

Advancements in Microfluidic Platforms for Glioblastoma Research


Abstract
1. Introduction
2. Understanding GBM
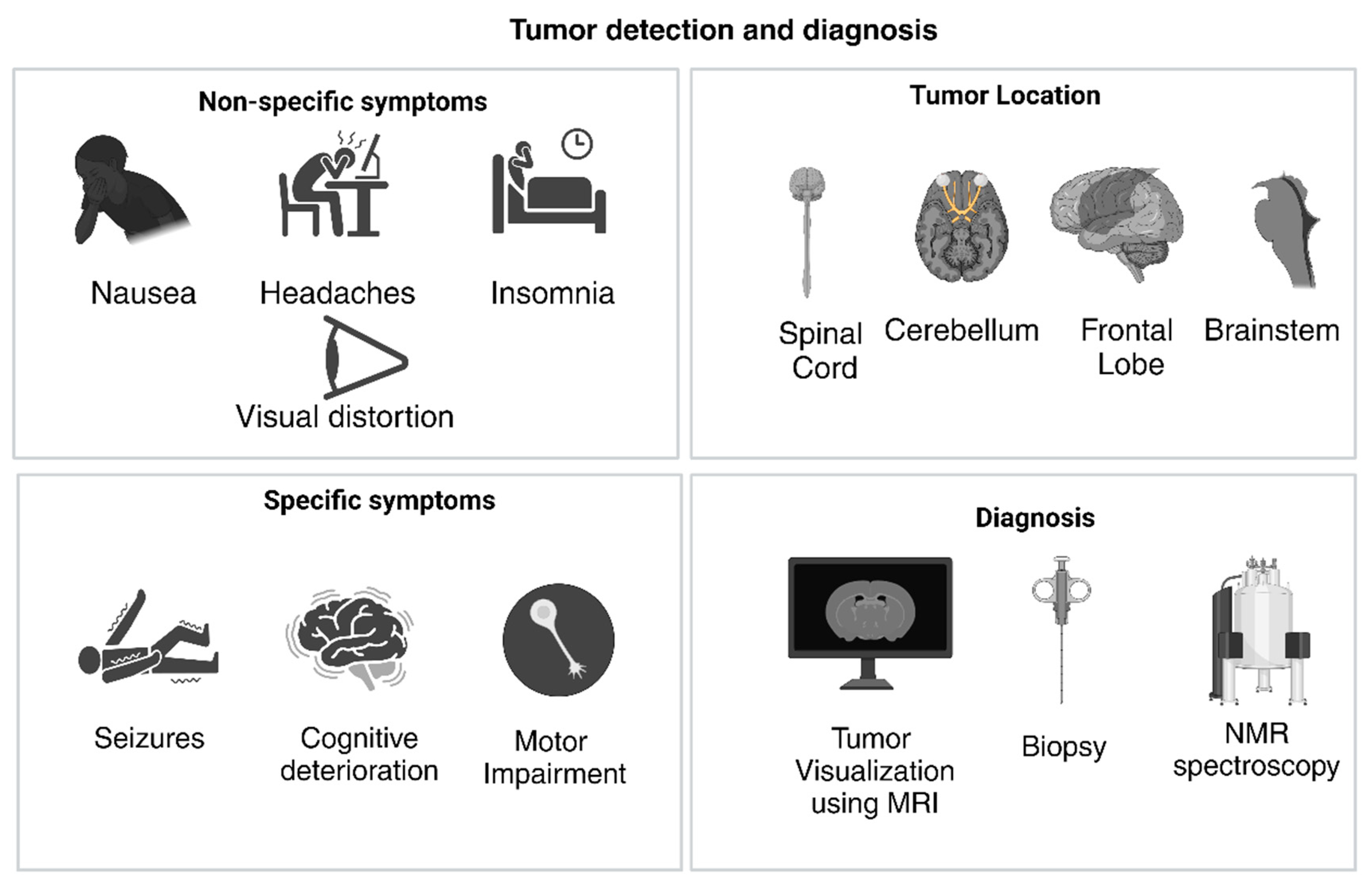
2.1. Physical Presentation of GBM
2.2. Tumor Structure in Glioblastoma
2.3. Tumor Microenvironment in Glioblastoma
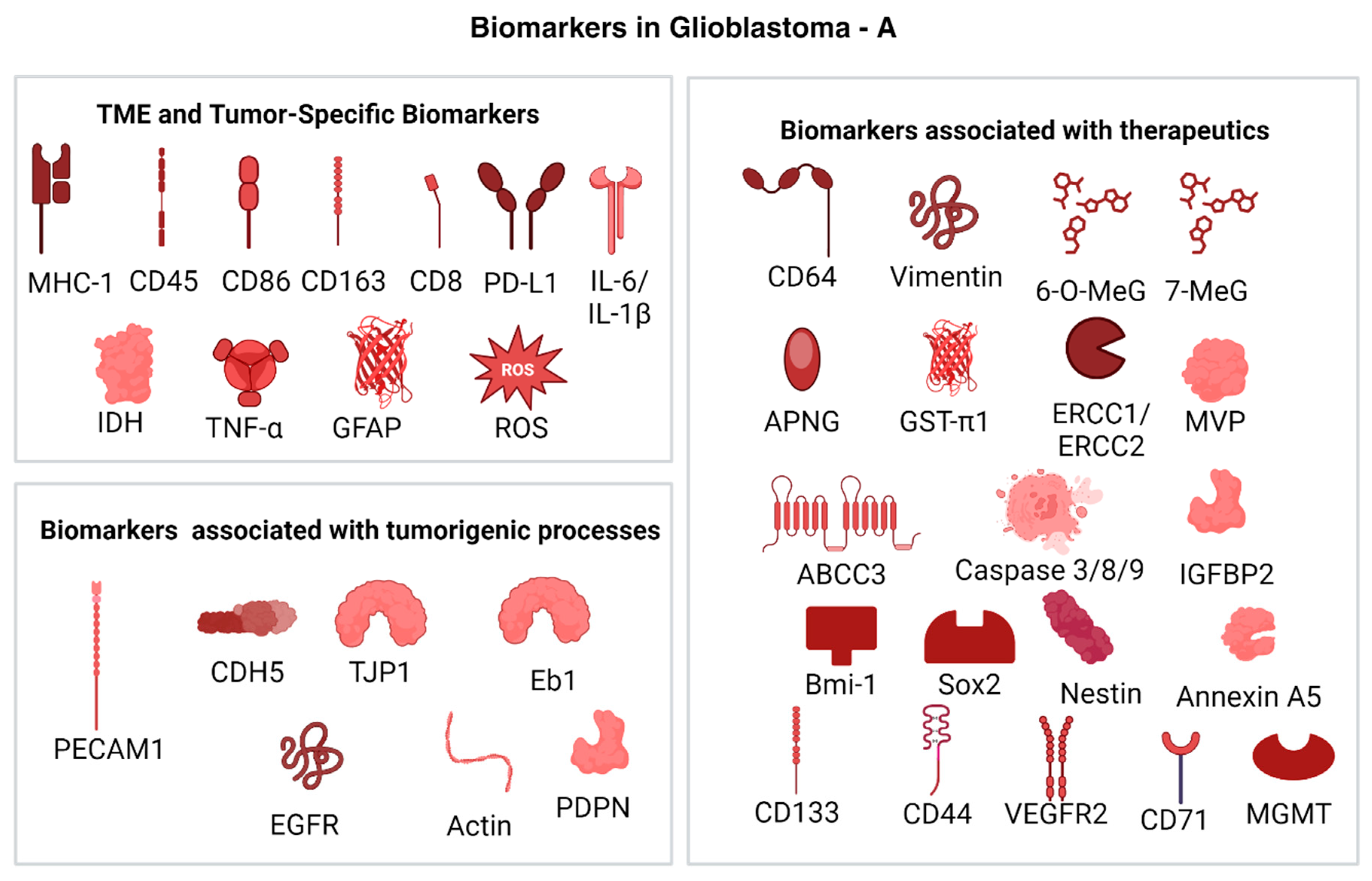
2.4. Biomarkers in Glioblastoma
2.4.1. TME and Tumor-Specific Biomarkers
2.4.2. Biomarkers Associated with Therapeutics
2.4.3. Biomarkers Associated with Tumorigenic Processes
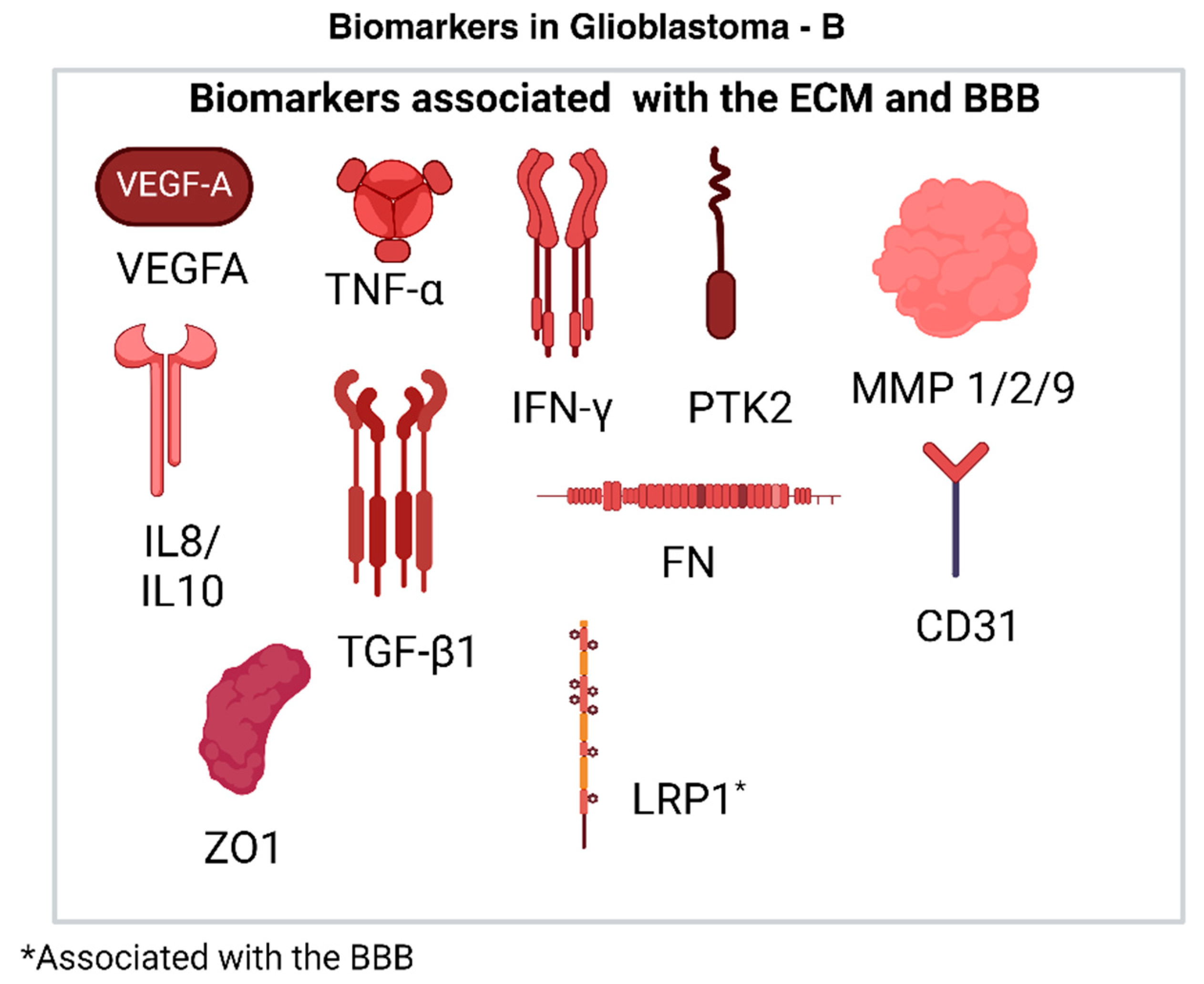
2.4.4. Biomarkers Associated with the ECM
2.4.5. Biomarkers Associated with the BBB
2.5. Challenges Associated with Detection and Treatment
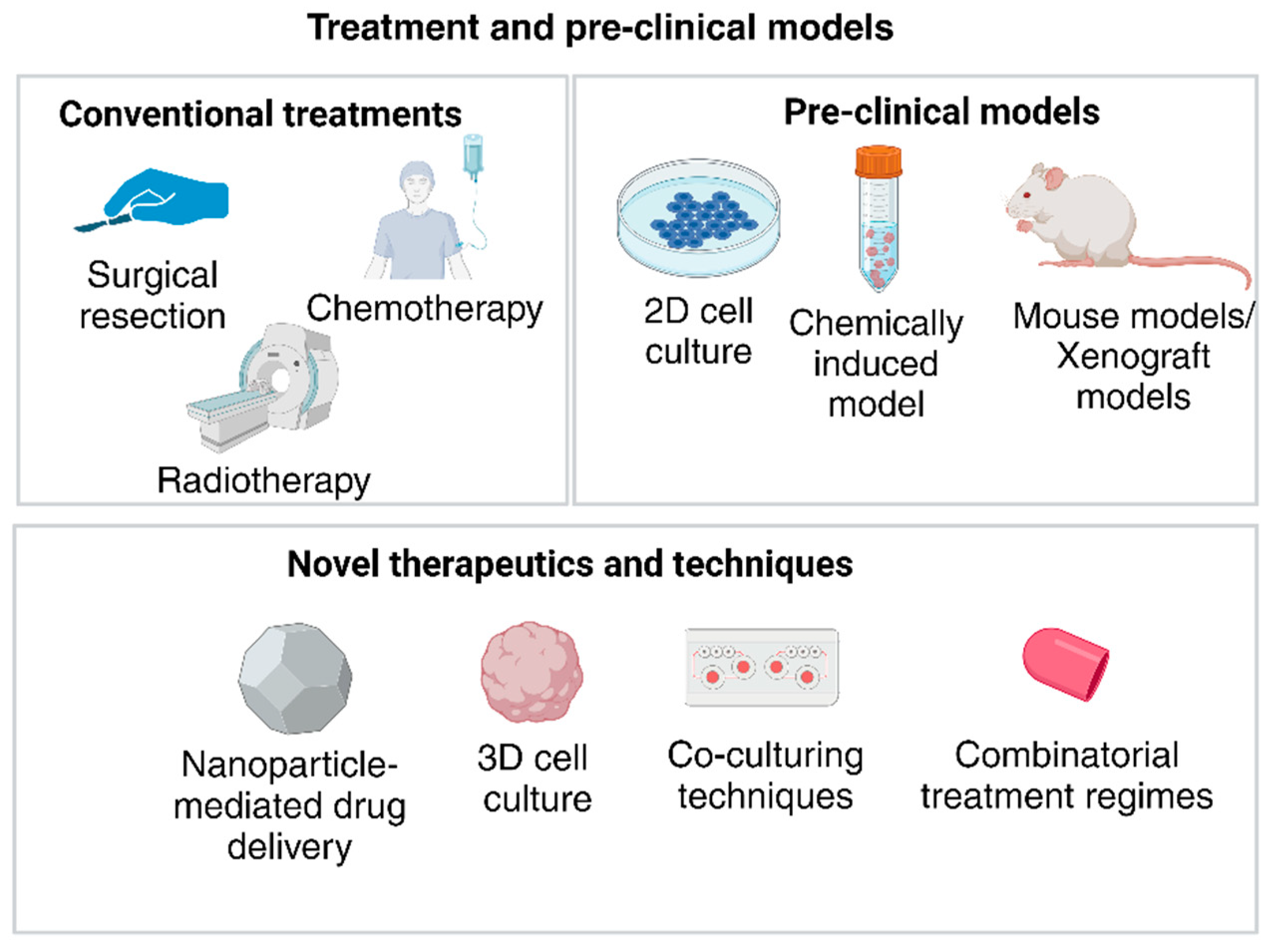
2.6. Challenges with Drug Development for GBM
2.6.1. Disadvantages of Current Pre-Clinical Models for Drug Development
2.6.2. Emerging Role of Organoids in GBM
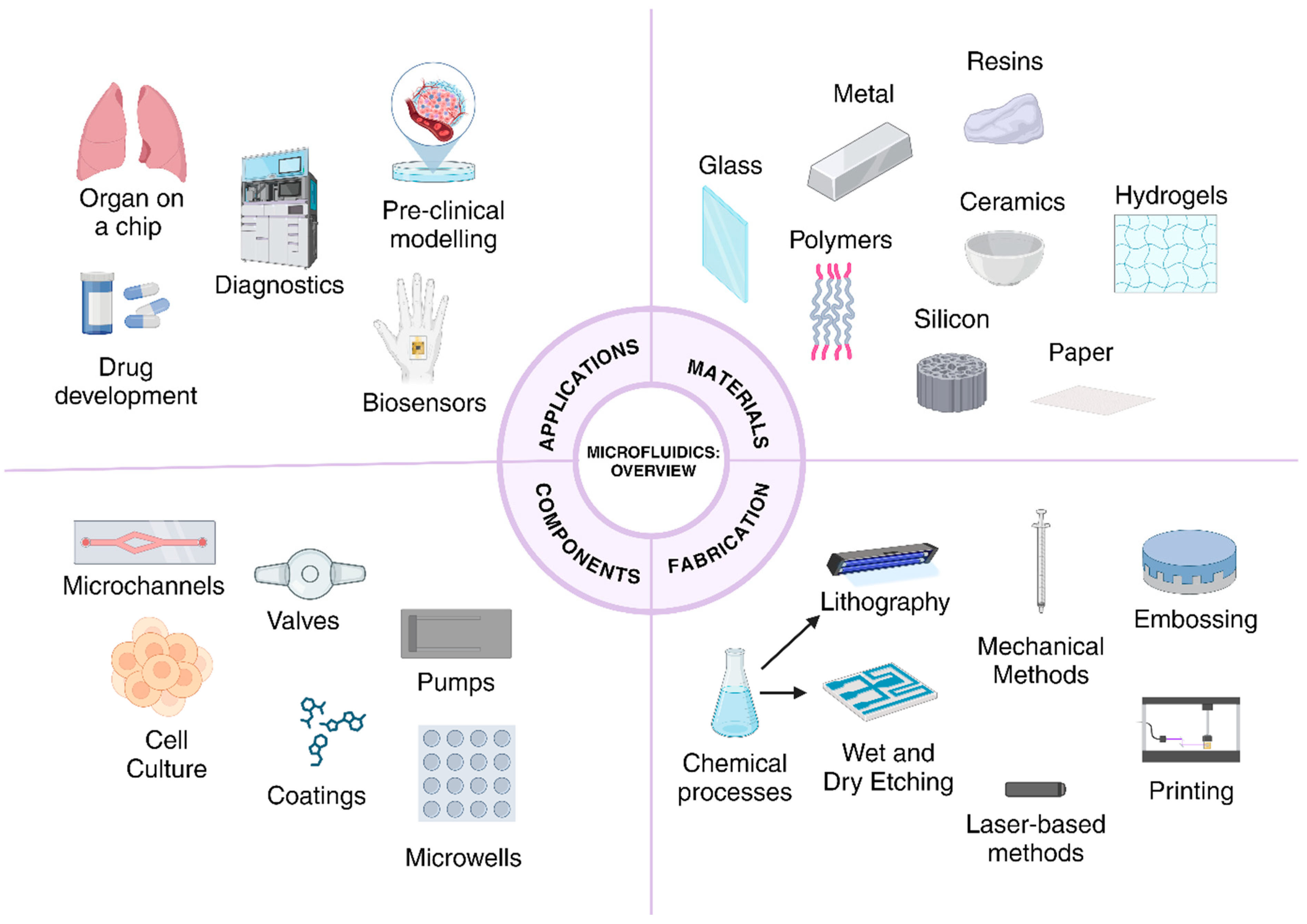
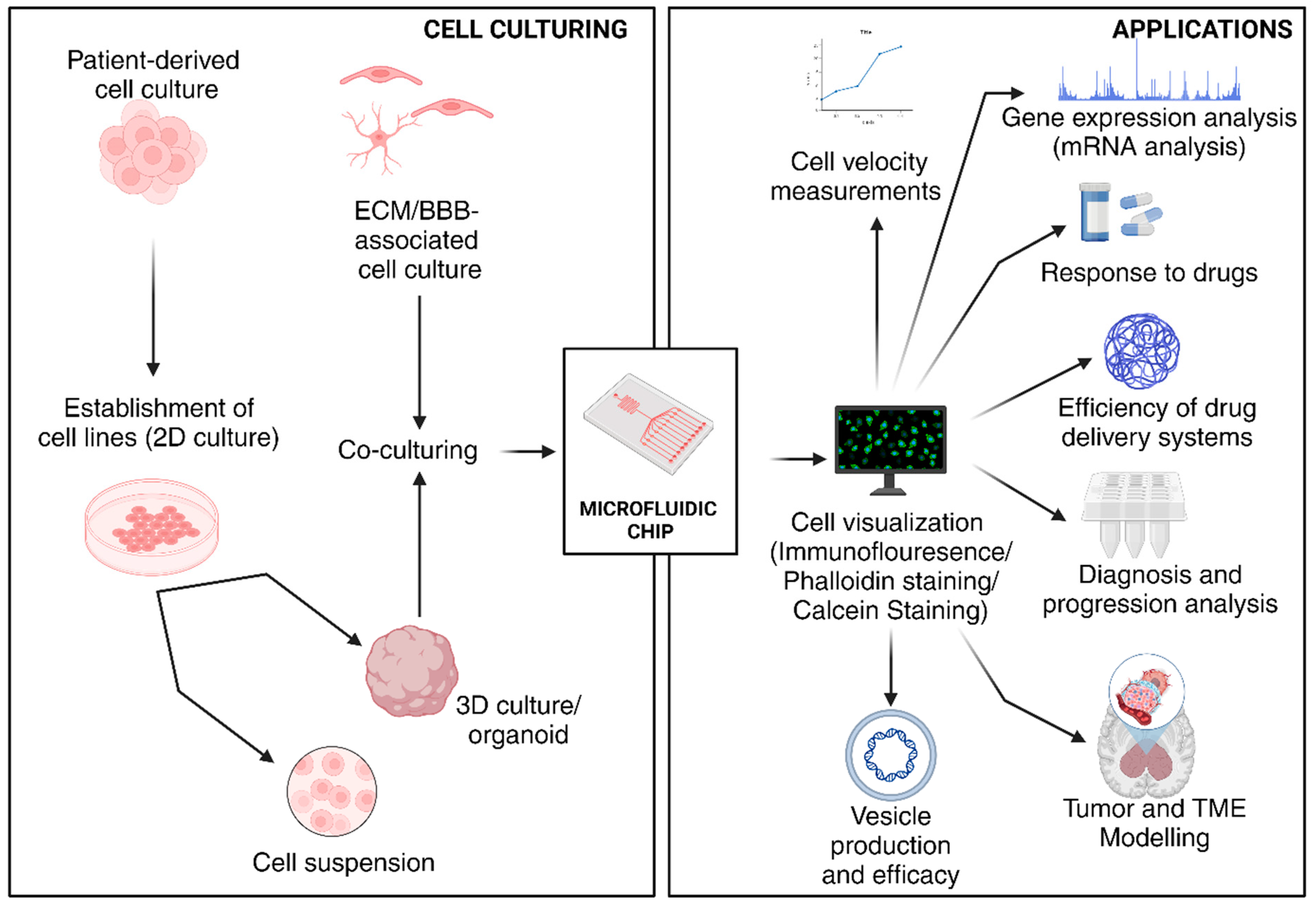
3. Microfluidics-Based Systems for the Study of Tumors/Microfluidics in Cancer
3.1. General Features of Microfluidic Devices Used in Cancer Studies
3.2. Progression of Microfluidics in Disease Modeling, Diagnosis, and Treatment
3.3. Microfluidic Platforms for the Study of GBM TME and Tumor Modeling
3.4. Microfluidic Platforms for Drug Development
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Malloggi, F. Microfluidics: From Basic Principles to Applications. In Soft Matter at Aqueous Interfaces; Lang, P., Liu, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 515–546. [Google Scholar]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Hajam, M.I.; Khan, M.M. Microfluidics: A concise review of the history, principles, design, applications, and future outlook. Biomater. Sci. 2023, 12, 218–251. [Google Scholar] [CrossRef] [PubMed]
- Yeo, L.Y.; Chang, H.C.; Chan, P.P.Y.; Friend, J.R. Microfluidic devices for bioapplications. Small 2011, 7, 12–48. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.G.; Chircov, C.; Bîrcă, A.C.; Grumezescu, A.M. Fabrication and applications of microfluidic devices: A review. Int. J. Mol. Sci. 2021, 22, 2011. [Google Scholar] [CrossRef] [PubMed]
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12, e8195. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef]
- Weber, C.E.; Kuo, P.C. The tumor microenvironment. Surg. Oncol. 2012, 21, 172–177. [Google Scholar] [CrossRef]
- Gilard, V.; Tebani, A.; Dabaj, I.; Laquerrière, A.; Fontanilles, M.; Derrey, S.; Marret, S.; Bekri, S. Diagnosis and management of glioblastoma: A comprehensive perspective. J. Pers. Med. 2021, 11, 258. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.-X.; Huang, G.H.; Xiang, Y.; Yang, L.; Pei, Y.C.; Yang, W.; Lv, S.-Q. A systematic review of multifocal and multicentric glioblastoma. J. Clin. Neurosci. 2021, 83, 71–76. [Google Scholar] [CrossRef]
- Regmi, S.; Poudel, C.; Adhikari, R.; Luo, K.Q. Applications of Microfluidics and Organ-on-a-Chip in Cancer Research. Biosensors 2022, 12, 459. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting Organ-Level Lung Functions on a Chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Białkowska, K.; Komorowski, P.; Bryszewska, M.; Miłowska, K. Spheroids as a type of three-dimensional cell cultures—Examples of methods of preparation and the most important application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef] [PubMed]
- Ravi, M.; Paramesh, V.; Kaviya, S.R.; Anuradha, E.; Paul Solomon, F.D. 3D Cell Culture Systems: Advantages and Applications. J. Cell. Physiol. 2015, 230, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Liu, X.; Bi, Y.; Wang, Y.; Antony, A.; Lee, D.; Huntoon, K.; Jeong, S.; Ma, Y.; Li, X.; et al. Adaptive design of mRNA-loaded extracellular vesicles for targeted immunotherapy of cancer. Nat. Commun. 2023, 14, 6610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Mao, S.; Li, W.; Huang, Q.; Feng, S.; Hong, Z.; Lin, J. Microfluidic adhesion analysis of single glioma cells for evaluating the effect of drugs. Sci. China Chem. 2020, 63, 865–870. [Google Scholar] [CrossRef]
- Samiei, E.; Seyfoori, A.; Toyota, B.; Ghavami, S.; Akbari, M. Investigating programmed cell death and tumor invasion in a three-dimensional (3d) microfluidic model of glioblastoma. Int. J. Mol. Sci. 2020, 21, 3162. [Google Scholar] [CrossRef]
- Yi, H.-G.; Jeong, Y.H.; Kim, Y.; Choi, Y.; Moon, H.; Park, S.; Kang, K.; Bae, M.; Jang, J.; Youn, H. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Ma, J.; Li, N.; Wang, Y.; Wang, L.; Wei, W.; Shen, L.; Sun, Y.; Jiao, Y.; Chen, W.; Liu, J. Engineered 3D tumour model for study of glioblastoma aggressiveness and drug evaluation on a detachably assembled microfluidic device. Biomed. Microdevices 2018, 20, 80. [Google Scholar] [CrossRef]
- Lin, C.; Lin, L.; Mao, S.; Yang, L.; Yi, L.; Lin, X.; Wang, J.; Lin, Z.; Lin, J. Reconstituting Glioma Perivascular Niches on a Chip for Insights into Chemoresistance of Glioma. Anal. Chem. 2018, 90, 10326–10333. [Google Scholar] [CrossRef]
- Akay, M.; Hite, J.; Avci, N.G.; Fan, Y.; Akay, Y.; Lu, G.; Zhu, J. Drug Screening of Human GBM Spheroids in Brain Cancer Chip. Sci. Rep. 2018, 8, 15423. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jie, M.; He, Z.; Li, H.; Lin, J. Study of antioxidant effects on malignant glioma cells by constructing a tumor-microvascular structure on microchip. Anal. Chim. Acta 2017, 978, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jie, M.; Mao, S.; Liu, H.; He, Z.; Li, H.; Lin, J. Evaluation of drug combination for glioblastoma based on an intestine–liver metabolic model on microchip. Analyst 2017, 142, 3629–3638. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.S.; Shah, S.R.; Yankaskas, C.L.; Bajpai, V.; Wu, P.; Chin, D.; Ifemembi, B.; ReFaey, K.; Schiapparelli, P.; Zheng, X.; et al. A microfluidic cell-migration assay for the prediction of progression-free survival and recurrence time of patients with glioblastoma. Nat. Biomed. Eng. 2021, 5, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Straehla, J.P.; Hajal, C.; Safford, H.C.; Offeddu, G.; Boehnke, N.; Dacoba, T.; Wyckoff, J.; Kamm, R.; Hammond, P. A predictive microfluidic model of human glioblastoma to assess trafficking of blood–brain barrier-penetrant nanoparticles. Proc. Natl. Acad. Sci. 2022, 119, e2118697119. [Google Scholar] [CrossRef]
- Müller Bark, J.; Kulasinghe, A.; Hartel, G.; Leo, P.; Warkiani, M.; Jeffree, R.; Chua, B.; Day, B.; Punyadeera, C. Isolation of Circulating Tumour Cells in Patients with Glioblastoma Using Spiral Microfluidic Technology—A Pilot Study. Front. Oncol. 2021, 11, 681130. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Monge, R.; Martínez-González, A.; Virumbrales-Muñoz, M.; Llamazares, G.; Berganzo, J.; Hernández-Laín, A.; Santolaria, J.; Doblaré, M.; Hubert, C.; et al. Glioblastoma on a microfluidic chip: Generating pseudopalisades and enhancing aggressiveness through blood vessel obstruction events. Neuro Oncol. 2017, 19, 503–513. [Google Scholar] [CrossRef]
- Liu, W.; Sun, P.; Yang, L.; Wang, J.; Li, L.; Wang, J. Assay of glioma cell responses to an anticancer drug in a cell-based microfluidic device. Microfluid. Nanofluidics 2010, 9, 717–725. [Google Scholar] [CrossRef]
- Adjei-Sowah, E.A.; O’Connor, S.A.; Veldhuizen, J.; Lo Cascio, C.; Plaisier, C.; Mehta, S.; Nikkhah, M. Investigating the Interactions of Glioma Stem Cells in the Perivascular Niche at Single-Cell Resolution using a Microfluidic Tumor Microenvironment Model. Adv. Sci. 2022, 9, 2201436. [Google Scholar] [CrossRef]
- Mamani, J.B.; Marinho, B.S.; Rego, G.N.; Nucci, M.P.; Alvieri, F.; Santos, R.S.; Ferreira, J.V.; Oliveira, F.A.; Gamarra, L.F. Magnetic hyperthermia therapy in glioblastoma tumor on-a-Chip model. Einstein 2020, 18, eAO4954. [Google Scholar] [CrossRef]
- Krol, I.; Castro-Giner, F.; Maurer, M.; Gkountela, S.; Szczerba, B.; Scherrer, R.; Coleman, N.; Carreira, S.; Bachmann, F.; Anderson, S.; et al. Detection of circulating tumour cell clusters in human glioblastoma. Br. J. Cancer 2018, 119, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Ye, J.; Gao, X.; Chen, H.; Chen, M.; Lian, J.; Ma, J.; Wang, H. Evaluation of nanoparticle albumin-bound paclitaxel loaded macrophages for glioblastoma treatment based on a microfluidic chip. Front. Bioeng. Biotechnol. 2024, 12, 1361682. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Ma, C.; Vasudevaraja, V.; Serrano, J.; Tong, J.; Peng, Y.; Delorenzo, M.; Shen, G.; Frenster, J.; Morales, R.; et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. Elife 2020, 9, e52253. [Google Scholar] [CrossRef] [PubMed]
- Sengul, E.; Elitas, M. Long-term migratory velocity measurements of single glioma cells using microfluidics. Analyst 2021, 146, 5143–5149. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Chung, J.; Lee, K.; Balaj, L.; Min, C.; Carter, B.; Hochberg, F.; Breakerfield, X.; Lee, H.; Weissleder, R. Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun. 2015, 6, 6999. [Google Scholar] [CrossRef]
- Peeters, M.C.M.; Dirven, L.; Koekkoek, J.A.F.; Gortmaker, E.G.; Fritz, L.; Vos, M.J.; Taphoorn, M.J.B. Prediagnostic symptoms and signs of adult glioma: The patients’ view. J. Neurooncol. 2020, 146, 293–301. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestier, O.D.; Cavenee, W.; Ellison, D.; Figarella-Branger, D.; Perry, A.; Reifenberger, G.; Von Deimling, A. WHO Classification of Tumours of the Central Nervous System. Neurol. Med. Chir. 2016, 57, 301–311. [Google Scholar]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, S.; Brown, D.; Parney, I. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neurooncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.; Miller, C.; Ding, L.; Golub, T.; Mesirov, J.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.G.; Sahebjam, S.; Mason, W.P. Emerging biomarkers in glioblastoma. Cancers 2013, 5, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Hein, T.; Krammer, P.H.; Weyd, H. Molecular analysis of Annexin expression in cancer. BMC Cancer 2022, 22, 994. [Google Scholar] [CrossRef] [PubMed]
- Gerke, V.; Moss, S.E. Annexins: From Structure to Function. Physiol. Rev. 2002, 82, 331–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huo, L.; Jin, H.; Han, Y.; Wang, J.; Zhang, Y.; Lai, X.; Le, Z.; Zhang, J.; Hua, Z. Anti-cancer activity of Annexin V in murine melanoma model by suppressing tumor angiogenesis. Oncotarget 2017, 8, 42602–42612. [Google Scholar] [CrossRef]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed. Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef]
- Fianco, G.; Mongiardi, M.P.; Levi, A.; De Luca, T.; Desideri, M.; Trisciuoglio, D.; Bufalo, D.; Cinà, I.; Benedetto, A.; Mottolese, M. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. Elife 2017, 6, e22593. [Google Scholar] [CrossRef]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and therapeutic biomarkers in glioblastoma: Current status and future perspectives. Biomed. Res. Int. 2017, 2017, 8013575. [Google Scholar] [CrossRef]
- Boyé, K.; Pujol, N.; DAlves, I.; Chen, Y.; Daubon, T.; Lee, Y.; Dedieu, S.; Constantin, M.; Bello, L.; Rossi, M.; et al. The role of CXCR3/LRP1 cross-talk in the invasion of primary brain tumors. Nat. Commun. 2017, 8, 1571. [Google Scholar] [CrossRef]
- Hosseini, A.; Ashraf, H.; Rahimi, F.; Alipourfard, I.; Alivirdiloo, V.; Hashemi, B.; Yazdani, Y.; Ghazi, F.; Eslami, M.; Ameri Shah Reza, M.; et al. Recent advances in the detection of glioblastoma, from imaging-based methods to proteomics and biosensors: A narrative review. Cancer Cell Int. 2023, 23, 98. [Google Scholar] [CrossRef]
- Flores Ledur, P.; Onzi, G.R.; Zong, H.; Lenz, G. Oncotarget 69185. Culture Conditions Defining Glioblastoma Cells Behavior: What Is the Impact for Novel Discoveries? Available online: www.impactjournals.com/oncotarget (accessed on 1 July 2024).
- Glas, M.; Rath, B.H.; Simon, M.; Reinartz, R.; Schramme, A.; Trageser, D.; Eisenreich, R.; Leinhaas, A.; Keller, M.; Schildhaus, H.; et al. Residual tumor cells are unique cellular targets in glioblastoma. Ann. Neurol. 2010, 68, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Ganipineni, L.P.; Danhier, F.; Préat, V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J. Control. Release 2018, 281, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Akter, F.; Simon, B.; de Boer, N.L.; Redjal, N.; Wakimoto, H.; Shah, K. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. Biochim. Et Biophys. Acta (BBA)—Rev. Cancer 2021, 1875, 188458. [Google Scholar] [CrossRef]
- Varna, M.; Bertheau, P.; Legres, L. Tumor Microenvironment in Human Tumor Xenografted Mouse Models. J. Anal. Oncol. 2014, 3, 159–166. [Google Scholar] [CrossRef]
- Xu, C.; Yuan, X.; Hou, P.; Li, Z.; Wang, C.; Fang, C.; Tan, Y. Development of glioblastoma organoids and their applications in personalized therapy. Cancer Biol. Med. 2023, 20, 353–368. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Y.; Zhang, D.Y.; Ming, G.; Song, H. Glioblastoma modeling with 3D organoids: Progress and challenges. Oxf. Open Neurosci. 2023, 2, kvad008. [Google Scholar] [CrossRef]
- Rybin, M.J.; Ivan, M.E.; Ayad, N.G.; Zeier, Z. Organoid Models of Glioblastoma and Their Role in Drug Discovery. Front. Cell. Neurosci. 2021, 15, 605255. [Google Scholar] [CrossRef]
- Trujillo-de Santiago, G.; Flores-Garza, B.G.; Tavares-Negrete, J.A.; Lara-Mayorga, I.; González-Gamboa, I.; Zhang, Y.; Rojas-Martínez, A.; Ortiz-López, R.; Álvarez, M.M. The tumor-on-chip: Recent advances in the development of microfluidic systems to recapitulate the physiology of solid tumors. Materials 2019, 12, 2945. [Google Scholar] [CrossRef]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.; Ko, J.; Rho, H.; Chen, Z.; Habibovic, P.; Jeon, N.; Takayama, S.; et al. A guide to the organ-on-a-chip. Nat. Rev. Methods Primers 2022, 2, 33. [Google Scholar] [CrossRef]
- Tevlek, A.; Kecili, S.; Ozcelik, O.S.; Kulah, H.; Tekinm, H. Spheroid Engineering in Microfluidic Devices. ACS Omega 2022, 8, 3630–3649. [Google Scholar] [CrossRef]
- Preetam, S.; Nahak, B.K.; Patra, S.; Toncu, D.; Park, S.; Syväjärvi, M.; Orive, G.; Tiwari, A. Emergence of microfluidics for next generation biomedical devices. Biosens. Bioelectron. X 2022, 10, 100106. [Google Scholar] [CrossRef]
- Prakash, S.; Kumar, S. Fabrication of microchannels: A review. Proc. Inst. Mech. Eng. B J. Eng. Manuf. 2014, 229, 1273–1288. [Google Scholar] [CrossRef]
- Cao, U.M.; Zhang, Y.; Chen, J.; Sayson, D.; Pillai, S.; Tran, S.D. Microfluidic Organ-on-A-chip: A Guide to Biomaterial Choice and Fabrication. Int. J. Mol. Sci. 2023, 24, 3232. [Google Scholar] [CrossRef] [PubMed]
- Tajeddin, A.; Mustafaoglu, N. Design and fabrication of organ-on-chips: Promises and challenges. Micromachines 2021, 12, 1443. [Google Scholar] [CrossRef] [PubMed]
- Rasponi, M. Organ-on-a-Chip Methods and Protocols; Spinger: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Miri, A.K.; Mostafavi, E.; Khorsandi, D.; Hu, S.K.; Malpica, M.; Khademhosseini, A. Bioprinters for organs-on-chips. Biofabrication 2019, 11, 042002. [Google Scholar] [CrossRef] [PubMed]
- Chliara, M.A.; Elezoglou, S.; Zergioti, I. Bioprinting on Organ-on-Chip: Development and Applications. Biosensors 2022, 12, 1135. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef]
- Beier, C.P.; Kumar, P.; Meyer, K.; Leukel, P.; Bruttel, V.; Aschenbrenner, I.; Riemenschneider, M.; Fragoulis, A.; Rümmele, P.; Lamszus, K.; et al. The cancer stem cell subtype determines immune infiltration of Glioblastoma. Stem Cells Dev. 2012, 21, 2753–2761. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.; Fuller, C.; Hamner, B.; Oh, E.; Gaber, M.; Finkelstein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Liu, J.; Yang, F.; Hu, J.; Zhang, X. Nanoparticles for efficient drug delivery and drug resistance in glioma: New perspectives. CNS Neurosci. Ther. 2024, 30, e14715. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C. Temozolomide: Therapeutic limitations in the treatment of adult high-grade gliomas. Expert. Rev. Neurother. 2010, 10, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Manini, I.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Loreto, C.; Beltrami, A.; Cesselli, D. Role of microenvironment in glioma invasion: What we learned from in vitro models. Int. J. Mol. Sci. 2018, 19, 147. [Google Scholar] [CrossRef] [PubMed]
- van der Helm, M.W.; van der Meer, A.D.; Eijkel, J.C.T.; van den Berg, A.; Segerink, L. Microfluidic organ-on-chip technology for blood-brain barrier research. Tissue Barriers 2016, 4, e1142493. [Google Scholar] [CrossRef] [PubMed]
- Brandalise, F.; Ramieri, M.; Pastorelli, E.; Priori, E.; Ratto, D.; Venuti, M.; Roda, E.; Talpo, F.; Rossi, P. Role of Na+/Ca2+ Exchanger (NCX) in Glioblastoma Cell Migration (In Vitro). Int. J. Mol. Sci. 2023, 24, 12673. [Google Scholar] [CrossRef]
- Zhang, Y. Ion Channels and Transporters in the Regulation of Cancer Cell Migration and Metastasis. Doctoral Dissertation, Johns Hopkins University, Baltimore, MD, USA, 2022. [Google Scholar]
- Hu, H.-J.; Wang, S.-S.; Wang, Y.-X.; Liu, Y.; Feng, X.; Shen, Y.; Zhu, L.; Chen, H.; Song, M. Blockade of the forward Na+/Ca2+ exchanger suppresses the growth of glioblastoma cells through Ca2+-mediated cell death. Br. J. Pharmacol. 2019, 176, 2691–2707. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purpose of Device | Type of Device | Drugs | Cellular Biomarker/Metrics for Staining/Detection | Advantages | Disadvantages | Ref |
|---|---|---|---|---|---|---|
| Drug delivery system design and component manufacturing | Electroporation | miRNAs | mRNA, vesicle proteins, CD64, CD71, MHC1, PD-L1 | High throughput, low cost Enhanced specificity for targeting cancer cells | Lack of research validating applications in GBM | [16] |
| Modeling suitable environments for drug delivery | Microfluidic probe | TMZ | Calcein-AM/PI double straining | Trypsin–TMZ model showed potential to measure extent of cell adhesion in vitro Addition of lactic acid and ECM materials provided a more accurate model environment | N/A | [17] |
| Modeling 3D and monolayer cultures to test drug mechanisms | Multichannel microfluidic device and hydrogel scaffolds | TMZ and Simva | Vimentin, cell viability, cell invasion | Direct modification and quantification of 3D model-based parameters Presence of ECM and 3D model Homogeneous cell distribution for viability and pathway analyses, and non-homogeneous cell distribution for modeling invasion. Addition of HA to stimulate acidic TME | Absence of BBB modelling Lower cytotoxicity observed in 3D models | [18] |
| TME modeling and drug development | 3D-printed tumor environment on a chip, eye shaped layout with one inlet and one outlet | CCRT and TMZ | VEGFA and IL8 (angiogenesis), PTK2, FN, MMP1, MMP2 and MMP9 (ECM re-modeling), PECAM1, CDH5 and TJP1 (cell junction), CD31, oxygen concentration and Ki-67 (cell migration) | Modeling patient-specific TME and developing customized treatment courses Presence of BdECM enhances cell proliferation Use of bioink increases drug sensitivity Fast rate of tumor formation and drug testing | N/A | [19] |
| Drug development | Detachable microfluidic device with upstream channels and downstream capsules | Resveratrol and TMZ | Immunofluorescence, Ki67 (proliferation), vimentin and MMP2 (invasiveness) | Compartmentalization of multiple cell spheroids to test different drug combinations and concentrations simultaneously Lower cell volumes and ease of operation | Contamination of downstream capsules with upstream components | [20] |
| Drug development and TME analysis | Artificial perivascular niche on a chip | TMZ | 6-O-MeG and 7-MeG Sox2 mRNA and Bmi-1 gene (chemoresistance and cell proliferation) | Presence of endothelial cells enhanced the biological similarity between in vitro and in vivo models | N/A | [21] |
| Tumor growth modeling, drug efficacy testing and development of personalized treatments | Capsule and channel model to grow multiple spheroids and test drug combinations | TMZ and BEV | Nestin, VEGFR2, and GFAP | Presence of diffusion gaps that prevent culture contamination during drug testing, genetic fidelity of the primary tumors was maintained | Maintaining cell cultures for prolonged periods of testing may be more expensive as opposed to traditional 2D cultures | [22] |
| TME stimulation (vasculature), drug development | Flat chip with TG–gelatin–PEG hydrogel | α-lipoic acid, ascorbic acid and catechins | GSH and ROS levels | Use of TG–gelatin–PEG hydrogel enhanced cell culture maintenance | N/A | [23] |
| TME stimulation (drug metabolism), drug development | 3D chip with separate chambers for liver and GBM co-culture and microporous tubules | CPT-11, TMZ and CP | ROS and GSH | Elucidates effect of drug combination, models a more realistic environment, presence of micropores enhances drug travel | N/A | [24] |
| Diagnostic device | Series of Y-shaped channels to measure aggregation and migration characteristics | N/A | Ki-67, MGMT and IDH1 | Composite MAqCI score provides an accurate picture of patient outcomes, results are more reliable as patient-specific factors (age, etc.) are not related to the MAqCI score | IDH1 as a marker is more commonly associated with lower-grade gliomas and is prone to mutations | [25] |
| Tumor modeling and drug development | Two-component microfluidic chip with central area of organoid integration | AP2, CDDP | LRP1 (BBB), Annexin V, Caspase 3, Caspase 7 (spheroid) | Accurate in vitro stimulation of the BBB (permeability is very close to in vivo models) and other associated cellular processes Detects small tumors to provide early therapeutic options | Lack of continuous flow, device is limited to culturing microscopic tumors | [26] |
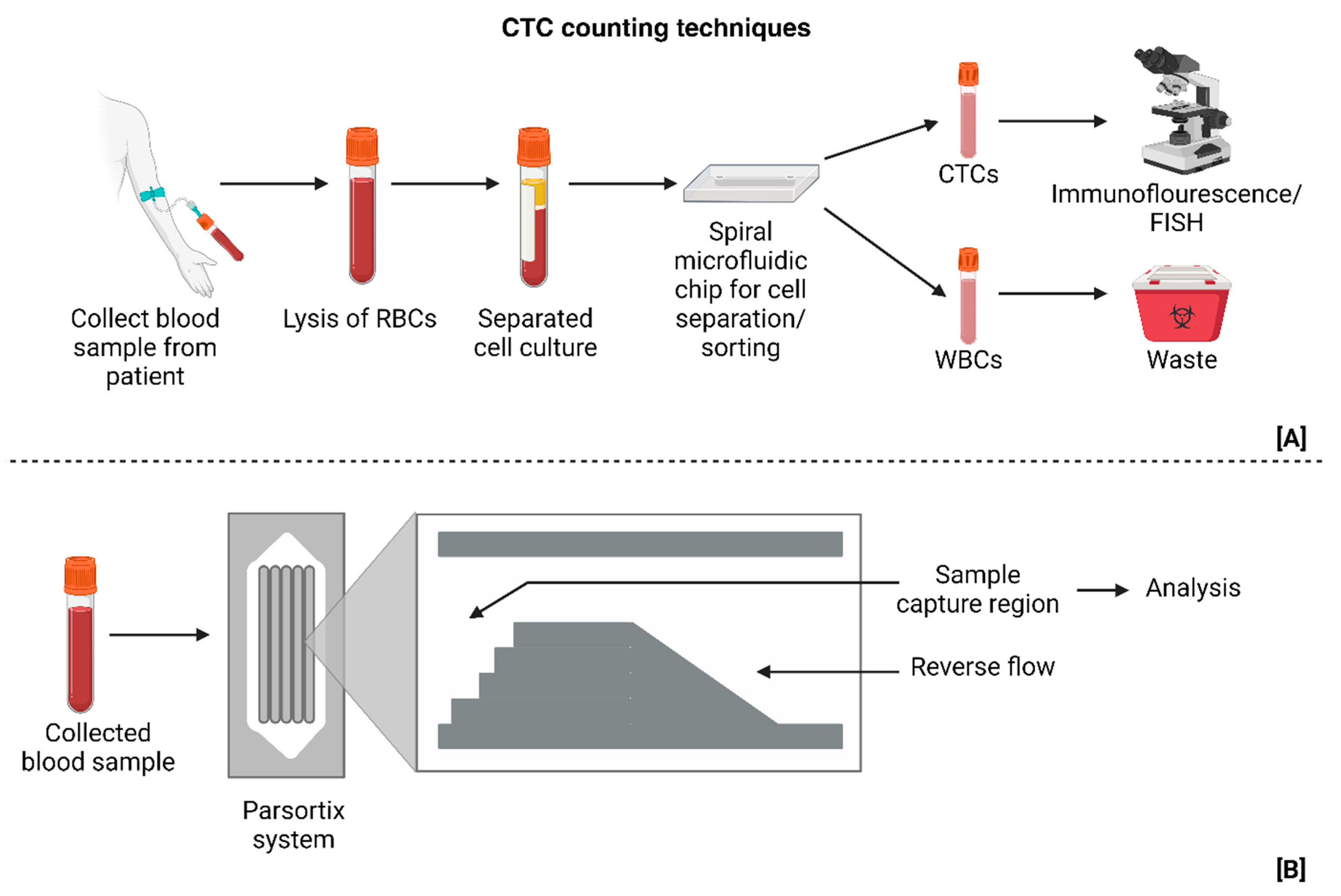
| Tumor modeling | Spiral Microfluidic technology | N/A | EGFR, DAPI positive (nuclei staining), GFAP and CSV | Established clinical significance of CTC counts in GBM studies | Small patient cohort | [27] |
| TME/Tumor modeling | Lateral microchannels with a central chamber for tumor movement | N/A | Glucose gradient (NBDG), Ki-67, oxygen profiles | Mathematical models quantify and correlate in vitro and in vivo measurements for hypoxia, migration, and re-oxygenation Direct observation of GBM invasion and pseudopalisade formation under controlled conditions | N/A | [28] |
| Drug development | Parallel cell culture chambers with separate inlets and outlets | Colchicine | N/A | Prevented cellular erosion/damage due to nutrient medium introduction by applying hydrostatic pressure and regulatory supplying new cells (intermittent dynamic culture) | N/A | [29] |
| TME modeling | Concentric microchannel layouts (triculture model) | N/A | CD31 and phalloidin (vasculogenesis), EdU and Ki-67 (cell proliferation), CD44, Nestin, and SOX2 (GSCs) | Integrates multiple cell types to create a biologically active tumor niche | N/A | [30] |
| Cancer therapy | Central chamber with parallel microchannels | Magnetic nanoparticles coated with aminosilane | N/A | Reduces cellular proliferation and less toxic | Lack of vascular network associated with tumor tissue | [31] |
| Tumor modeling and identification of cellular targets for treatment | Parsortix® PC1 system | BAL101553 (microtubule inhibitor) | EGFR, Ki-67, CD45 (-) and EB1 | Proves that CTCs move across the BBB and contribute to metastasis | Small patient cohort (no established relation between MRI volume and CTC count) | [32] |
| Drug development | Multiple consecutive channels with loading sites for spheroid formation | Nanoparticle albumin-bound paclitaxel (nab-PTX)- nab-PTX/MΦ | IL-1β, IL-6, and TNF-α, CD86 (M1 specific), ZO-1 (HUVECs), | The carriers (macrophages) were not damaged by addition of NPs, macrophage-associated cultures showed higher rates of cell death | Acquisition of sufficient quantities of MΦ poses a challenge due to their status as terminally differentiated cells, challenges in tumor maintenance, prolonging circulation half-life of macrophages | [33] |
| Drug development, immunotherapy | Three inlet and outlet channels with a central, concentric culture environment | BLZ945 (CSF-1R inhibitor), Nivolumab | IL-10, TGF-β1, IFN-γ and TNF-α (cytokine quantification), Caspase 3/7 (cell apoptosis), CD163, CD154 and CD69 (Cytotoxic T-cell) | Multidimensional readout of patient-specific responses to different immunotherapy regimens ex vivo | Allogeneic immune and stromal cells used in the current proof-of-concept GBM model may limit the clinical significance of the findings for patient-specific immunotherapy screening, absence of BBB, no proper ECM | [34] |
| TME modeling | Platform to map cell migration velocity | N/A (No drugs were tested in this model) | Cell velocity | Use of macrophage-depleted medium and a regular growth medium ensured that the influence of different factors could be inferred | Lack of concrete conclusions, absence of the ECM and BB | [35] |
| Drug development | iMER platform (exosomal RNA analysis platform) | TMZ | CD63, EGFR and EGFRvIII, PDPN, EPHA2, MGMT, APNG, GSTπ1, ERCC1, ERCC2, MVP, ABCC3, CASP8, IGFBP2 | Tumor-derived exosomes have the advantages that they represent the heterogeneity of the tumor, are very abundant in blood, highly stable, readily pass the blood–brain barrier and can be analyzed in small volumes in serum/plasma samples, highly sensitive measurements, rapid turnaround time | Expanded marker panel, more chambers for diversified diagnostics, small patient cohort | [36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raman, R.; Prabhu, V.; Kumar, P.; Mani, N.K. Advancements in Microfluidic Platforms for Glioblastoma Research. Chemistry 2024, 6, 1039-1062. https://doi.org/10.3390/chemistry6050060
Raman R, Prabhu V, Kumar P, Mani NK. Advancements in Microfluidic Platforms for Glioblastoma Research. Chemistry. 2024; 6(5):1039-1062. https://doi.org/10.3390/chemistry6050060
Chicago/Turabian StyleRaman, Rachana, Vijendra Prabhu, Praveen Kumar, and Naresh Kumar Mani. 2024. "Advancements in Microfluidic Platforms for Glioblastoma Research" Chemistry 6, no. 5: 1039-1062. https://doi.org/10.3390/chemistry6050060
APA StyleRaman, R., Prabhu, V., Kumar, P., & Mani, N. K. (2024). Advancements in Microfluidic Platforms for Glioblastoma Research. Chemistry, 6(5), 1039-1062. https://doi.org/10.3390/chemistry6050060