
Recent Developments in Enantioselective Scandium-Catalyzed Transformations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
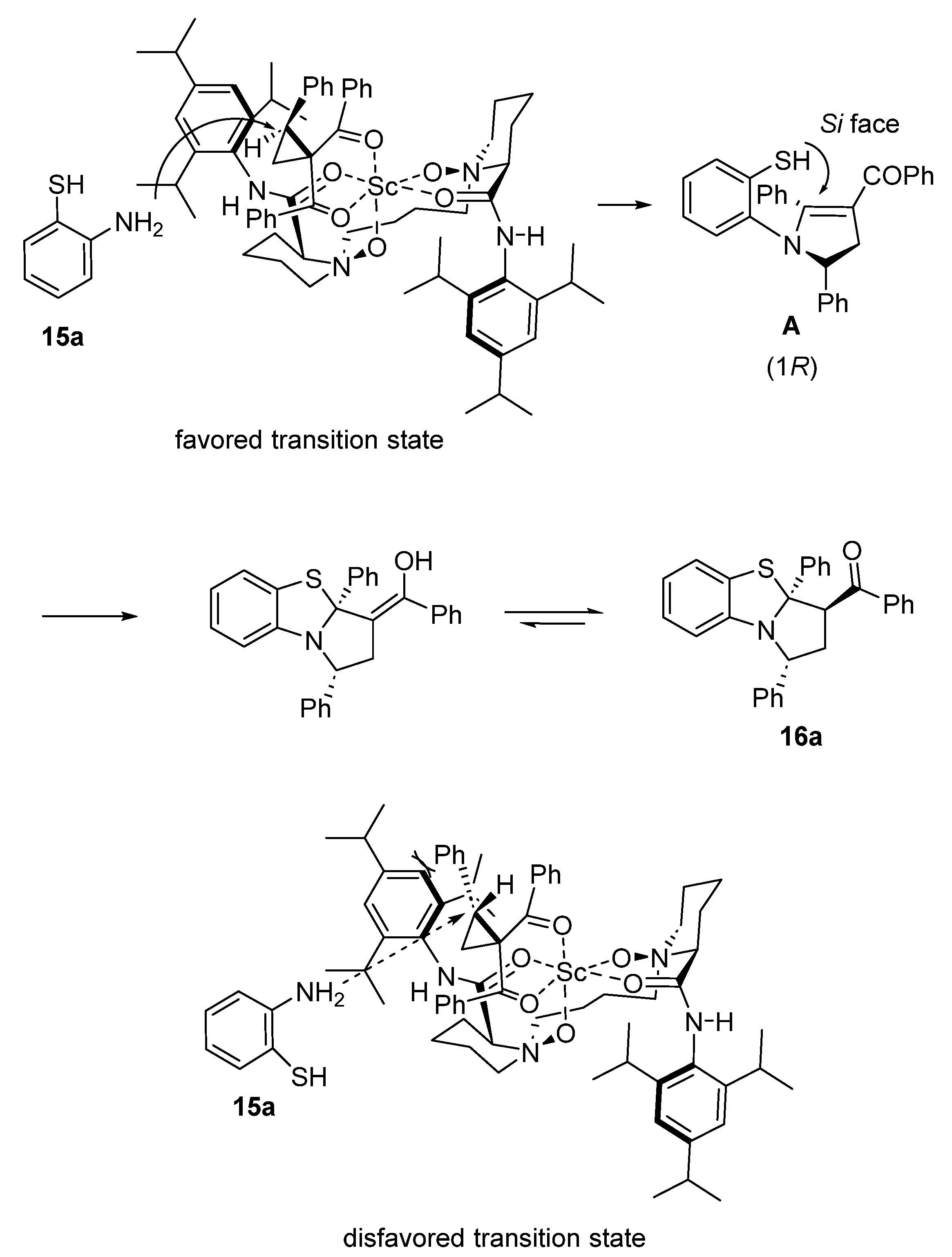{kind=link}
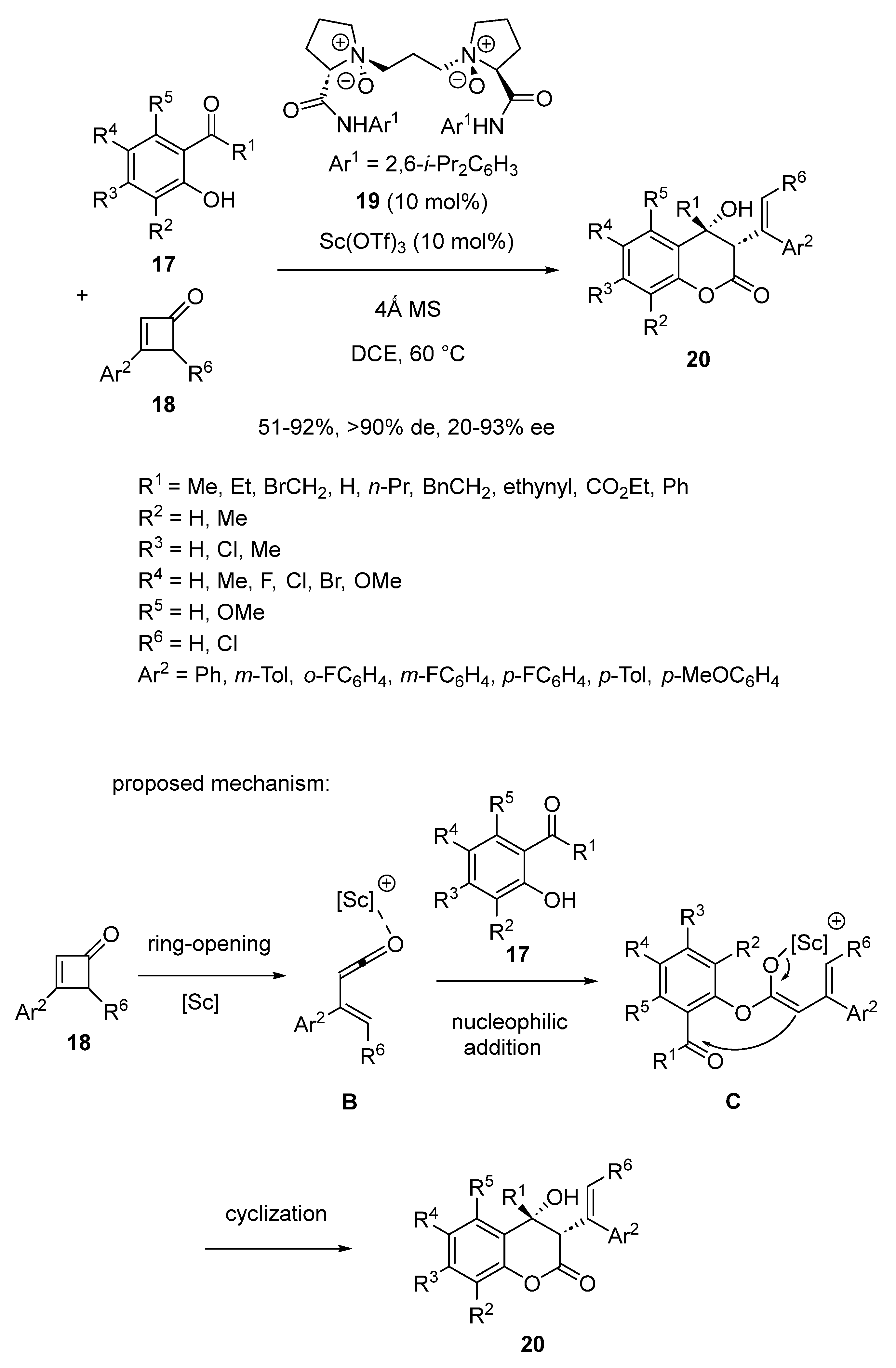{kind=link}
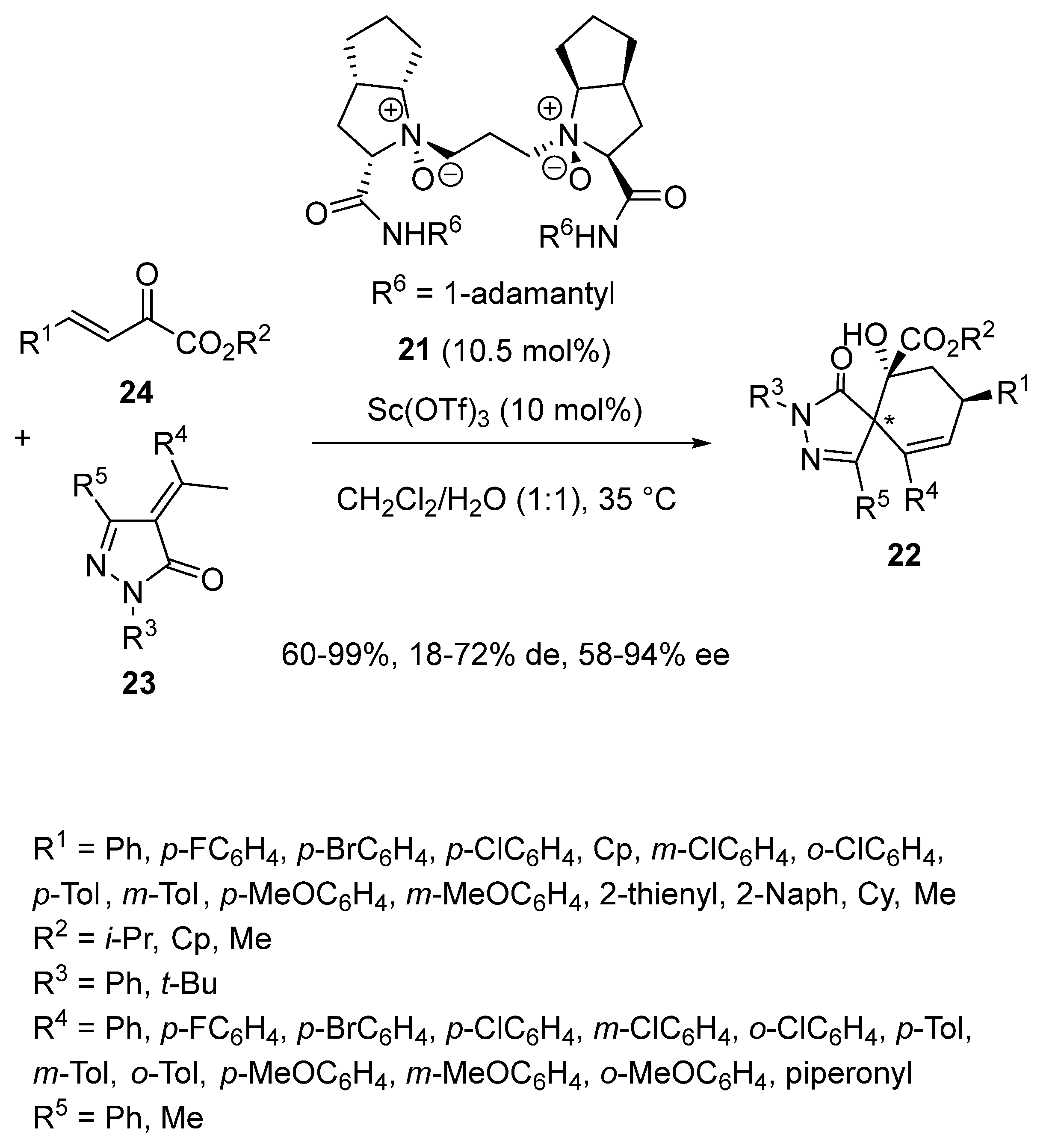{kind=link}
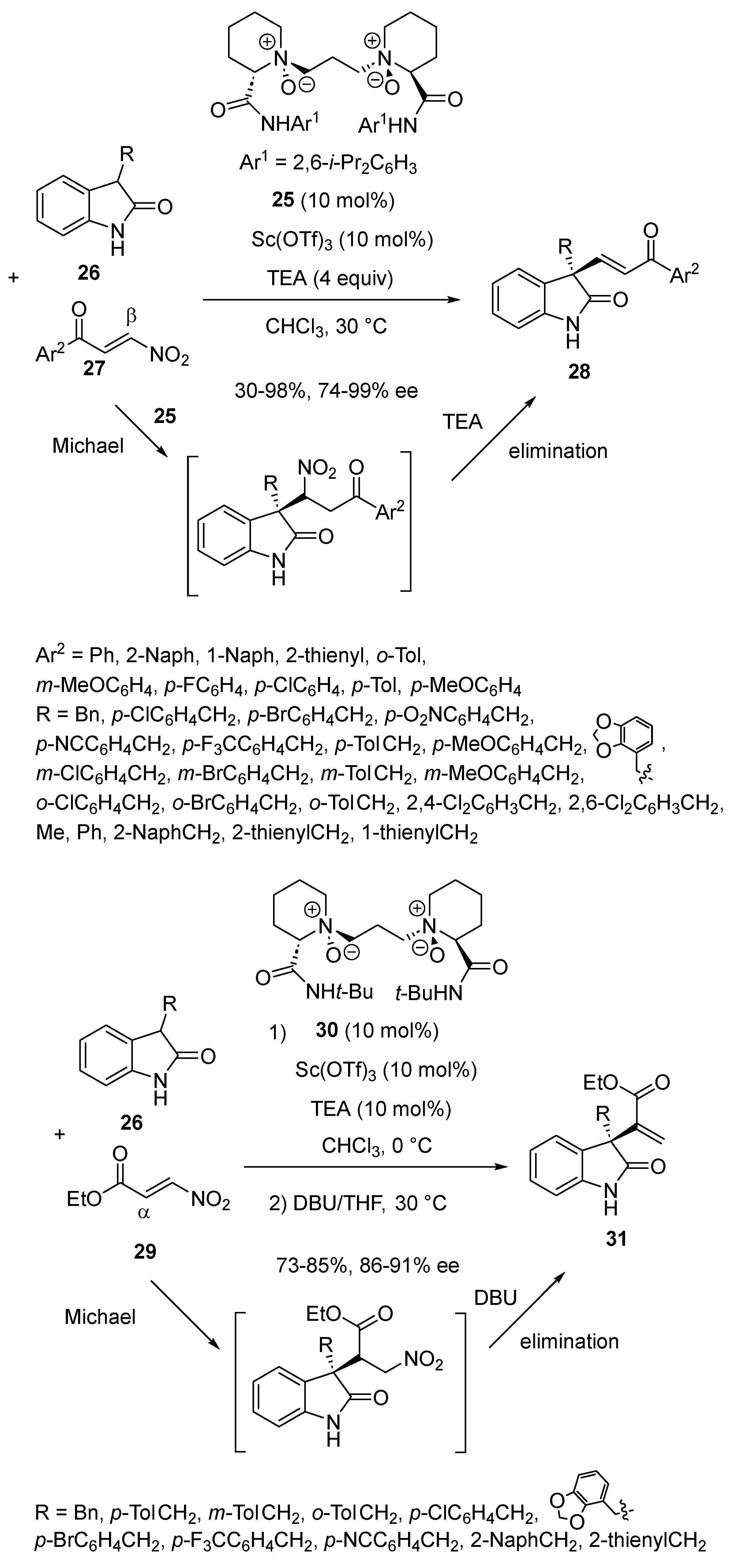{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
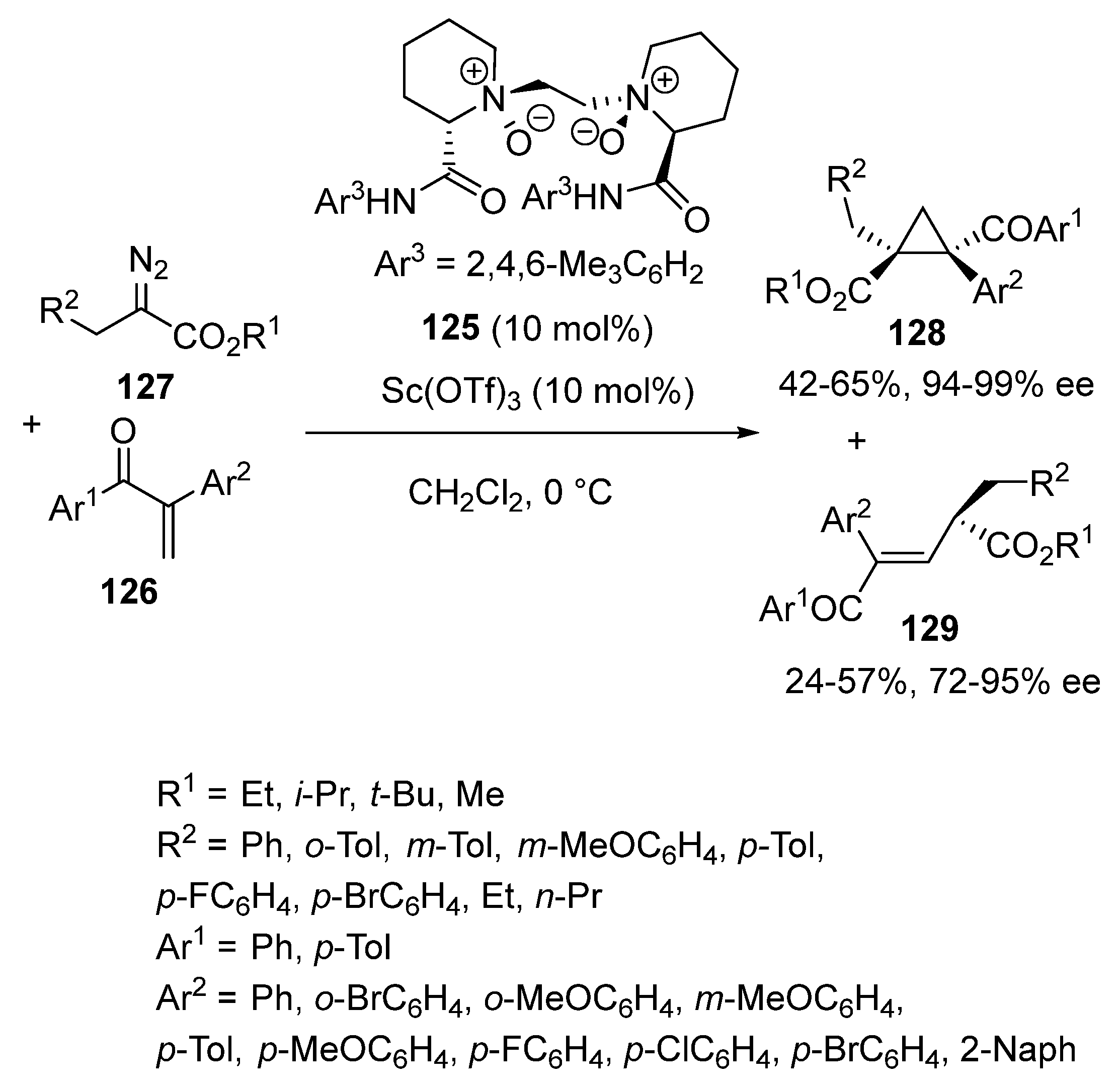{kind=link}
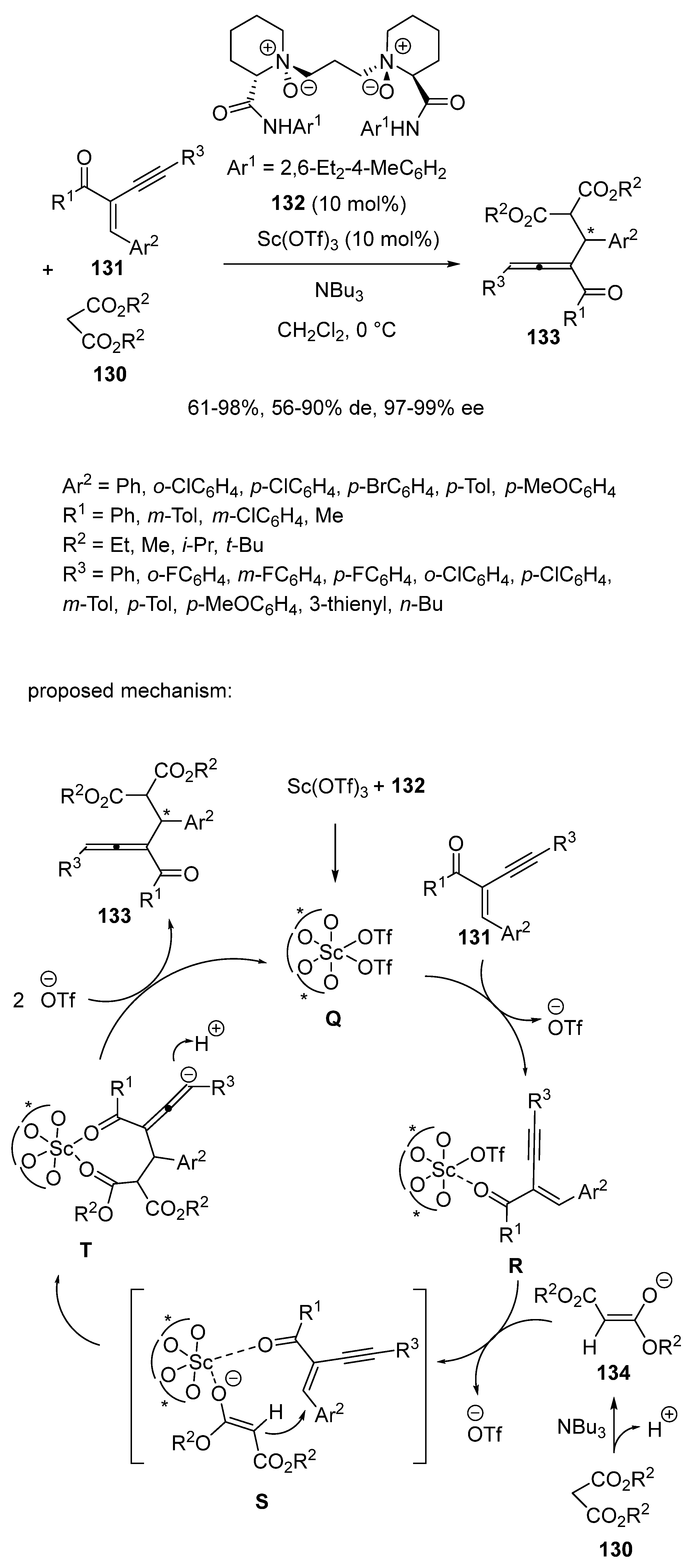{kind=link}
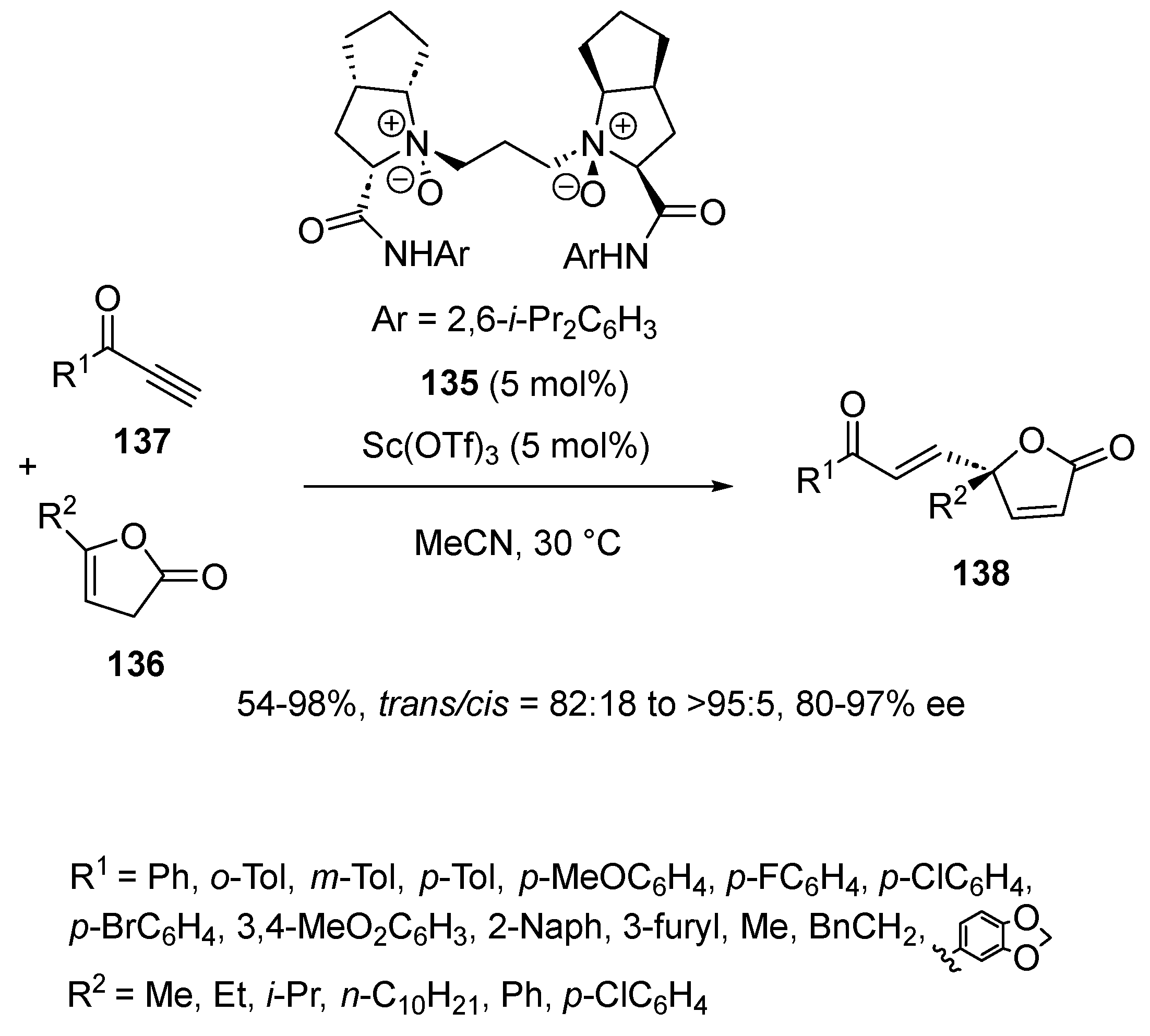{kind=link}
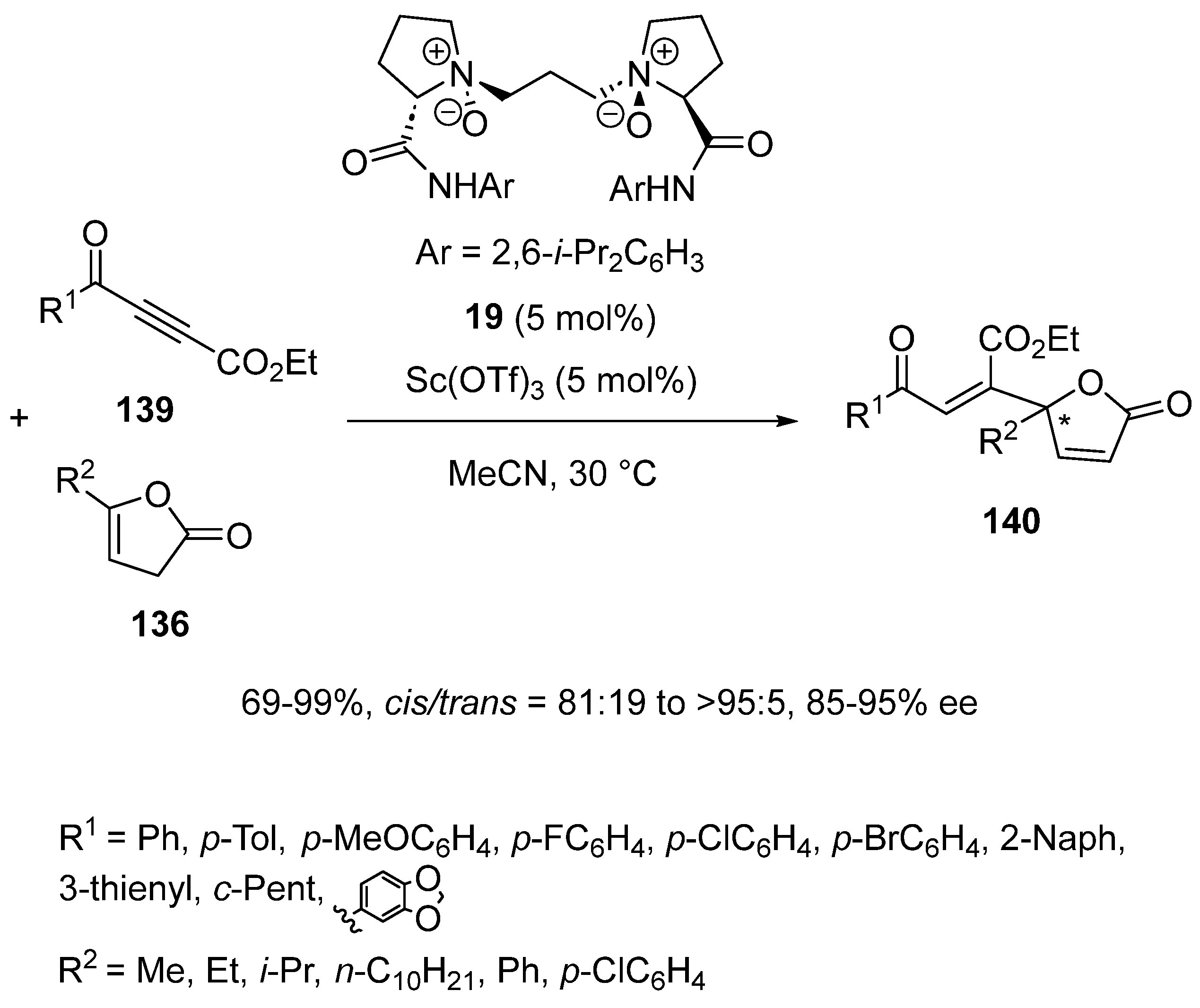{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
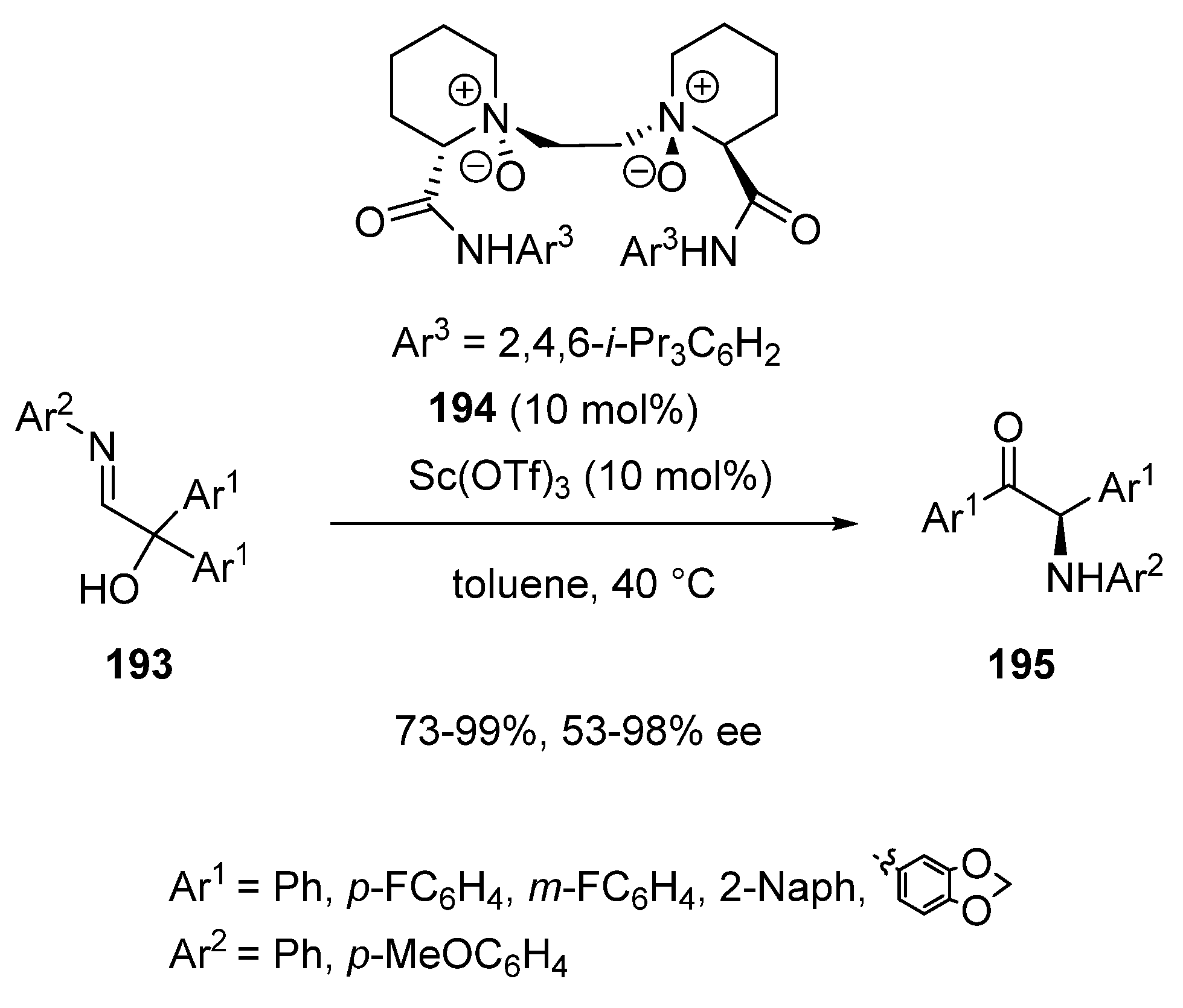{kind=link}
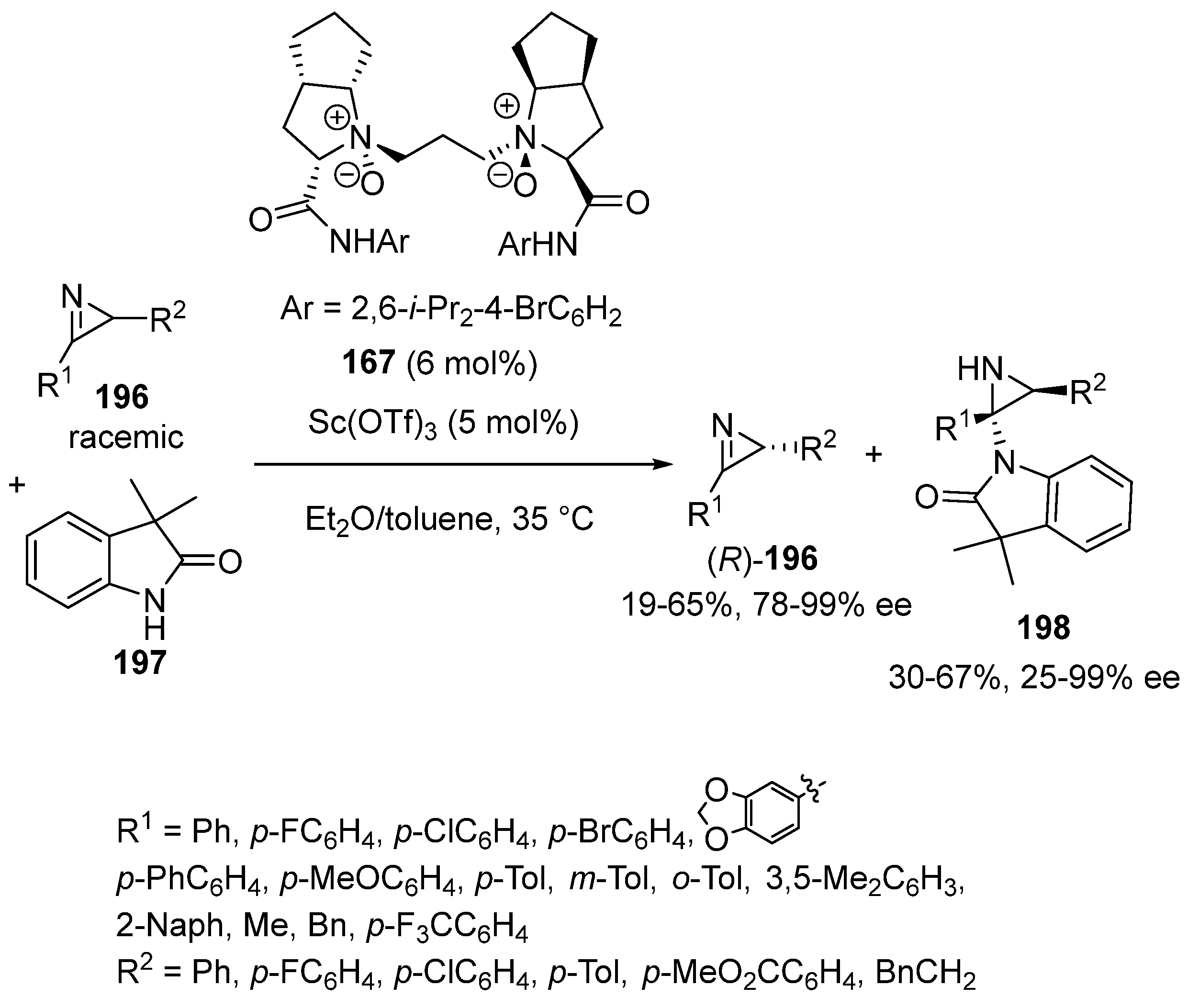{kind=link}
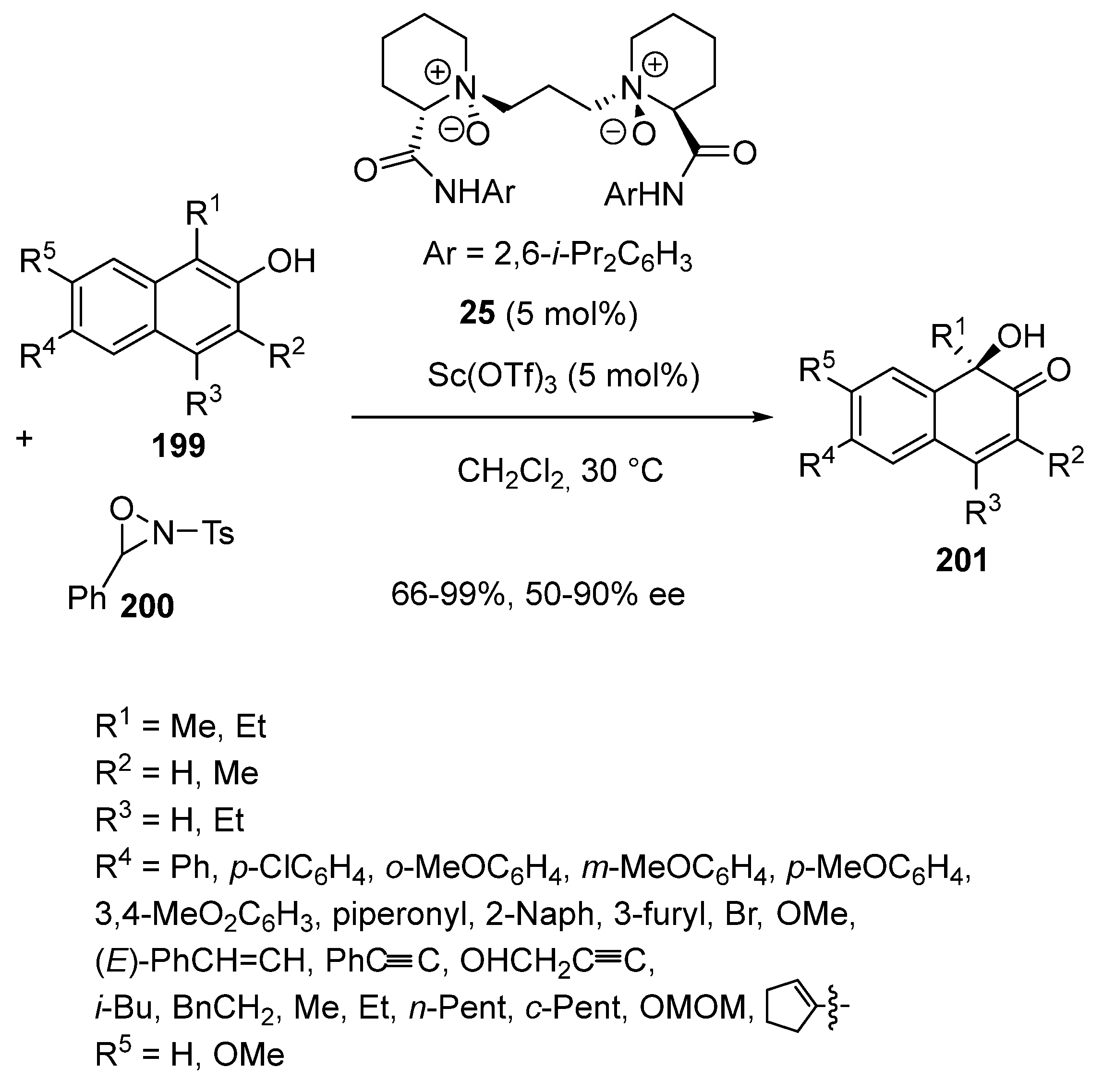{kind=link}
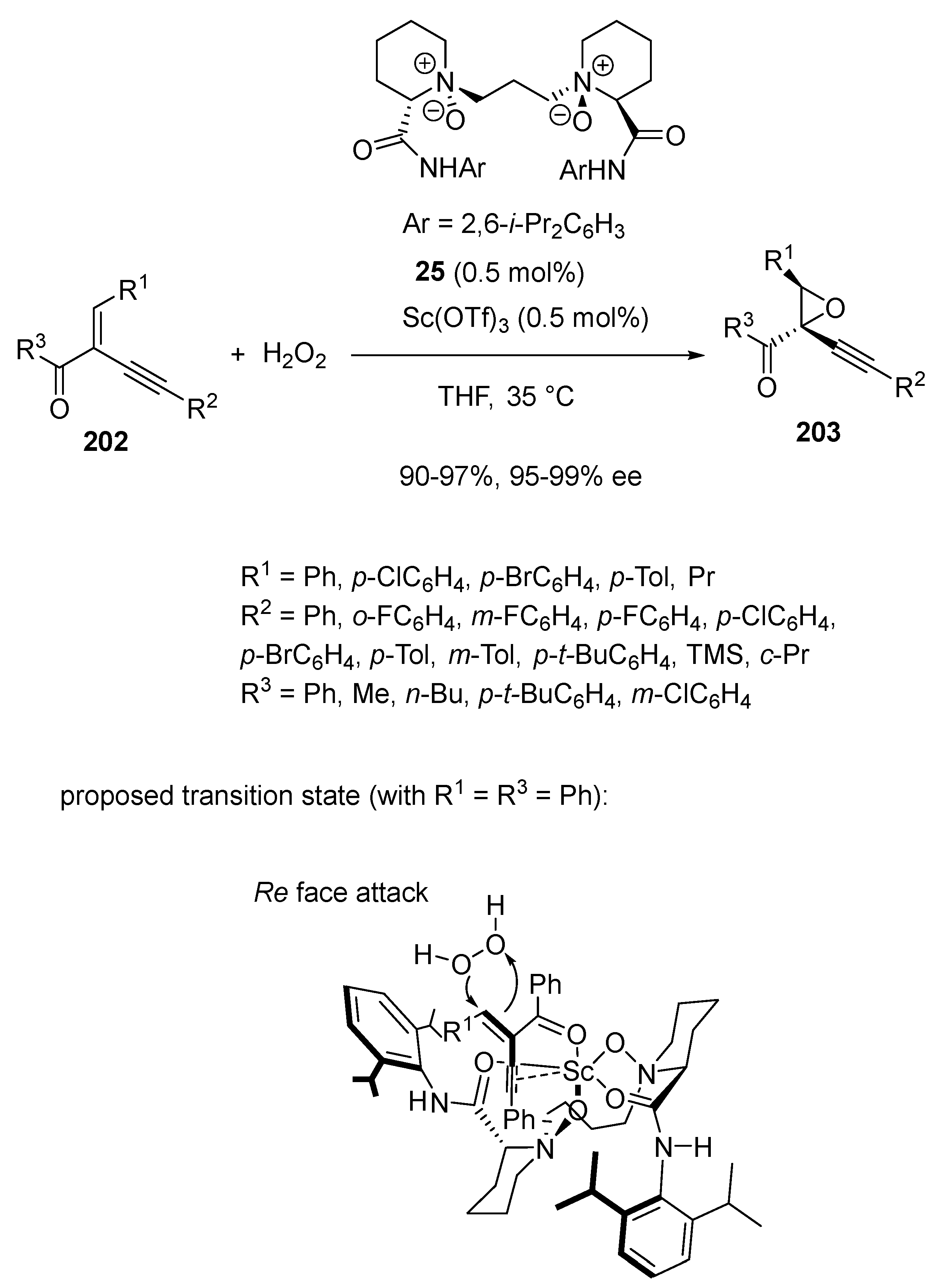{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Enantioselective Scandium-Catalyzed Domino and Tandem Reactions
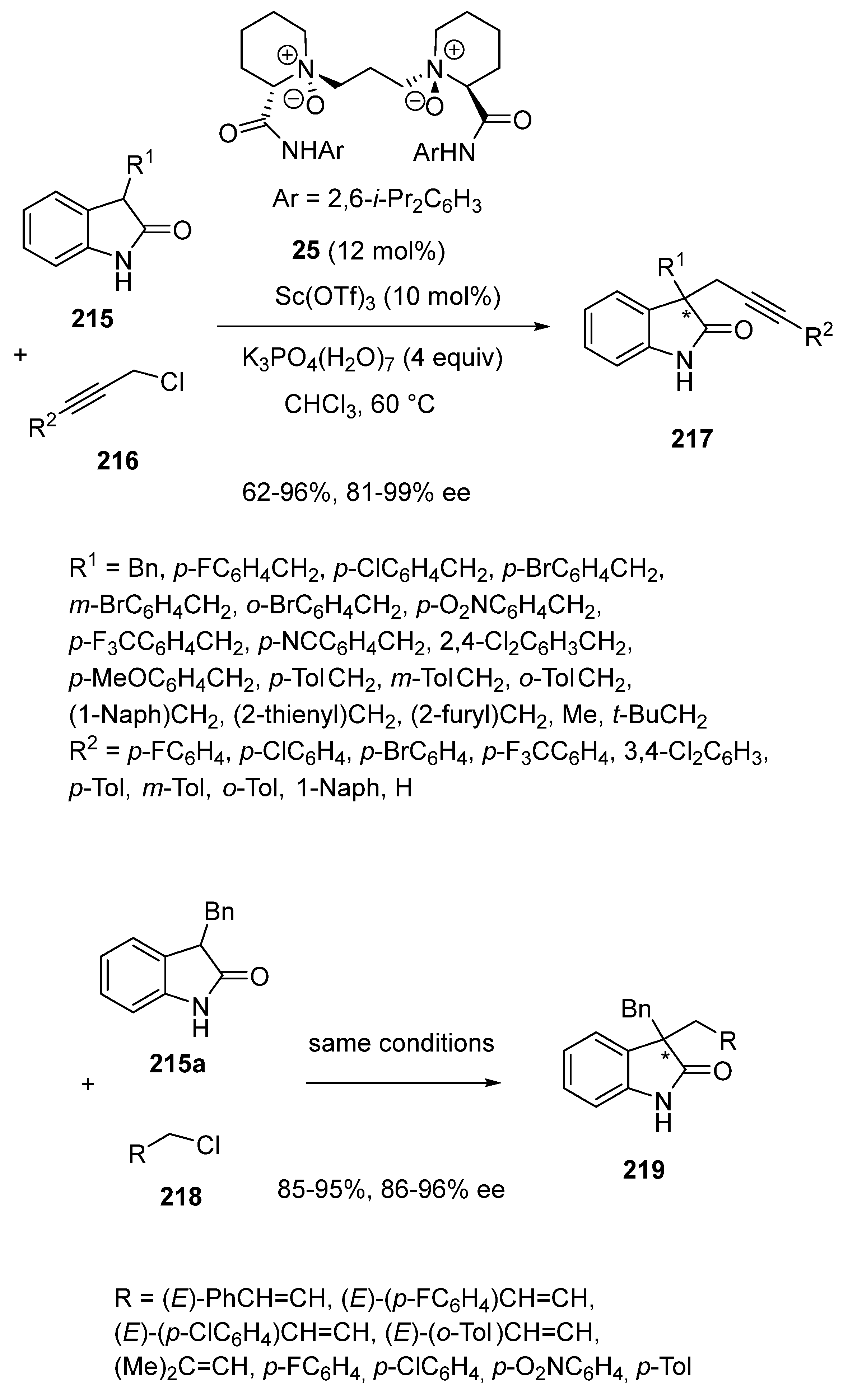
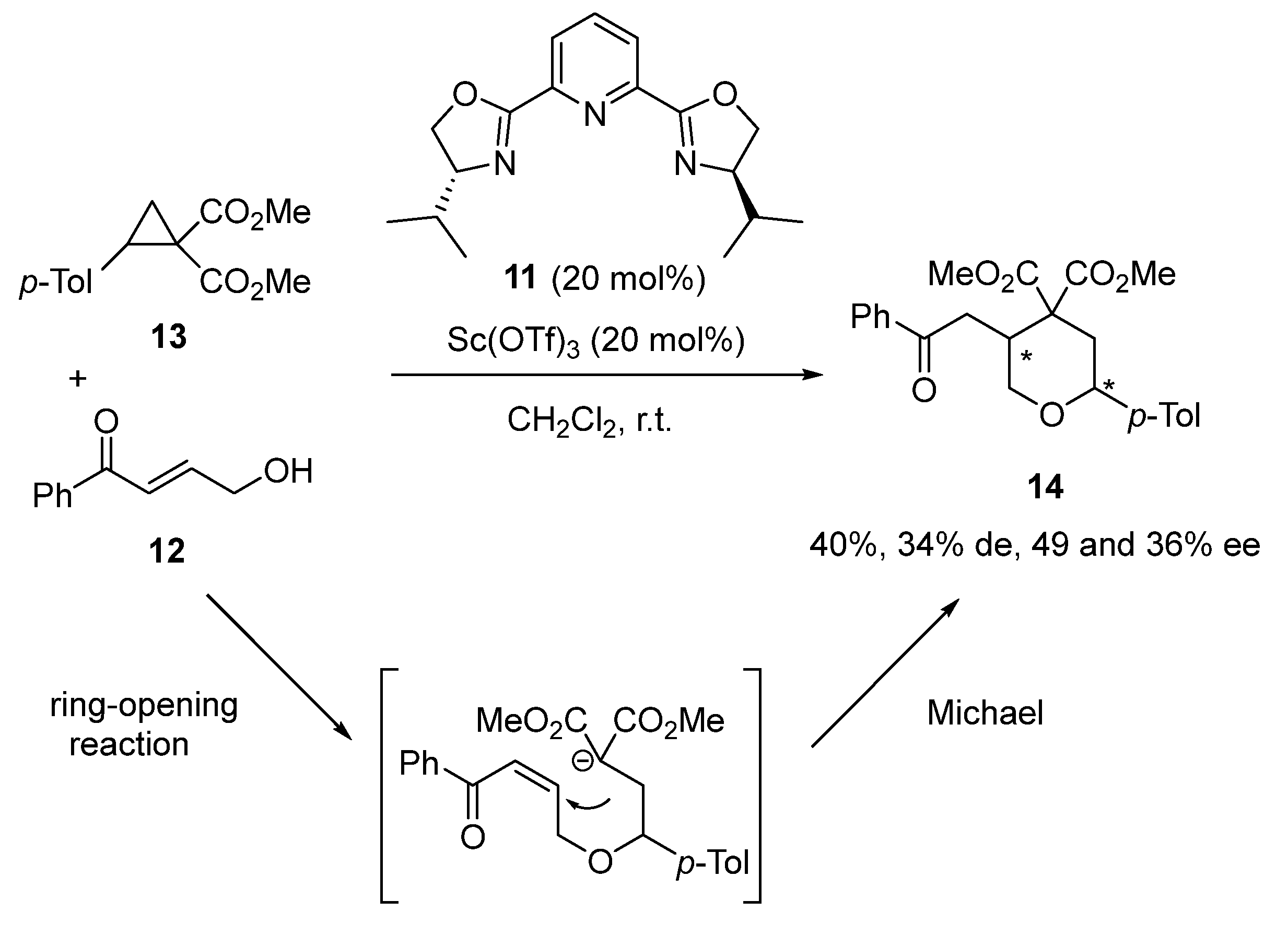
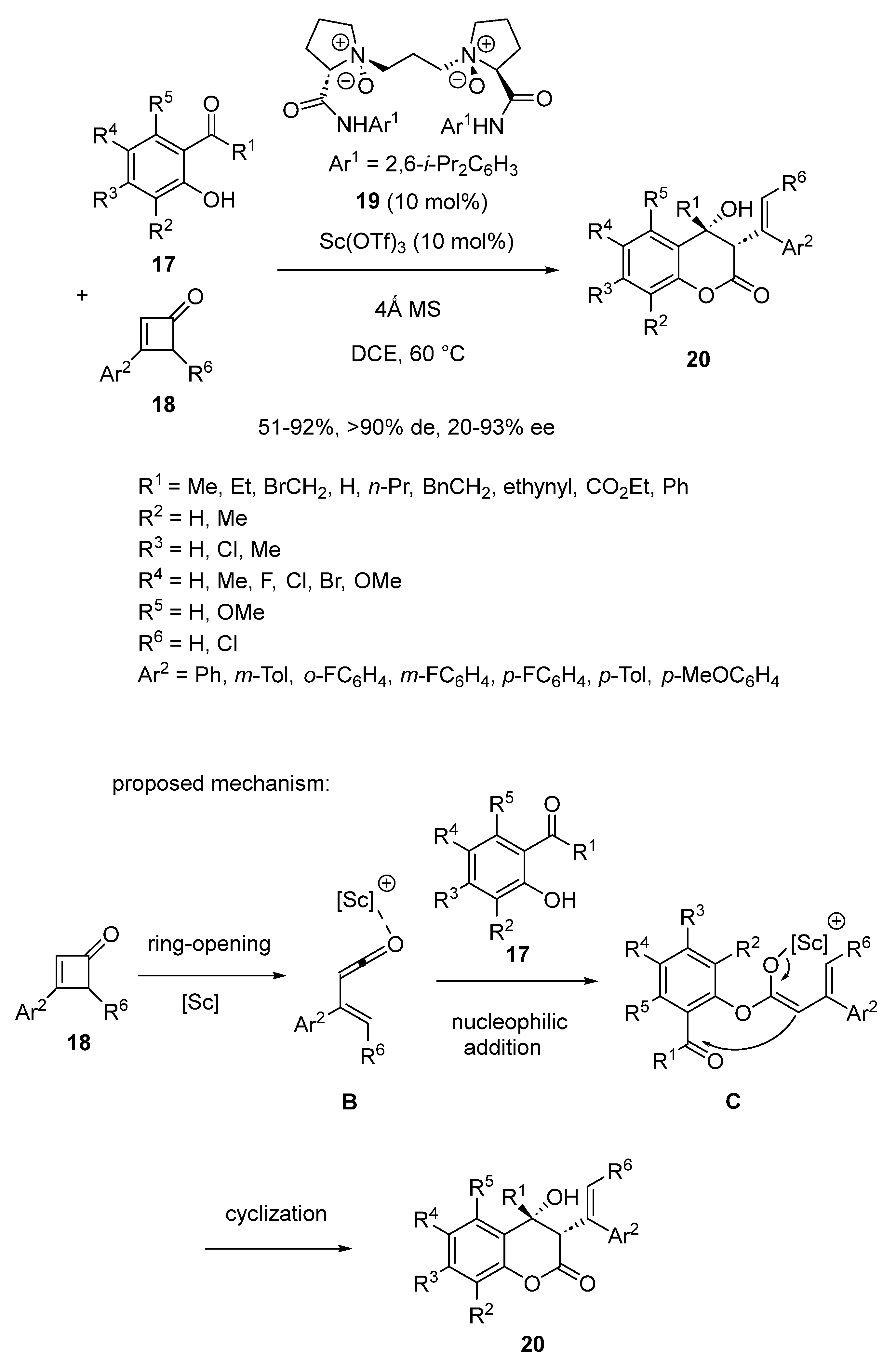
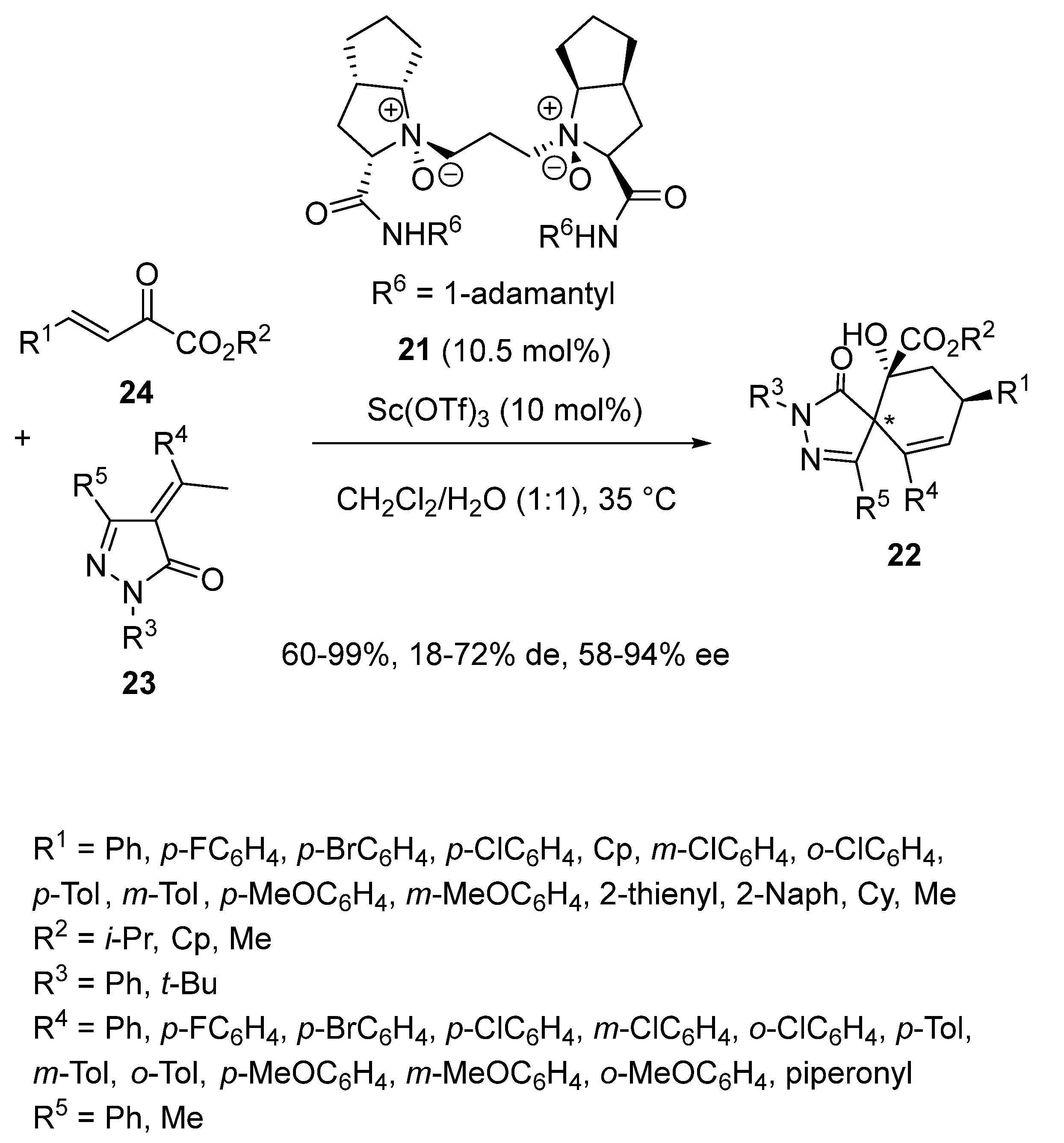
2.1. Ring-Opening-Initiated Domino Reactions
2.2. Michael-Initiated Domino and Tandem Reactions
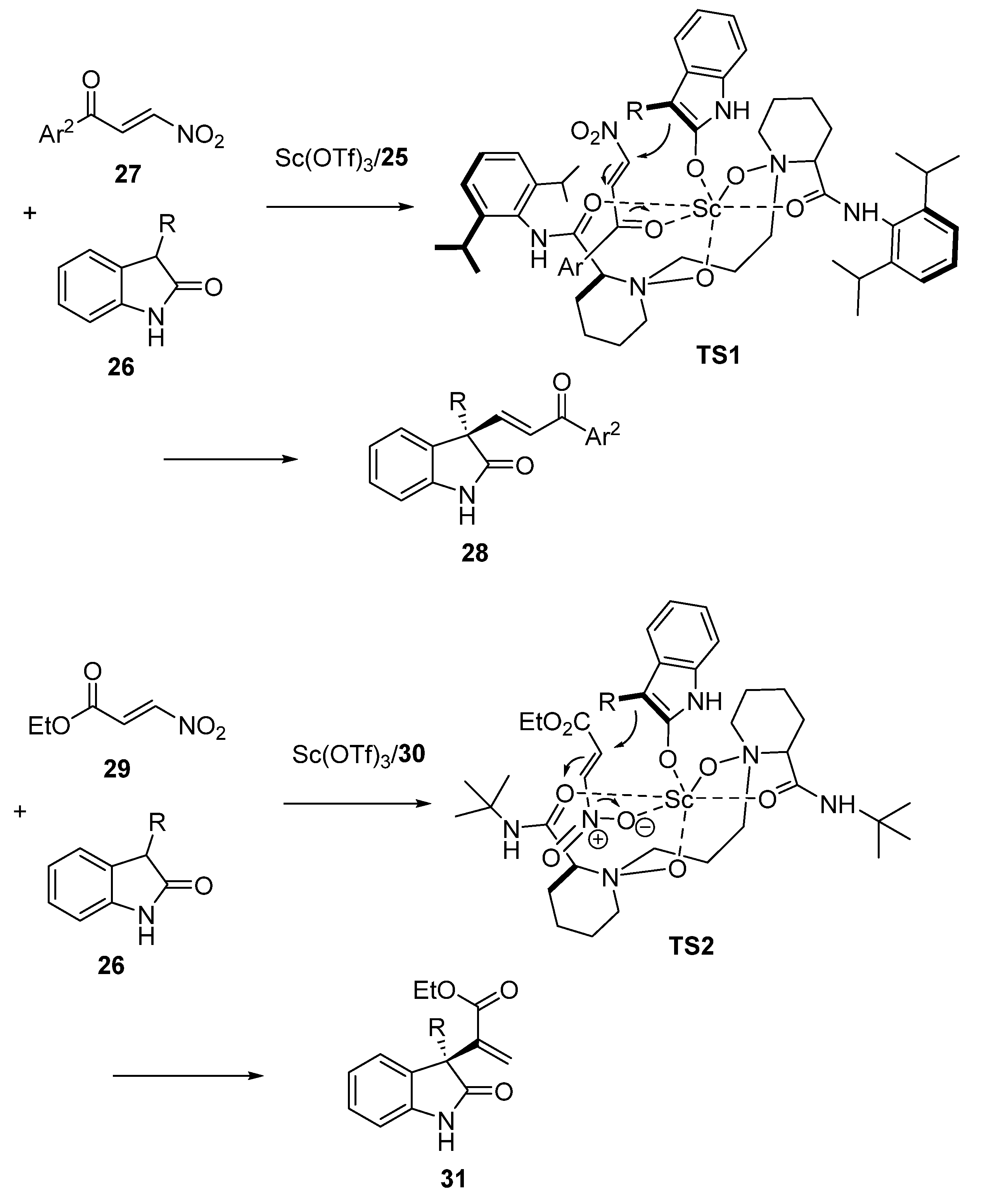
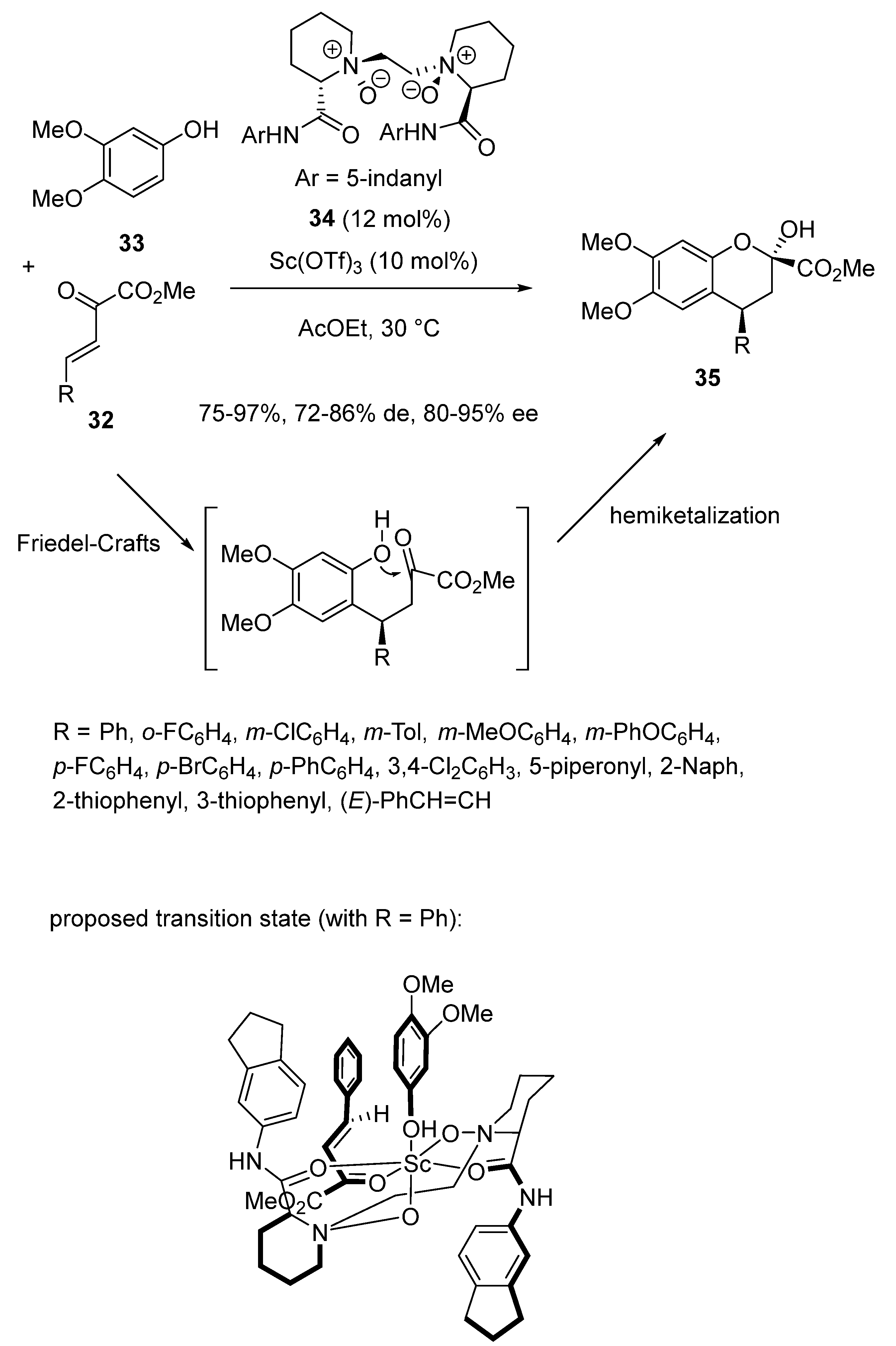
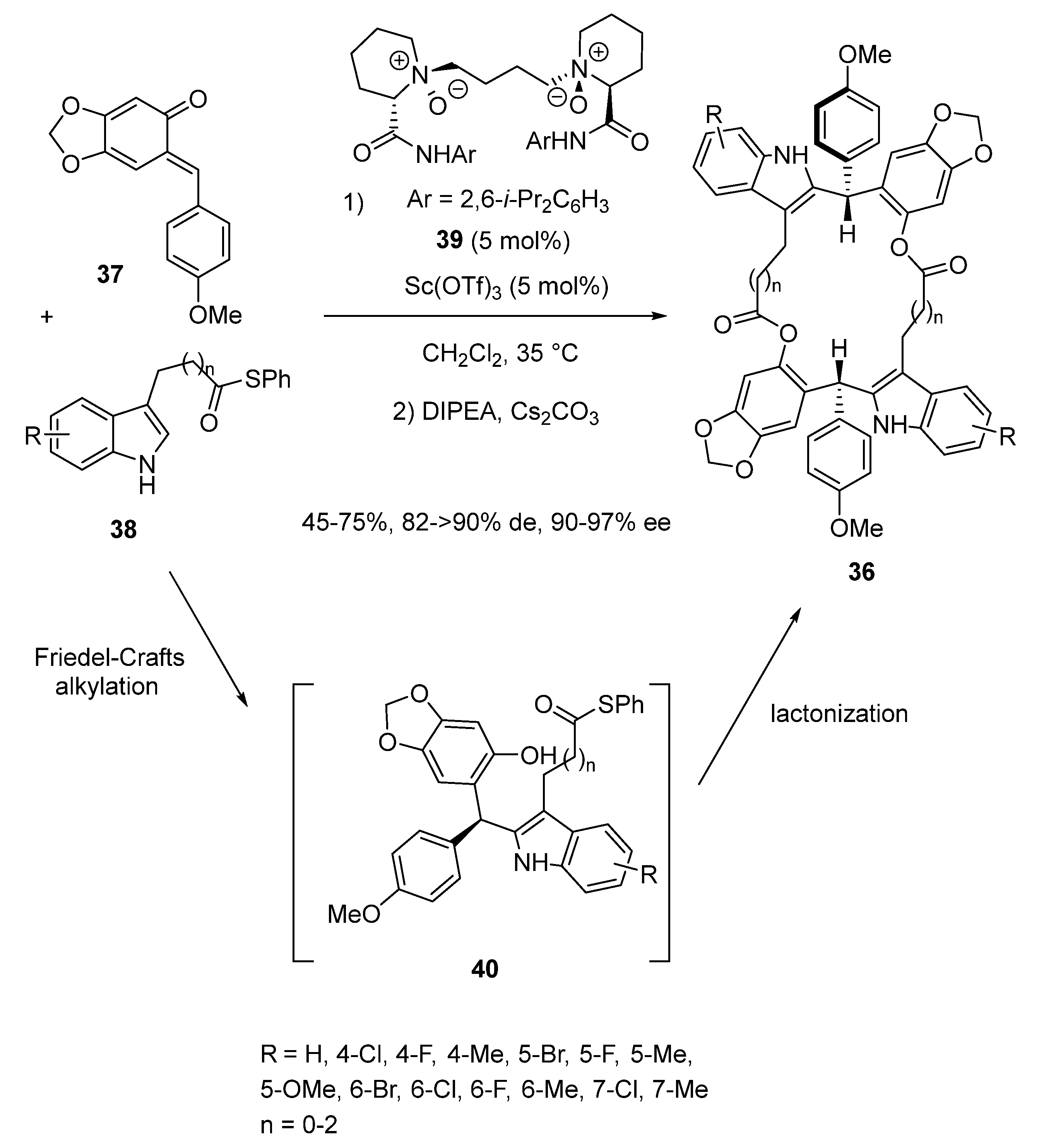
2.3. Friedel-Crafts-Initiated Domino and Tandem Reactions
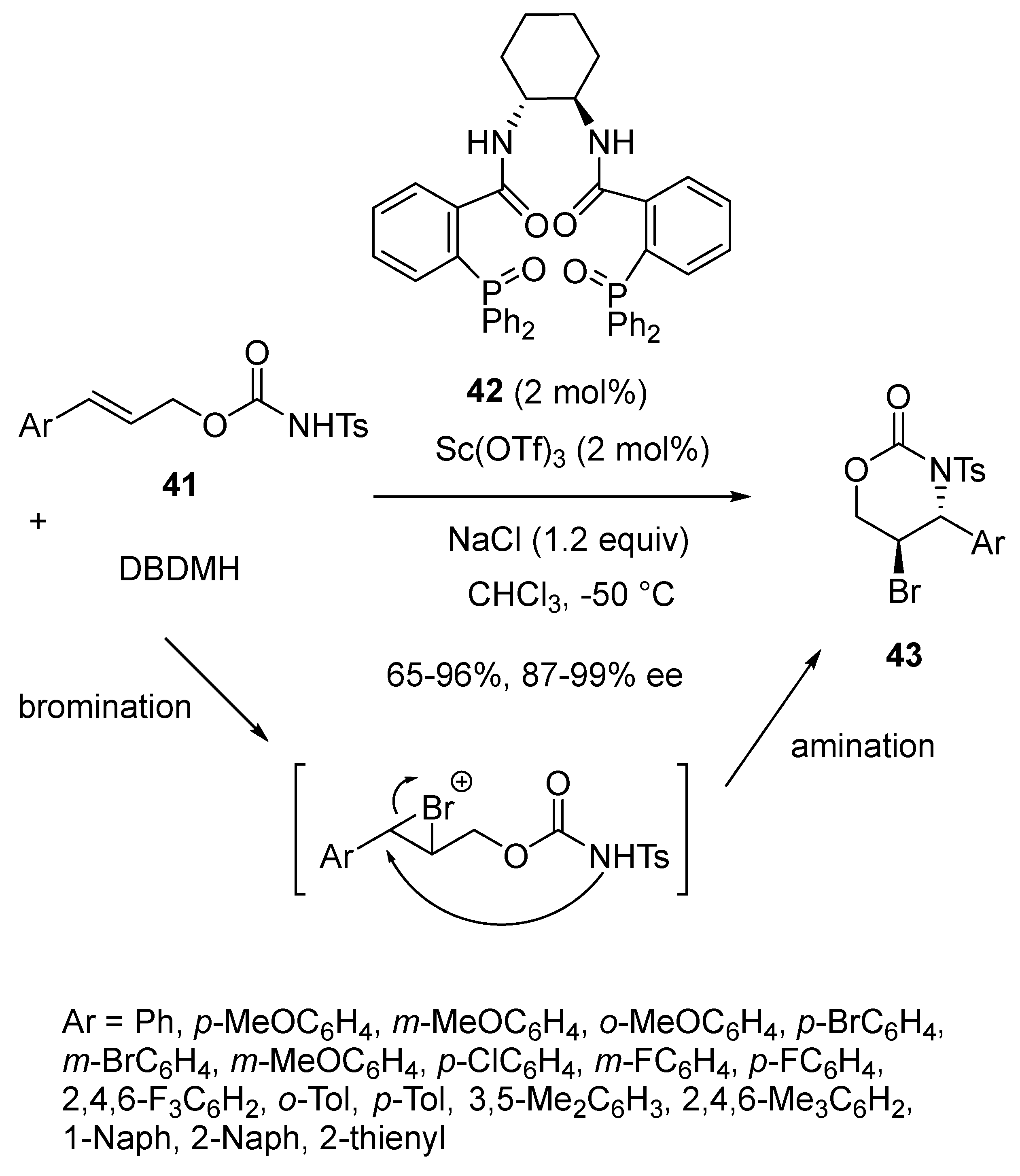
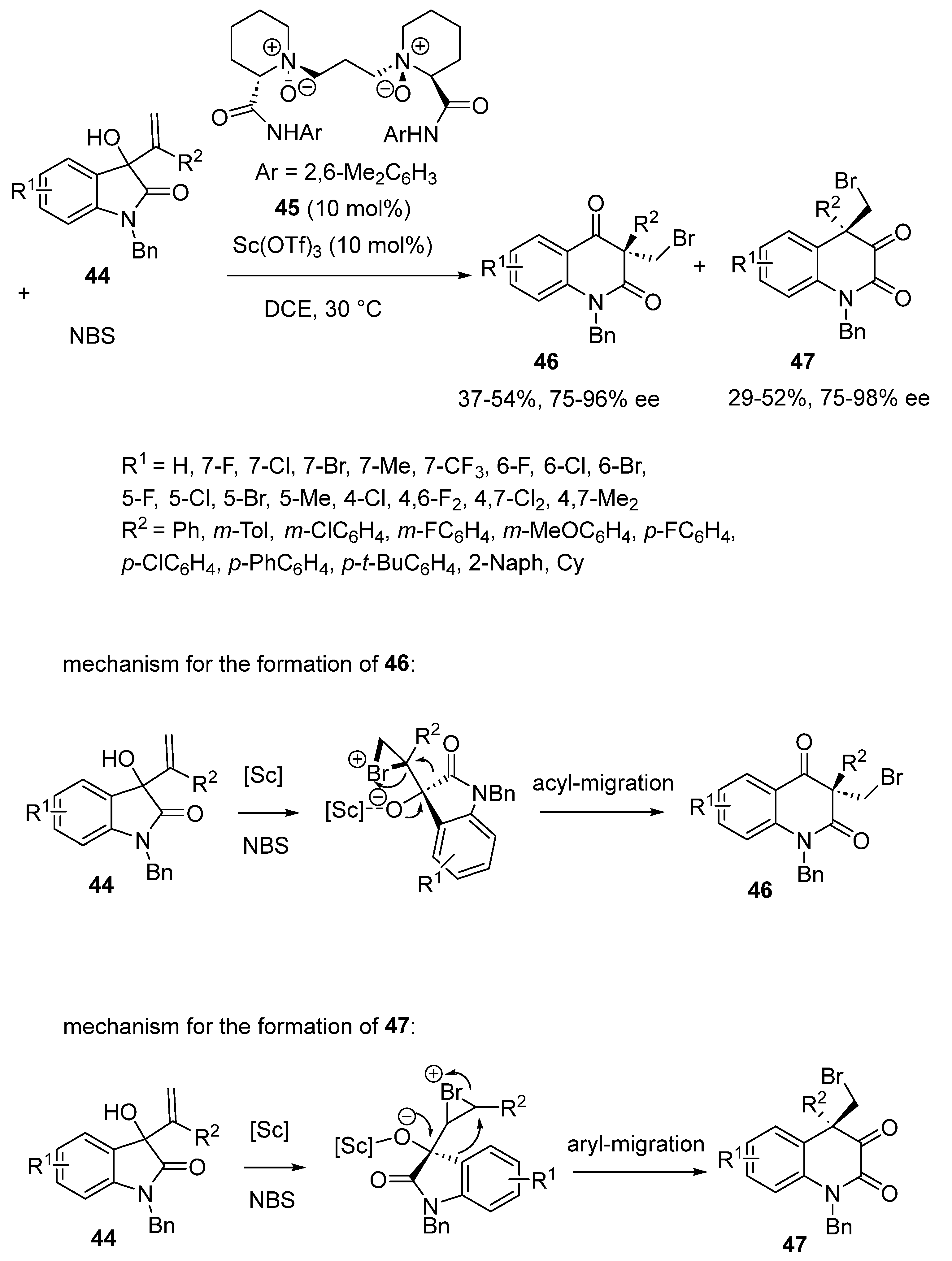
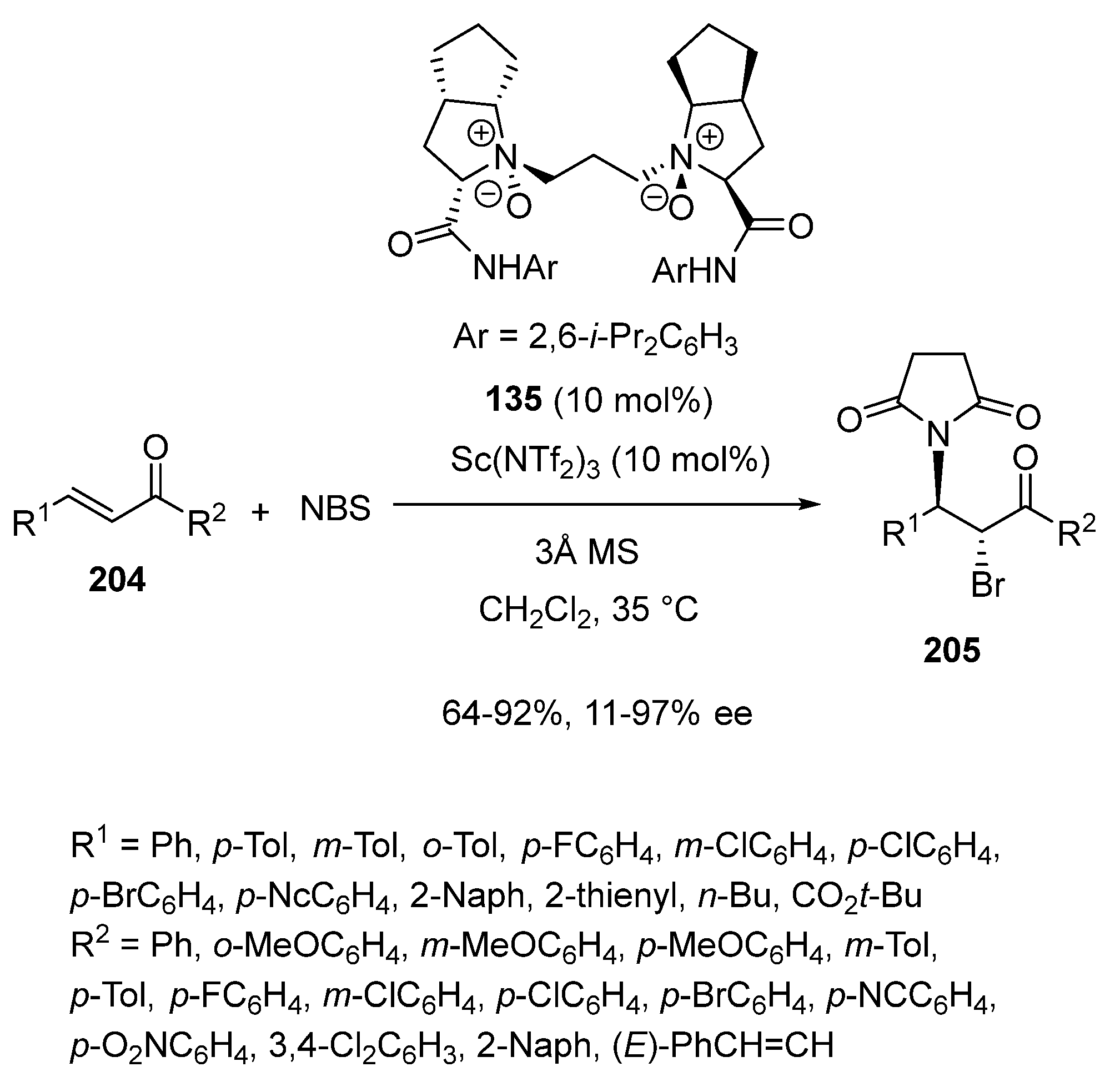
2.4. Bromination-Initiated Domino Reactions
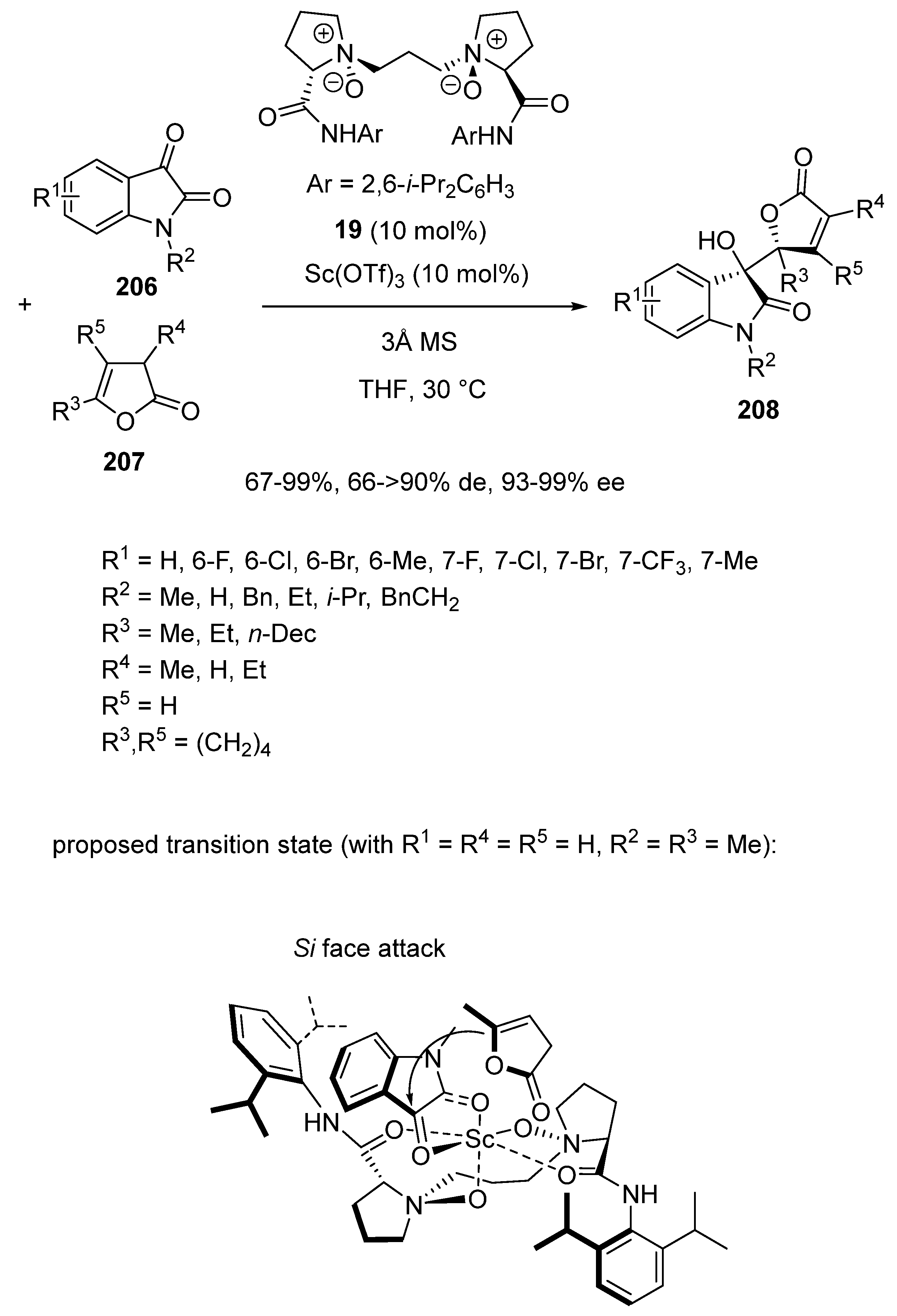
2.5. Three-Component Domino Reactions
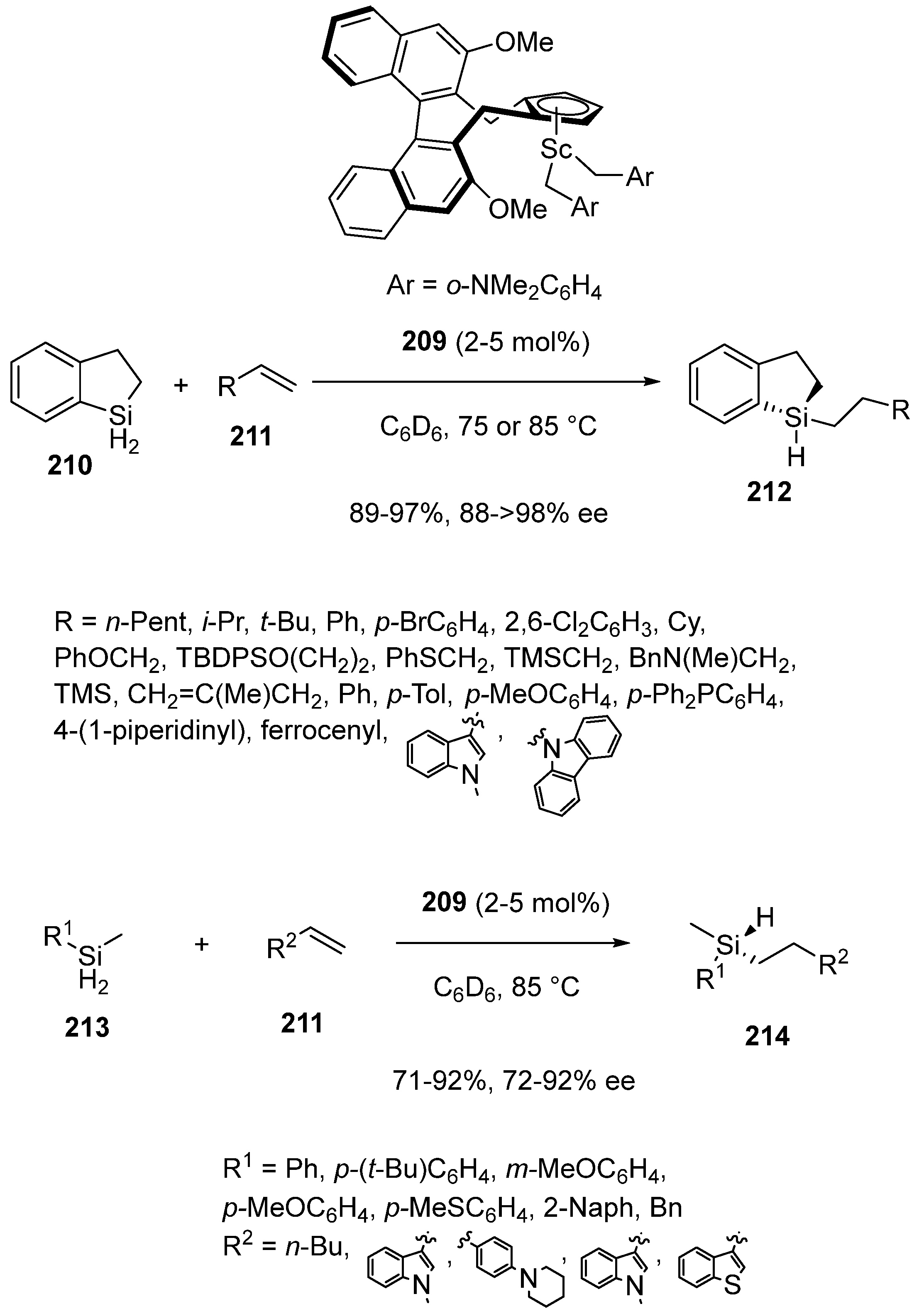
2.6. Miscellaneous Domino and Tandem Reactions
3. Enantioselective Scandium-Catalyzed Cycloadditions
3.1. [3 + 2] Cycloadditions
3.2. (Hetero)-Diels–Alder Reactions
3.3. [2 + 1] Cycloadditions
4. Enantioselective Scandium-Catalyzed Michael Additions
5. Enantioselective Scandium-Catalyzed Ring-Opening Reactions
6. Enantioselective Scandium-Catalyzed Friedel-Crafts Reactions
7. Enantioselective Scandium-Catalyzed Ring-Expansion Reactions
8. Enantioselective Scandium-Catalyzed Rearrangement Reactions
9. Enantioselective Scandium-Catalyzed Miscellaneous Reactions
10. Conclusions
Funding
Conflicts of Interest
References
- Noyori, R. Asymmetric Catalysts in Organic Synthesis; Wiley-VCH: New York, NY, USA, 1994. [Google Scholar]
- Beller, M.; Bolm, C. Transition Metals for Organic Synthesis; Wiley-VCH: Weinheim, Germany, 1998; Volumes I and II. [Google Scholar] [CrossRef]
- Ojima, I. Catalytic Asymmetric Synthesis, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar] [CrossRef]
- Negishi, E. Handbook of Organopalladium Chemistry for Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002; Volume 2, pp. 1689–1705. [Google Scholar] [CrossRef]
- Beller, M.; Bolm, C. Transition Metals for Organic Synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar] [CrossRef]
- Tietze, L.F.; Hiriyakkanavar, I.; Bell, H.P. Enantioselective Palladium-Catalyzed Transformations. Chem. Rev. 2004, 104, 3453–3516. [Google Scholar] [CrossRef] [PubMed]
- Ramon, D.J.; Yus, M. In the Arena of Enantioselective Synthesis, Titanium Complexes Wear the Laurel Wreath. Chem. Rev. 2006, 106, 2126–2208. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H.; Clavier, H. Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev. 2014, 114, 2775–2823. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective scandium-catalyzed transformations. Coord. Chem. Rev. 2016, 313, 1–37. [Google Scholar] [CrossRef]
- Pellissier, H. Enantioselective Silver-Catalyzed Transformations. Chem. Rev. 2016, 116, 14868–14917. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Recent developments in enantioselective lanthanide-catalyzed transformations. Coord. Chem. Rev. 2017, 336, 96–151. [Google Scholar] [CrossRef]
- Pellissier, H. Enantioselective Magnesium-Catalyzed Transformations. Org. Biomol. Chem. 2017, 15, 4750–4782. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective cobalt-catalyzed transformations. Coord. Chem. Rev. 2018, 360, 122–168. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Enantioselective Iron-Catalyzed Transformations. Coord. Chem. Rev. 2019, 386, 1–31. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Enantioselective Vanadium-Catalyzed Transformations. Coord. Chem. Rev. 2020, 418, 213395. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective zinc-catalyzed transformations. Coord. Chem. Rev. 2021, 439, 213926. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective titanium-catalyzed transformations. Coord. Chem. Rev. 2022, 463, 214537. [Google Scholar] [CrossRef]
- Kobayashi, S.; Araki, M.; Hachiya, I. A Chiral Scandium Catalyst for Enantioselective Diels-Alder Reactions. J. Org. Chem. 1994, 59, 3758–3759. [Google Scholar] [CrossRef]
- Yao, Y.; Nie, K. Homogeneous Catalysis. In The Rare Earth Elements; Wiley-VCH: Weinheim, Germany, 2012; pp. 459–474. [Google Scholar]
- Mori, Y.; Kobayashi, S. Organic Synthesis. In The Rare Earth Elements; Wiley-VCH: Weinheim, Germany, 2012; pp. 437–457. [Google Scholar]
- Feng, X.; Liu, X. Scandium: Compounds, Productions, and Applications; Nova Science Publishers: New York, NY, USA, 2011; pp. 1–47. [Google Scholar]
- Roesky, P.W. Molecular Catalysis of Rare Earth Elements; Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Brennan, J.G.; Sella, A. Organometallic Chemistry; Royal Society of Chemistry: Cambridge, UK, 2010; Volume 36, p. 121. [Google Scholar]
- Ogawa, C.; Gu, Y.; Boudou, M.; Kobayashi, S. Acid Catalysis in Modern Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2008; p. 589. [Google Scholar]
- Zeimentz, P.M.; Arndt, S.; Elvidge, B.R.; Okuda, J. Cationic Organometallic Complexes of Scandium, Yttrium, and the Lanthanoids. Chem. Rev. 2006, 106, 2404–2433. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Sugiura, M.; Kitagawa, H.; Lam, W.W.-L. Rare-earth metal triflates in organic synthesis. Chem. Rev. 2002, 102, 2227–2302. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S. Scandium Triflate in Organic Synthesis. Eur. J. Org. Chem. 1999, 1999, 15–27. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Loup, J.; Dhawa, U.; Pesciaioli, F.; Wencel-Delord, J.; Ackermann, L. Enantioselective C-H Activation with Earth-Abundant 3d Transition Metals. Angew. Chem. Int. Ed. 2019, 58, 12803–12818. [Google Scholar] [CrossRef]
- Tietze, L.F.; Beifuss, U. Sequential Transformations in Organic Chemistry: A Synthetic Strategy with a Future. Angew. Chem. Int. Ed. 1993, 32, 131–163. [Google Scholar] [CrossRef]
- Tietze, L.F. Domino Reactions in Organic Synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef]
- Tietze, L.F.; Brasche, G.; Gericke, K. Domino Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Pellissier, H. Asymmetric domino reactions. Part B: Reactions based on the use of chiral catalysts and biocatalysts. Tetrahedron 2006, 62, 2143–2173. [Google Scholar] [CrossRef]
- Clavier, H.; Pellissier, H. Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions. Adv. Synth. Catal. 2012, 354, 3347–3403. [Google Scholar] [CrossRef]
- Pellissier, H. Stereocontrolled Domino Reactions. Chem. Rev. 2013, 113, 442–524. [Google Scholar] [CrossRef]
- Tietze, L.F. Domino Reactions—Concepts for Efficient Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Zhu, J.; Wang, Q.; Wang, M. Multicomponent Reactions in Organic Synthesis; Wiley: Weinheim, Germany, 2014. [Google Scholar]
- Pellissier, H. Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions. Adv. Synth. Catal. 2016, 358, 2194–2259. [Google Scholar] [CrossRef]
- Snyder, S.A. Science of Synthesis. Applications of Domino Transformations in Organic Synthesis; Thieme Verlag: Stuttgart, Germany, 2016; Volumes 1 and 2. [Google Scholar]
- Pellissier, H. Recent developments in enantioselective metal-catalyzed domino reactions. Adv. Synth. Catal. 2019, 361, 1733–1755. [Google Scholar] [CrossRef]
- Pellissier, H. The Use of Domino Reactions for the Synthesis of Chiral Rings. Synthesis 2020, 52, 3837–3854. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric Zinc Catalysis in Green One-Pot Processes. Curr. Org. Chem. 2021, 25, 857–875. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Enantioselective Domino Reactions. Part A: Noble Metal Catalysts. Adv. Synth. Catal. 2023, 365, 620–681. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Enantioselective Domino Reactions. Part B First Row Metal Catalysts. Adv. Synth. Catal. 2023, 365, 768–819. [Google Scholar] [CrossRef]
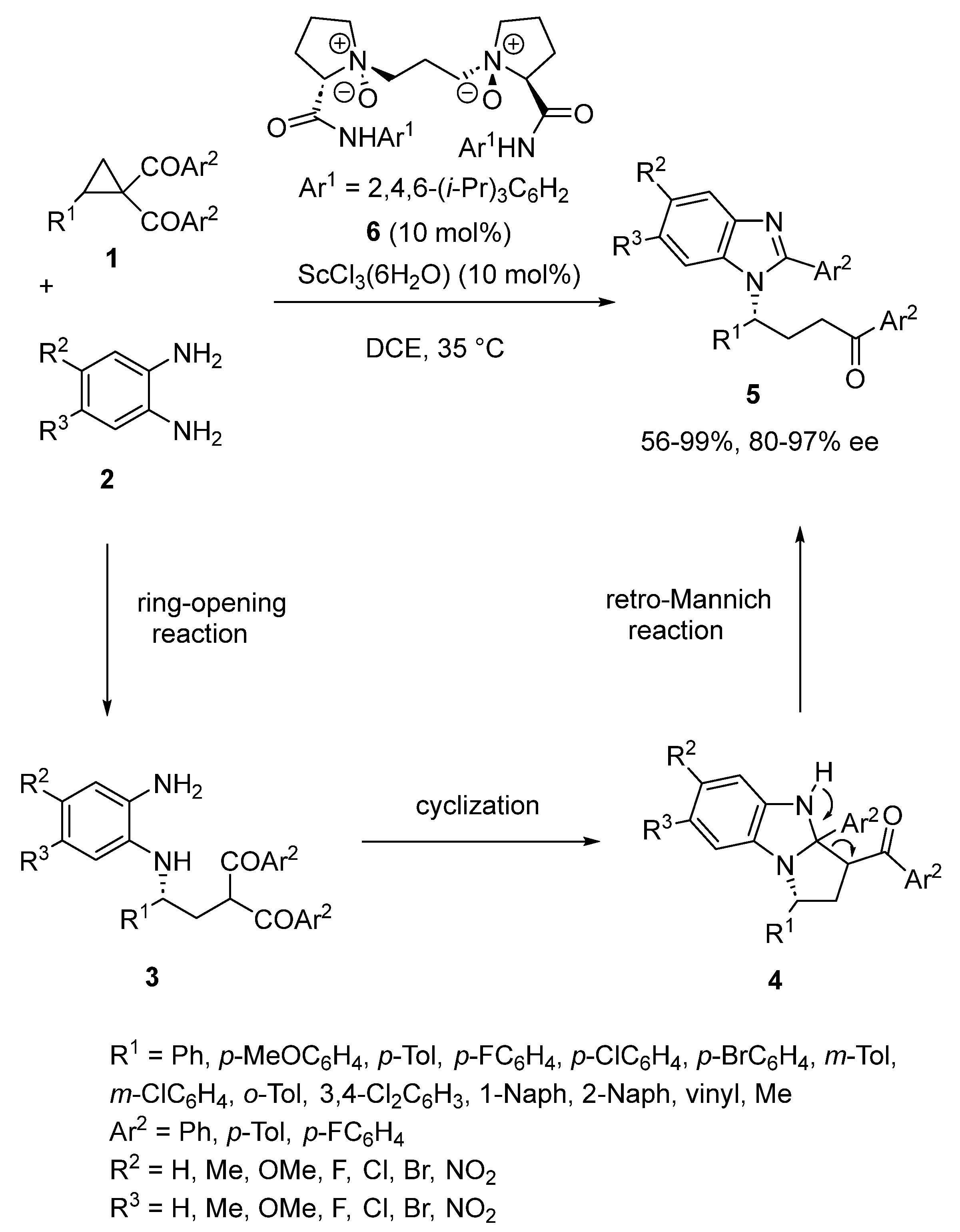
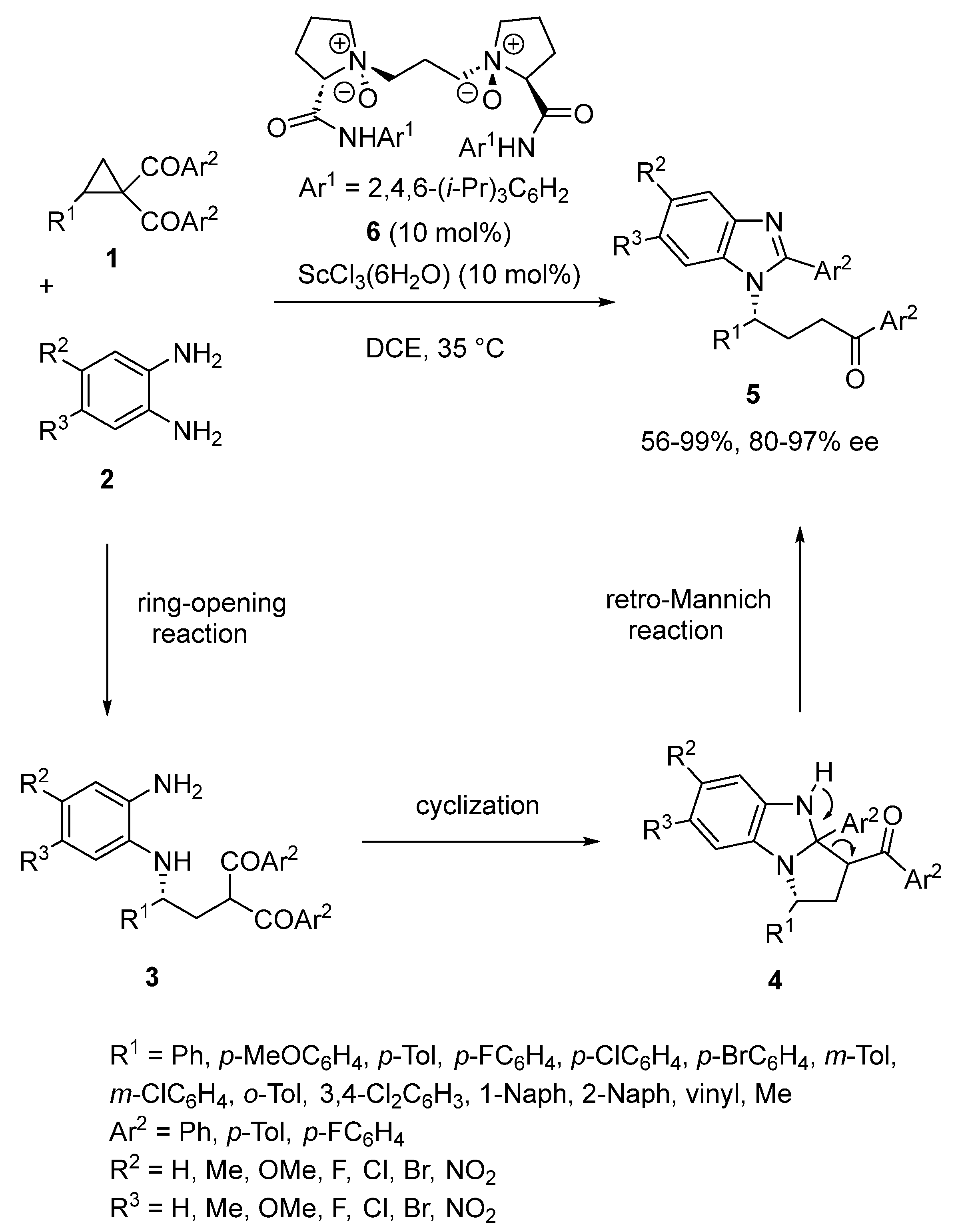
- Xia, Y.; Lin, L.; Chang, F.; Liao, Y.; Liu, X.; Feng, X. Asymmetric Ring Opening/Cyclization/Retro-Mannich Reaction of Cyclopropyl Ketones with Aryl 1,2-Diamines for the Synthesis of Benzimidazole Derivatives. Angew. Chem. Int. Ed. 2016, 55, 12228–12232. [Google Scholar] [CrossRef]
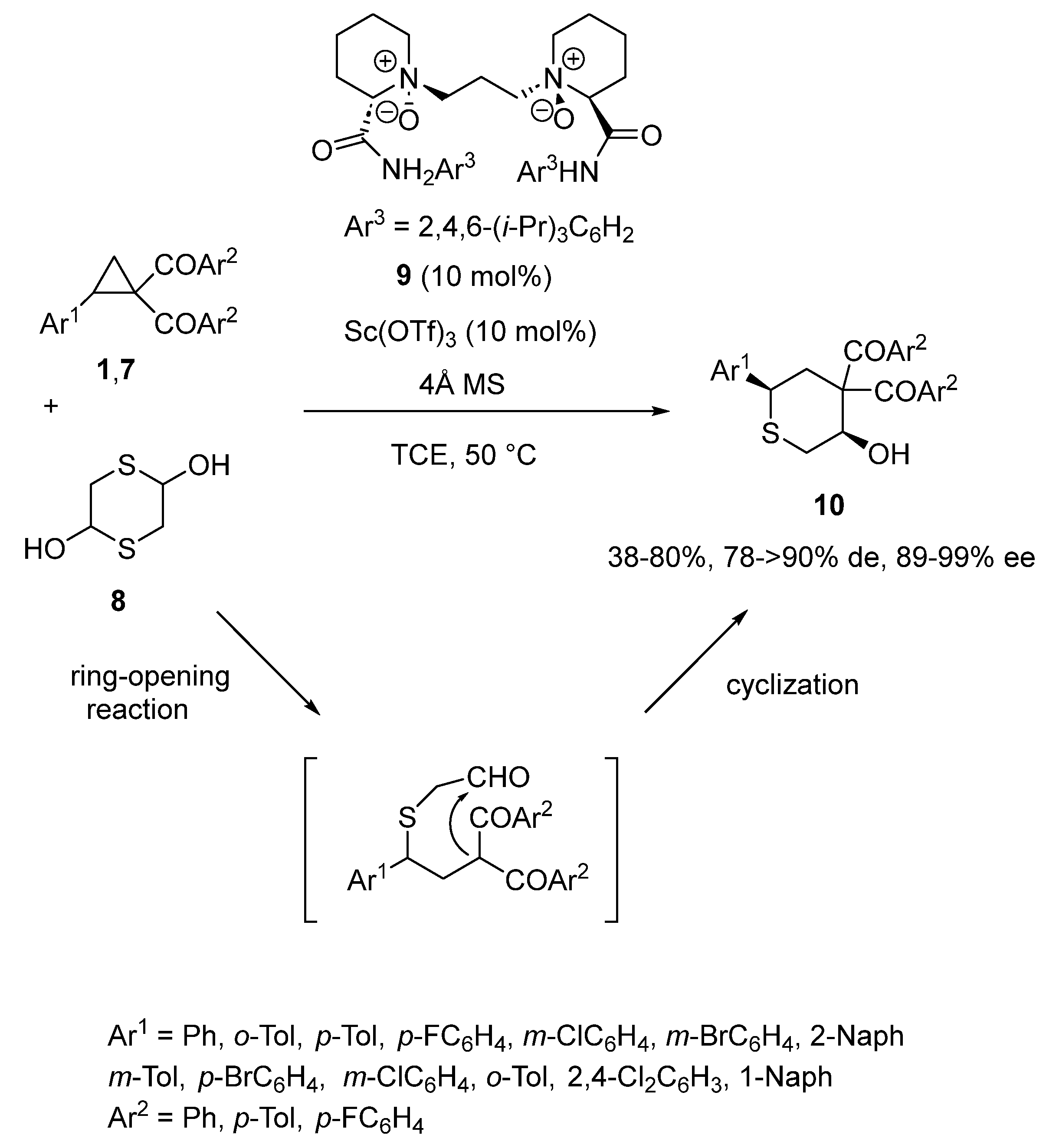
- Fu, X.; Lin, L.; Xia, Y.; Zhou, P.; Liu, X.; Feng, X. Catalytic asymmetric [3 + 3] annulation of cyclopropanes with mercaptoacetaldehyde. Org. Biomol. Chem. 2016, 14, 5914–5917. [Google Scholar] [CrossRef]
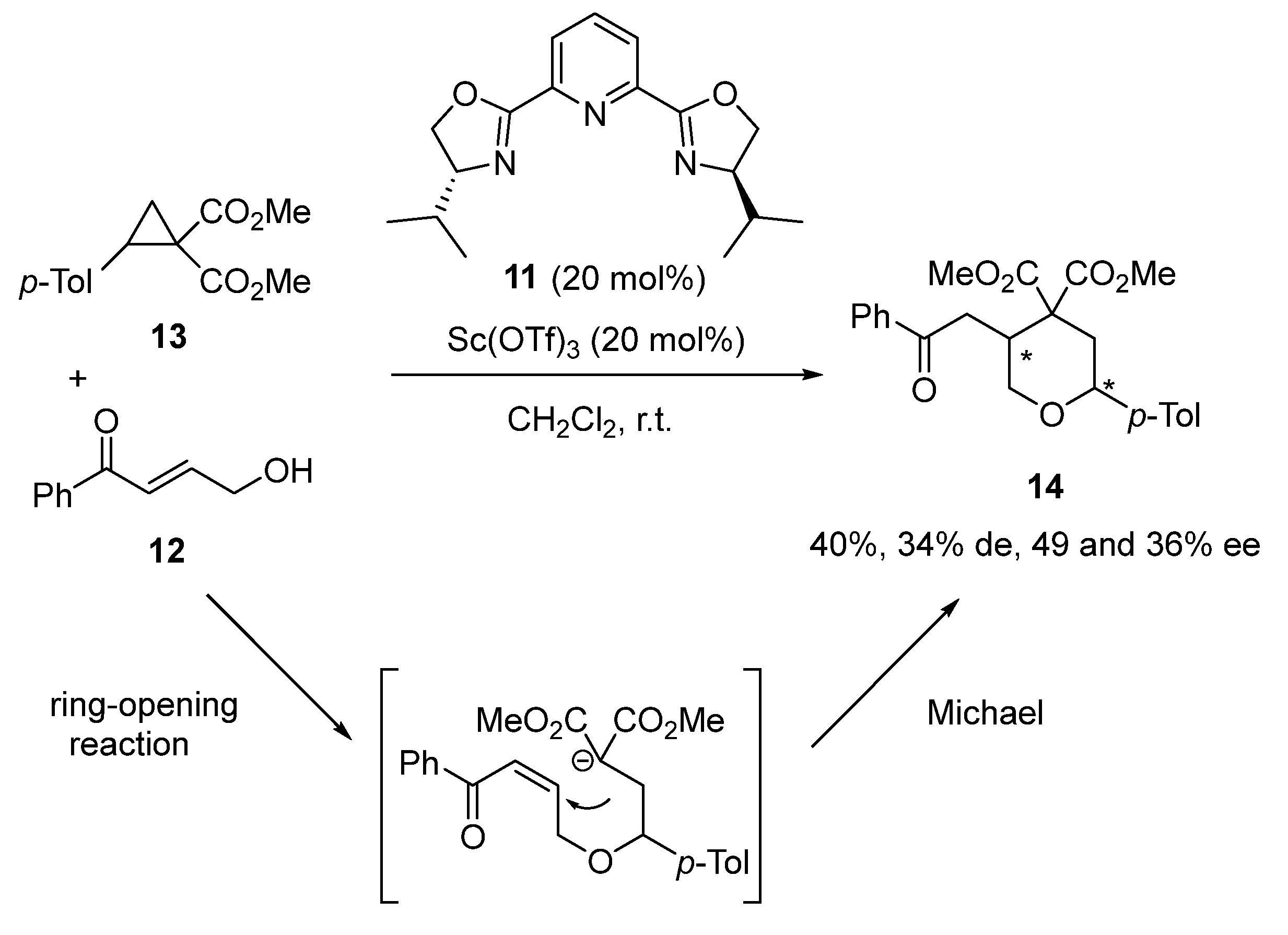
- Mondal, K.; Pan, S.C. Lewis Acid Catalyzed [3+3] Annulation of Donor–Acceptor Cyclopropanes with γ-Hydroxyenones: Access to Highly Functionalized Tetrahydropyrans. Eur. J. Org. Chem. 2017, 2017, 534–537. [Google Scholar] [CrossRef]
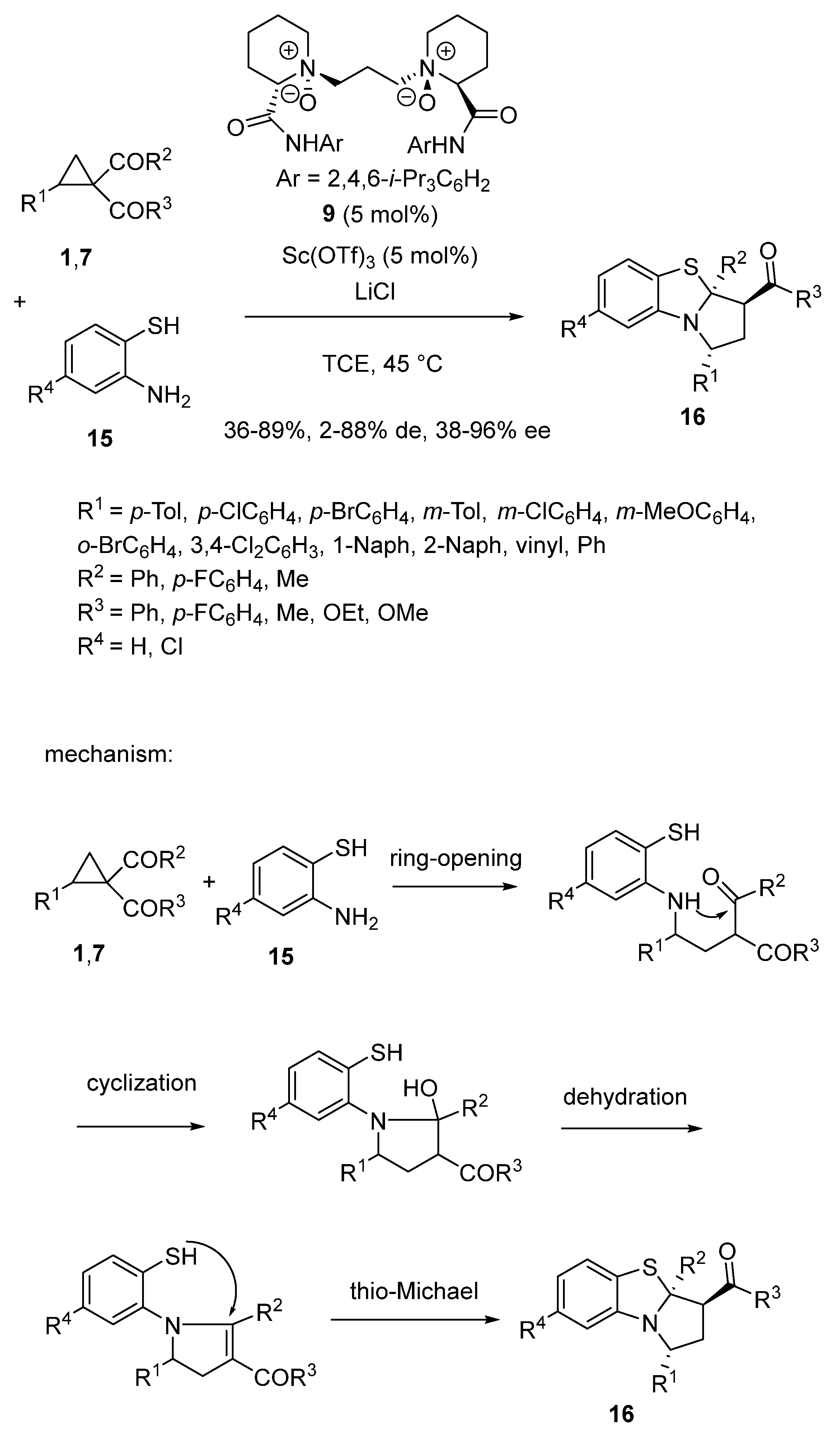
- Chang, F.; Shen, B.; Wang, S.; Lin, L.; Feng, X. Lewis acid catalysed asymmetric cascade reaction of cyclopropyl ketones: Concise synthesis of pyrrolobenzothiazoles. Chem. Commun. 2020, 56, 13429–13432. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, Y.; Li, D.; Yao, Q.; Dong, S.; Liu, X.; Feng, X. Enantioselective Synthesis of 4-Hydroxy-dihydrocoumarins via Catalytic Ring Opening/Cycloaddition of Cyclobutenones. Org. Lett. 2019, 21, 2388–2392. [Google Scholar] [CrossRef]
- Xu, J.; Hu, L.; Hu, H.; Ge, S.; Liu, X.; Feng, X. Enantioselective Vinylogous Michael–Aldol Reaction to Synthesize Spirocyclohexene Pyrazolones in Aqueous Media. Org. Lett. 2019, 21, 1632–1636. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Tang, X.; He, X.; Zhou, Y.; Liu, X.; Feng, X. Regio- and enantioselective conjugate addition of β-nitro α,β-unsaturated carbonyls to construct 3-alkenyl disubstituted oxindoles. Chin. Chem. Lett. 2023, 34, 107487. [Google Scholar] [CrossRef]
- Hao, X.; Lin, L.; Tan, F.; Ge, S.; Liu, X.; Feng, X. Asymmetric synthesis of chromans via the Friedel–Crafts alkylation–hemiketalization catalysed by an N,N′-dioxide scandium(iii) complex. Org. Chem. Front. 2017, 4, 1647–1650. [Google Scholar] [CrossRef]
- Xiao, W.; Mo, Y.; Guo, J.; Su, Z.; Dong, S.; Feng, X. Catalytic enantioselective synthesis of macrodiolides and their application in chiral recognition. Chem. Sci. 2021, 12, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
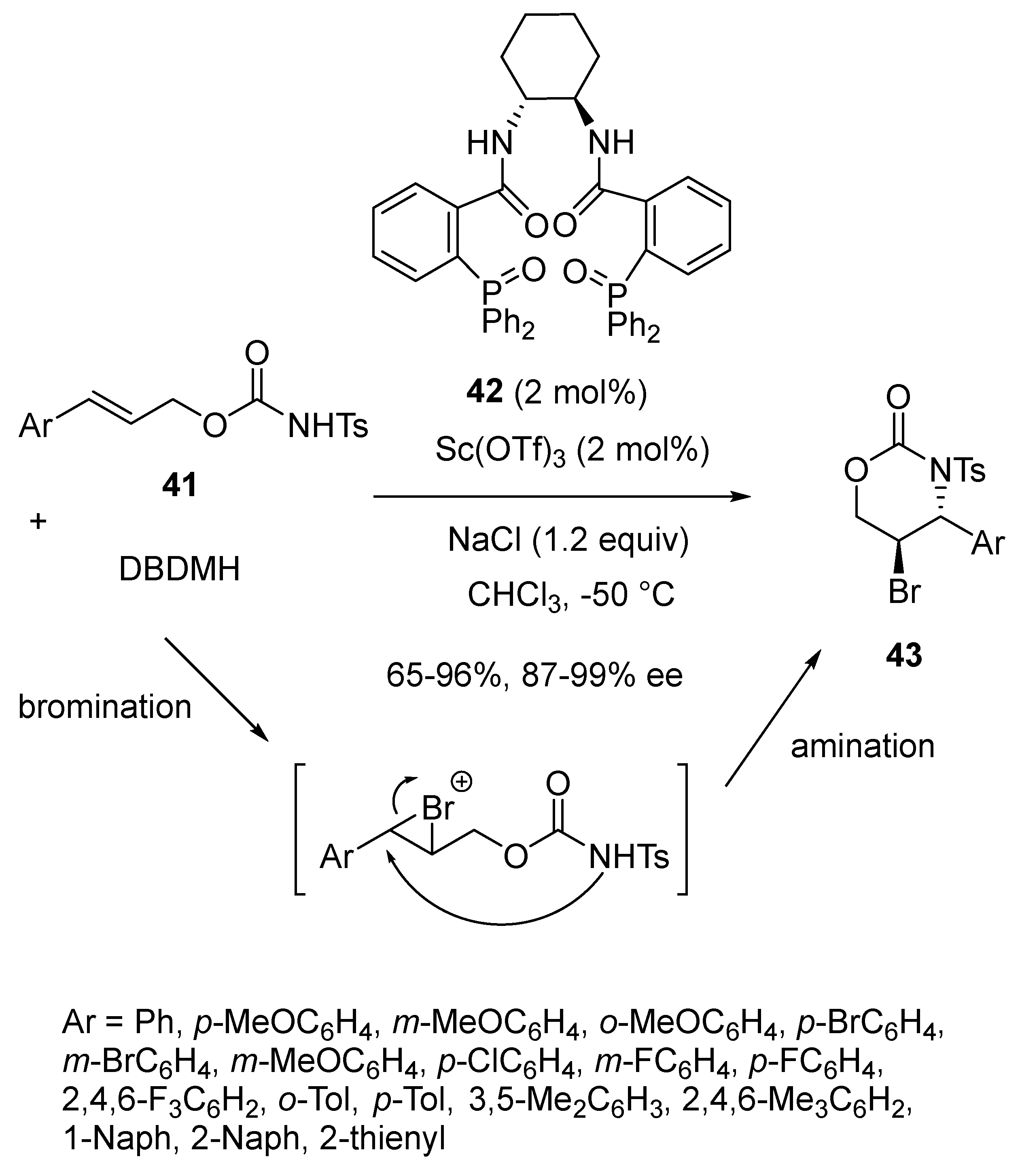
- Pan, H.; Huang, H.; Liu, W.; Tian, H.; Shi, Y. Phosphine Oxide–Sc(OTf)3 Catalyzed Highly Regio- and Enantioselective Bromoaminocyclization of (E)-Cinnamyl Tosylcarbamates. An Approach to a Class of Synthetically Versatile Functionalized Molecules. Org. Lett. 2016, 18, 896–899. [Google Scholar] [CrossRef]
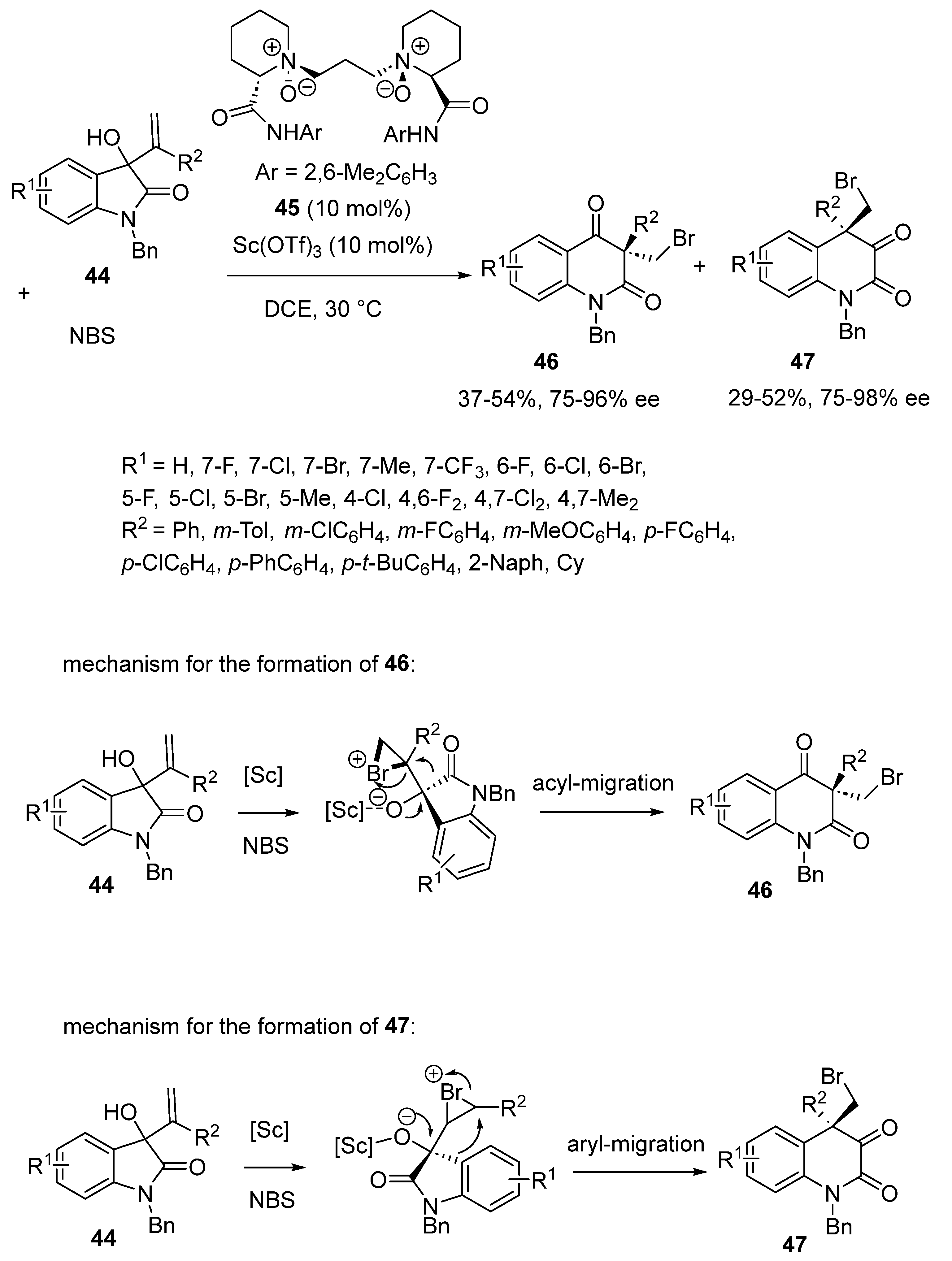
- Dai, L.; Liu, W.; Zhou, Y.; Zeng, Z.; Hu, X.; Cao, W.; Feng, X. Catalytic Asymmetric Halogenation/Semipinacol Rearrangement of 3-Hydroxyl-3-vinyl Oxindoles: A Stereodivergent Kinetic Resolution Process. Angew. Chem. Int. Ed. 2021, 60, 26599–26603. [Google Scholar] [CrossRef]
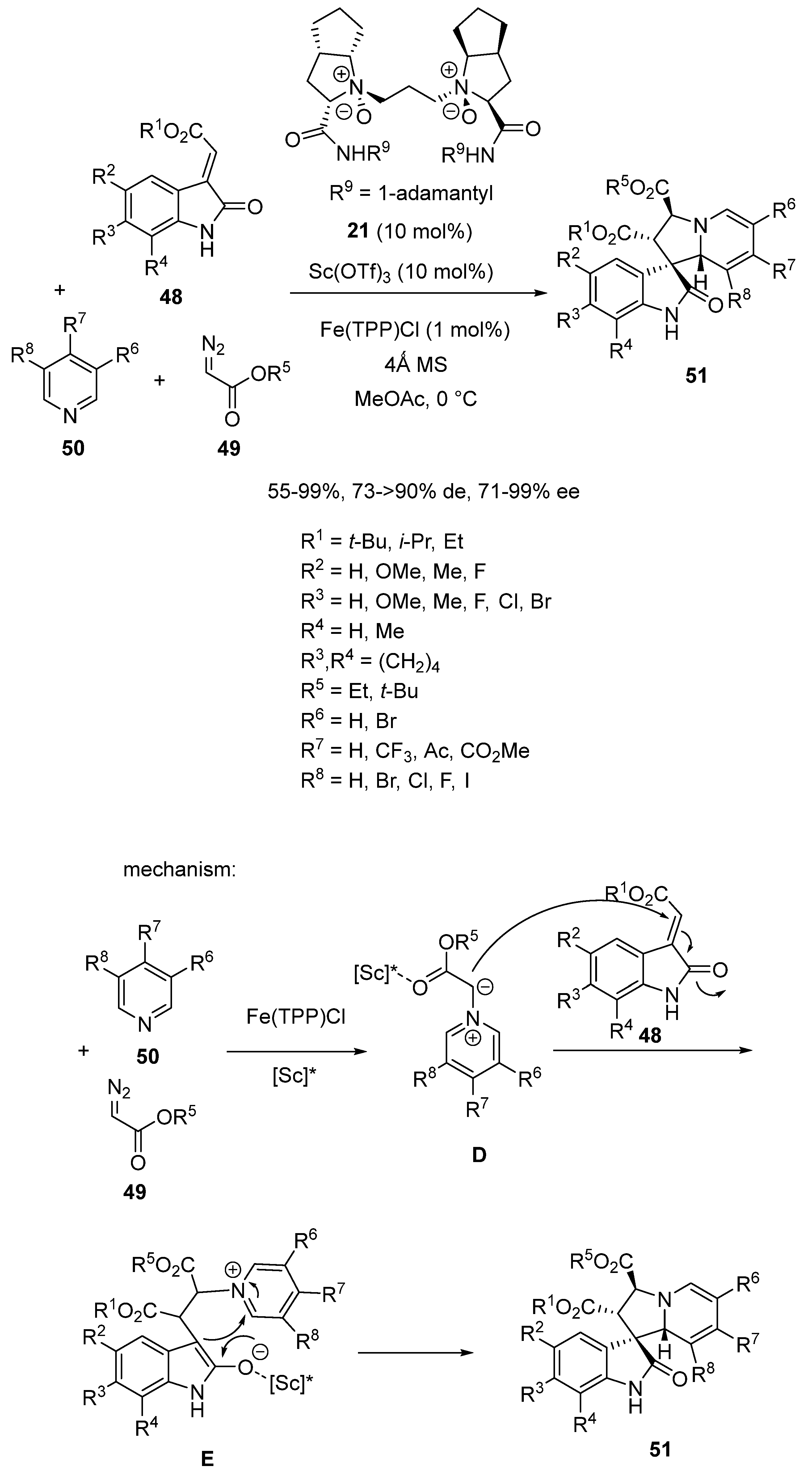
- Zhang, D.; Lin, L.; Yang, J.; Liu, X.; Feng, X. Asymmetric Synthesis of Tetrahydroindolizines by Bimetallic Relay Catalyzed Cycloaddition of Pyridinium Ylides. Angew. Chem. Int. Ed. 2018, 57, 12323–12327. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, M.; Zhan, T.; Cao, W.; Feng, X. Catalytic Asymmetric Three-component Hydroacyloxylation/1,4-Conjugate Addition of Ynamides. Chem. Asian J. 2020, 15, 1953–1956. [Google Scholar] [CrossRef] [PubMed]
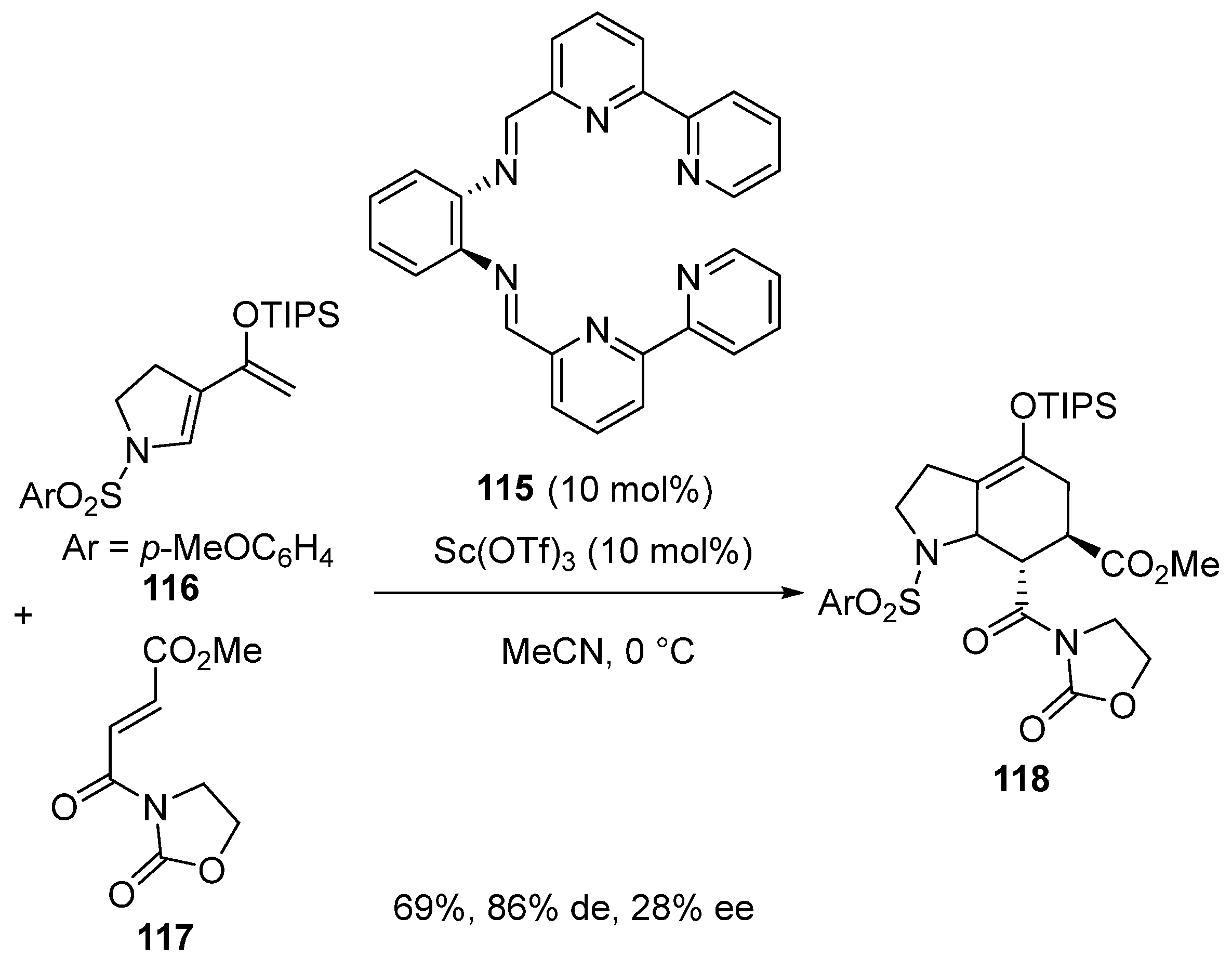
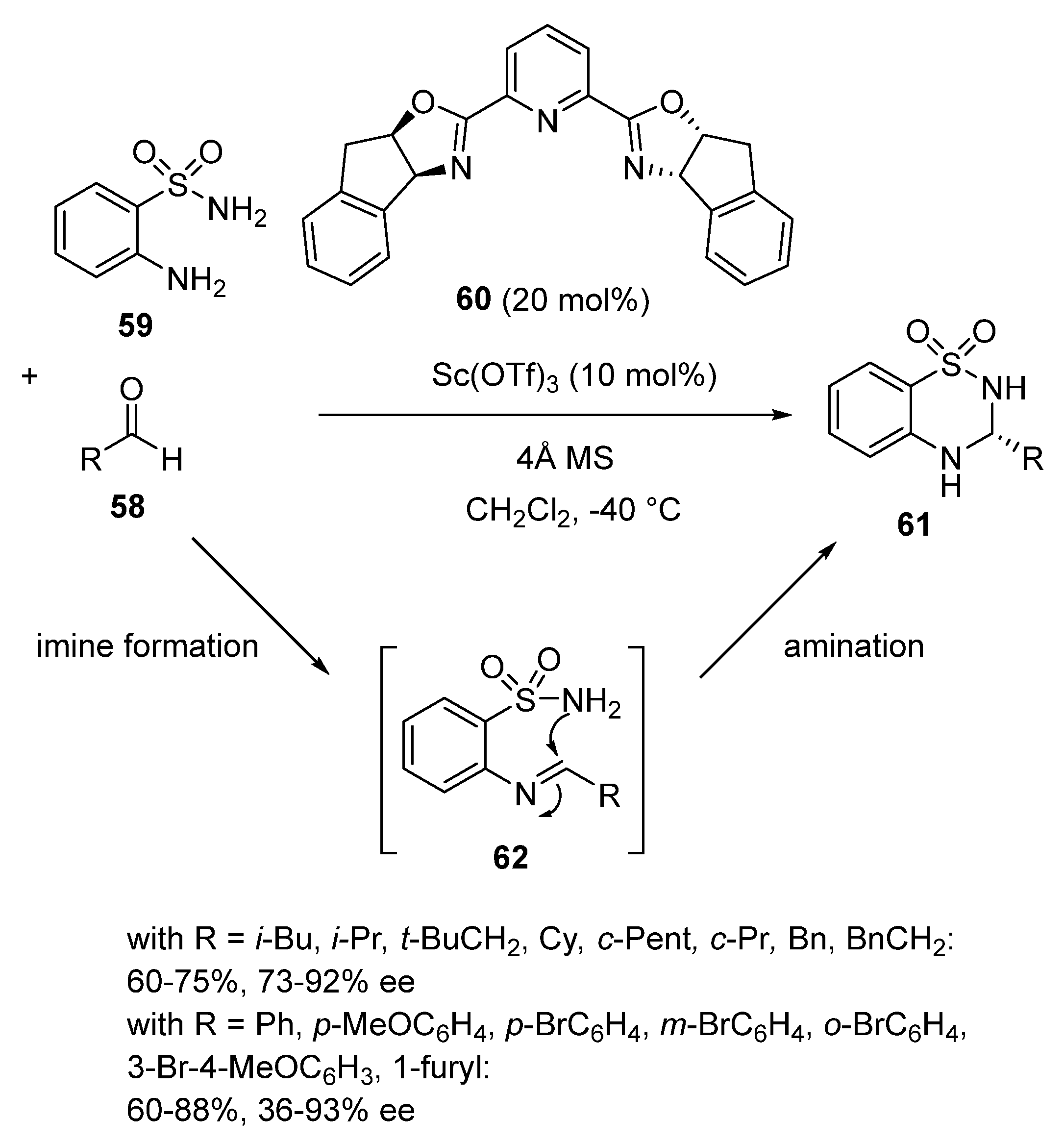
- Du, P.; Zhou, H.; Sui, Y.; Liu, Q.; Zou, K. Asymmetric synthesis of 3,4-dihydro-2H-1,2,4-benzothiadiazine-1,1- dioxides catalyzed by scandium(III)-inda-Pybox. Tetrahedron 2016, 72, 1573–1578. [Google Scholar] [CrossRef]
- Aillerie, A.; Rodriguez-Ruiz, V.; Carlino, R.; Bourdreux, F.; Guillot, R.; Bezzenine-Lafollée, S.; Gil, R.; Prim, D.; Hannedouche, J. Asymmetric Assisted Tandem Catalysis: Hydroamination followed by Asymmetric Friedel–Crafts Reaction from a Single Chiral N,N,N′,N′-Tetradentate Pyridylmethylamine-Based Ligand. ChemCatChem 2016, 8, 2455–2460. [Google Scholar] [CrossRef]
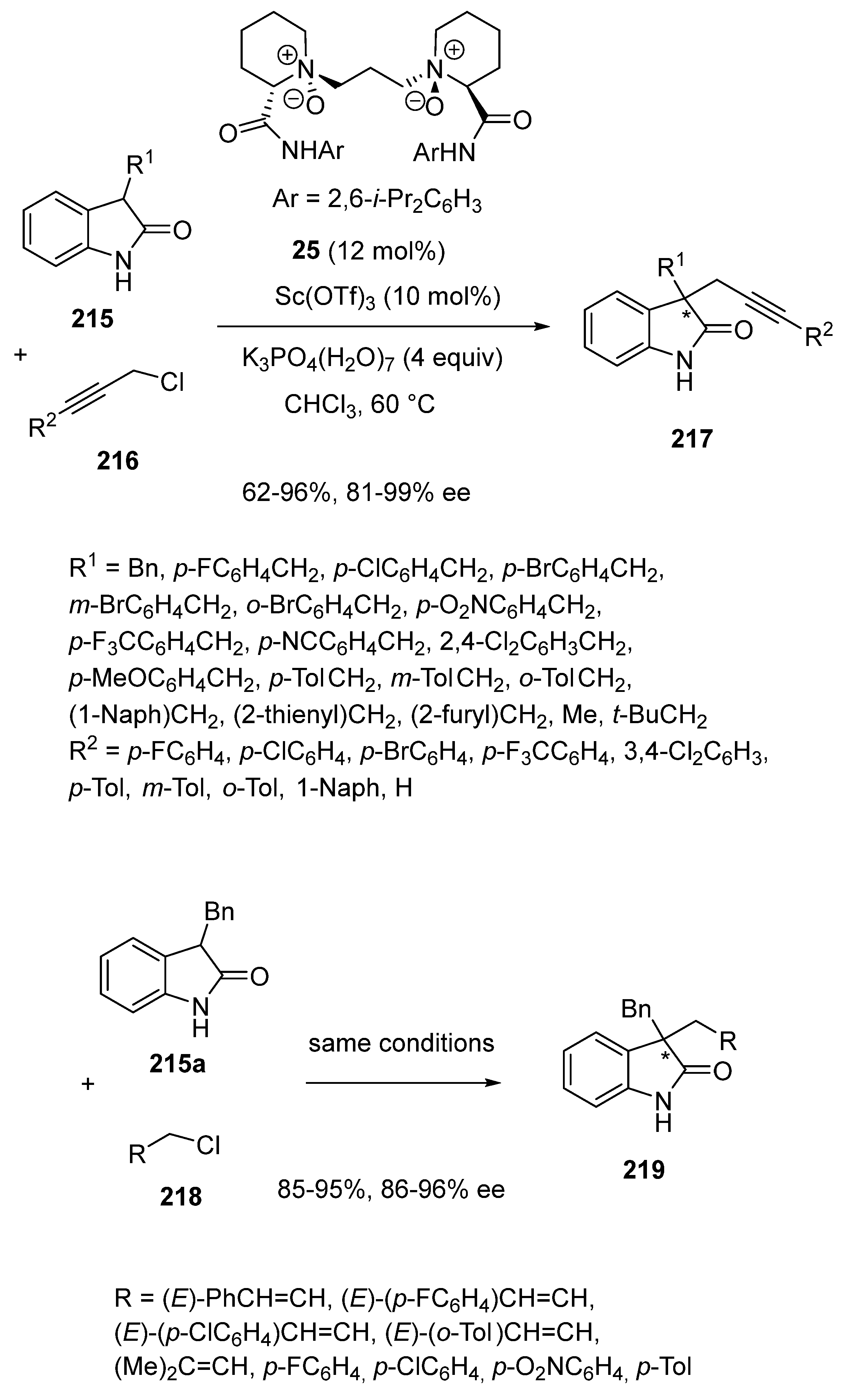
- Zhu, S.; Chen, C.; Duan, K.; Sun, Y.-M.; Li, S.-S.; Liu, Q.; Xiao, J. Cascade [1,5]-Hydride Transfer/Cyclization for Synthesis of [3,4]-Fused Oxindoles. J. Org. Chem. 2019, 84, 8440–8448. [Google Scholar] [CrossRef] [PubMed]
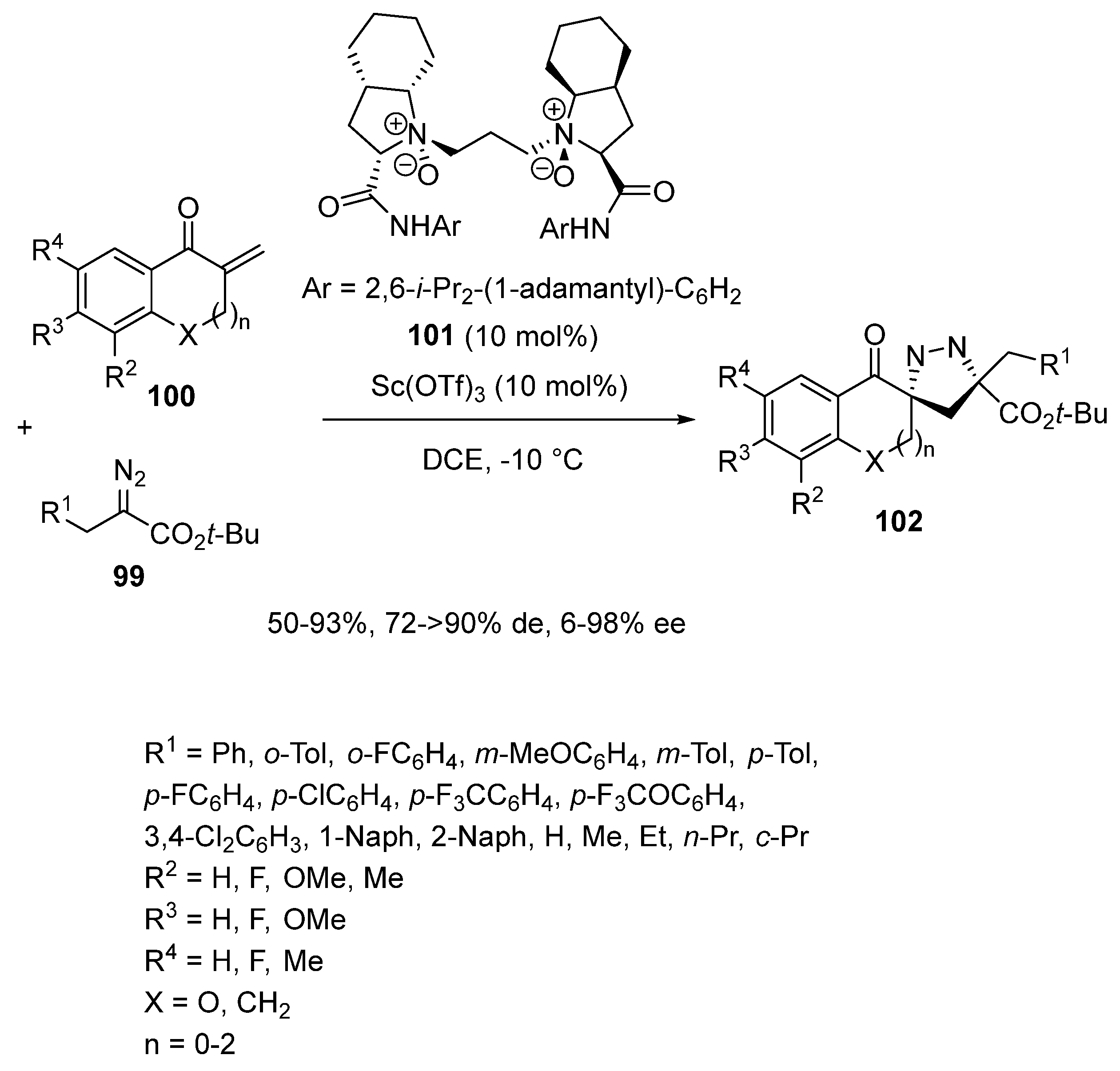
- Tan, F.; Pu, M.; He, J.; Li, J.; Yang, J.; Dong, S.; Liu, X.; Wu, Y.-D.; Feng, X. Catalytic Asymmetric Homologation of Ketones with α-Alkyl α-Diazo Esters. J. Am. Chem. Soc. 2021, 143, 2394–2402. [Google Scholar] [CrossRef]
- Dong, S.; Liu, X.; Feng, X. Asymmetric Catalytic Rearrangements with α-Diazocarbonyl Compounds. Acc. Chem. Res. 2022, 55, 415–428. [Google Scholar] [CrossRef]
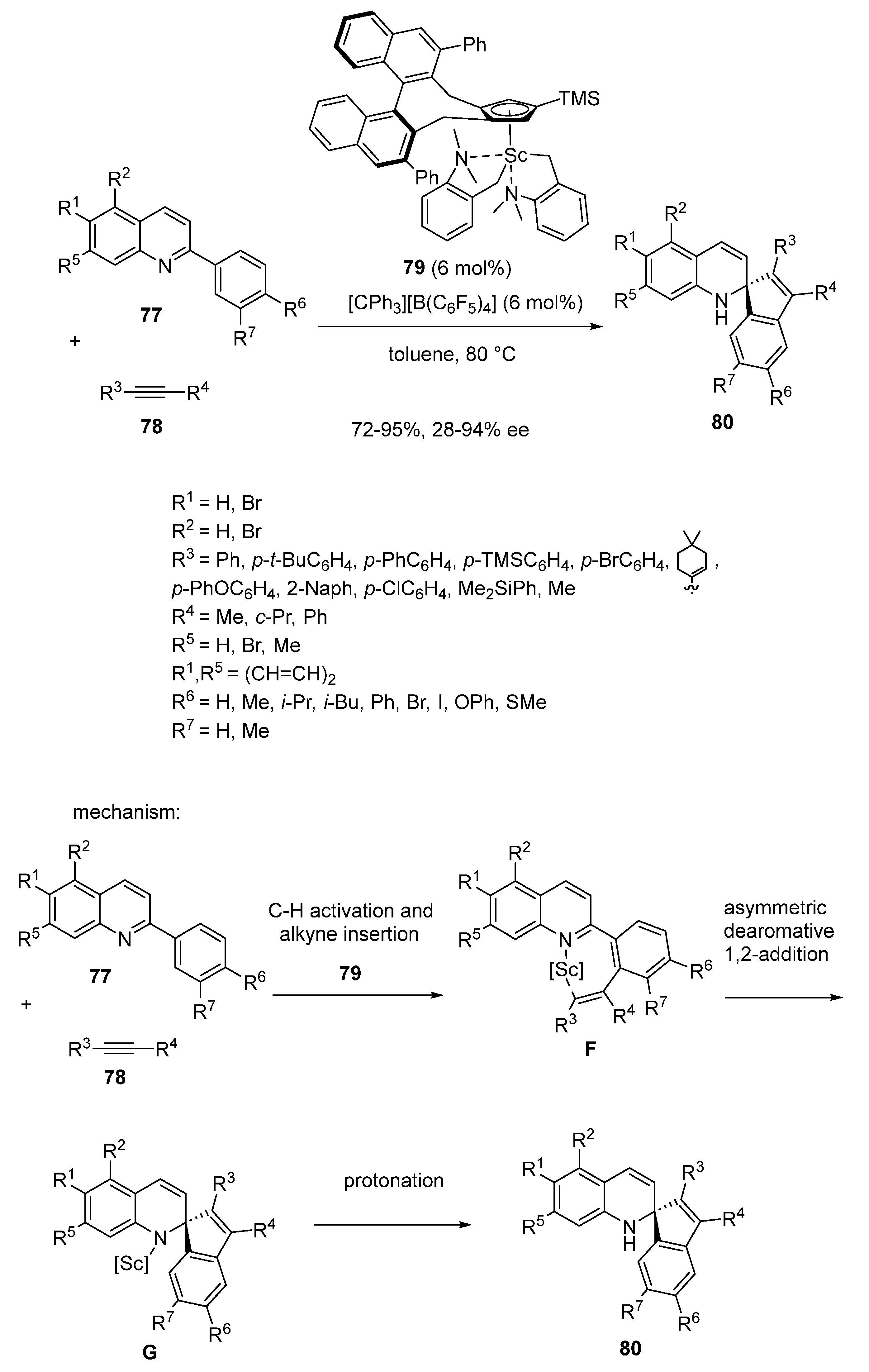
- Lou, S.-J.; Luo, G.; Yamaguchi, S.; An, K.; Nishiura, M.; Hou, Z. Modular Access to Spiro-dihydroquinolines via Scandium-Catalyzed Dearomative Annulation of Quinolines with Alkynes. J. Am. Chem. Soc. 2021, 143, 20462–20471. [Google Scholar] [CrossRef]
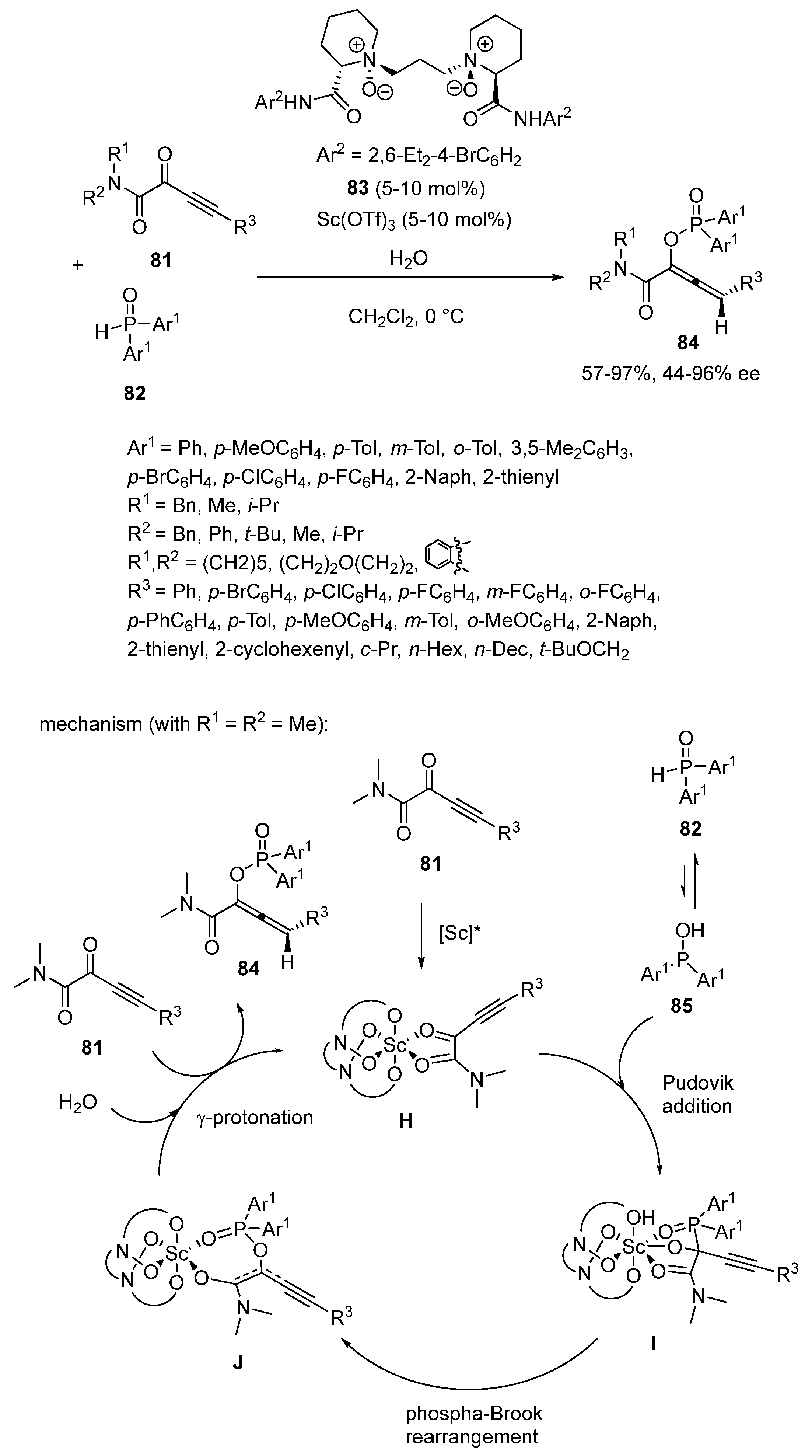
- Lin, Q.; Zheng, S.; Chen, L.; Wu, J.; Li, J.; Liu, P.; Dong, S.; Liu, X.; Peng, Q.; Feng, X. Catalytic Regio- and Enantioselective Protonation for the Synthesis of Chiral Allenes: Synergistic Effect of the Counterion and Water. Angew. Chem. Int. Ed. 2022, 61, e202203650. [Google Scholar] [CrossRef]
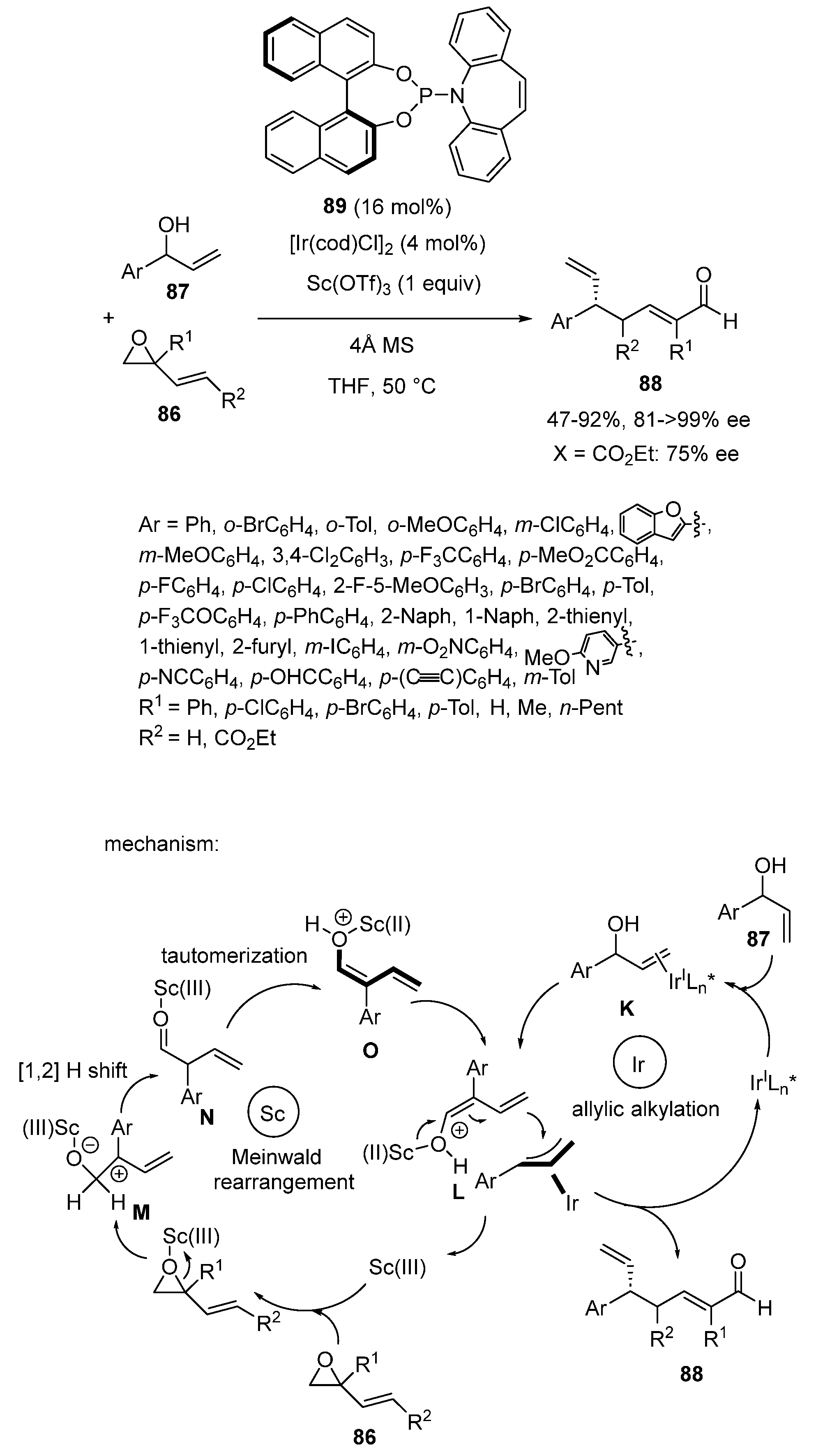
- Zhang, J.; Yang, W.-L.; Zheng, H.; Wang, Y.; Deng, W.-P. Regio- and Enantioselective γ-Allylic Alkylation of In Situ-Generated Free Dienolates via Scandium/Iridium Dual Catalysis. Angew. Chem. Int. Ed. 2021, 61, e202117079. [Google Scholar] [CrossRef]
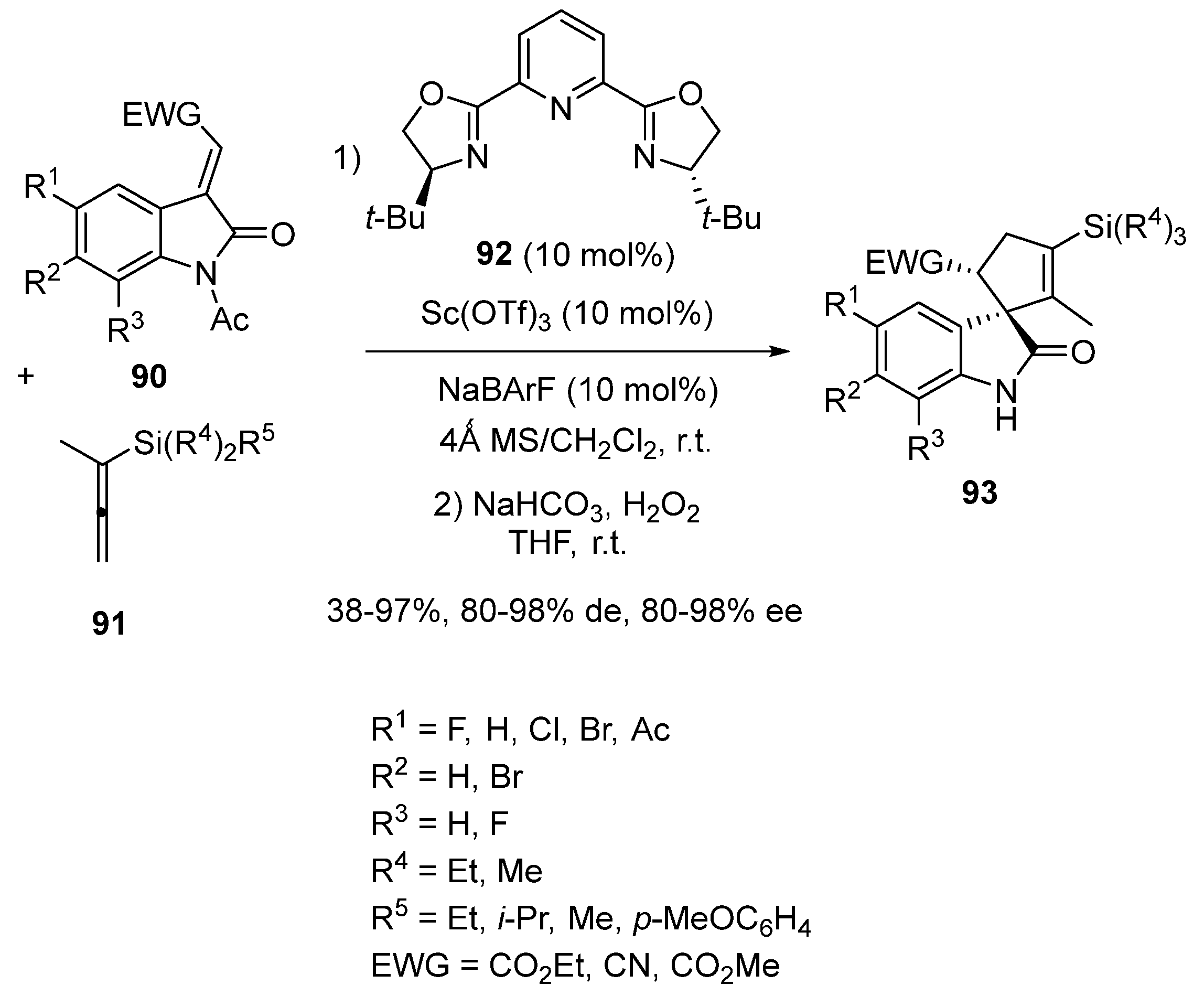
- Cobo, A.A.; Armstrong, B.M.; Fettinger, J.C.; Franz, A.K. Catalytic Asymmetric Synthesis of Cyclopentene-spirooxindoles Bearing Vinylsilanes Capable of Further Transformations. Org. Lett. 2019, 21, 8196–8200. [Google Scholar] [CrossRef]
- Ball-Jones, N.R.; Cobo, A.A.; Armstrong, B.M.; Wigman, B.; Fettinger, J.C.; Hein, J.E.; Franz, A.K. Ligand-Accelerated Catalysis in Scandium(III)-Catalyzed Asymmetric Spiroannulation Reactions. ACS Catal. 2022, 12, 3524–3533. [Google Scholar] [CrossRef]
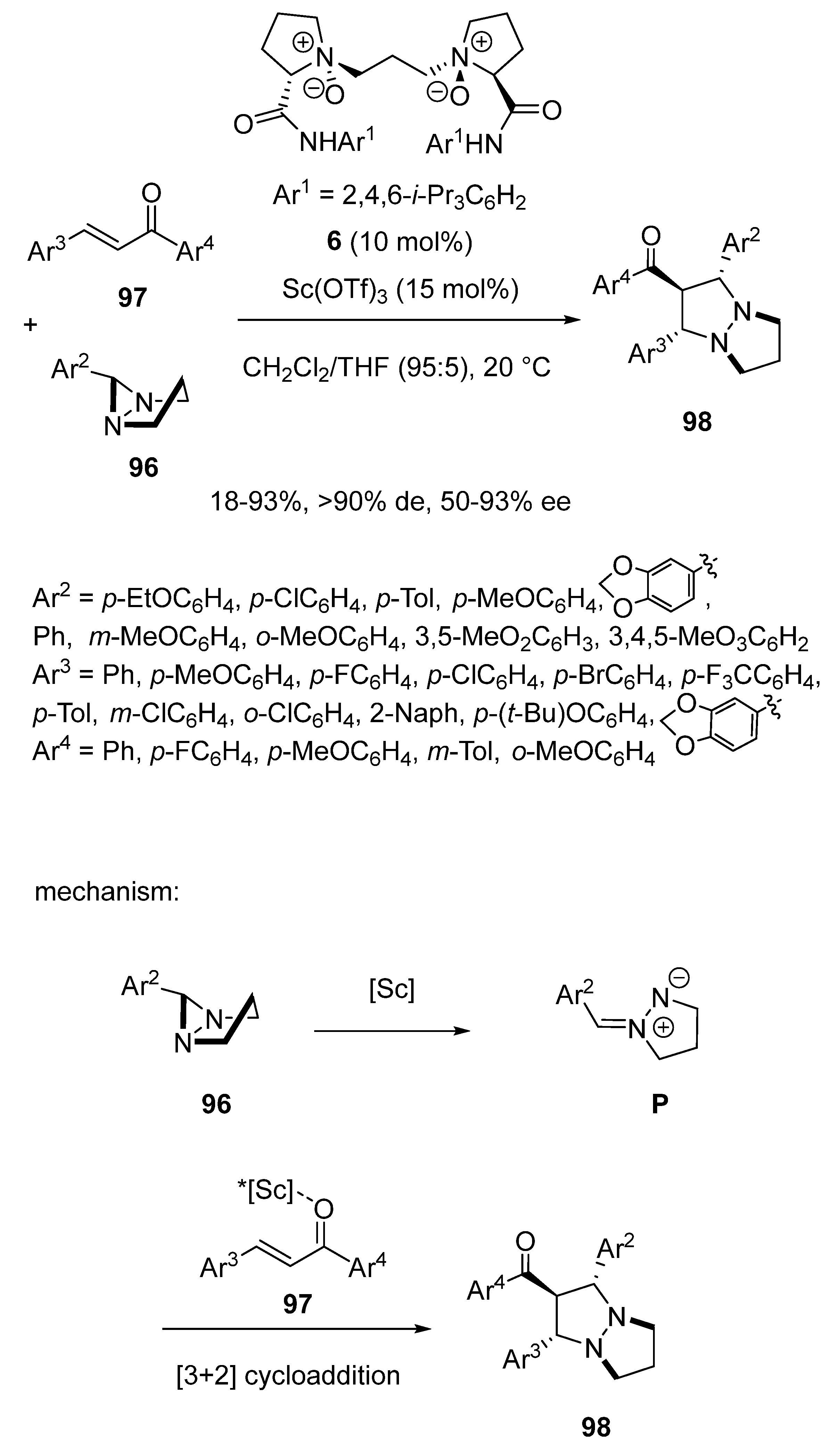
- Hu, H.; Xu, J.; Wang, F.; Dong, S.; Liu, X.; Feng, X. Chiral ScIII–N,N′-Dioxide-Catalyzed 1,3-Dipolar Cycloaddition of Diaziridines with Chalcones. Org. Lett. 2020, 22, 93–97. [Google Scholar] [CrossRef]
- Zhao, P.; Li, Z.; He, J.; Liu, X.; Feng, X. Asymmetric catalytic 1,3-dipolar cycloaddition of α-diazoesters for synthesis of 1-pyrazoline-based spirochromanones and beyond. Sci. China Chem. 2021, 64, 1355–1360. [Google Scholar] [CrossRef]
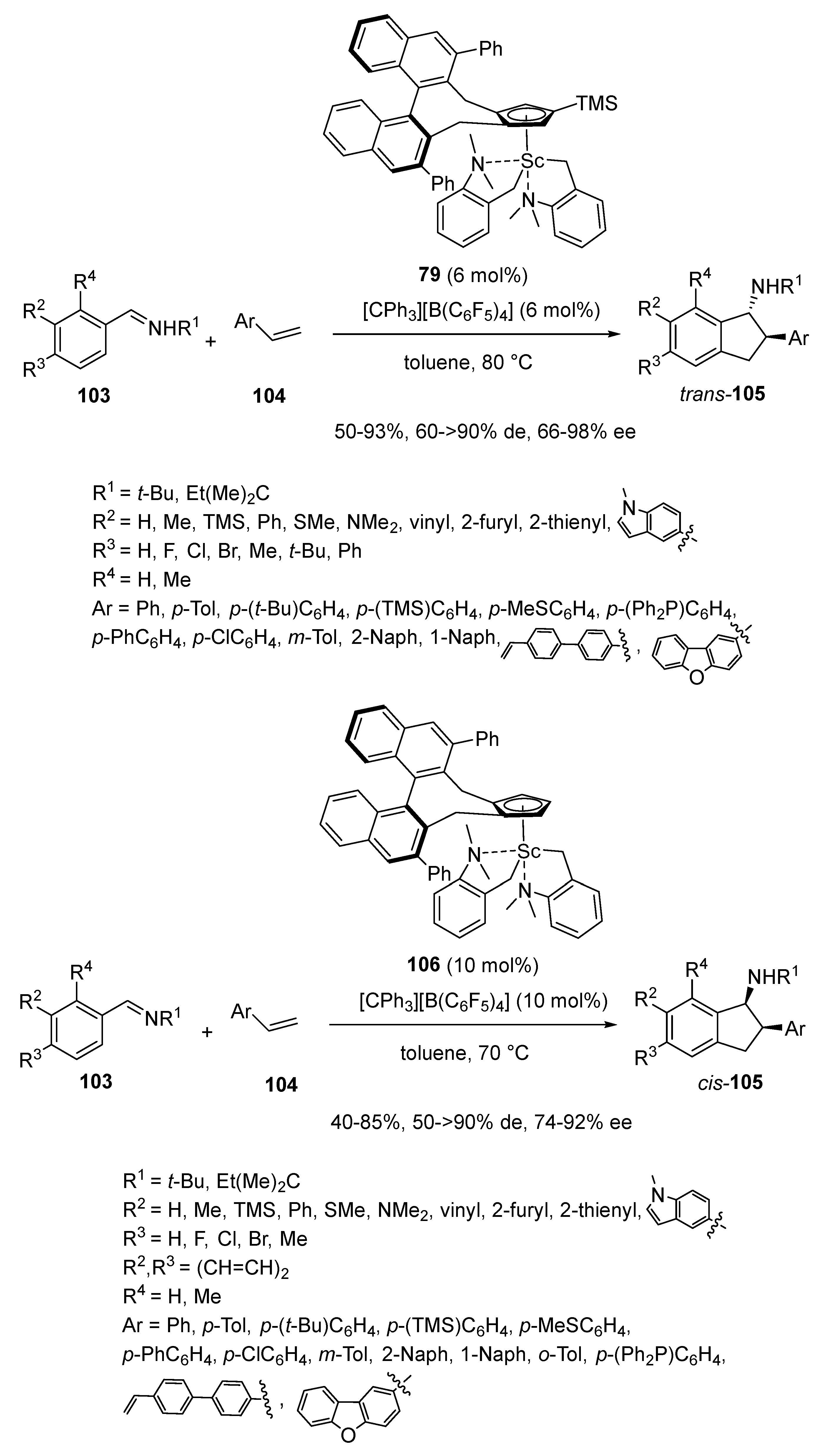
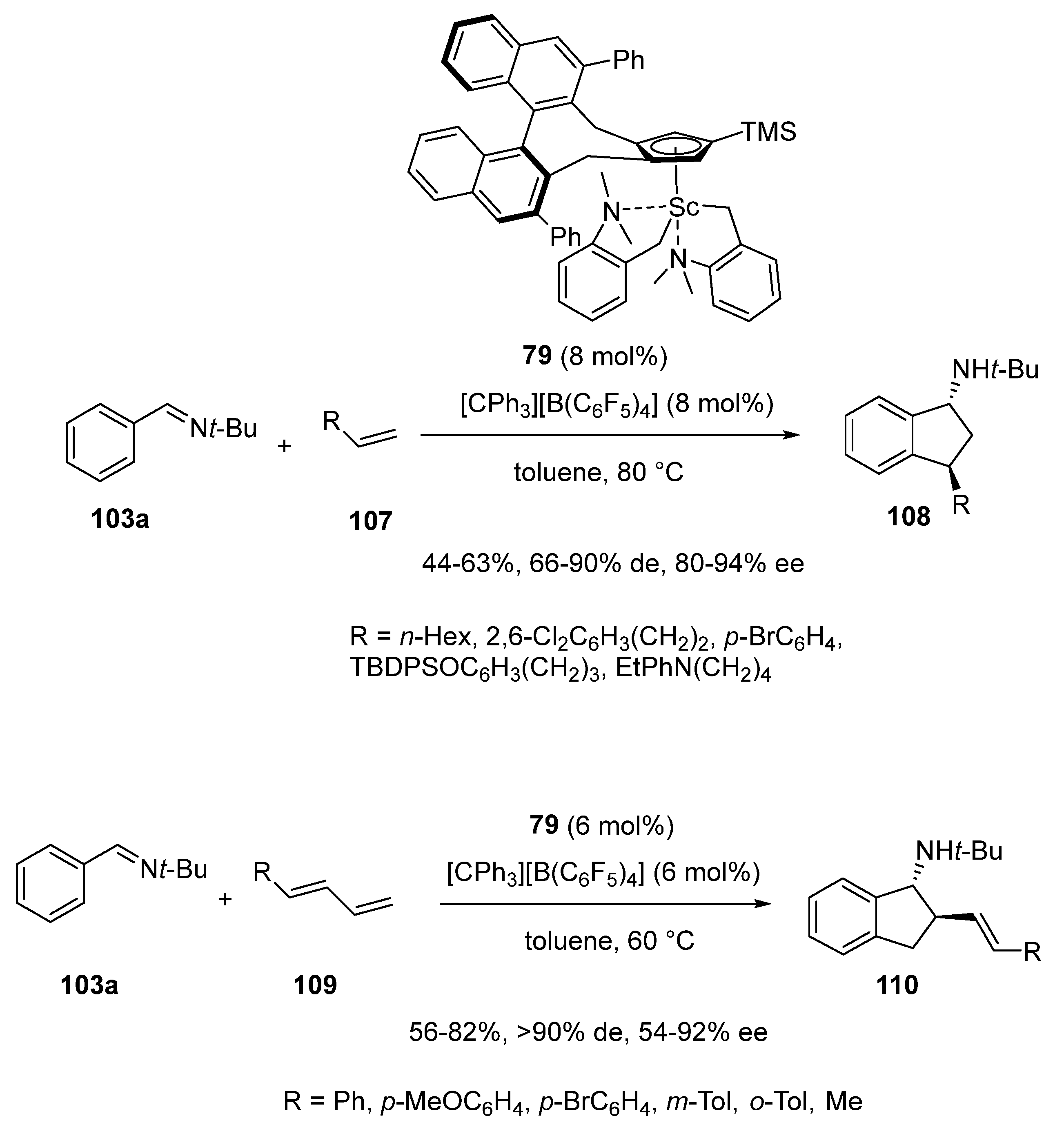
- Mishra, A.; Cong, X.; Nishiura, M.; Hou, Z. Enantioselective Synthesis of 1-Aminoindanes via [3 + 2] Annulation of Aldimines with Alkenes by Scandium-Catalyzed C–H Activation. J. Am. Chem. Soc. 2023, 145, 17468–17477. [Google Scholar] [CrossRef] [PubMed]
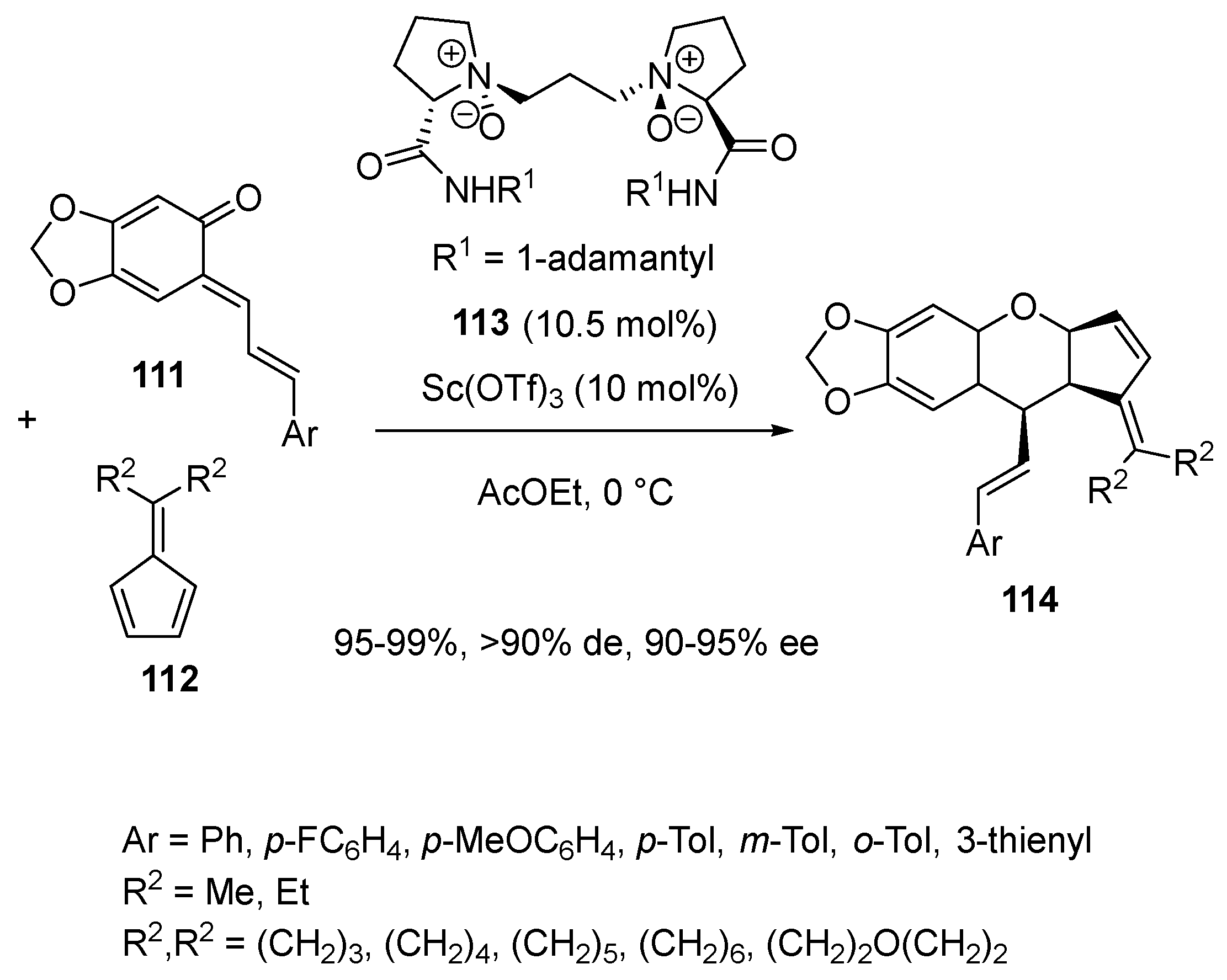
- Zhang, J.; Len, L.; He, C.; Xiong, Q.; Liu, X.; Feng, X. Chiral scandium-complex-catalyzed asymmetric inverse-electron-demand oxa-Diels–Alder reaction of o-quinone methides with fulvenes. Chem. Commun. 2018, 54, 74–77. [Google Scholar] [CrossRef]
- Harada, S.; Nakashima, S.; Sekino, S.; Oishi, W.; Nishida, A. Optically Active Helical Lanthanide Complexes: Storable Chiral Lewis Acidic Catalysts for Enantioselective Diels–Alder Reaction of Siloxydienes. Chem. Asian J. 2020, 15, 483–486. [Google Scholar] [CrossRef] [PubMed]
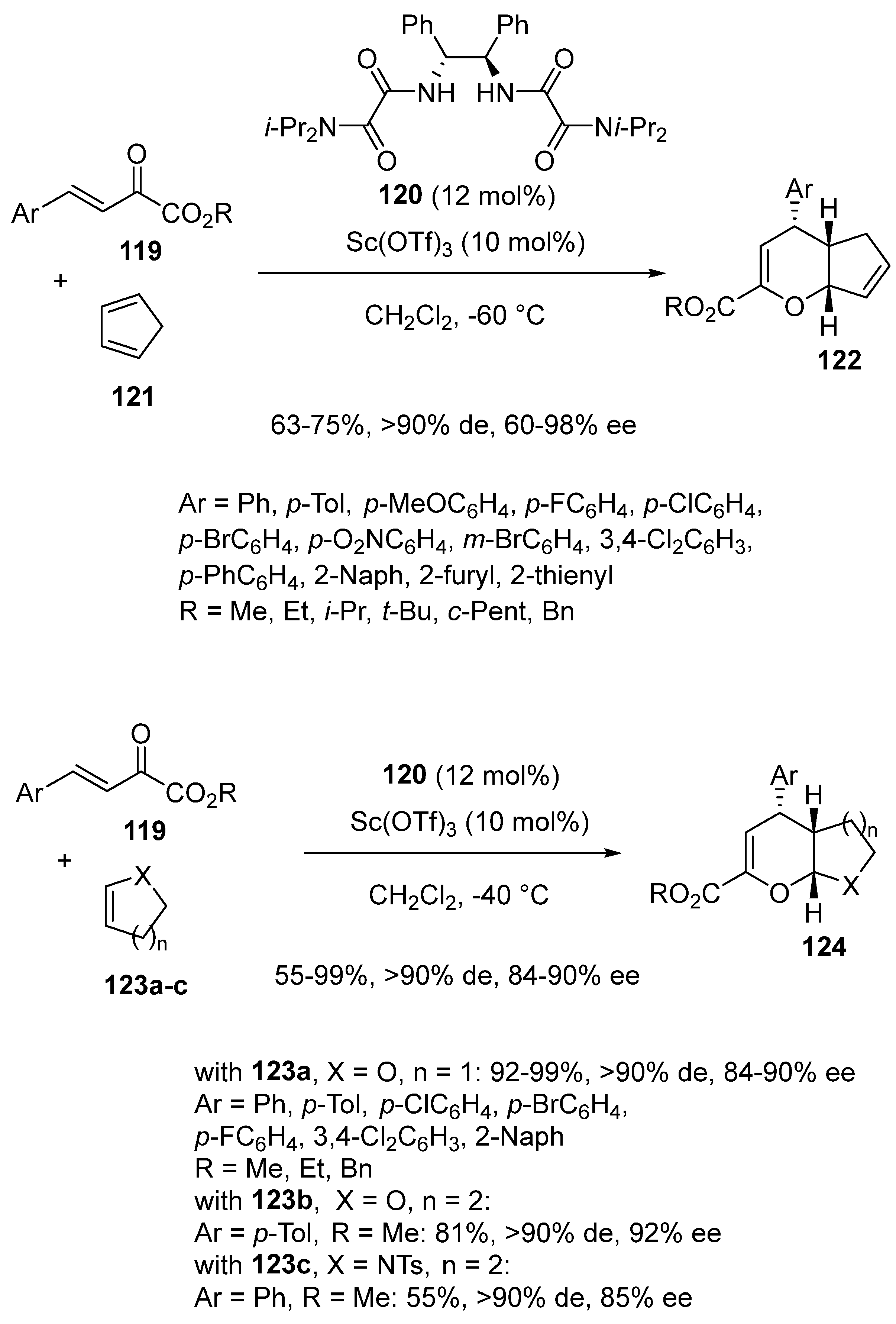
- Yang, H.-D.; Chen, J.-B.; Peng, C.; Liu, W.-K.; Zhou, S.-S.; Song, J.-X.; Qi, Z.-H.; Wang, Y.; Wang, X.-W. Chiral Bis-Oxalamide-Rare Earth Complex Catalyzed Inverse- Electron-Demand Asymmetric oxa-Hetero-Diels-Alder Reaction for Optically Active Dihydropyran Core Structures. Adv. Synth. Catal. 2022, 364, 4347–4362. [Google Scholar] [CrossRef]
- Zhao, P.; Wu, S.; Ke, C.; Liu, X.; Feng, X. Chiral Lewis acid-catalyzed enantioselective cyclopropanation and C–H insertion reactions of vinyl ketones with α-diazoesters. Chem. Commun. 2018, 54, 9837–9840. [Google Scholar] [CrossRef]
- Yao, Q.; Liao, Y.; Lin, L.; Lin, X.; Ji, J.; Liu, X.; Feng, X. Efficient Synthesis of Chiral Trisubstituted 1,2-Allenyl Ketones by Catalytic Asymmetric Conjugate Addition of Malonic Esters to Enynes. Angew. Chem. Int. Ed. 2016, 55, 1859–1863. [Google Scholar] [CrossRef]
- Ji, J.; Tang, Q.; Kang, T.; Liu, X.; Feng, X. First-Principles Selection of Solute Elements for Er-Stabilized Bi2O3 Oxide-Ion Conductor with Improved Long-Term Stability at Moderate Temperatures. ACS Catal. 2017, 7, 3763–3768. [Google Scholar] [CrossRef]
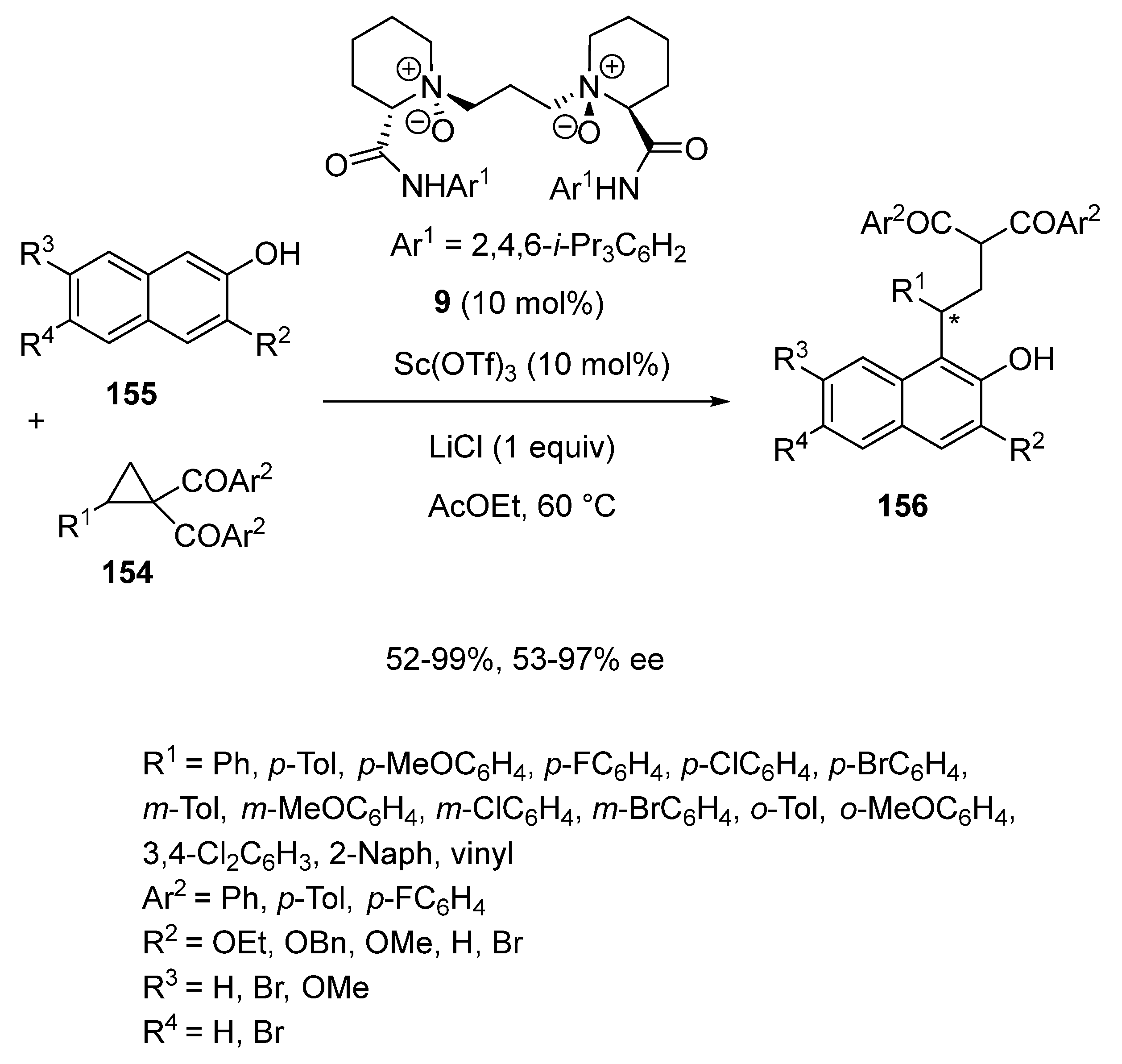
- Ge, S.; Kang, T.; Lin, L.; Zhang, X.; Zhao, P.; Liu, X.; Feng, X. Chiral N,N′-dioxide/Sc(OTf)3 complex-catalyzed asymmetric dearomatization of β-naphthols. Chem. Commun. 2017, 53, 11759–11762. [Google Scholar] [CrossRef]
- Rout, S.; Das, A.; Singh, V.K. Metal-Controlled Switching of Enantioselectivity in the Mukaiyama–Michael Reaction of α,β-Unsaturated 2-Acyl Imidazoles Catalyzed by Chiral Metal–Pybox Complexes. J. Org. Chem. 2018, 83, 5058–5071. [Google Scholar] [CrossRef] [PubMed]
- Rout, S.; Dasa, A.; Singh, V.K. An asymmetric vinylogous Mukaiyama–Michael reaction of a,b-unsaturated 2-acyl imidazoles catalyzed by chiral Sc(III)– or Er(III)–pybox complexes. Chem. Commun. 2017, 53, 5143–5146. [Google Scholar] [CrossRef] [PubMed]
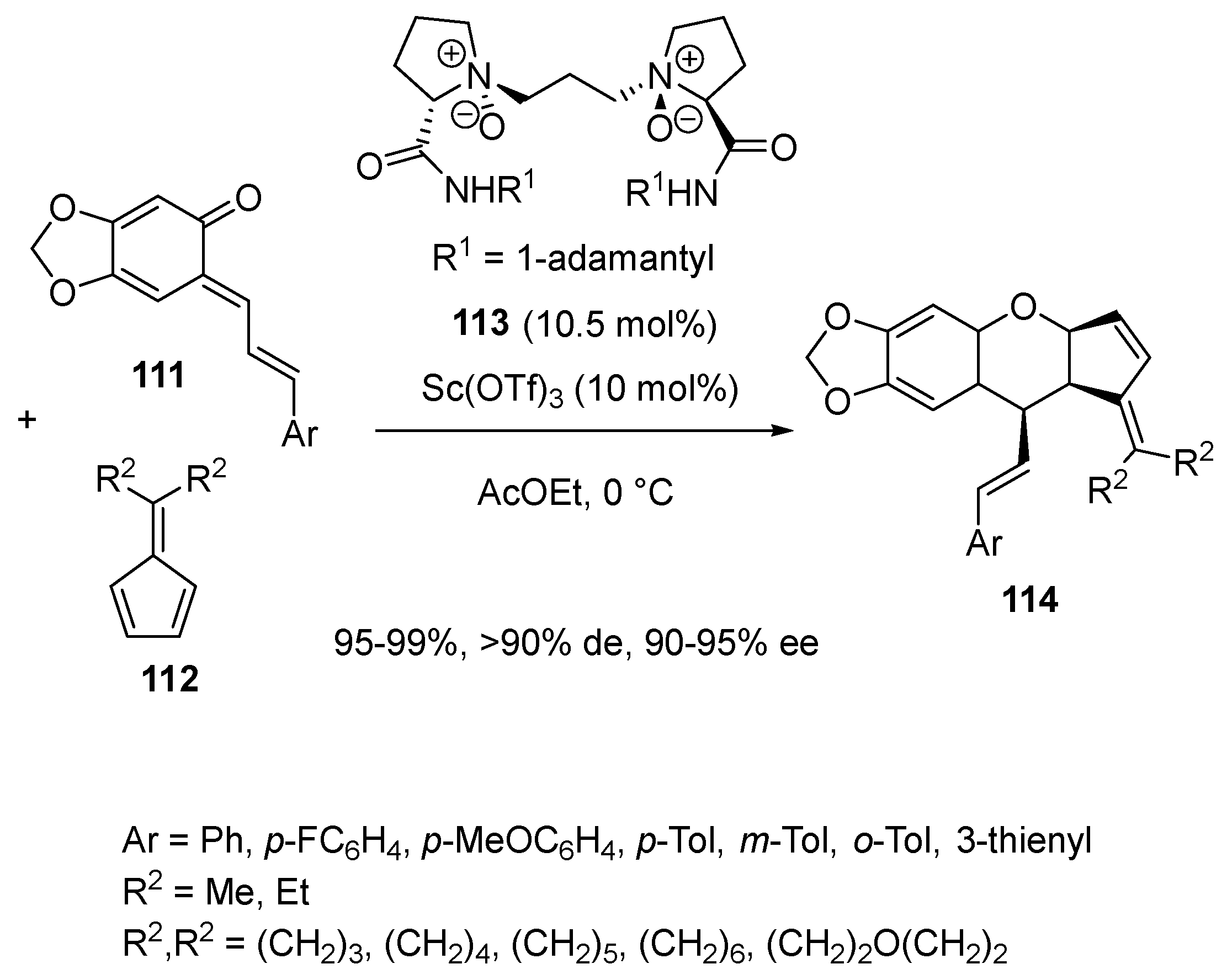
- Riehl, P.S.; Richardson, A.D.; Sakamoto, T.; Schindler, C.S. Eight-Step Enantiodivergent Synthesis of (+)- and (−)-Lingzhiol. Org. Lett. 2020, 22, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Chang, L.; Lin, L.; Xu, Y.; Liu, X.; Feng, X. Asymmetric ring-opening of cyclopropyl ketones with β-naphthols catalyzed by a chiral N,N′-dioxide–scandium(iii) complex. Org. Chem. Front. 2018, 5, 1293–1296. [Google Scholar] [CrossRef]
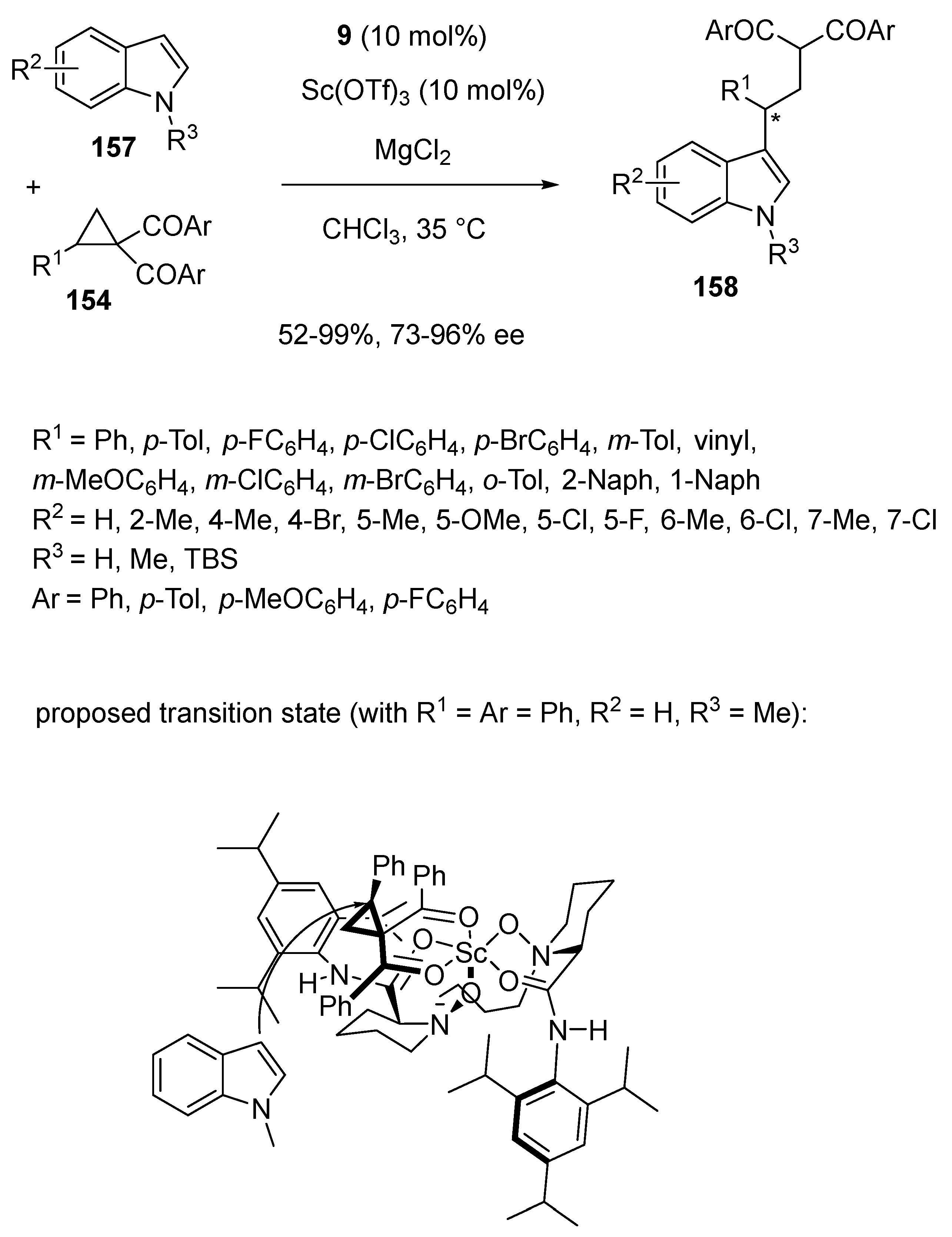
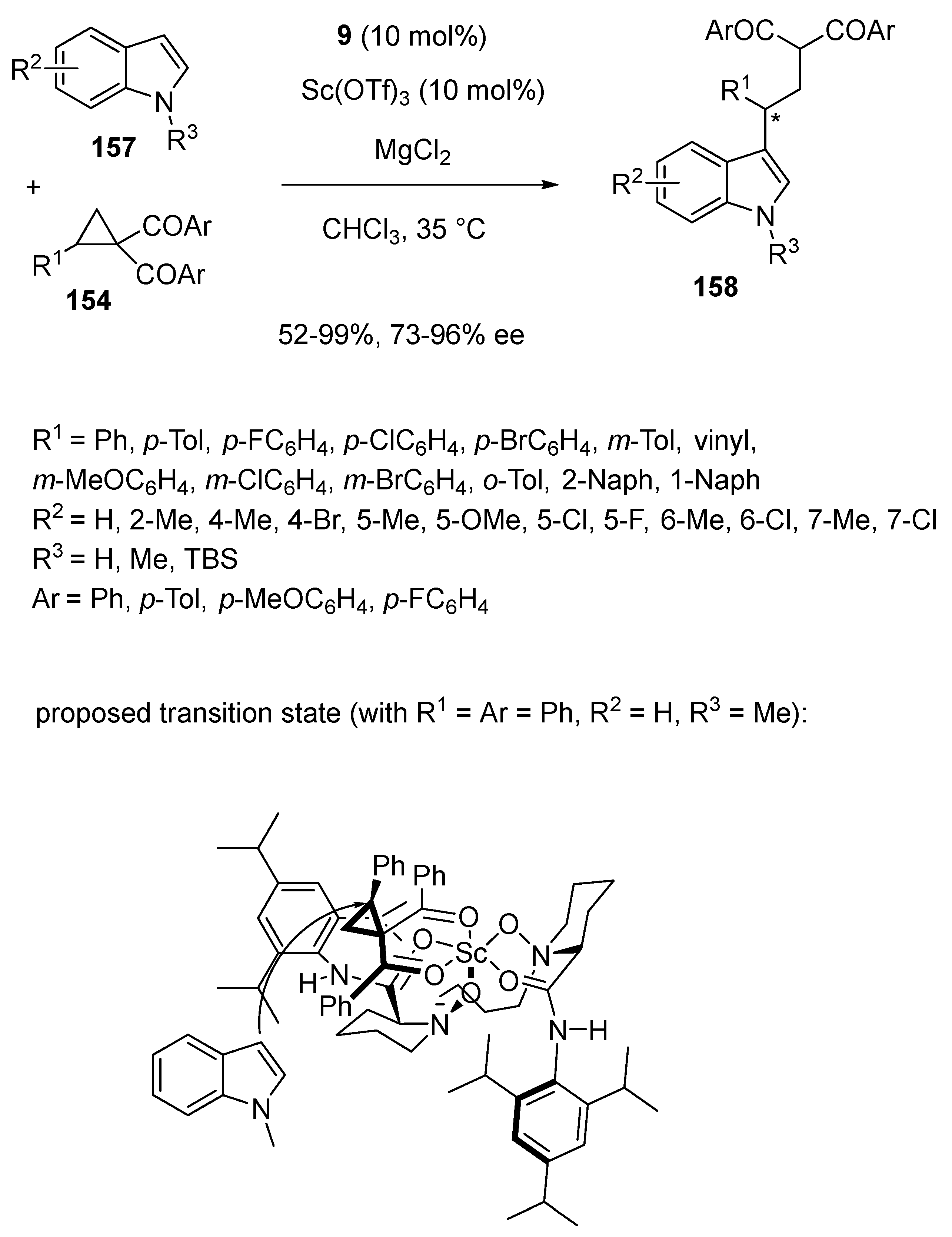
- Chang, F.; Lin, L.; Xia, Y.; Zhang, H.; Dong, S.; Liu, X.; Feng, X. Scandium-Catalyzed Enantioselective Ring-Opening of Cyclopropyl Ketones with Indoles. Adv. Synth. Catal. 2018, 360, 2608–2612. [Google Scholar] [CrossRef]
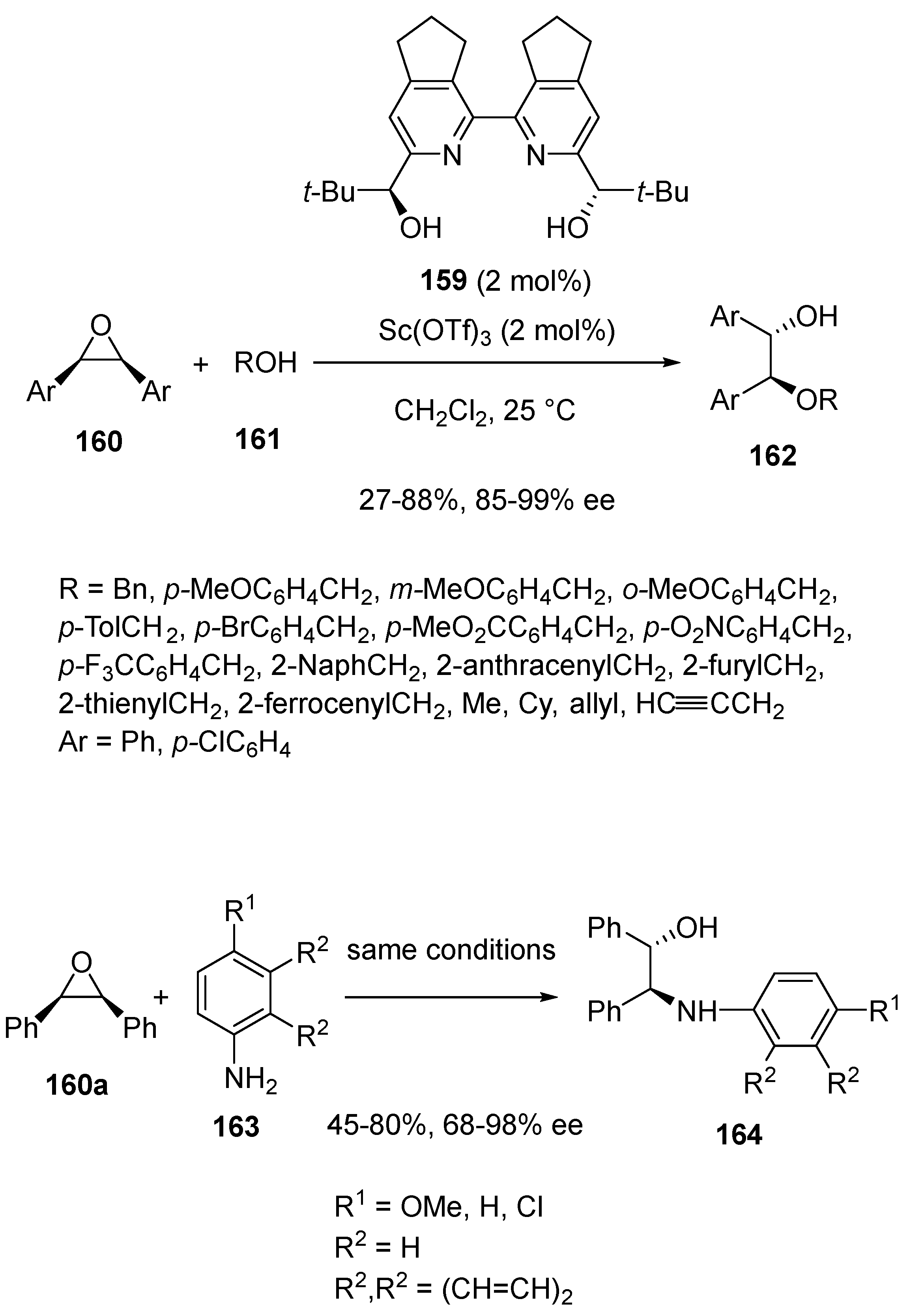
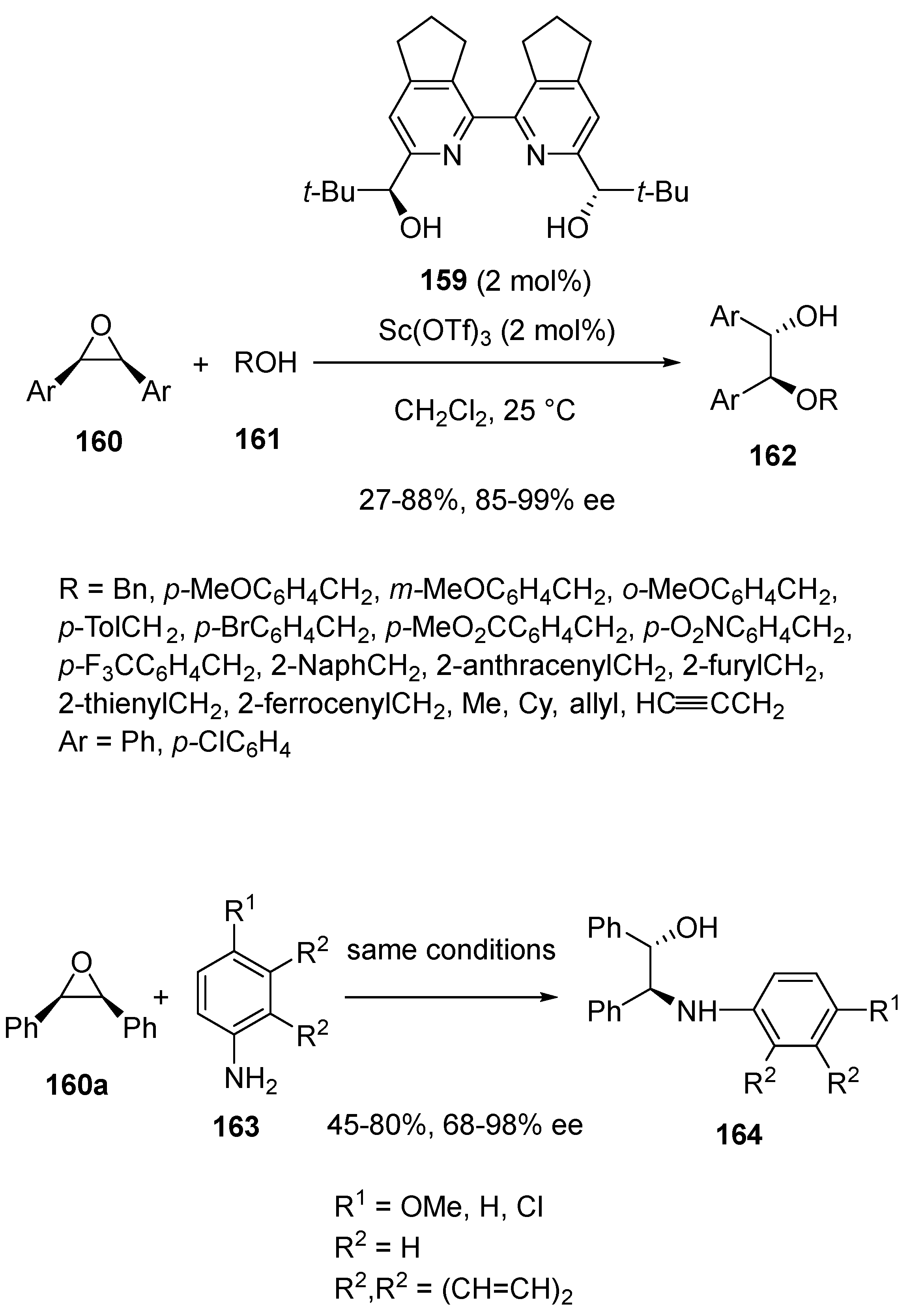
- Malatinec, S.; Bednařova, E.; Tanaka, H.; Kotora, M. Highly Enantioselective Ring-Opening of meso-Epoxides with O- and N-Nucleophiles Catalyzed by a Chiral Sc(III)/bipyridine Complex. Eur. J. Org. Chem. 2021, 2021, 1249–1257. [Google Scholar] [CrossRef]
- Kitanosono, T.; Lu, F.; Masuda, K.; Yamashita, Y.; Kobayashi, S. Efficient Recycling of Catalyst-Solvent Couples from Lewis Acid-Catalyzed Asymmetric Reactions in Water. Angew. Chem. Int. Ed. 2022, 61, e202202335. [Google Scholar] [CrossRef]
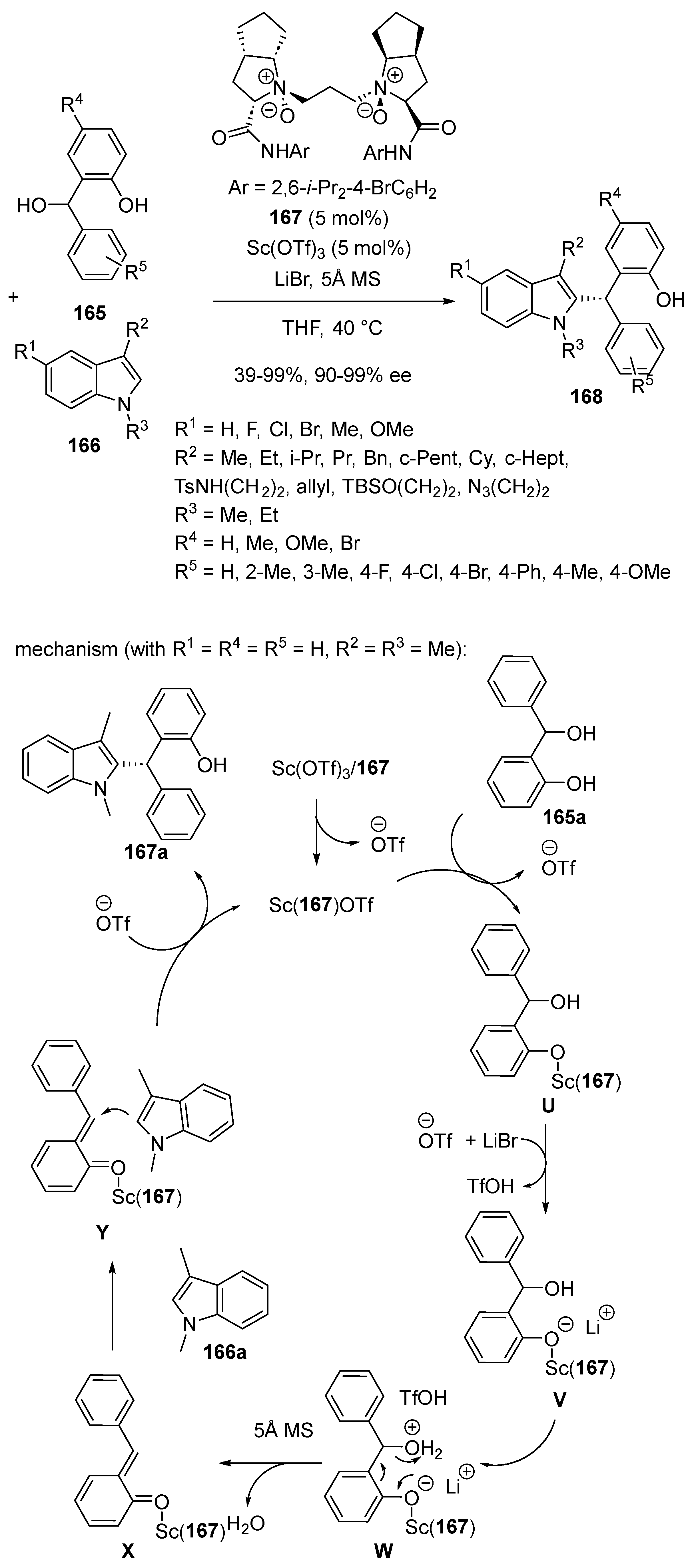
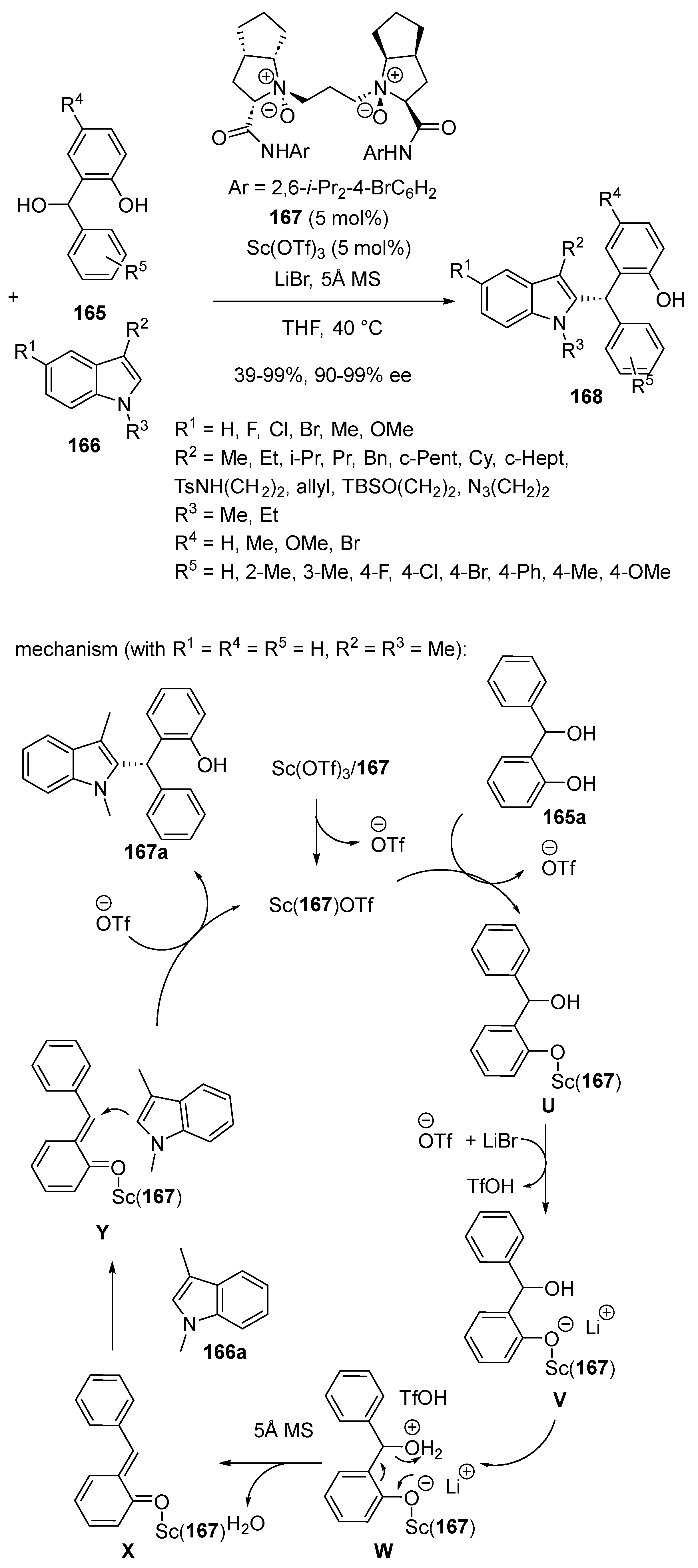
- Zheng, J.; Lin, L.; Dai, L.; Yuan, X.; Liu, X.; Feng, X. Chiral N,N′-Dioxide–Scandium(III) Complex-Catalyzed Asymmetric Friedel–Crafts Alkylation Reaction of ortho-Hydroxybenzyl Alcohols with C3-Substituted N-Protected Indoles. Chem. Eur. J. 2016, 22, 18254–18258. [Google Scholar] [CrossRef]
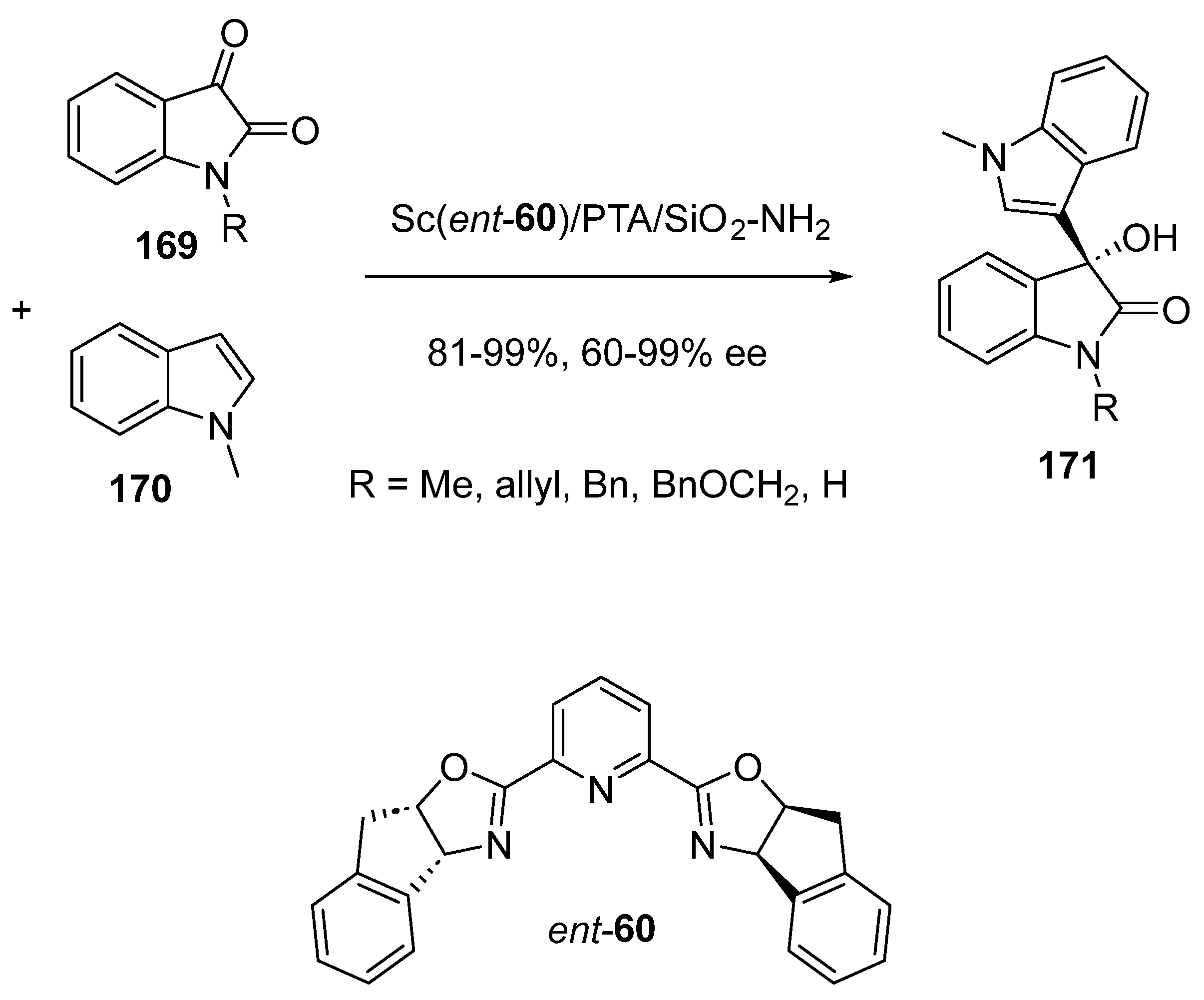
- Saito, Y.; Kobayashi, S. Chiral Heterogeneous Scandium Lewis Acid Catalysts for Continuous-Flow Enantioselective Friedel–Crafts Carbon–Carbon Bond-Forming Reactions. Angew. Chem. Int. Ed. 2021, 60, 26566–26570. [Google Scholar] [CrossRef]
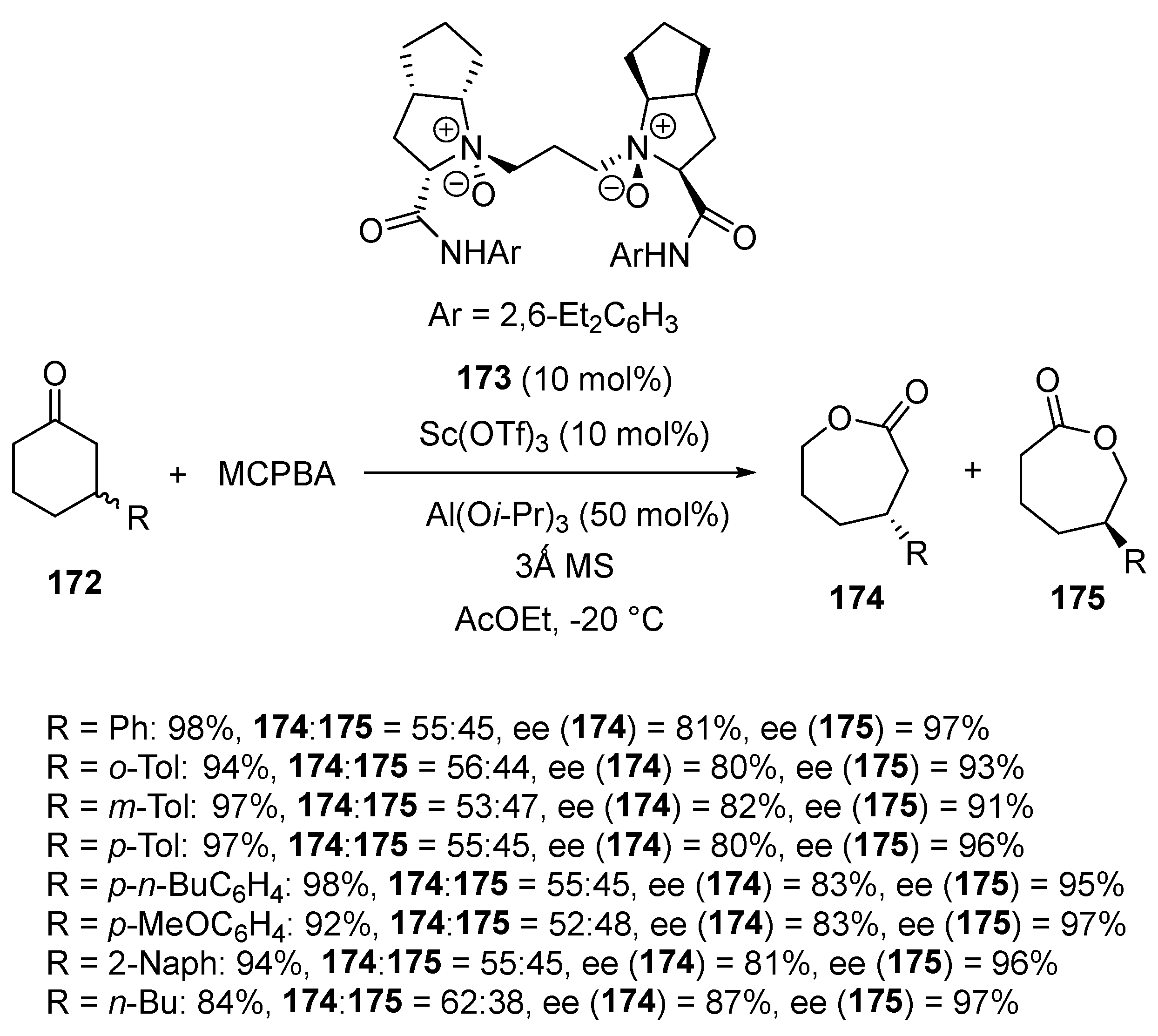
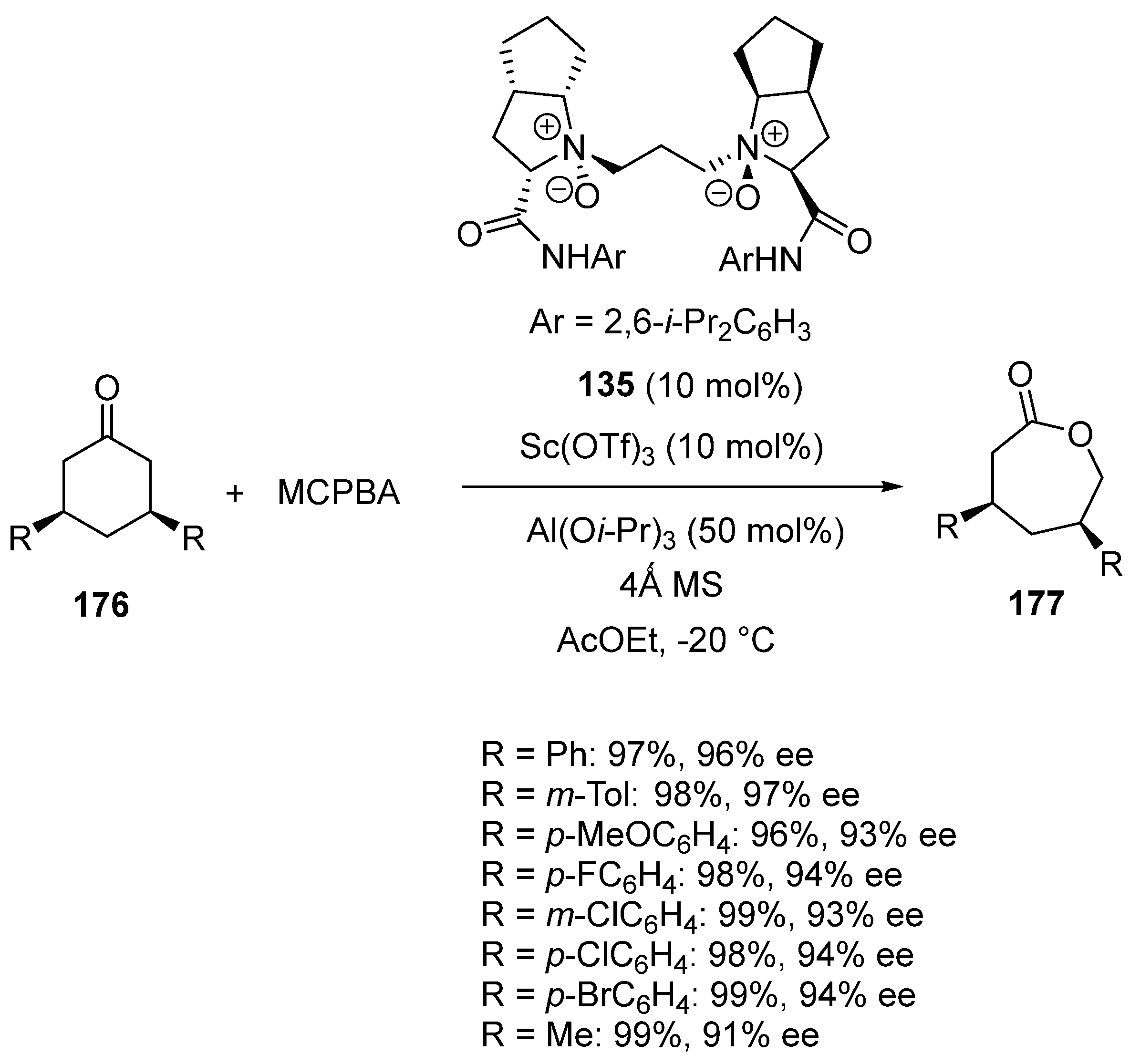
- Wu, W.; Cao, W.; Hu, L.; Su, Z.; Liu, X.; Feng, X. Asymmetric Baeyer–Villiger oxidation: Classical and parallel kinetic resolution of 3-substituted cyclohexanones and desymmetrization of meso-disubstituted cycloketones. Chem. Sci. 2019, 10, 7003–7008. [Google Scholar] [CrossRef]
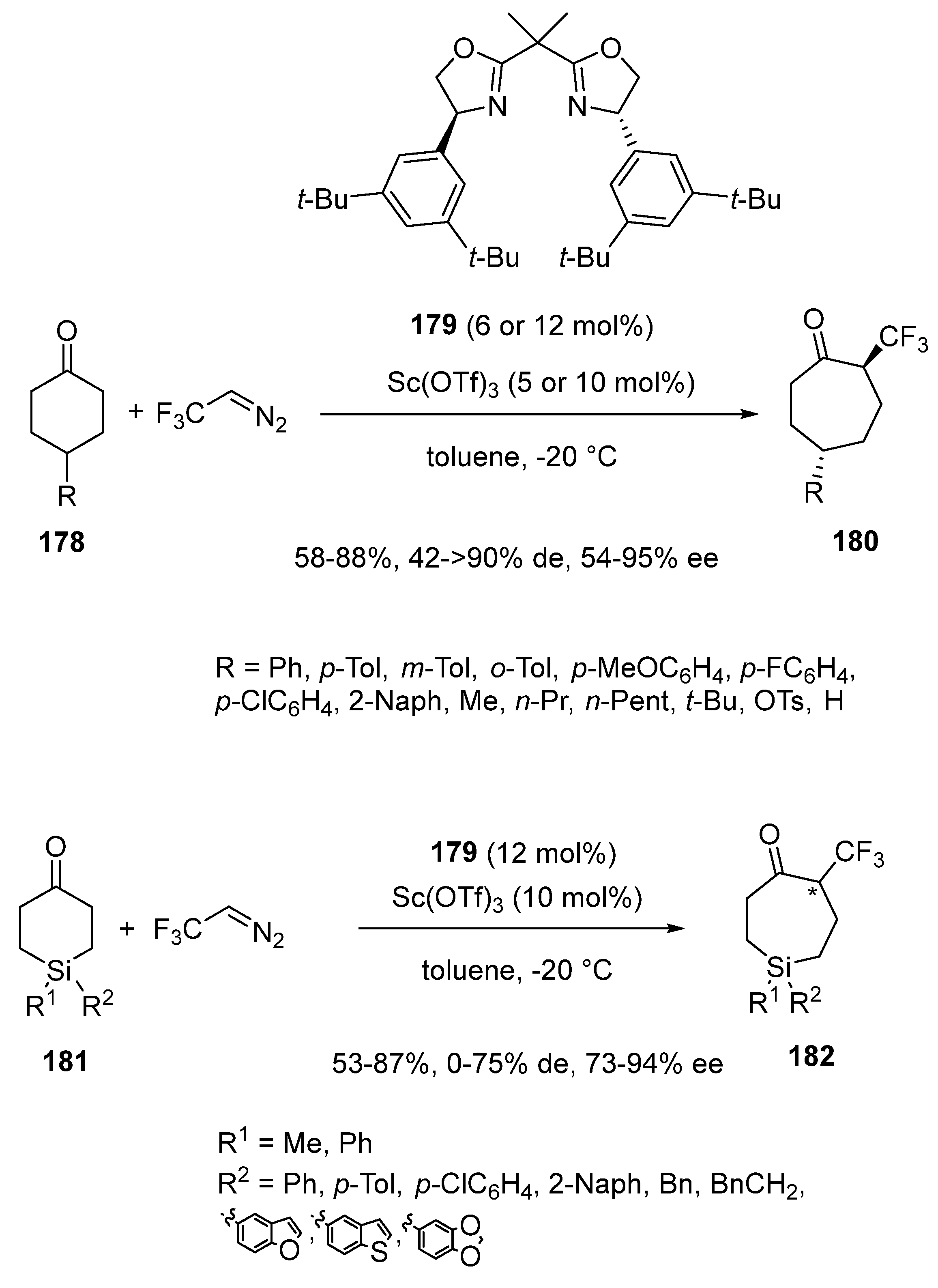
- Li, S.-S.; Sun, S.; Wang, J. Catalytic Asymmetric Homologation of 4-Substituted Cyclohexanones with CF3CHN2: Enantioselective Synthesis of α-Trifluoromethyl Cycloheptanones. Angew. Chem. Int. Ed. 2022, 61, e202115098. [Google Scholar] [CrossRef] [PubMed]
- Tenberge, M.; Wahl, J.M. Lewis Acid Catalysed Asymmetric One-Carbon Ring-Expansion of Prochiral Cyclobutanones. Synthesis 2023, 55, 892–898. [Google Scholar] [CrossRef]
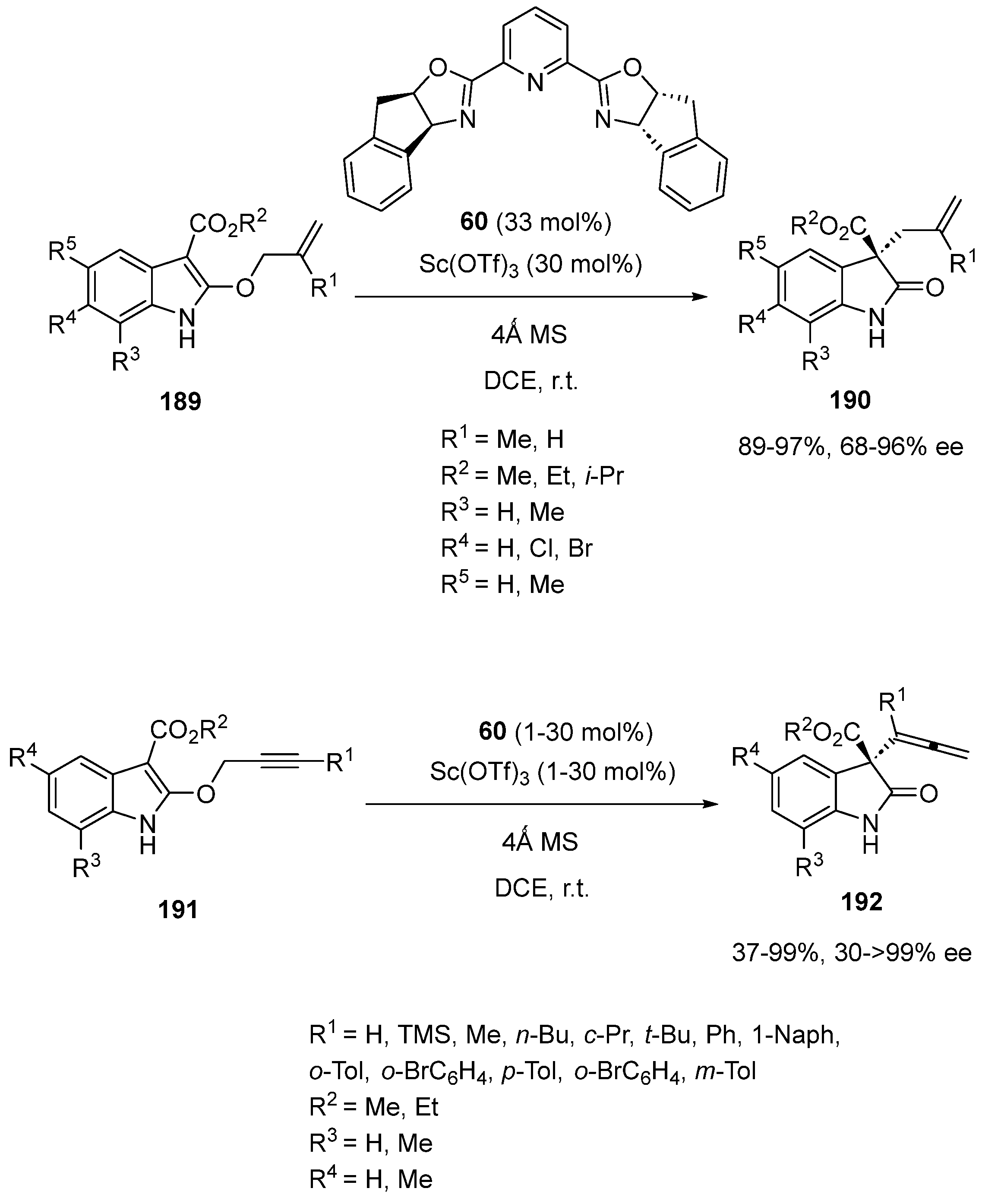
- Lai, Z.-W.; Liu, C.; Sun, H.; You, S.-L. Asymmetric Synthesis of 3-Allyloxindoles and 3-Allenyloxindoles by Scandium(III)-Catalyzed Claisen Rearrangement Reactions. Chin. J. Chem. 2017, 35, 1512–1516. [Google Scholar] [CrossRef]
- Dai, L.; Li, X.; Zeng, Z.; Dong, S.; Zhou, Y.; Liu, X.; Feng, X. Catalytic Asymmetric Acyloin Rearrangements of α-Ketols, α-Hydroxy Aldehydes, and α-Iminols by N,N′-Dioxide–Metal Complexes. Org. Lett. 2020, 22, 5041–5045. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Liu, Y.; Lin, L.; Zhang, Y.; Liu, X.; Feng, X. Kinetic Resolution of 2H-Azirines by Asymmetric Imine Amidation. Angew. Chem. Int. Ed. 2016, 55, 10098–10101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liao, Y.; Liu, X.; Xu, L.; Lin, L.; Feng, X. Catalytic asymmetric hydroxylative dearomatization of 2-naphthols: Synthesis of lacinilene derivatives. Chem. Sci. 2017, 8, 6645–6649. [Google Scholar] [CrossRef]
- Zhang, H.; Yao, Q.; Lin, L.; Xu, C.; Liu, X.; Feng, X. Catalytic Asymmetric Epoxidation of Electron-Deficient Enynes Promoted by Chiral N,N′-Dioxide-Scandium(III) Complex. Adv. Synth. Catal. 2017, 359, 3454–3459. [Google Scholar] [CrossRef]
- Wang, Z.; Lin, L.; Zhou, P.; Liua, X.; Feng, X. Chiral N,N′-dioxide-Sc(NTf2)3 complex-catalyzed asymmetric bromoamination of chalones with N-bromosuccinimide as both bromine and amide source. Chem. Commun. 2017, 56, 3462–3465. [Google Scholar] [CrossRef]
- Tang, Q.; Lin, L.; Ji, J.; Hu, H.; Liu, X.; Feng, X. Catalytic Asymmetric Direct Vinylogous Aldol Reaction of Isatins with β,γ-Unsaturated Butenolides. Chem. Eur. J. 2017, 23, 16447–16451. [Google Scholar] [CrossRef]
- Zhan, G.; Teng, H.-L.; Luo, Y.; Lou, S.-J.; Nishiura, M.; Hou, Z. Enantioselective Construction of Silicon-Stereogenic Silanes by Scandium-Catalyzed Intermolecular Alkene Hydrosilylation. Angew. Chem. Int. Ed. 2018, 57, 12342–12346. [Google Scholar] [CrossRef]
- He, C.; Cao, W.; Zhang, J.; Ge, S.; Feng, X. Chiral N,N′-Dioxide/Scandium(III)-Catalyzed Asymmetric Alkylation of N-Unprotected 3-Substituted Oxindoles. Adv. Synth. Catal. 2018, 360, 4301–4305. [Google Scholar] [CrossRef]
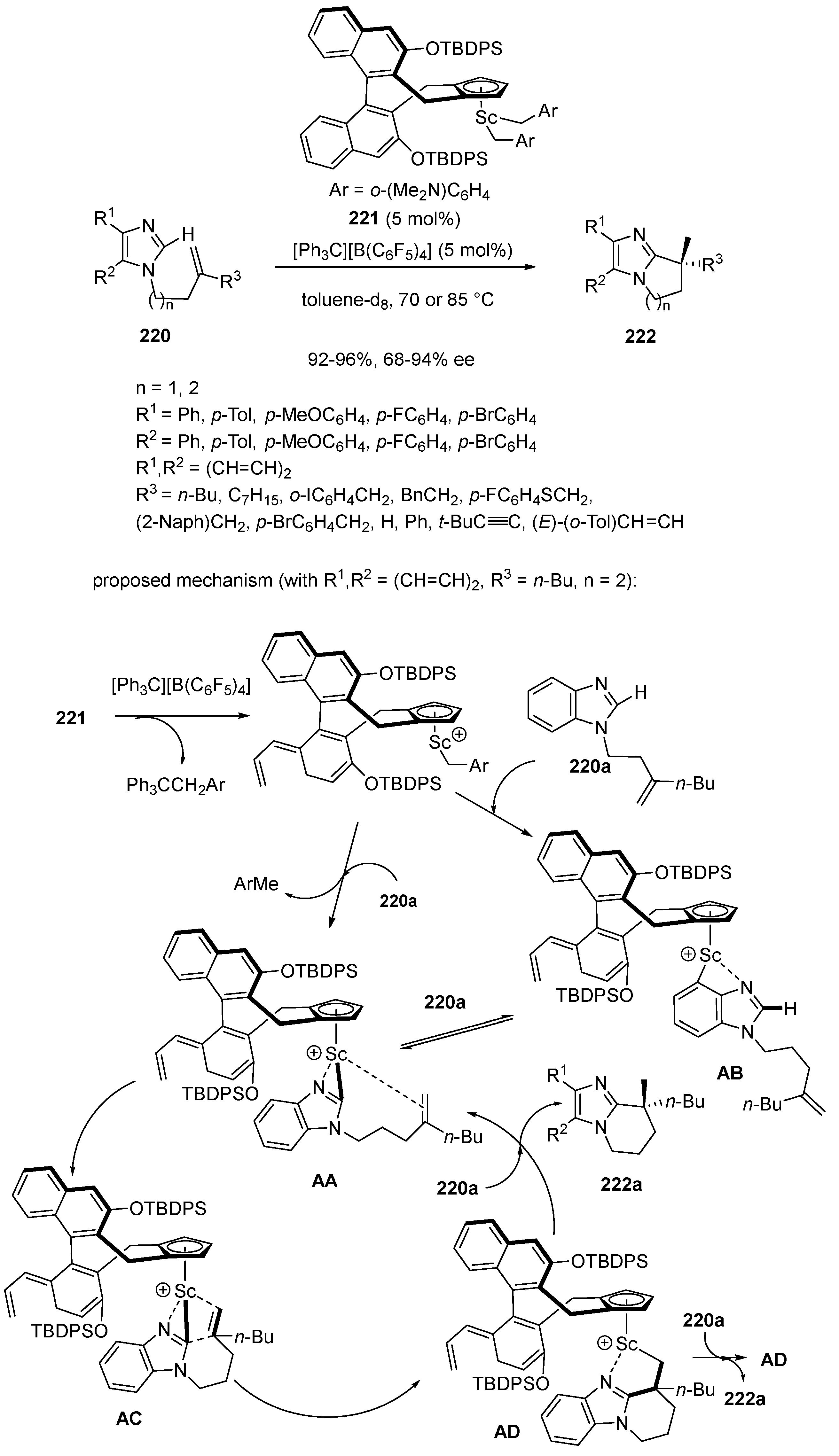
- Lou, S.-J.; Mo, Z.; Nishiura, M.; Hou, Z. Construction of All-Carbon Quaternary Stereocenters by Scandium-Catalyzed Intramolecular C–H Alkylation of Imidazoles with 1,1-Disubstituted Alkenes. J. Am. Chem. Soc. 2020, 142, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
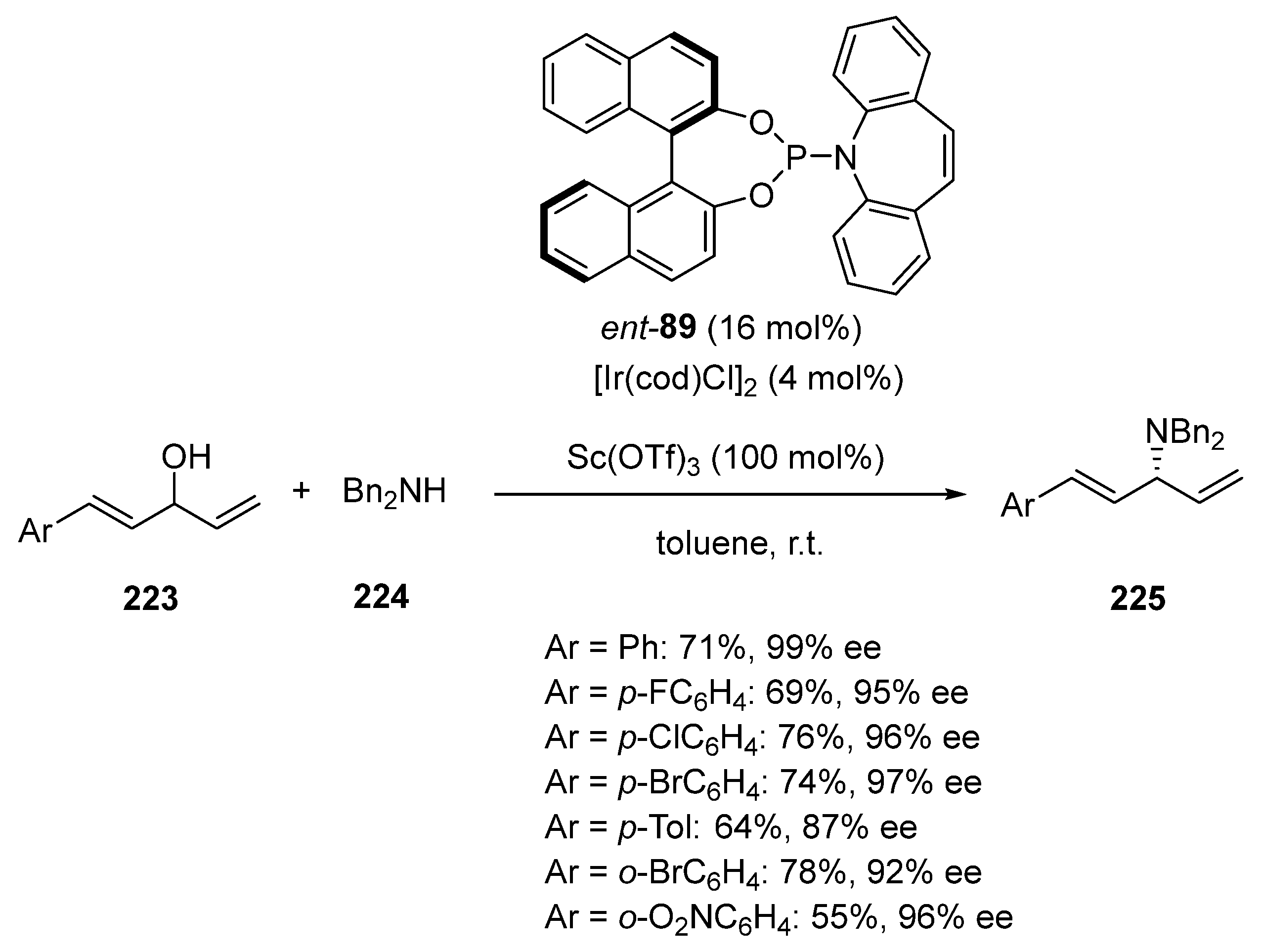
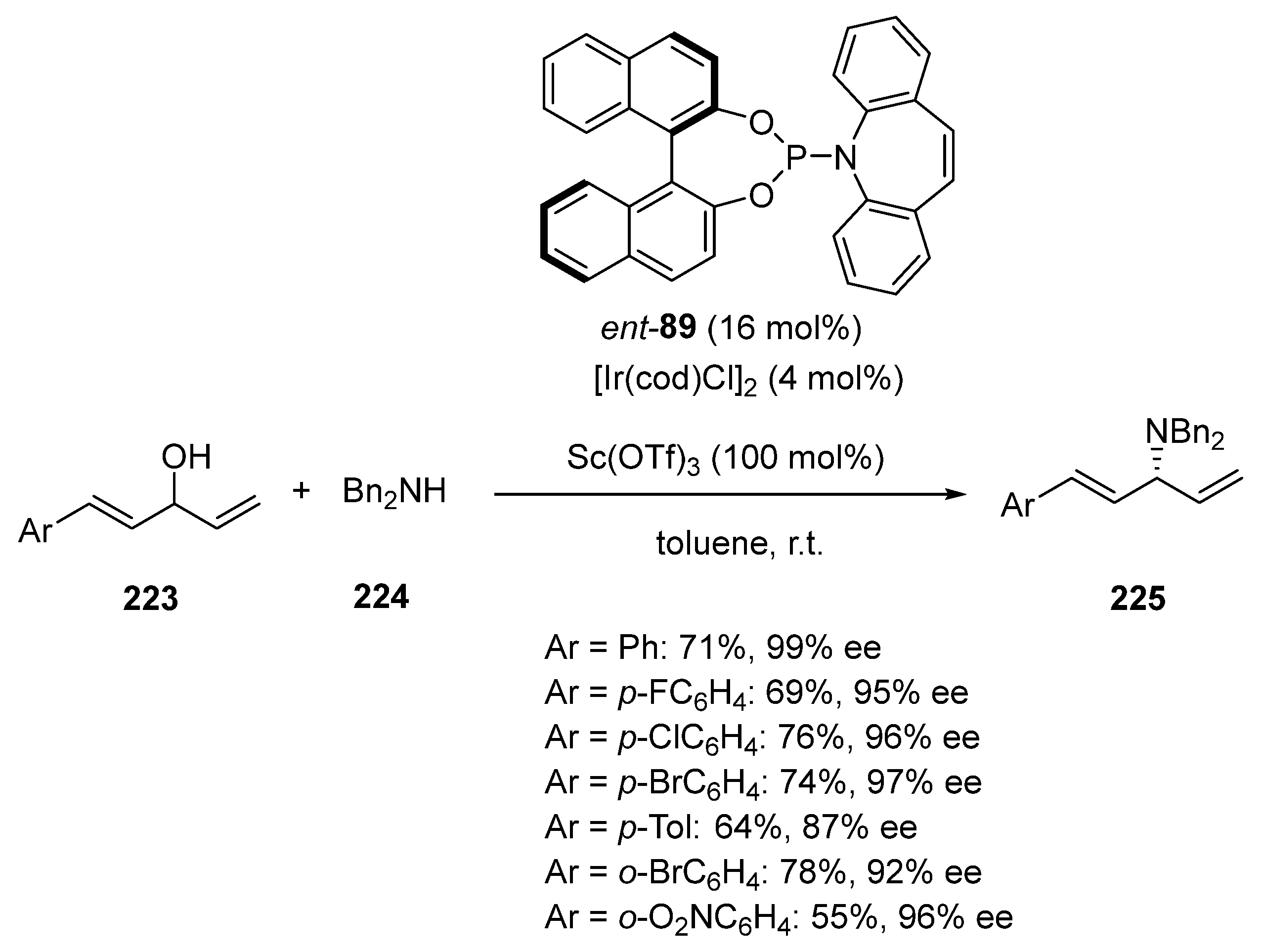
- Tang, S.; Li, Z.; Shao, Y.; Sun, J. Ir-Catalyzed Regiocontrolled Allylic Amination of Di-/Trienyl Allylic Alcohols with Secondary Amines. Ir-Catalyzed Regiocontrolled Allylic Amination of Di-/Trienyl Allylic Alcohols with Secondary Amines. Org. Lett. 2019, 21, 7228–7232. [Google Scholar] [CrossRef] [PubMed]
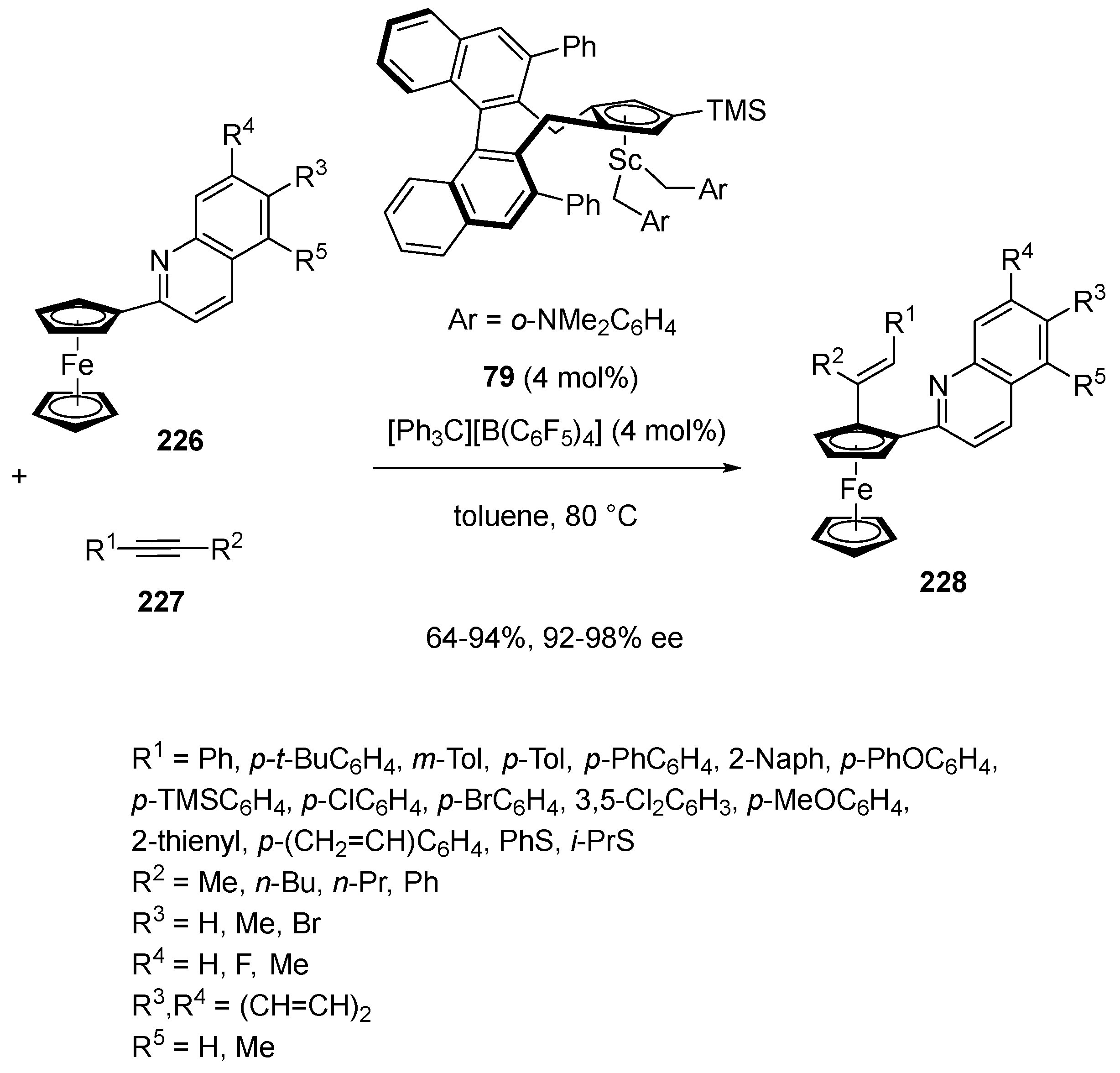
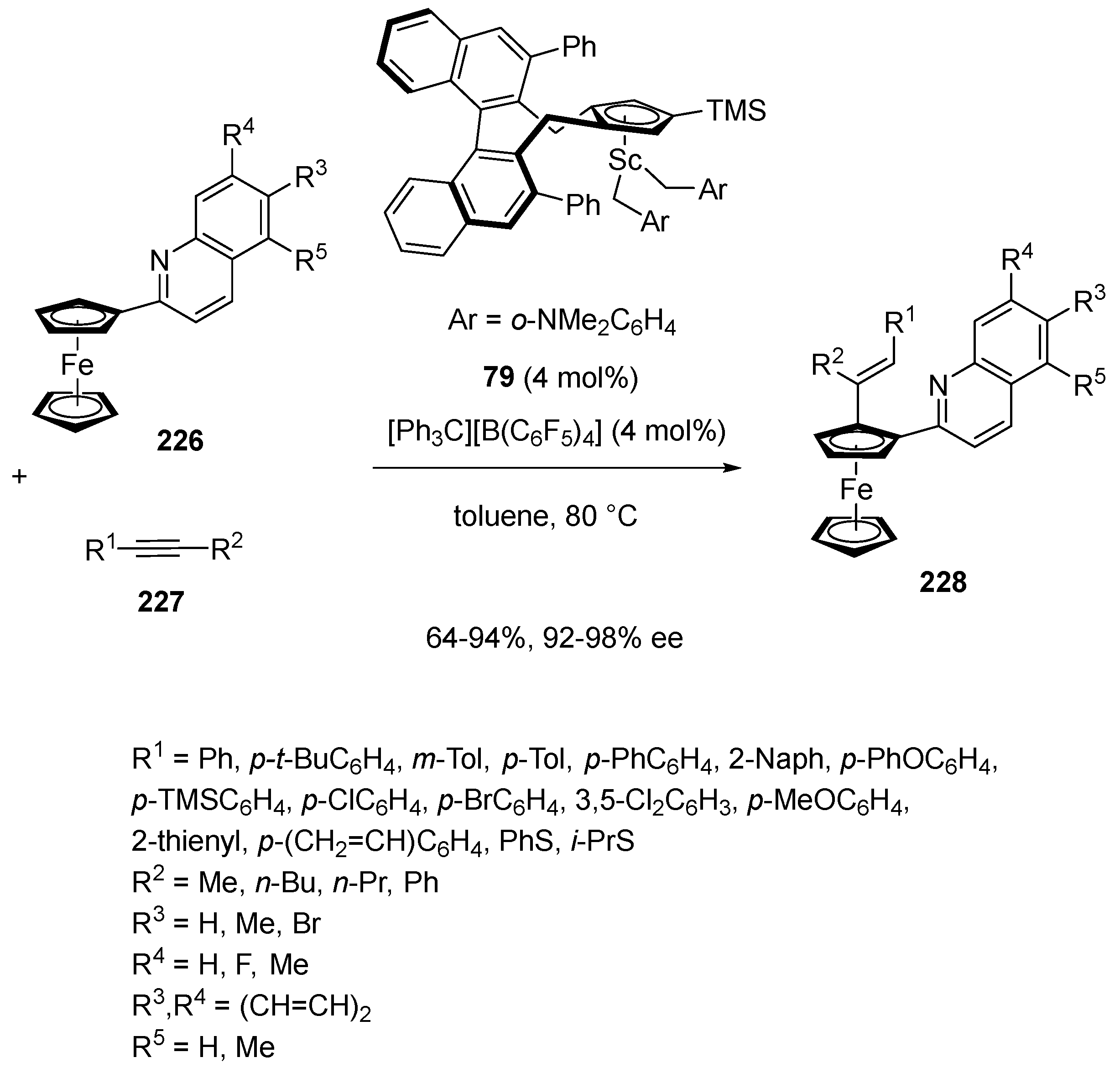
- Lou, S.-J.; Zhuo, Q.; Nishihura, M.; Luo, G.; Hou, Z. Enantioselective C–H Alkenylation of Ferrocenes with Alkynes by Half-Sandwich Scandium Catalyst. J. Am. Chem. Soc. 2021, 143, 2470–2476. [Google Scholar] [CrossRef]






















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellissier, H. Recent Developments in Enantioselective Scandium-Catalyzed Transformations. Chemistry 2024, 6, 98-152. https://doi.org/10.3390/chemistry6010007
Pellissier H. Recent Developments in Enantioselective Scandium-Catalyzed Transformations. Chemistry. 2024; 6(1):98-152. https://doi.org/10.3390/chemistry6010007
Chicago/Turabian StylePellissier, Hélène. 2024. "Recent Developments in Enantioselective Scandium-Catalyzed Transformations" Chemistry 6, no. 1: 98-152. https://doi.org/10.3390/chemistry6010007
APA StylePellissier, H. (2024). Recent Developments in Enantioselective Scandium-Catalyzed Transformations. Chemistry, 6(1), 98-152. https://doi.org/10.3390/chemistry6010007