholar]
- Rhodes, J.; Mighdoll, M.; Choi, I.Y.; Kopec, B. Methods and Treatment of Viral Infection with Substituted Furo-Pyrimidines. Patent WO2022261068, 8 June 2021. [Google Scholar]
- Kim, S.; Hong, J.H. Synthesis of Novel 4′-Trifluoromethyl-5′-Norcarbocyclic C-Nucleoside Phosphonic Acids as Potent Anti-Leukemic Agents. Nucleosides Nucleotides Nucleic Acids 2015, 34, 848–865. [Google Scholar] [CrossRef]
- Butora, G.; Olsen, D.B.; Carroll, S.S.; McMasters, D.R.; Schmitt, C.; Leone, J.F.; Stahlhut, M.; Burlein, C.; MacCoss, M. Synthesis and HCV Inhibitory Properties of 9-Deaza- and 7,9-Dideaza-7-Oxa-2′-C-Methyladenosine. Bioorg. Med. Chem. 2007, 15, 5219–5229. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The Anatomy of a Comprehensive Constrained, Restrained Refinement Program for the Modern Computing Environment—Olex2 Dissected. Acta Crystallogr. Sect. A 2015, 71, 59–75. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Theoclitou, M.-E.; Aquila, B.; Block, M.H.; Brassil, P.J.; Castriotta, L.; Code, E.; Collins, M.P.; Davies, A.M.; Deegan, T.; Ezhuthachan, J.; et al. Discovery of (+)-N-(3-Aminopropyl)-N-[1-(5-Benzyl-3-Methyl-4-Oxo-[1,2]Thiazolo[5,4-d]Pyrimidin-6-Yl)-2-Methylpropyl]-4-Methylbenzamide (AZD4877), a Kinesin Spindle Protein Inhibitor and Potential Anticancer Agent. J. Med. Chem. 2011, 54, 6734–6750. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, C.R.; Helm, M.D.; Rountree, J.S.S.; Flasz, J.T.; Arkoudis, E.; Miel, H.; Hewitt, P.R.; Jordan, L.; Barker, O.; Hughes, C.; et al. Identification and Structure-Guided Development of Pyrimidinone Based USP7 Inhibitors. ACS Med. Chem. Lett. 2018, 9, 238–243. [Google Scholar] [CrossRef]
- Zhang, D.; Li, W.; Huang, X.; Qin, W.; Liu, M. Removal of Ammonium in Surface Water at Low Temperature by a Newly Isolated Microbacterium Sp. Strain SFA13. Bioresour. Technol. 2013, 137, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Odo, K.; Ichikawa, E.; Shirai, K.; Sugino, K. Notes—A New Method for the Preparation of Formamidine. J. Org. Chem. 1957, 22, 1715. [Google Scholar] [CrossRef]
- Kamo, T.; Hiradate, S.; Fujii, Y. First Isolation of Natural Cyanamide as a Possible Allelochemical from Hairy Vetch Vicia Villosa. J. Chem. Ecol. 2003, 29, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kamo, T.; Sakurai, S.; Yamanashi, T.; Todoroki, Y. Cyanamide Is Biosynthesized from L-Canavanine in Plants. Sci. Rep. 2015, 5, 10527. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.J. A New Synthesis of Formamidine. J. Appl. Chem. 1952, 2, 202–203. [Google Scholar] [CrossRef]
- Pfrengle, F.; Reissig, H.-U. Amino Sugars and Their Mimetics via 1,2-Oxazines. Chem. Soc. Rev. 2010, 39, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Emmadi, M.; Kulkarni, S.S. Recent Advances in Synthesis of Bacterial Rare Sugar Building Blocks and Their Applications. Nat. Prod. Rep. 2014, 31, 870–879. [Google Scholar] [CrossRef]
- Skarbek, K.; Milewska, M.J. Biosynthetic and Synthetic Access to Amino Sugars. Carbohydr. Res. 2016, 434, 44–71. [Google Scholar] [CrossRef]
- Yang, J.; Xie, D.; Ma, X. Recent Advances in Chemical Synthesis of Amino Sugars. Molecules 2023, 28, 4724. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossa, T.A.; Neville, J.C.; Jun, S.P.; Söhnel, T.; Sperry, J. Expanding Heteroaromatic and 2-Aminosugar Chemical Space Accessible from the Biopolymer Chitin. Chemistry 2023, 5, 1998-2008. https://doi.org/10.3390/chemistry5030135
Rossa TA, Neville JC, Jun SP, Söhnel T, Sperry J. Expanding Heteroaromatic and 2-Aminosugar Chemical Space Accessible from the Biopolymer Chitin. Chemistry. 2023; 5(3):1998-2008. https://doi.org/10.3390/chemistry5030135
Chicago/Turabian StyleRossa, Thaís A., Jessica C. Neville, Seongmin Paul Jun, Tilo Söhnel, and Jonathan Sperry. 2023. "Expanding Heteroaromatic and 2-Aminosugar Chemical Space Accessible from the Biopolymer Chitin" Chemistry 5, no. 3: 1998-2008. https://doi.org/10.3390/chemistry5030135
APA StyleRossa, T. A., Neville, J. C., Jun, S. P., Söhnel, T., & Sperry, J. (2023). Expanding Heteroaromatic and 2-Aminosugar Chemical Space Accessible from the Biopolymer Chitin. Chemistry, 5(3), 1998-2008. https://doi.org/10.3390/chemistry5030135