
Ruthenium-p-Cymene Complexes Incorporating Substituted Pyridine–Quinoline Ligands with –Br (Br-Qpy) and –Phenoxy (OH-Ph-Qpy) Groups for Cytotoxicity and Catalytic Transfer Hydrogenation Studies: Synthesis and Characterization
 , , , , , and
, , , , , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Synthesis and Characterization
2.1.1. Synthesis of [Ru(η6-p-cymene)(Br-Qpy)Cl][Cl] (1a)
2.1.2. Synthesis of [Ru(η6-p-cymene)(Br-Qpy)Cl][PF6] (1b)
2.1.3. Synthesis of [Ru(η6-p-cymene)(OH-Ph-Qpy)Cl][Cl] (2a)
2.1.4. Synthesis of [Ru(η6-p-cymene)(OH-Ph-Qpy)Cl][PF6] (2b)
2.2. Single-Crystal X-ray Structural Determination
2.3. Biological Evaluation
2.3.1. Cell Lines
2.3.2. MTT Assay
2.4. Transfer Hydrogenation of Benzophenone Catalyzed by 1a, 1b
3. Results and Discussion
3.1. Chemistry
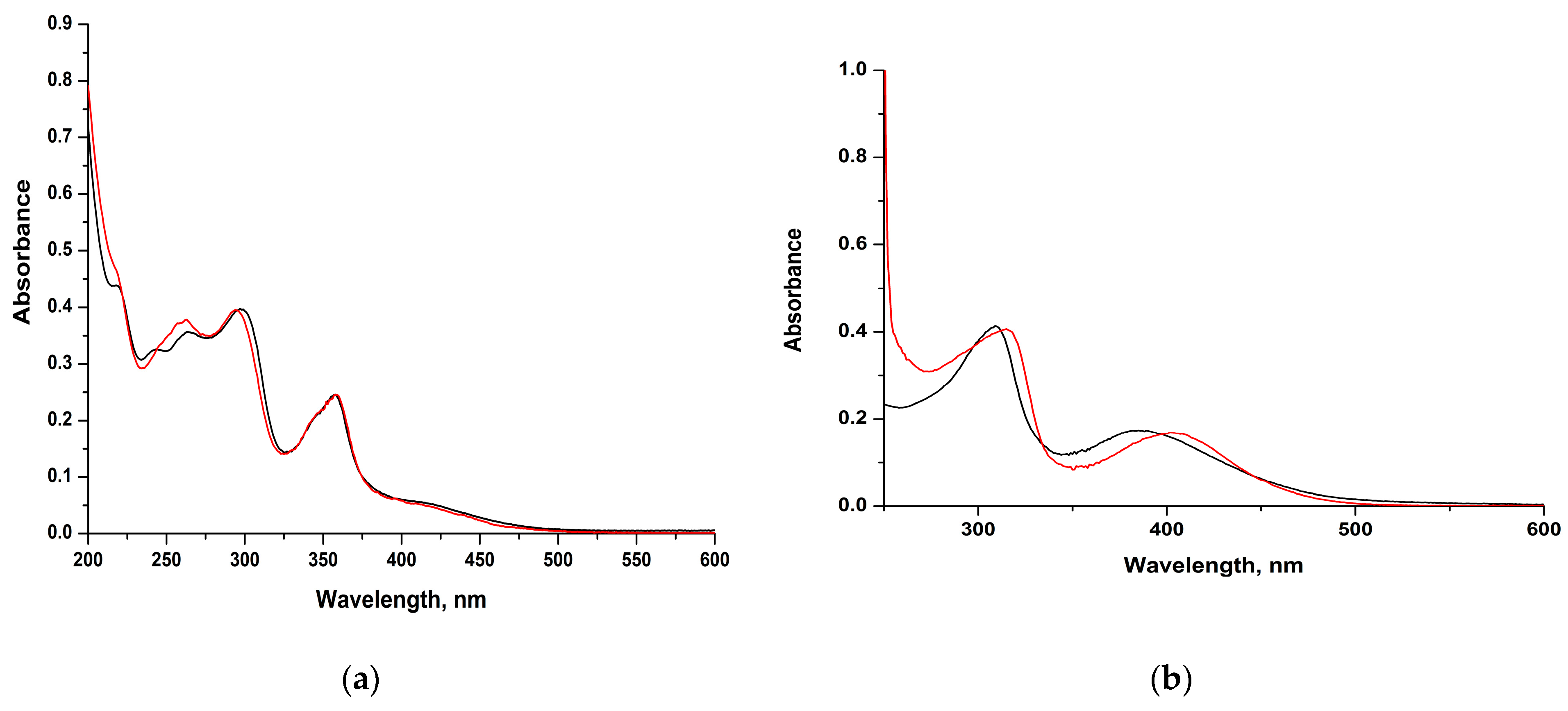
Electronic Spectra
3.2. Evaluation of Biological Activity
Cell Viability Assay
3.3. Catalytic Transfer Hydrogenation Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Madec, H.; Figueiredo, F.; Cariou, K.; Roland, S.; Sollogoub, M.; Gasser, G. Metal complexes for catalytic and photocatalytic reactions in living cells and organisms. Chem. Sci. 2023, 14, 409–442. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Zhao, Z.-Z.; Bo, H.-B.; Hao, X.-J.; Wang, J.-Q. Applications of Ruthenium Complex in Tumor Diagnosis and Therapy. Front. Pharmacol. 2018, 9, 1323. [Google Scholar] [CrossRef]
- Peacock, A.F.; Sadler, P.J. Medicinal organometallic chemistry: Designing metal arene complexes as anticancer agents. Chem. Asian J. 2008, 13, 1890–1899. [Google Scholar] [CrossRef]
- Nazarov, A.A.; Hartinger, C.G.; Dyson, P.J. Opening the lid on piano-stool complexes: An account of ruthenium(II)-arene complexes with medicinal applications. J. Organomet. Chem. 2014, 751, 251–260. [Google Scholar] [CrossRef]
- Bashir, M.; Mantoo, I.A.; Arjmand, F.; Tabassum, S.; Yousuf, I. An overview of advancement of organoruthenium(II) complexes as prospective anticancer agents. Coord. Chem. Rev. 2023, 487, 215169. [Google Scholar] [CrossRef]
- Chancha, S.; Sarkarb, S.; Mukhopadhyay, S. RutheniumIJII)–arene complexes as anti-metastatic agents, and related techniques. RSC Med. Chem. 2022, 13, 22–38. [Google Scholar] [CrossRef]
- Hafeez, J.; Bilal, M.; Rasool, N.; Hafeez, U.; Ali Shah, S.A.; Imran, S.; Zakaria, Z.A. Synthesis of ruthenium complexes and their catalytic applications: A review. Arab. J. Chem 2022, 15, 104165. [Google Scholar] [CrossRef]
- Samec, J.S.M.; Bäckvall, J.-E.; Andersson, P.G.; Brandt, P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006, 35, 237–248. [Google Scholar] [CrossRef]
- Muñoz-García, A.B.; Benesperi, I.; Boschloo, G.; Concepcion, J.J.; Delcamp, J.H.; Gibson, E.A.; Meyer, G.J.; Pavone, M.; Pettersson, H.; Hagfeldt, A.; et al. Dye-sensitized solar cells strike back. Chem. Soc. Rev. 2021, 50, 12450–12550. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Xu, G.; Li, C.; Chi, C.; Wu, L.Y.; Sun, Y.Y.; Zhao, J.; Xia, X.-H.; Gou, S.H. A supramolecular photosensitizer derived from an Arene-Ru(II) complex self-assembly for NIR activated photodynamic and photothermal therapy. Nat. Commun. 2022, 13, 3064. [Google Scholar] [CrossRef]
- Sánchez-González, P.D.; López-Hernández, F.J.; López-Novoa, J.M.; Morales, A.I. An integrative view of the pathophysiological events leading to cisplatin nephrotoxicity. Crit. Rev. Toxicol. 2011, 41, 803–821. [Google Scholar] [CrossRef]
- Bhhatarai, B.; Gramatica, P. Per- and Polyfluoro Toxicity (LC50 Inhalation) Study in Rat and Mouse Using QSAR Modeling. Chem. Res. Toxicol. 2010, 23, 528–539. [Google Scholar] [CrossRef]
- Cubeddu, L.X.; Hoffmann, I.S.; Fuenmayor, N.T.; Finn, A.L. Efficacy of Ondansetron (Gr 38032F) and the Role of Serotonin in Cisplatin-Induced Nausea and Vomiting. Engl. J. Med. 1990, 322, 810–816. [Google Scholar] [CrossRef]
- Adhikari, S.; Nath, P.; Das, A.; Datta, A.; Baildya, N.; Duttaroy, A.K.; Pathak, S. A review on metal complexes and its anti-cancer activities: Recent updates from in vivo studies. Biomed. Pharmacother. 2024, 171, 116211. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H. A Phase I and pharmacological study withimidazolium-trans-DMSO-imidazole-tetrachloro ruthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A new redox-active anticancer agent–preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2150. [Google Scholar] [CrossRef]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II Study with Ruthenium Compound NAMI-A and Gemcitabine in Patients with Non-Small Cell Lung Cancer after First Line Therapy. Invest. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef]
- Alessio, E. Thirty Years of the Drug Candidate NAMI-A and the Myths in the Field of Ruthenium Anticancer Compounds: A Personal Perspective. Eur. J. Inorg. Chem. 2017, 2017, 1549–1560. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Bergerbd, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, C.Y.; Nam, T.G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des Devel Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef]
- Allison, M.; Caramés-Méndez, P.; Hofmann, B.J.; Pask, C.M.; Phillips, R.M.; Lord, R.M.; McGowan, P.C. Cytotoxicity of Ruthenium(II) Arene Complexes Containing Functionalized Ferrocenyl β-Diketonate Ligands. Organometallics 2023, 42, 1869–1881. [Google Scholar] [CrossRef]
- Mukherjee, A.; Acharya, S.; Purkait, K.; Chakraborty, K.; Bhattacharjee, A.; Mukherjee, A. Effect of N, N Coordination and Ru (II) Halide Bond in Enhancing Selective Toxicity of a Tyramine-Based Ru (II) p-cymene Complex. Inorg. Chem. 2020, 59, 6581–6594. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.L. [Ru(η6-p-cymene)Cl2 (pta)] (pta = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane): A water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun. 2001, 15, 1396–1397. [Google Scholar]
- Murray, S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Danopoulos, A.A.; Winston, S.; Motherwell, W.B. Stable N-Functionalised ‘Pincer’ Bis Carbene Ligands and Their Ruthenium Complexes; Synthesis and Catalytic Studies. Chem. Commun. 2002, 1376–1377. [Google Scholar]
- Canivet, J.; Labat, G.; Stoeckli-Evans, H.; Süss-Fink, G. Water Soluble Arene Ruthenium Complexes Containing a trans-1,2-Diaminocyclohexane Ligand as Enantioselective Transfer Hydrogenation Catalysts in Aqueous Solution. Eur. J. Inorg. Chem. 2005, 4493–4500. [Google Scholar] [CrossRef]
- Zacharopoulos, N.; Koukoulakis, K.; Bakeas, E.; Philippopoulos, A.I. A 2-(2′-pyridyl)quinoline ruthenium(II) complex as an active catalyst for the transfer hydrogenation of ketones. Open Chemistry 2016, 14, 308–315. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-Catalyzed C–H Bond Activation and Functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, K.; Yu, Z. Ruthenium(III)-Catalyzed β-Alkylation of Secondary Alcohols with Primary Alcohols. Organometallics 2016, 35, 1251–1256. [Google Scholar] [CrossRef]
- Copéret, C.; Berkson, Z.J.; Chan, K.W.; Silva, J.J.; Gordon, C.P.; Pucino, M.; Zhizhko, P.A. Olefin metathesis: What have we learned about homogeneous and heterogeneous catalysts from surface organometallic chemistry? Chem. Sci. 2021, 12, 3092–3115. [Google Scholar] [CrossRef] [PubMed]
- Selvi, G.; Ozdemir, F.A.; Aykutoglu, G.; Özdemir, N.; Şerbetçi, Z.; Dinçer, M.; Dayan, O. (2020). Synthesis, catalytic, cytotoxic, and antibacterial properties of new Ru(II) and Pd(II) complexes bearing bidentate Schiff base ligand. Inorg. Nano-Met. Chem. 2020, 51, 1697–1706. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric Transfer Hydrogenation Catalyzed by chiral Ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Baidilov, D.; Hayrapetyan, D.; Khalimon, A.Y. Recent advances in homogeneous base-metal-catalyzed transfer hydrogenation reactions. Tetrahedron 2021, 98, 132435. [Google Scholar] [CrossRef]
- Taleb, B.; Jahjah, R.; Cornu, D.; Bechelany, M.; Al Ajami, M.; Kataya, G.; Hijazi, A.; El-Dakdouki, M.H. Exploring Hydrogen Sources in Catalytic Transfer Hydrogenation: A Review of Unsaturated Compound Reduction. Molecules 2023, 28, 7541. [Google Scholar] [CrossRef]
- Tyagi, N.; Borah, G.; Patel, P.; Ramaiah, D. Ruthenium–An Element Loved by Researchers. In Recent Advances in Ru Catalyzed Transfer Hydrogenation and Its Future Perspectives, 1st ed.; Ishida, H., Ed.; IntechOpen: London, UK, 2022; pp. 1–24. [Google Scholar] [CrossRef]
- Matveevskaya, V.V.; Pavlov, D.I.; Sukhikh, T.S.; Gushchin, A.L.; Ivanov, A.Y.; Tennikova, T.B.; Sharoyko, V.V.; Baykov, S.V.; Benassi, E.; Potapov, A.S. Arene-Ruthenium(II) Complexes Containing 11H-Indeno [1,2-b]quinoxalin-11-one Derivatives and Tryptanthrin-6-oxime: Synthesis, Characterization, Cytotoxicity, and Catalytic Transfer Hydrogenation of Aryl Ketones. ACS Omega 2020, 5, 11167–11179. [Google Scholar] [CrossRef]
- Peppas, A.; Papadaki, E.; Schnakenburg, G.; Magrioti, V.; Philippopoulos, A.I. Heteroleptic copper(I) complexes incorporating sterically demanding diazabutadiene ligands (DABs). Synthesis, spectroscopic characterization and solid state structural analysis. Polyhedron 2019, 171, 412–422. [Google Scholar] [CrossRef]
- Zacharopoulos, N.; Kolovou, E.; Peppas, A.; Koukoulakis, K.; Bakeas, E.; Schnakenburg, G.; Philippopoulos, A.I. Pyridyl based ruthenium(II) catalyst precursors and their dihydride analogues as the catalytically active species for the transfer hydrogenation of ketones. Polyhedron 2018, 154, 27–38. [Google Scholar] [CrossRef]
- Margariti, A.; Papakonstantinou, V.D.; Stamatakis, G.M.; Demopoulos, C.A.; Schnakenburg, G.; Andreopoulou, A.K.; Giannopoulos, G.; Kallitsis, J.K.; Philippopoulos, A.I. Substituted pyridine-quinoline ligands as building blocks for neutral rhodium(III) complexes. Synthesis, structural characterization studies and anti-platelet activity towards the Platelet-Activating Factor (PAF). Polyhedron 2020, 178, 114336. [Google Scholar] [CrossRef]
- Peppas, A. Synthesis and Characterization of Homoleptic Copper(I) Complexes. Application in Third Generation Solar Cells (Gratzel Type). Master’s Thesis, National and Kapodistrian University of Athens, Athens, Greece, 2015. [Google Scholar]
- Peppas, A.; Sokalis, D.; Perganti, D.; Schnakenburg, G.; Falaras, P.; Philippopoulos, A.I. Sterically demanding pyridine-quinoline anchoring ligands as building blocks for copper(I)-based dye-sensitized solar cell (DSSC) complexes. Dalton Trans. 2022, 51, 15049–15066. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.A.; Huang, T.N.; Matheson, T.W.; Smith, K. Inorganic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 1982; Volume 21, pp. 74–77. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallorg. C. Struct Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallorg. A 2008, 64, 112. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Cryst. 1990, A46, 194–201. [Google Scholar] [CrossRef]
- Chen, X.; Qiu, D.; Ma, L.; Cheng, Y.; Geng, Y.; Xie, Z.; Wang, L. Synthesis, Crystal Structure, Spectroscopy and Electroluminescence of Zinc(II) Complexes Containing Bidentate 2-(2-pyridyl)quinoline Derivative Ligands. Transit. Met. Chem. 2006, 31, 639–644. [Google Scholar] [CrossRef]
- Laguna, E.M.; Olsen, P.M.; Sterling, M.D.; Eicher, J.F.; Rheingold, A.L.; Larsen, C.H. Structure and Properties of Neutral and Cationic Gold(III) Complexes from Substituted 2-(2′-Pyridyl)quinoline Ligands. Inorg. Chem. 2014, 53, 12231–12233. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, P.; Lu, R.; Qian, Q.; Lei, X.; Xiao, Q.; Huang, S.; Liu, L.; Huang, C.; Su, W. Synthesis, X-ray Diffraction Study, and Cytotoxicity of a Cationic p-Cymene Ruthenium Chloro Complex Containing a Chelating Semicarbazone Ligand. Z Anorg. Allg. Chem. 2013, 639, 943–946. [Google Scholar] [CrossRef]
- Dai, F.; Zhuang, Q.; Huang, G.; Deng, H.; Zhang, X. Infrared Spectrum Characteristics and Quantification of OH Groups in Coal. ACS Omega. 2023, 8, 17064–17076. [Google Scholar] [CrossRef]
- Rüther, T.; Woodward, C.P.; Jones, T.W.; Campbell, J.C.; Hebtin, Y.; Cordiner, R.L.; Dawson, R.E.; Diane, E.J.E.; Robinson Wilson, G.J. Synthesis, characterisation, and properties of p-cymene Ruthenium(II) tetracarboxylate bipyridine complexes [(η6-p-cymene)Ru(Rn,Rn′-tcbpy)Cl][Cl]. J. Organomet. Chem. 2016, 823, 136–146. [Google Scholar] [CrossRef]
- Fedor, A.M.; Toda, M.J. Investigating Hydrogen Bonding in Phenol Using Infrared Spectroscopy and Computational Chemistry. J. Chem. Educ. 2014, 91, 2191–2194. [Google Scholar] [CrossRef]
- Tsierkezos, N.G.; Ritter, U.; Philippopoulos, A.I.; Schröder, D. Electrochemical studies of the bis (triphenyl phosphine) ruthenium(II) complex, cis-[RuCl2(L)(PPh3)2], with L = 2-(2′-pyridyl)quinoxaline. J. Coord. Chem. 2010, 63, 3517–3530. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Chen, F.; Romero-Canelón, I.; Soldevila-Barreda, J.J.; Song, J.-I.; Coverdale, J.P.C.; Clarkson, G.J.; Kasparkova, J.; Habtemariam, A.; Wills, M.; Brabec, V.; et al. Transfer Hydrogenation and Antiproliferative Activity of Tethered Half-Sandwich Organoruthenium Catalysts. Organometallics 2018, 37, 1555–1566. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Omondi, B.; Friedrich, H.B. Crystal structure of η6-p-cymene-iodido-(N-isopropyl-1-(pyridin-2-yl)methanimine-κ2N,N′)ruthenium(II) hexafluorophosphate(V), C19H26IN2F6Ru. Z. Kristallogr. NCS 2020, 235, m485–m487. [Google Scholar] [CrossRef]
- Dömötör, O.; Pape, V.F.S.; May, N.V.; Szakacs, G.; Enyedy, E.A. Comparative solution equilibrium studies of antitumor ruthenium(η6 -p-cymene) and rhodium(η5-C5Me5) complexes of 8- hydroxyquinolines. Dalton Trans. 2017, 46, 4382–4396. [Google Scholar] [PubMed]
- Bratsos, I.; Urankar, D.; Zangrando, E.; Genova-Kalou, P.; Kosmrlj, J.; Alessio, E.; Turel, I. 1-(2-Picolyl)-substituted 1,2,3-triazole as novel chelating ligand for the preparation of ruthenium complexes with potential anticancer activity. Dalton Trans. 2011, 40, 5188. [Google Scholar] [CrossRef]
- Pujante-Galián, M.A.; Pérez, S.A.; Montalbán, M.G.; Carissimi, G.; Fuster, M.G.; Víllora, G.; García, G. p-Cymene Complexes of Ruthenium(II) as Antitumor Agents. Molecules 2020, 25, 5063. [Google Scholar] [CrossRef] [PubMed]
- Fuster, M.G.; Moulefera, I.; Montalbán, M.G.; Pérez, J.; Víllora, G.; García, G. Synthesis and Characterization of New Ruthenium (II) Complexes of Stoichiometry [Ru(p-Cymene)Cl2L] and Their Cytotoxicity against HeLa-Type Cancer Cells. Molecules 2022, 27, 7264. [Google Scholar] [CrossRef]
- Paitandi, R.P.; Sharma, V.; Singh, V.D.; Dwivedi, B.K.; Mobin, S.M.; Pandey, D.S. Pyrazole Appended Quinoline-BODIPY Based Arene Ruthenium Complexes: Their Anticancer Activity and Potential Applications in Cellular Imaging. Dalton Trans. 2018, 47, 17500–17514. [Google Scholar] [CrossRef]
- Rafols, L.; Josa, D.; Aguilà, D.; Barrios, L.A.; Roubeau, O.; Cirera, J.; Soto-Cerrato, V.; Pérez-Tomás, R.; Martínez, M.; Grabulosa, A. Piano-Stool Ruthenium(II) Complexes with Delayed Cytotoxic Activity: Origin of the Lag Time. Inorg. Chem. 2021, 60, 7974–7990. [Google Scholar] [CrossRef] [PubMed]
- Muralisankar, M.; Chen, J.-R.; Haribabu, J.; Ke, S.-C. Effective and Selective Ru(II)-Arene Complexes Containing 4,4′-Substituted 2,2′ Bipyridine Ligands Targeting Human Urinary Bladder Cancer Cells. Int. J. Mol. Sci. 2023, 24, 11896. [Google Scholar] [CrossRef]
- Zacharopoulos, N. Syhthesis and characterization of Ru complexes suitable for transfer hydrogenation and α,β alkylation reactions. Doctoral Dissertation, National and Kapodistrian University of Athens, Athens, Greece, 2023. Available online: https://pergamos.lib.uoa.gr/uoa/dl/object/2973928 (accessed on 4 May 2024).
- Yan, Y.K.; Melchart, M.; Habtemariam, A.; Dyson, P.J. Organometallic chemistry, biology and medicine: Ruthenium arene anticancer complexes. Chem. Commun. 2005, 4764–4776. [Google Scholar] [CrossRef]
- Moore, C.M.; Bark, B.; Szymczak, N.K. Simple Ligand Modifications with Pendent OH Groups Dramatically Impact the Activity and Selectivity of Ruthenium Catalysts for Transfer Hydrogenation: The Importance of Alkali Metals. ACS Catal. 2016, 6, 1981–1990. [Google Scholar] [CrossRef]
- Toubiana, J.; Medina, L.; Sasson, Y. The Nature of the True Catalyst in Transfer Hydrogenation with Alcohol Donors Using (arene)2Ru2Cl4(II)/TsDPEN Precursor. Mod. Res. Catal. 2014, 3, 68–88. [Google Scholar] [CrossRef]
- Štefane, B.; Požgan, F. Metal-Catalysed Transfer Hydrogenation of Ketones. Top. Curr. Chem. 2016, 374, 18. [Google Scholar] [CrossRef]
- Chelucci, G.; Baldino, S.; r Baratta, W. Ruthenium and osmium complexes containing 2-(aminomethyl)pyridine (Ampy)-based ligands in catalysis. Coord. Chem. Rev. 2015, 300, 29–85. [Google Scholar] [CrossRef]
- Joseph, M.C.; Swarts, A.J.; Mapolie, S.F. Cationic half-sandwich ruthenium (II) complexes ligated by pyridyl-triazole ligands: Transfer hydrogenation and mechanistic studies. Polyhedron 2022, 212, 115579. [Google Scholar] [CrossRef]
- Bäckvall, J.E. Transition metal hydrides as active intermediates in hydrogen transfer reactions. J. Organomet. Chem. 2002, 652, 105–111. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part B: Applications in Coordination, Organometallic and Bioinorganic Chemistry, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2008; ISBN 9780470405888. [Google Scholar] [CrossRef]
- Carrión, M.C.; Sepúlveda, F.; Jalón, F.A.; Manzano, B.R.; Rodríguez, A.M. Rodríguez, Base-Free Transfer Hydrogenation of Ketones Using Arene Ruthenium(II) Complexes. Organometallics 2009, 28, 3822–3833. [Google Scholar] [CrossRef]
- Grabulosa, A.; Mannu, A.; Alberico, E.; Denurra, S.; Gladiali, S.; Muller, G. Neutral η6-arene ruthenium complexes with monodentate P-donor ligands: Activation in the transfer hydrogenation reaction. J. Mol. Catal. A Chem. 2012, 49, 363–364. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 2b |
|---|---|
| Color, habit | Orange, Plate |
| Size/mm | 0.411 × 0.127 × 0.072 |
| Empirical formula | C36H32ClF6N2OPRu × 0.25C3H6O |
| FW | 807.11 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a/Å | 13.6107(5) |
| b/Å | 10.5278(3) |
| c/Å | 25.8896(11) |
| α/° | 90 |
| β/° | 92.702(2) |
| γ/° | 90 |
| V/Å3 | 3705.6(2) |
| Z | 4 |
| μ/mm−1 | 0.628 |
| T/K | 100 |
| θmin/max/full (°) | 2.089/28.294/25.242 |
| Completeness to θmax/full (%) | 99.9/99.9 |
| Reflections Total/ Independent | 95,535/8017 |
| Parameters/restraints | 726/340 |
| Rint | 0.0517 |
| Final R1, wR2 | 0.0586/0.1787 |
| Goodness-of-fit | 1.077 |
| Largest peak, hole/e.Å−3 | 1.9/−0.9 |
| ρcalc/g.cm−3 | 1.447 |
| Compounds | HEK293T (IC50 in μM) | HeLa (IC50 in μM) |
|---|---|---|
| Br-Qpy | 13.7 ± 0.3 | 108.5 ± 3.6 |
| OH-Ph-Qpy | 23.4 ± 0.7 | 84.5 ± 2.2 |
| [Ru(p-cymene)Cl2]2 | 48.3 ± 1.7 | 48.9 ± 1.7 |
| 1a | 102.9 ± 3.4 | 106.9 ± 1.1 |
| 1b | 36.2 ± 1.2 | 109.6 ± 1.5 |
| 2a | 35.2 ± 4.6 | 75.0 ± 1.0 |
| 2b | 22.7 ± 1.2 | 85.1 ± 1.8 |
| cisplatin | 20.8 ± 0.0 | 9.5 ± 0.0 |
| Catalyst | Conversion (%) b | Time (h) | TOF (h−1) |
|---|---|---|---|
| 1a * | 73 | 1 | 73 |
| 76 | 3 | 25 | |
| 92 | 24 | 4 | |
| 1a ** | 81 | 1 | 324 |
| 94 | 3 | 125 | |
| 2a * | 45 | 1 | 45 |
| 70 | 3 | 23 | |
| 94 | 24 | 4 | |
| 2a ** | 42 | 1 | 168 |
| 73 | 3 | 97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dritsopoulos, A.; Zacharopoulos, N.; Peyret, A.-E.; Karampella, E.; Tsoureas, N.; Cheilari, A.; Machalia, C.; Emmanouilidou, E.; Andreopoulou, A.K.; Kallitsis, J.K.; et al. Ruthenium-p-Cymene Complexes Incorporating Substituted Pyridine–Quinoline Ligands with –Br (Br-Qpy) and –Phenoxy (OH-Ph-Qpy) Groups for Cytotoxicity and Catalytic Transfer Hydrogenation Studies: Synthesis and Characterization. Chemistry 2024, 6, 773-793. https://doi.org/10.3390/chemistry6040046
Dritsopoulos A, Zacharopoulos N, Peyret A-E, Karampella E, Tsoureas N, Cheilari A, Machalia C, Emmanouilidou E, Andreopoulou AK, Kallitsis JK, et al. Ruthenium-p-Cymene Complexes Incorporating Substituted Pyridine–Quinoline Ligands with –Br (Br-Qpy) and –Phenoxy (OH-Ph-Qpy) Groups for Cytotoxicity and Catalytic Transfer Hydrogenation Studies: Synthesis and Characterization. Chemistry. 2024; 6(4):773-793. https://doi.org/10.3390/chemistry6040046
Chicago/Turabian StyleDritsopoulos, Alexandros, Nikolaos Zacharopoulos, Aigli-Eleonora Peyret, Eftychia Karampella, Nikolaos Tsoureas, Antigoni Cheilari, Christina Machalia, Evangelia Emmanouilidou, Aikaterini K. Andreopoulou, Joannis K. Kallitsis, and et al. 2024. "Ruthenium-p-Cymene Complexes Incorporating Substituted Pyridine–Quinoline Ligands with –Br (Br-Qpy) and –Phenoxy (OH-Ph-Qpy) Groups for Cytotoxicity and Catalytic Transfer Hydrogenation Studies: Synthesis and Characterization" Chemistry 6, no. 4: 773-793. https://doi.org/10.3390/chemistry6040046