
Non-Coding RNAs in the Cardiac Action Potential and Their Impact on Arrhythmogenic Cardiac Diseases




Abstract
:1. The Electrical Components of the Adult Heart
2. Non-Coding RNAs, Classification, and Function
3. Role of ncRNAs in the Cardiac Action Potential
3.1. ncRNAs in the Upstroke Phase (INa Currents)
3.2. ncRNAs in the Upstroke Phase (If Current)
3.3. ncRNAs in Sodium Channel Interacting Proteins
4. Role of ncRNAs in Cardiac Repolarization
4.1. ncRNAs in the Early Repolarization (ITO Transient Outward K+ Current)
4.2. ncRNAs in the Plateau Phase and Terminal Repolarization (IKr, IKs, IKur K+ Current)
4.3. ncRNAs Modulating the Ultra-Rapid Delayed Rectifier K+ Current (IKur)
4.4. ncRNAs Modulating the Rapid Delayed Rectifier K+ Current (IKr)
4.5. ncRNAs Modulating the Slow Delayed Rectifier K + Current (IKs)
4.6. ncRNAs in the Resting Membrane Potential (IK1 Current and Na,K ATPase)
5. Role of ncRNAs in Conduction Contraction Coupling
5.1. ncRNAs in Calcium Currents (ICa,L Current)
5.2. The Role of ncRNAs in Calcium-Induced Calcium Release
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Zipes, D.P.; Wellens, H.J.J. Sudden Cardiac Death. Circulation 1998, 98, 2334–2351. [Google Scholar] [CrossRef]
- Van Weerd, J.H.; Christoffels, V.M. The formation and function of the cardiac conduction system. Development 2016, 143, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Shih, H.T. Anatomy of the action potential in the heart. Texas Heart Inst. J. 1994, 21, 30–41. [Google Scholar]
- Nerbonne, J.M.; Kass, R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartos, D.C.; Grandi, E.; Ripplinger, C.M. Ion channels in the heart. Compr. Physiol. 2015, 5, 1423–1464. [Google Scholar] [PubMed] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Endo, M. Calcium-induced release of calcium from the sarcoplasmic reticulum. Am. J. Physiol. Cell Physiol. 2007, 592, 275–285. [Google Scholar]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef] [PubMed]
- Milani-Nejad, N.; Janssen, P.M.L. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharmacol. Ther. 2014, 141, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Vornanen, M.; Hassinen, M. Zebrafish heart as a model for human cardiac electrophysiology. Channels 2016, 10, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J. Evidences of the gender-related differences in cardiac repolarization and the underlying mechanisms in different animal species and human. Fundam. Clin. Pharmacol. 2006, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, D.; Rosati, B. Transmural gradients in ion channel and auxiliary subunit expression. Prog. Biophys Mol. Biol 2016, 122, 165–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, D.M.; Nerbonne, N.J. Myocardial potassium channels: Electrophysiological and molecular diversity. Annu. Rev. Physiol. 1996, 58, 363–394. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Namekata, I.; Nouchi, H.; Shigenobu, K.; Kawanishi, T.; Takahara, A. New aspects for the treatment of cardiac diseases based on the diversity of functional controls on cardiac muscles: Diversity in the excitation-contraction mechanisms of the heart. J. Pharmacol. Sci. 2009, 109, 327–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ber, D.M. Species Differences and the Role of Sodium-Calcium Exchange in Cardiac Muscle Relaxation. Ann. N. Y. Acad. Sci. 1991, 639, 375–385. [Google Scholar]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, C.; Wells, R.; et al. Molecular biology: The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [PubMed] [Green Version]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1764. [Google Scholar] [CrossRef] [Green Version]
- García-Padilla, C.; Aránega, A.; Franco, D. The role of long non-coding RNAs in cardiac development and disease. AIMS Genet. 2018, 5, 124–140. [Google Scholar] [CrossRef]
- Expósito-Villén, A.; Aránega, A.E.; Franco, D. Functional role of non-coding RNAs during epithelial-to-mesenchymal transition. Non-Coding RNA 2018, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Sun, M.; Liu, H.; Yao, Y.; Song, Y. Long non-coding RNAs: A new frontier in the study of human diseases. Cancer Lett. 2013, 339, 159–166. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.S.; Ono, M. From snoRNA to miRNA: Dual function regulatory non-coding RNAs. Biochimie 2011, 93, 1987–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNA Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.R.; Rameshwar, P. MicroRNA in Development and in the Progression of Cancer; Springer Science & Business: Berlin, Germany, 2014; ISBN1 978-1-4899-8064-9. ISBN2 978-1-4899-8065-6. (eBook). [Google Scholar]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef]
- Mathieu, È.L.; Belhocine, M.; Dao, L.T.M.; Puthier, D.; Spicuglia, S. Functions of lncRNA in development and diseases. Med. Sci. 2014, 30, 790–796. [Google Scholar]
- García-Padilla, C.; Domínguez, J.N.; Aránega, A.E.; Franco, D. Differential chamber-specific expression and regulation of long non-coding RNAs during cardiac development. BBA Gene Regul. Mech. 2019, 1862, 194435. [Google Scholar] [CrossRef]
- Luo, S.; Lu, J.Y.; Liu, L.; Yin, Y.; Chen, C.; Han, X.; Bohou, W.; Xu, R.; Liu, W.; Yan, P.; et al. Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell 2016, 18, 637–652. [Google Scholar] [CrossRef]
- Amin, A.S.; Tan, H.L.; Wilde, A.A.M. Cardiac ion channels in health and disease. Heart Rhythm 2010, 7, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Remme, C.A. Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. J. Physiol. 2013, 591, 4099–4116. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.S.; Asghari-Roodsari, A.; Tan, H.L. Cardiac sodium channelopathies. Pflugers Arch. Eur. J. Physiol. 2010, 460, 223–237. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Agustín, A.; Pinsach-Abuin, M.L.; Pagans, S. Role of non-coding variants in brugada syndrome. Int. J. Mol. Sci. 2020, 21, 8556. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; et al. Spectrum of mutations in Long-QT Syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3624763/pdf/nihms412728.pdf (accessed on 25 June 2021). [CrossRef]
- Gando, I.; Williams, N.; Fishman, G.I.; Sampson, B.A.; Tang, Y.; Coetzee, W.A. Functional characterization of SCN10A variants in several cases of sudden unexplained death. Forensic Sci. Int. 2019, 301, 289–298. [Google Scholar] [CrossRef]
- Delaney, J.T.; Muhammad, R.; Shi, Y.; Schildcrout, J.S.; Blair, M.; Short, L.; Roden, D.M.; Darbar, D. Common SCN10A variants modulate PR interval and heart rate response during atrial fibrillation. Europace 2014, 16, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Savio-Galimberti, E.; Weeke, P.; Muhammad, R.; Blair, M.; Ansari, S.; Short, L.; Atack, T.C.; Kor, K.; Vanoye, C.G.; Olesen, M.S.; et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc. Res. 2014, 104, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monasky, M.M.; Micaglio, E.; Vicedomini, G.; Locati, E.T.; Ciconte, G.; Giannelli, L.; Giordano, F.; Crisa, S.; Vecchi, M.; Borrelli, V.; et al. Comparable clinical characteristics in Brugada syndrome patients harboring SCN5A or novel SCN10A variants. Europace 2019, 21, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Van Den Boogaard, M.; Smemo, S.; Burnicka-Turek, O.; Arnolds, D.E.; Van De Werken, H.J.G.; Klous, P.; McKean, D.; Muehlschlegel, J.D.; Moosmann, J.; Toka, O.; et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Investig. 2014, 124, 1844–1852. [Google Scholar] [CrossRef] [Green Version]
- Daimi, H.; Lozano-Velasco, E.; Haj Khelil, A.; Chibani, J.B.E.; Barana, A.; Amorós, I.; Gonzáles de la Fuente, M.; Caballero, R.; Aranega, A.; Franco, D. Regulation of SCN5A by microRNAs: miR-219 modulates SCN5A transcript expression and the effects of flecainide intoxication in mice. Heart Rhythm 2015, 12, 1333–1342. Available online: http://www.ncbi.nlm.nih.gov/pubmed/25701775 (accessed on 15 April 2016). [CrossRef] [PubMed]
- Zhao, Y.; Huang, Y.; Li, W.; Wang, Z.; Zhan, S.; Zhou, M.; Yao, Y.; Zen, Z.; Hou, Y.; Chen, Q.; et al. Post-transcriptional regulation of cardiac sodium channel gene SCN5A expression and function by miR-192-5p. Biochim. Biophys. Acta 2015, 1852, 2024–2034. [Google Scholar] [CrossRef] [Green Version]
- Chinchilla, A.; Daimi, H.; Lozano-Velasco, E.; Dominguez, J.N.; Caballero, R.; Delpo, E.; Tamargo, J.; Cinca, J.; Hove-Madsen, L.; Aranega, A.; et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 2011, 4, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Poon, E.N.Y.; Hao, B.; Guan, D.; Li, M.J.; Lu, J.; Yang, Y.; Wu, B.; Wu, S.C.M.; Webb, S.E.; Liang, Y.; et al. Integrated transcriptomic and regulatory network analyses identify microRNA-200c as a novel repressor of human pluripotent stem cell-derived cardiomyocyte differentiation and maturation. Cardiovasc. Res. 2018, 114, 894–906. [Google Scholar] [CrossRef]
- Li, J.; Xu, C.; Liu, Y.; Li, Y.; Du, S.; Zhang, R.; Sun, Y.; Zhang, R.; Wang, Y.; Xue, H.; et al. Fibroblast growth factor 21 inhibited ischemic arrhythmias via targeting miR-143/EGR1 axis. Basic Res. Cardiol. 2020, 115, 9. [Google Scholar] [CrossRef]
- Zhang, X.; Yoon, J.Y.; Morley, M.; McLendon, J.M.; Mapuskar, K.A.; Gutmann, R.; Mehdi, H.; Bloom, H.L.; Dudley, S.C.; Ellinor, P.T.; et al. A common variant alters SCN5A-miR-24 interaction and associates with heart failure mortality. J. Clin. Investig. 2018, 128, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Daimi, H.; Khelil, A.H.; Neji, A.; Ben Hamda, K.; Maaoui, S.; Aranega, A.; Chibani, J.B.; Franco, D. Role of SCN5A coding and non-coding sequences in Brugada syndrome onset: What’s behind the scenes? Biomed. J. 2019, 42, 252–260. [Google Scholar] [CrossRef]
- Tan, Y.X.; Hong, Y.; Jiang, S.; Lu, M.N.; Li, S.; Chen, B.; Zhang, L.; Hu, T.; Mao, R.; Mei, R.; et al. MicroRNA-449a regulates the progression of brain aging by targeting SCN2B in SAMP8 mice. Int. J. Mol. Med. 2020, 45, 1091–1102. [Google Scholar] [CrossRef]
- Brandenburger, T.; Johannsen, L.; Prassek, V.; Kuebart, A.; Raile, J.; Wohlfromm, S.; Kohrer, K.; Huhn, R.; Hollmann, M.W.; Hermanns, H. MiR-34a is differentially expressed in dorsal root ganglia in a rat model of chronic neuropathic pain. Neurosci. Lett. 2019, 708, 134365. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, X.; Cui, L.; He, G.; Wang, X.; Wang, F.; Duan, S.; He, L.; Li, Q.; Yu, X.; et al. Genetic regulatory subnetworks and key regulating genes in rat hippocampus perturbed by prenatal malnutrition: Implications for major brain disorders. Aging 2020, 12, 8434–8458. [Google Scholar] [CrossRef] [PubMed]
- Norcini, M.; Choi, D.; Lu, H.; Cano, M.; Piskoun, B.; Hurtado, A.; Sideris, A.; Blanck, T.J.J.; Recio-Pinto, E. Intrathecal Injection of miR-133b-3p or miR-143-3p Prevents the Development of Persistent Cold and Mechanical Allodynia Following a Peripheral Nerve Injury in Rats. Neuroscience 2018, 386, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Salunkhe, V.A.; Esguerra, J.L.S.; Ofori, J.K.; Mollet, I.G.; Braun, M.; Stoffel, M.; Wendt, A.; Eliasson, L. Modulation of microRNA-375 expression alters voltage-gated Na+ channel properties and exocytosis in insulin-secreting cells. Acta Physiol. 2015, 213, 882–892. [Google Scholar] [CrossRef]
- Huang, H.; Qing, X.Y.; Zhou, Q.; Li, H.D.; Hu, Z.Y. Silencing of microRNA-3175 represses cell proliferation and invasion in prostate cancer by targeting the potential tumor-suppressor SCN4B. Kaohsiung J. Med. Sci. 2021, 37, 20–26. [Google Scholar] [CrossRef]
- Dai, W.; Zhou, J.; Wang, H.; Zhang, M.; Yang, X.; Song, W. miR-424-5p promotes the proliferation and metastasis of colorectal cancer by directly targeting SCN4B. Pathol Res. Pract. 2020, 216, 152731. [Google Scholar] [CrossRef]
- Zhao, J.; Lee, M.C.; Momin, A.; Cendan, C.M.; Shepherd, S.T.; Baker, M.D.; Asante, C.; Bee, L.; Berthy, A.; Perkins, J.R.; et al. Small RNAs control sodium channel expression, nociceptor excitability, and pain thresholds. J. Neurosci. 2010, 30, 10860–10871. [Google Scholar] [CrossRef]
- Yan, J.; Yu, H.; Shen, J.; Han, C.; Li, C.; Shen, X.; Li, B. Early over-expressing of microRNA-145 effectively precludes the development of neuropathic mechanical hyperalgesia via suppressing Nav1.8 in diabetic rats. Pain Physician 2020, 23, E673–E686. [Google Scholar]
- D’Souza, A.; Pearman, C.M.; Wang, Y.; Nakao, S.; Logantha, S.J.R.J.; Cox, C.; Bennett, H.; Zhang, Y.; Johnsen, A.B.; Linscheid, N.; et al. Targeting miR-423-5p Reverses Exercise Training-Induced HCN4 Channel Remodeling and Sinus Bradycardia. Circ. Res. 2017, 121, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Yanni, J.; D’Souza, A.; Wang, Y.; Li, N.; Hansen, B.J.; Zakharkin, S.O.; Smith, M.; Hayward, C.; Whitson, B.; Mohler, P.J.; et al. Silencing miR-370-3p rescues funny current and sinus node function in heart failure. Sci. Rep. 2020, 10, 11279. [Google Scholar] [CrossRef] [PubMed]
- Petkova, M.; Atkinson, A.J.; Yanni, J.; Stuart, L.; Aminu, A.J.; Ivanova, A.D.; Pustovit, K.B.; Geragthy, C.; Feather, A.; Li, N.; et al. Identification of Key Small Non-Coding MicroRNAs Controlling Pacemaker Mechanisms in the Human Sinus Node. J. Am. Heart Assoc. 2020, 9, e016590. [Google Scholar] [CrossRef]
- Li, Y.D.; Hong, Y.F.; Yusufuaji, Y.; Tang, B.P.; Zhou, X.H.; Xu, G.J.; Li, J.X.; Sun, L.; Zhang, J.H.; Xin, Q.; et al. Altered expression of hyperpolarization-activated cyclic nucleotide-gated channels and microRNA-1 and -133 in patients with age-associated atrial fibrillation. Mol. Med. Rep. 2015, 12, 3243–3248. [Google Scholar] [CrossRef]
- Suffredini, S.; Stillitano, F.; Comini, L.; Bouly, M.; Brogioni, S.; Ceconi, C.; Ferrati, R.; Mugelli, A.; Cerbai, E. Long-term treatment with ivabradine in post-myocardial infarcted rats counteracts f-channel overexpression. Br. J. Pharmacol. 2012, 165, 1457–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.D.; Xia, S.; Zha, C.Q.; Deng, S.B.; Du, J.L.; She, Q. Spironolactone Regulates HCN Protein Expression Through Micro-RNA-1 in Rats with Myocardial Infarction. J. Cardiovasc. Pharmacol. 2015, 65, 587–592. [Google Scholar] [CrossRef] [Green Version]
- D’souza, A.; Bucchi, A.; Johnsen, A.B.; Logantha, S.J.R.J.; Monfredi, O.; Yanni, J.; Prehar, S.; Hart, G.; Cartwright, E.; Wisloff, U.; et al. Exercise training reduces resting heart rate via downregulation of the funny channel HCN4. Nat. Commun. 2014, 5, 3775. [Google Scholar] [CrossRef]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Y.; Du, W.; Liang, H.; He, H.; Zhang, L.; Pan, Z.; Li, X.; Xu, C.; Zhou, Y.; et al. MiR-223-3p as a Novel MicroRNA Regulator of Expression of Voltage-Gated K + Channel Kv4.2 in Acute Myocardial Infarction. Cell Physiol. Biochem. 2016, 39, 102–114. [Google Scholar] [CrossRef]
- Nassal, D.M.; Wan, X.; Liu, H.; Maleski, D.; Ramirez-Navarro, A.; Moravec, C.S.; Ficker, E.; LAurita, K.; Deschenes, I. KChIP2 is a core transcriptional regulator of cardiac excitability. Elife 2017, 6, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Yang, M.; Ren, H.; Shen, G.; Chen, J.; Zhang, J.; Liu, J.; Sun, C. Long noncoding RNA MALAT1 downregulates cardiac transient outward potassium current by regulating miR-200c/HMGB1 pathway. J. Cell Biochem. 2018, 119, 10239–10249. [Google Scholar] [CrossRef] [PubMed]
- Mondejar-Parreño, G.; Callejo, M.; Barreira, B.; Morales-Cano, D.; Esquivel-Ruiz, S.; Moreno, L.; Cogolludo, A.; Perez-Vizcaino, F. miR-1 is increased in pulmonary hypertension and downregulates Kv1.5 channels in rat pulmonary arteries. J. Physiol. 2019, 597, 1185–1197. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Guo, J.; Huang, X.; Yang, X.; Huang, G.; Mao, H.; Sun, H.H.; Ba, Y.; Zhou, J. miRNAs Regulate hERG. J. Cardiovasc. Electrophysiol. 2016, 27, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, C.M.; Xi, Y.; Wu, G.; Shelat, H.; Gao, S.; Cheng, J.; Geng, Y.J. MicroRNA-1/133 targeted dysfunction of potassium channels KCNE1 and KCNQ1 in human cardiac progenitor cells with simulated hyperglycemia. IJC 2013, 167, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zheng, S.; Xie, X.; Zhang, Y.; Wang, W.; Wang, Z.; Zhang, Y.; Wang, J.; Gao, M.; Hou, Y. MicroRNA-1 accelerates the shortening of atrial effective refractory period by regulating KCNE1 and KCNB2 expression: An atrial tachypacing rabbit model. PLoS ONE 2013, 8. Available online: https://pubmed.ncbi.nlm.nih.gov/24386485/ (accessed on 11 December 2020). [CrossRef] [Green Version]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef]
- Girmatsion, Z.; Biliczki, P.; Bonauer, A.; Wimmer-Greinecker, G.; Scherer, M.; Moritz, A.; Bukowska, A.; Goetter, A.; Nattel, S.; Hohnloser, S.H.; et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm 2009, 6, 1802–1809. [Google Scholar] [CrossRef]
- Li, X.; Hu, H.; Wang, Y.; Xue, M.; Li, X.; Cheng, W.; Xuan, Y.; Yin, J.; Yang, N.; Yan, S. Valsartan ameliorates KIR2.1 in rats with myocardial infarction via the NF-κB-miR-16 pathway. Gene 2016, 590, 201–209. [Google Scholar] [CrossRef]
- Luo, X.; Pan, Z.; Shan, H.; Xiao, J.; Sun, X.; Wang, N.; Lin, X.; Xiao, L.; Maguy, A.; Qi, X.Y.; et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. JCI 2013, 123, 1939–1951. Available online: https://pubmed.ncbi.nlm.nih.gov/23543060/ (accessed on 11 December 2020). [CrossRef]
- Qi, X.Y.; Huang, H.; Ordog, B.; Luo, X.; Naud, P.; Sun, Y.; Wu, C.T.; Dawson, K.; Tadevosyan, A.; Chen, Y.; et al. Fibroblast inward-rectifier potassium current upregulation in profibrillatory atrial Remodeling. Circ. Res. 2015, 116, 836–845. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; Van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Goldoni, D.; Yarham, J.M.; McGahon, M.K.; O’Connor, A.; Guduric-Fuchs, J.; Edgar, K.; McDonald, D.M.; Simpson, D.A.; Collins, A. A novel dual-fluorescence strategy for functionally validating microRNA targets in 3′ untranslated regions: Regulation of the inward rectifier potassium channel Kir2.1 by miR-212. Biochem. J. 2012, 448, 103–113. [Google Scholar] [CrossRef]
- Zhang, X.J.; Liao, C.X.; Sun, K.J.; Liu, L.L.; Xu, D.Y. A soluble epoxide hydrolase inhibitor upregulated kcnj12 and kcnip2 by downregulating microrna-29 in a mouse model of myocardial infarction. Heart Surg. Forum. 2020, 23, E579–E585. [Google Scholar] [CrossRef]
- Binas, S.; Knyrim, M.; Hupfeld, J.; Kloeckner, U.; Rabe, S.; Mildenberger, S.; Quarch, K.; Stratz, N.; Misiak, D.; Gekle, M.; et al. miR-221 and -222 target CACNA1C and KCNJ5 leading to altered cardiac ion channel expression and current density. CLMS 2019, 77, 903–918. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, Y.; Wang, N.; Pan, Z.; Gao, X.; Zhang, F.; Zhang, Y.; Shan, H.; Luo, X.; Bai, Y.; et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 2010, 122, 2378–2387. [Google Scholar] [CrossRef]
- Barana, A.; Matamoros, M.; Dolz-Gaitón, P.; Pérez-Hernández, M.; Amorós, I.; Núñez, M.; Sacristan, S.; Pedraz, A.; Pinto, A.; Fernandez-Aviles, F.; et al. Chronic atrial fibrillation increases MicroRNA-21 in human atrial myocytes decreasing L-type calcium current. Circ. Arrhythmia Electrophysiol. 2014, 7, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cañón, S.; Caballero, R.; Herraiz-Martínez, A.; Pérez-Hernández, M.; López, B.; Atienza, F.; Jalife, J.; Hove-Madsen, L.; Delpon, E.; Bernard, A. miR-208b upregulation interferes with calcium handling in HL-1 atrial myocytes: Implications in human chronic atrial fibrillation. J. Mol. Cell Cardiol. 2016, 99, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yuan, Y.; Qiu, C. Underexpression of CACNA1C caused by overexpression of microRNA-29a underlies the pathogenesis of atrial fibrillation. Med. Sci. Monit. 2016, 22, 2175–2181. [Google Scholar] [CrossRef] [Green Version]
- Ling, T.Y.; Wang, X.L.; Chai, Q.; Lu, T.; Stulak, J.M.; Joyce, L.D.; Daly, R.C.; Greason, K.L.; Wu, L.Q.; Shen, W.K.; et al. Regulation of cardiac CACNB2 by microRNA-499: Potential role in atrial fibrillation. BBA Clin. 2017, 7, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Li, A.; Chen, X.; Qin, Y.; Sun, X.; Li, Y.; Yue, E.; Wang, C.; Ding, X.; Yan, Y.; et al. Overexpression of miR-135b attenuates pathological cardiac hypertrophy by targeting CACNA1C. Int. J. Cardiol. 2018, 269, 235–241. [Google Scholar] [CrossRef]
- Liu, Z.; Tao, B.; Fan, S.; Pu, Y.; Xia, H.; Xu, L. MicroRNA-145 protects against myocardial ischemia reperfusion injury via CaMKII-mediated antiapoptotic and anti-inflammatory pathways. Oxid. Med. Cell Longev. 2019, 10, 8948657. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Kongchan, N.; Beavers, D.L.; Alsina, K.M.; Voigt, N.; Neilson, J.R.; Jakob, H.; Martin, J.F.; Dobrev, D.; Wehrens, X.H.T.; et al. Loss of MicroRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ. Arrhythmia Electrophysiol. 2014, 7, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Qin, M.; Tan, Q.; Li, T.; Gu, Z.; Huang, P.; Ren, L. MicroRNA-129-1-3p protects cardiomyocytes from pirarubicin-induced apoptosis by down-regulating the GRIN2D-mediated Ca2+ signalling pathway. J. Cell Mol. Med. 2020, 24, 2260–2271. [Google Scholar] [CrossRef] [Green Version]
- Belevych, A.E.; Sansom, S.E.; Terentyeva, R.; Ho, H.T.; Nishijima, Y.; Martin, M.M.; Jindal, H.K.; Rochira, J.A.; Kunimoto, Y.; Abdellatif, M.; et al. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS ONE 2011, 6, e28324. [Google Scholar] [CrossRef]
- Terentyev, D.; Belevych, A.E.; Terentyeva, R.; Martin, M.M.; Malana, G.E.; Kuhn, D.E.; Abdellatif, M.; Feldman, D.S.; Elton, T.S.; Gyorke, S. MiR-1 Overexpression Enhances Ca2+ release and Promotes Cardiac Arrhythmogenesis by Targeting PP2A Regulatory Subunit B56α and Causing CaMKII-Dependent Hyperphosphorylation of RyR2. Circ Res. 2009, 104, 514–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.C.; Tao, J.; Guo, Y.B.; Wu, H.D.; Liu, R.F.; Bai, Y.; LV, Z.Z.; Luo, G.Z.; Li, L.L.; Wang, M.; et al. In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ. Res. 2013, 112, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-Y.; Shen, H.; Yang, Q.; Min, J.; Wang, Q.; Xi, W.; Yin, L.; Le, S.G.; Zhang, Y.F.; Xiao, J.; et al. LncRNA-LINC00472 contributes to the pathogenesis of atrial fibrillation (Af) by reducing expression of JP2 and RyR2 via miR-24. Biomed. Pharmacother. 2019, 120, 109364. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Yoo, J.; Lee, P.; Kepreotis, S.V.; Lee, A.; Wahlquist, C.; Brown, B.D.; Kho, C.; Mercola, M.; Hajjar, R.J. miR-25 Tough Decoy Enhances Cardiac Function in Heart Failure. Mol. Ther. 2018, 7, 718–729. [Google Scholar] [CrossRef]
- Wahlquist, C.; Jeong, D.; Rojas-Muñoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; van Mil, A.; Park, W.J.; Sluijter, J.P.G.; Doevendans, P.A.F.; et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014, 508, 531–535. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Gao, X.; Zhang, R.; Zhang, Y.; Liang, H.; Xu, C.; Du, W.; Zhang, Y.; Liu, X.; et al. MicroRNA-328 as a regulator of cardiac hypertrophy. Int. J. Cardiol. 2014, 173, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Hu, X.; Ge, T.; Li, M.; Shi, M.; Luo, J.; Lai, H.; Nie, T.; Li, F.; Li, H. MicroRNA-328 is involved in the effect of selenium on hydrogen peroxide-induced injury in H9c2 cells. J. Biochem. Mol. Toxicol. 2017, 31, 3–9. [Google Scholar] [CrossRef]
- Williams, A.L.; Walton, C.B.; MacCannell, K.A.; Avelar, A.; Shohet, R.V. HIF1 regulation of miR-29c impairs SERCA2 expression and cardiac contractility. AJPheart. 2019, 53, 21–25. Available online: http://doi.org/10.1152/ajpheart.00617.2018 (accessed on 25 June 2021).
- Mayourian, J.; Ceholski, D.K.; Gorski, P.A.; Mathiyalagan, P.; Murphy, J.F.; Salazar, S.I.; Stillitano, F.; Hare, J.M.; Sahoo, S.; Hajjar, R.J.; et al. Exosomal microRNA-21-5p Mediates Mesenchymal Stem Cell Paracrine Effects on Human Cardiac Tissue Contractility. Circ. Res. 2018, 122, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Gurha, P.; Abreu-Goodger, C.; Wang, T.; Ramirez, M.O.; Drumond, A.L.; Van Dongen, S.; Chen, Y.; Bartonicek, N.; Enright, A.J.; Lee, B.; et al. Targeted deletion of MicroRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation 2012, 125, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.F.S.; Barauna, V.G.; Neves, V.J.; Fernandes, T.; Lara, L.d.S.; Mazzotti, D.R. Exercise training restores the cardiac microRNA-1 and -214 levels regulating Ca2+ handling after myocardial infarction. BMC Cardiovasc. Disord. 2015, 15, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Chiasson, V.; Takano, A.P.C.; Guleria, R.S.; Gupta, S. Deficiency of MicroRNA miR-1954 Promotes Cardiac Remodeling and Fibrosis. J. Am. Heart Assoc. 2019, 8, 1–13. [Google Scholar] [CrossRef]
- Mishra, P.K.; Metreveli, N.; Tyagi, S.C. MMP-9 gene ablation and TIMP-4 mitigate PAR-1-mediated cardiomyocyte dysfunction: A plausible role of dicer and miRNA. Cell Biochem. Biophys. 2010, 57, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Soller, K.J.; Yang, J.; Veglia, G.; Bowser, M.T. Reversal of phospholamban inhibition of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) using short, protein-interacting RNAs and oligonucleotide analogs. J. Biol. Chem. 2016, 291, 21510–21518. [Google Scholar] [CrossRef] [Green Version]
- Bedada, F.B.; Martindale, J.J.; Arden, E.; Metzger, J.M. Molecular inotropy mediated by cardiac miR-based PDE4D/PRKAR1α/phosphoprotein signaling. Sci. Rep. 2016, 6, 36803. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Lee, J.; Seo, H.H.; Lee, C.Y.; Yoo, K.J.; Kim, S.M.; Lee, S.; Hwang, K.C.; Choi, E. Na+-Ca2+ exchanger targeting miR-132 prevents apoptosis of cardiomyocytes under hypoxic condition by suppressing Ca2+ overload. Biochem. Biophys. Res. Commun. 2015, 460, 931–937. [Google Scholar] [CrossRef]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincen, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Mourad, N.A.-R. Microrna Regulation of Herg-Related Current: Potential Role in Heart Failure-Associated Arrhythmias [Internet]. Purdue University, 2015. Available online: https://docs.lib.purdue.edu/dissertations/AAI10188989/ (accessed on 25 June 2021).
- Takumi, T.; Ohkubo, H.; Nakanishi, S. Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science 1988, 242, 1042–1045. [Google Scholar] [CrossRef]
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. K(V)LQT1 and IsK (minK) proteins associate to form the I(Ks) Cardiac Potassium Current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M. Coaseembly of KvLQT1 and minK (IsK) proteins to form cardiac Iks potassium channel. Lett. Nat. 1996, 384, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Xue, H.; Jin, Q.H.; Guo, J.; Chen, Y.D. Increased expression of ryanodine receptor type-2 during atrial fibrillation by miR-106-25 cluster independent mechanism. Exp. Cell Res. 2019, 375, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wang, Y.; Wang, Y.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Zhang, J.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges miR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, E108–E125. [Google Scholar] [CrossRef]
- DiFrancesco, D.; Borer, J.S. The Funny Current. Drugs 2007, 67 (Suppl. S2), 15–24. [Google Scholar] [CrossRef]
- Yu, S.Y.; Na, J.Y.; Lee, Y.J.; Kim, K.T.; Park, J.T.; Kim, H.S. Forensic application of microRNA-706 as a biomarker for drowning pattern identification. Forensic Sci. Int. 2015, 255, 96–101. [Google Scholar] [CrossRef]
- Abriel, H. Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. J. Mol. Cell Cardiol. 2010, 48, 2–11. [Google Scholar] [CrossRef]
- Shy, D.; Gillet, L.; Abriel, H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: The multiple pool model. Biochim. Biophys. Acta 2013, 1833, 886–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chazin, W.J.; Johnson, C.N. Calmodulin mutations associated with heart arrhythmia: A status report. Int. J. Mol. Sci. 2020, 21, 1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takla, M.; Huang, C.L.H.; Jeevaratnam, K. The cardiac CaMKII-Nav1.5 relationship: From physiology to pathology. J. Mol. Cell Cardiol. 2020, 139, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, M.; Patel, H.H.; Roth, D.M. Caveolins and HEart Diseases, Caveolins and Caveolae. In Caveolins and Caveolae Advances in Experimental Medicine and Biology; Springer Science+Business Media: Berlin, Germany, 2012; Volume 729, pp. 145–156. ISBN 978-1-4614-1221-2. [Google Scholar]
- Balijepalli, R.C.; Kamp, T.J. Caveolae, Ion Channels and Cardiac Arrhythmias Ravi. Prog. Biophys. Mol. Biol. 2008, 98, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Greer-Short, A.; Musa, H.; Alsina, K.M.; Ni, L.; Word, T.A.; Reynolds, J.O.; Gratz, D.; Lane, C.; El-Refaey, M.; Unudurthu, S.; et al. Calmodulin kinase II regulates atrial myocyte late sodium current, calcium handling, and atrial arrhythmia. Heart Rhythm 2020, 17, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Vermij, S.H.; Sottas, V.; Shestak, A.; Ross-Kaschitza, D.; Zaklyazminskaya, E.V.; Hudmon, A.; Pitt, G.S.; Rougier, J.S.; Abriel, H. Calmodulin binds to the N-terminal domain of the cardiac sodium channel Nav1.5. Channels 2020, 14, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Gabelli, S.B.; Boto, A.; Kuhns, V.H.; Bianchet, M.A.; Farinelli, F.; Aripirala, S.; Yoder, J.; Jakoncic, J.; Tomaselli, G.F.; Amzel, L.M. Regulation of the NaV 1.5 cytoplasmic domain by calmodulin. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabelli, S.B.; Yoder, J.B.; Tomaselli, G.F.; Amzel, L.M. Calmodulin and Ca2+ control of voltage gated Na+ channels. Channels 2016, 10, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.N.; Pattanayek, R.; Potet, F.; Rebbeck, R.T.; Blackwell, D.J.; Nikolaienko, R.; Sequeria, V.; Le Meur, R.; Radwanski, P.B.; Davis, J.P.; et al. The CaMKII inhibitor KN93-calmodulin interaction and implications for calmodulin tuning of NaV1.5 and RyR2 function. Cell Calcium 2019, 82, 102063. [Google Scholar] [CrossRef]
- Gardill, B.R.; Rivera-Acevedo, R.E.; Tung, C.C.; Van Petegem, F. Crystal structures of Ca2+—Calmodulin bound to NaV C-terminal regions suggest role for EF-hand domain in binding and inactivation. Proc Natl Acad Sci. USA 2019, 166, 10763–10772. [Google Scholar] [CrossRef] [Green Version]
- Koval, O.M.; Snyder, J.S.; Wolf, R.M.; Pavlovicz, R.E.; Glynn, P.; Curran, J.; Leymaster, N.D.; Dun, W.; Wright, P.J.; Cardona, N.; et al. Ca2+/calmodulin-dependent protein kinase ii-based regulation of voltage-gated na+ channel in cardiac disease. Circulation 2012, 126, 2084–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 2012, 287, 19856–19869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol. Cell Biol. 2009, 29, 2193–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Sun, F.; Luo, S.; Zhao, W.; Yang, T.; Zhang, G.; Gao, M.; Lu, R.; Shu, Y.; Mu, W.; et al. Let-7a is an antihypertrophic regulator in the heart via targeting calmodulin. Int. J. Biol. Sci. 2017, 13, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, K.; Chen, H. MiR-625-5p Inhibits Cardiac Hypertrophy through Targeting STAT3 and CaMKII. Hum. Gene Ther. Clin. Dev. 2019, 30, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hou, Y.; Gao, F.M.; Xiao, J.; Li, C.W.; Tang, Y.C. lncRNA GAS5 regulates myocardial infarction by targeting the miR-525-5p/CALM2 axis. J. Cell Biochem. 2019, 120, 18678–18688. [Google Scholar] [CrossRef]
- Li, K.; Lin, Y.; Li, C. MiR-338-5p ameliorates pathological cardiac hypertrophy by targeting CAMKIIδ. Arch Pharm. Res. 2019, 42, 1071–1080. [Google Scholar] [CrossRef]
- Kim, J.O.; Song, D.W.; Kwon, E.J.; Hong, S.E.; Song, H.K.; Min, C.K.; Kim, D.H. MiR-185 plays an anti-hypertrophic role in the heart via multiple targets in the calcium-signaling pathways. PLoS ONE 2015, 13, e0122509. [Google Scholar] [CrossRef]
- Cha, M.J.; Jang, J.K.; Ham, O.; Song, B.W.; Lee, S.Y.; Lee, C.Y.; Park, J.H.; Lee, J.; Seo, H.H.; Choi, E.; et al. MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIδ. Biochem. Biophys. Res. Commun. 2013, 435, 720–726. [Google Scholar] [CrossRef]
- He, J.; Jiang, S.; Li, F.L.; Zhao, X.J.; Chu, E.F.; Sun, M.N.; Chen, M.Z.; Li, H. MicroRNA-30b-5p is involved in the regulation of cardiac hypertrophy by targeting CaMKIIδ. J. Investig. Med. 2013, 61, 604–612. [Google Scholar] [CrossRef]
- Liu, P.Y.; Tian, Y.; Xu, S.Y. Mediated protective effect of electroacupuncture pretreatment by miR-214 on myocardial ischemia/reperfusion injury. J. Geriatr. Cardiol. 2014, 11, 303–310. [Google Scholar]
- Qi, X.Y.; Hassani, F.V.; Hoffmann, D.; Xiao, J.; Xiong, F.; Villeneuve, L.R.; Ljubojevic-Holzer, S.; Kamler, M.; Abu-Taha, I.; Heijman, J.; et al. Inositol Trisphosphate Receptors and Nuclear Calcium in Atrial Fibrillation. Circ. Res. 2021, 128, 619–635. [Google Scholar] [CrossRef]
- Vatta, M.; Ackerman, M.J.; Ye, B.; Makielski, J.C.; Ughanze, E.E.; Taylor, E.W.; Tester, D.J.; Balijepali, R.C.; Foell, J.D.; Li, Z.; et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 2006, 114, 2104–2112. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Valdivia, C.R.; Vaidyanathan, R.; Balijepalli, R.C.; Ackerman, M.J.; Makielski, J.C. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. JMCC 2013, 61, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Qi, Y.; Gao, C. Cardiac myocyte-protective effect of microRNA-22 during ischemia and reperfusion through disrupting the caveolin-3/eNOS signaling. Int. J. Clin. Exp. Pathol. 2015, 8, 4614–4626. [Google Scholar] [PubMed]
- Zhang, L.; Yin, H.; Jiao, L.; Liu, T.; Gao, Y.; Shao, Y.; Zhang, Y.; Shan, H.; Zhang, Y.; Yang, B. Abnormal Downregulation of Caveolin-3 Mediates the Pro-Fibrotic Action of MicroRNA-22 in a Model of Myocardial Infarction. Cell Physiol. Biochem. 2018, 45, 1641–1653. [Google Scholar] [CrossRef]
- Casini, S.; Marchal, G.A.; Kawasaki, M.; Nariswari, F.A.; Portero, V.; van den Berg, N.W.E.; Guan, K.; Driessen, A.H.G.; Veldkamp, M.W.; Mengarelli, I.; et al. Absence of Functional Nav1.8 Channels in Non-diseased Atrial and Ventricular Cardiomyocytes. Cardiovasc. Drugs Ther. 2019, 33, 649–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.; Hu, Y.; Wang, Z.; Cheng, C.; Wang, P.; Liang, L.; Xiong, H.; Luo, C.; Xu, C.; Chen, Q.; et al. UBC9 regulates cardiac sodium channel Na v 1.5 ubiquitination, degradation and sodium current density. JMCC 2019, 129, 79–91. [Google Scholar] [CrossRef]
- Minegishi, S.; Ishigami, T.; Kawamura, H.; Kino, T.; Chen, L.; Nakashima-Sasaki, R.; Doi, H.; Azushima, K.; Wakui, H.; Chiba, Y.; et al. An isoform of Nedd4-2 plays a pivotal role in electrophysiological cardiac abnormalities. Int. J. Mol. Sci. 2017, 18, 1268. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Heidersbach, A.; Kathiriya, I.S.; Garay, B.I.; Ivey, K.N.; Srivastava, D.; Han, Z.; King, I.N. The E3 ubiquitin ligase Nedd4/Nedd4L is directly regulated by microRNA 1. Development 2017, 144, 866–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Laurita, K.R.; Rosenbaum, D.S.; Rudy, Y. Two Components of the Delayed Rectifier K+ Current in Ventricular Myocytes of the Guinea Pig Type. Circ. Res. 1995, 77, 140–152. [Google Scholar] [CrossRef]
- Wettwer, E.; Amos, G.; Gath, J.; Zerkowski, H.R.; Reidemeister, J.C.; Ravens, U. Transient outward current in human and rat ventricular myocytes. Cardiovasc. Res. 1993, 27, 1662–1669. [Google Scholar] [CrossRef]
- Grant, A.O. Cardiac ion channels. Circ. Arrhythmia Electrophysiol. 2009, 2, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, N.; Nerbonne, J.M. Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J. Mol. Cell Cardiol. 2010, 48, 12–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, S.; Calloe, K.; Thomsen, M.B. Impact of KChiP2 on cardiac electrophysiology and the progression of heart failure. Front. Physiol. 2012, 3, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Xu, D.; Wang, Z.; Nattel, S. Ultrarapid delayed rectifier current inactivation in human atrial myocytes: Properties and consequences. Am. J. Physiol. Heart Circ. Physiol. 1998, 275, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Rampe, D.; Wang, Z.; Fermini, B.; Wible, B.; Dage, R.C.; Nattel, S. Voltage- and time-dependent block by perhexiline of K+ currents in human atrium and in cells expressing a Kv1.5-type cloned channel. J. Pharmacol. Exp. Ther. 1995, 274, 444–449. [Google Scholar] [PubMed]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquird cardiac arrthytmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Warmke, J.W.; Ganetzky, B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci. USA 1994, 91, 3438–3442. [Google Scholar] [CrossRef] [Green Version]
- Abbott, G.W.; Sesti, F.; Splawski, I.; Buck, M.E.; Lehmann, M.H.; Timothy, K.W.; Keating, M.T.; Goldstein, S.A. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999, 97, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, L.; Yin, C.; An, B.; Hao, Y.; Wei, T.; Li, L.; Song, G. Arsenic trioxide inhibits breast cancer cell growth via microRNA-328/hERG pathway in MCF-7 cells. Mol. Med. Rep. 2015, 12, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Assiri, A.A.; Mourad, N.; Shao, M.; Kiel, P.; Liu, W.; Skaar, T.C.; Overholser, B.R. MicroRNA 362-3p reduces hERG-related current and inhibits breast cancer cells proliferation. Cancer Genom. Proteom. 2019, 16, 433–442. [Google Scholar] [CrossRef]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef]
- Brahmajothi, M.V.; Morales, M.J.; Liu, S.; Rasmusson, R.L.; Campbell, D.L.; Strauss, H.C. In Situ Hybridization Reveals Extensive Diversity of K+ Channel mRNA in Isolated Ferret Cardiac Myocytes. Circ. Res. 1996, 76, 1083–1089. [Google Scholar] [CrossRef]
- Chen, L.; Sampson, K.J.; Kass, R.S. Cardiac Delayed Rectifier Potassium Channels in Health and Disease. Card. Electrophysiol. Clin. 2016, 8, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Tristani-Firouzi, M.; Chen, J.; Mitcheson, J.S.; Sanguinetti, M.C. Molecular biology of K+ channels and their role in cardiac arrhythmias. Am. J. Med. 2001, 110, 50–59. [Google Scholar] [CrossRef]
- Dhamoon, A.S.; Jalife, J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm 2005, 2, 316–324. [Google Scholar] [CrossRef]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.B.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwinger, R.H.G.; Bundgaard, H.; Muller-ehmsen, J.; Kjeldsen, K. The Na, K-ATPase in the failing human heart. Card. Res. 2003, 57, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Lichtstein, D. Na+, K(+)-ATPase and heart excitability. Adv. Exp. Med. Biol. 1995, 382, 23–30. [Google Scholar]
- Bundgaard, H.; Kjeldsen, K. Human myocardial Na,K-ATPase concentration in heart failure. Mol. Cell Biochem. 1996, 163–164, 277–283. [Google Scholar] [CrossRef]
- Liu, L.; Wu, J.; Kennedy, D.J. Regulation of cardiac remodeling by cardiac Na+/K+-ATPase isoforms. Front. Physiol. 2016, 7, 382. [Google Scholar] [CrossRef] [Green Version]
- Shao, Q.; Ren, B.; Elimban, V.; Tappia, P.S.; Takeda, N.; Dhalla, N.S. Modification of sarcolemmal Na+-K+-ATPase and Na+/Ca2+ exchanger expression in heart failure by blockade of renin-angiotensin system. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, 2637–2646. [Google Scholar] [CrossRef] [Green Version]
- Ostadal, P.; Elmoselhi, A.B.; Zdobnicka, I.; Lukas, A.; Chapman, D.; Dhalla, N.S. Ischemia-reperfusion alters gene expression of Na+-K+ ATPase isoforms in rat heart. Biochem. Biophys. Res. Commun. 2003, 306, 457–462. [Google Scholar] [CrossRef]
- Yalcin, Y.; Carman, D.; Shao, Y.; Ismail-Beigi, F.; Klein, I.; Ojamaa, K. Regulation of Na/K-ATPase gene expression by thyroid hormone and hyperkalemia in the heart. Thyroid 1999, 9, 53–59. [Google Scholar] [CrossRef]
- Schwinger, R.H.G.; Wang, J.; Frank, K.; Müller-Ehmsen, J.; Brixius, K.; McDonough, A.A.; Erdmann, E. Reduced sodium pump α1, α3, and β1-isoform protein levels and Na+, K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 1999, 99, 2105–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaremba, L.S.; Smolenski, W.H. Optimal portfolio choice under a liability constraint. Ann. Oper. Res. 2000, 97, 131–141. [Google Scholar] [CrossRef]
- Vér, Á.; Szántó, I.; Bányász, T.; Csermely, P.; Végh, E.; Somogyi, J. Changes in the expression of Na+/K+-ATPase isoenzymes in the left ventricle of diabetic rat hearts: Effect of insulin treatment. Diabetologia 1997, 40, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Drummond, C.A.; Fan, X.; Haller, S.T.; Kennedy, D.J.; Liu, J.; Tian, J. Na/K-ATPase signaling mediates miR-29b-3p regulation and cardiac fibrosis formation in mice with chronic kidney disease. PLoS ONE 2018, 13, e0197688. [Google Scholar] [CrossRef] [Green Version]
- Drummond, C.A.; Hill, M.C.; Shi, H.; Fan, X.; Xie, J.X.; Haller, S.T.; KEnnedy, D.J.; Liu, J.; Garrett, M.R.; Xie, Z.; et al. Na/K-ATPase signaling regulates collagen synthesis through microRNA-29b-3p in cardiac fibroblasts. Physiol. Genom. 2016, 48, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, L.; Zhang, Y.; Liang, H.; Li, X.; Cai, R.; Wang, L.; Du, W.; Zhang, R.; Li, J.; et al. Overexpression of microRNA-1 causes atrioventricular block in rodents. Int. J. Biol. Sci. 2013, 9, 445–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wei, F.; Ding, L.; Wang, L.; Zhang, X.; Yu, L.; Liu, R.; Kuang, X.; Jiao, B.; Yang, B.; et al. MicroRNA-1976 regulates degeneration of the sinoatrial node by targeting Cav1.2 and Cav1.3 ion channels. J. Mol. Cell Cardiol. 2019, 134, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tao, B.; Fan, S.; Cui, S.; Pu, Y.; Qiu, L.; Xia, H.; Xu, L. Over-expression of microRNA-145 drives alterations in β-adrenergic signaling and attenuates cardiac remodeling in heart failure post myocardial infarction. Aging 2020, 12, 11603–11622. [Google Scholar] [CrossRef]
- Boštjančič, E.; Zidar, N.; Glavač, D. MicroRNAs and cardiac sarcoplasmic reticulum calcium ATPase-2 in human myocardial infarction: Expression and bioinformatic analysis. BMC Genomics 2012, 15, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumarswamy, R.; Lyon, A.R.; Volkmann, I.; Mills, A.M.; Bretthauer, J.; Pahuja, A.; Geers-Knorr, C.; Kraft, T.; Hajjar, R.J.; Macleod, K.T.; et al. SERCA2a gene therapy restores microRNA-1 expression in heart failure via an Akt/FoxO3A-dependent pathway. Eur. Heart J. 2012, 33, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, A.; Formisano, L.; Cerullo, P.; Guida, N.; Cuomo, O.; Esposito, A.; Di Renzo, G.; Annunziato, L.; Pignataro, G. MicroRNA-103-1 selectively downregulates brain NCX1 and its inhibition by anti-miRNA ameliorates stroke damage and neurological deficits. Mol. Ther. 2014, 22, 1829–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.H. MicroRNA regulation of cardiac conduction and arrhythmias. Transl. Res. 2013, 161, 381–392. [Google Scholar] [CrossRef] [Green Version]



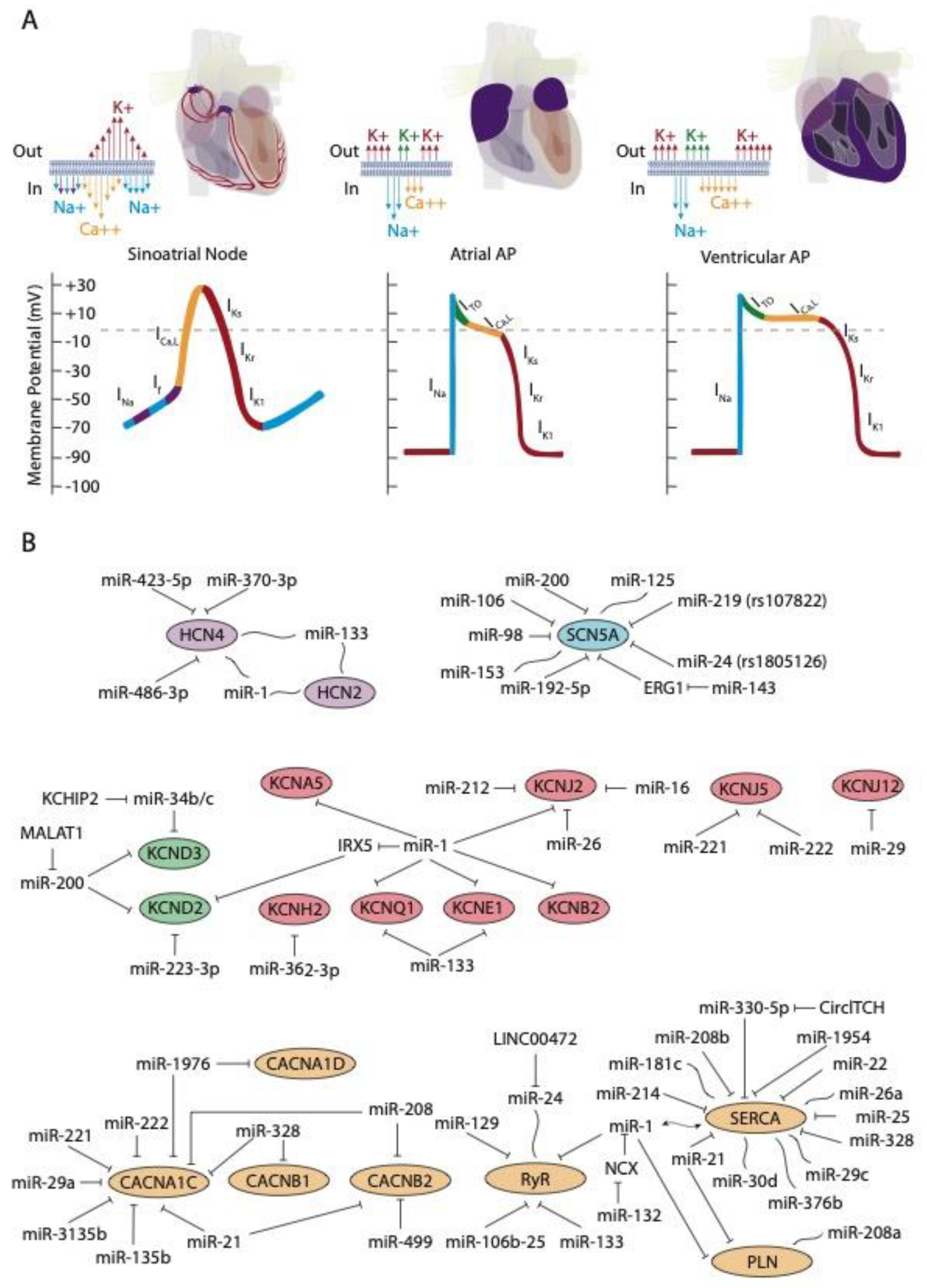{kind=link}
{kind=link}
| Current | microRNA | Gene | Function | Reference |
|---|---|---|---|---|
| INa | miR-98, miR-106, miR-200, miR-219, miR-125, miR-153 | SCN5A | INa ↑/INa ↓ | [47,53] |
| miR-192-5p | INa ↓ | [48] | ||
| miR-200c | - | [50] | ||
| miR-143 | INa ↓ | [51] | ||
| miR-24 | INa ↓ | [52] | ||
| If | miR-423-5p | HCN4 | If ↓ | [63] |
| miR-370-3p | If ↓ | [64] | ||
| miR-486-3p | If ↓ | [65] | ||
| miR-1, miR-133 | If ↑ | [66,67,68,69] | ||
| miR-1, miR-133 | HCN2 | If ↑ | [66,67,68,69] | |
| ITO | miR-1 | KCND2 | ITO ↓ | [70] |
| miR-223-3p | ITO ↓ | [71] | ||
| miR-34b/c | ITO = | [72] | ||
| miR-200 | ITO ↓ | [73] | ||
| miR-200 | KCND3 | ITO ↓ | [73] | |
| IKur | miR-1 | KCNA5 | IKur ? | [74] |
| IKr | miR-134, miR-103a-1, miR-143, miR-3619 | hERG | IKr ↓ | [75] |
| IKS | miR-1, miR-133 | KCNE1 | IKS ↓ | [76,77] |
| miR-1, miR-133 | KCNQ1 | IKS ↓ | [76] | |
| miR-1, miR-133 | KCNB2 | IKS ↓ | [77] | |
| IK1 | miR-1 | KCNJ2 | IK1 ↓/IK1 ↑ | [49,78,79] |
| miR-16 | IK1 ↓ | [80] | ||
| miR-26 | IK1 ↑ | [81,82] | ||
| miR-212 | IK1 ↓ | [83,84] | ||
| miR-29 | KCNJ12 | IK1 ↓ | [85] | |
| miR-221/222 | KCNJ5 | IK1 ↓ | [86] | |
| ICa,L | miR-328 | CACNA1C | ICa,L ↓ | [87] |
| miR-21, miR-208b | ICa,L ↓ | [88,89] | ||
| miR-20a, miR-3135b | ICa,L ↓ | [90] | ||
| miR-499 | ICa,L ↓ | [91] | ||
| miR-135b | ICa,L ↓ | [92] | ||
| miR-221/222 | ICa,L ↓ | [86] | ||
| miR-328 | CACNB1 | ICa,L ↓ | [87] | |
| miR-21, miR-208b | CACNB2 | ICa,L ↓ | [88,89] | |
| miR-499 | ICa,L ↓ | [91] | ||
| miR-329 | ICa,L ↓ | [91] | ||
| CICR | miR-106b | RYR2 | - | [93,94] |
| miR-129 | [95] | |||
| miR-1, miR-133 | [96,97] | |||
| miR-23 | [98,99] | |||
| miR-25 | SERCA2A | - | [100,101] | |
| miR-328 | [102,103] | |||
| miR-29c | [104] | |||
| miR-21 | [105] | |||
| miR-208b | [89] | |||
| miR-22 | [106] | |||
| miR-214 | [107] | |||
| miR-1954 | [108] | |||
| miR-376b, miR-1, miR-26a, miR-30d, miR-181 | [109] | |||
| miR-1, miR-21 | PLN | - | [110] | |
| miR-208a | [111] | |||
| miR-132 | NCX1 | - | [112] | |
| miR-1 | [107] |
| Gene | Disease | Alteration | Mir Related | Reference |
|---|---|---|---|---|
| SCN5A | Inherited arrhythmias and cardiomyopathy | Mutation | - | [35,36,37,38] |
| Sudden death | SNPs | - | [39,40] | |
| Brugada syndrome | SNPs/↓ expression | miR-219 | [47,53] | |
| Atrial fibrillation | ↓ expression | miR-192-5p | [48] | |
| Heart failure | SNPs/↓ expression | miR-24 | [52] | |
| SCN10A | Sudden death | Mutation | - | [41] |
| Atrial fibrillation | - | [42,43] | ||
| Brugada syndrome | - | [29,44,45] | ||
| HCN4 | Bradycardia | ↓ expression | miR-423-5p, miR-370-3p | [63,64] |
| Age atrial fibrillation | ↑ expression | miR-1, miR-133 | [66] | |
| Myocarial infarction | ↑ expression | miR-1, miR-133 | [67,68] | |
| HCN2 | Age atrial fibrillation | ↑ expression | miR-1, miR-133 | [66] |
| Myocardial infarction | ↑ expression | miR-1, miR-133 | [67,68] | |
| KCND2 | Sudden death | ↓ expression | miR-1 | [70] |
| Acute myocardial infarction | ↓ expression | miR-223-3p | [71] | |
| Myocardial infarction | ↓ expression | miR-200c | [73] | |
| KCNH2 | LQT syndrome (type 2) | Mutation | - | [113] |
| Heart failure | ↓ expression | miR362-3p | [114] | |
| KCNE2 | ||||
| LQT syndrome (type 6) | ||||
| LQT syndrome (type 1) | LQT syndrome (type 1) | ↓ expression | - | [115,116,117] |
| Atrial fibrillation | ↓ expression | miR-1 | [77] | |
| KCNB2 | ||||
| Atrial fibrillation | Myocardial infarction | ↓/↑ expression | miR-1, miR-16 | [78,81] |
| Atrial fibrillation | ↑ expression | miR-1, miR-26 | [49,79,82] | |
| Heart failure | ↓ expression | miR-212 | [84] | |
| KCNJ12 | Myocardial infarction | ↓ expression | miR-29 | [85] |
| KCNJ5 | Atrial fibrillation | ↓ expression | miR-221/222 | [86] |
| CACNA1C | Atrial fibrillation | ↓ expression | miR-221/222 | [86] |
| miR-328 | [87] | |||
| miR-21 | [88] | |||
| miR-208b | [89] | |||
| miR-29b, miR-3135b | [90] | |||
| CACNB2 | Atrial fibrillation | ↓ expression | miR-21 | [88] |
| miR-208b | [89] | |||
| miR-499, miR-329 | [91] | |||
| RYR2 | Atrial fibrillation | ↑ expression | miR-106b-25 | [93,94] |
| miR-106a, miR-93 | [94,118] | |||
| miR-129* | [95] | |||
| miR-1*, miR133* | [96,97] | |||
| miR-24* | [98,99] | |||
| SERCA2A | Atrial fibrillation | ↓ expression | miR-25 | [100,101] |
| miR-328 | [102,103] | |||
| miR-29c | [104] | |||
| miR-21*, miR-208b*, | [105,89] | |||
| miR-214*, miR-1954*, | [107,108] | |||
| miR-376b, miR-1*, | [109] | |||
| miR-26a*, miR-30d*, | [109] | |||
| miR-181c* | [109] | |||
| miR-330-5p* | [119] | |||
| PLN | Cardiac arrhythmias | - | miR-1, miR-21 | [98] |
| miR-208a* | [99] | |||
| NCX1 | Cardiac arrhythmias | - | miR-132 | [112] |
| miR-1 | [107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozano-Velasco, E.; Aranega, A.; Franco, D. Non-Coding RNAs in the Cardiac Action Potential and Their Impact on Arrhythmogenic Cardiac Diseases. Hearts 2021, 2, 307-330. https://doi.org/10.3390/hearts2030026
Lozano-Velasco E, Aranega A, Franco D. Non-Coding RNAs in the Cardiac Action Potential and Their Impact on Arrhythmogenic Cardiac Diseases. Hearts. 2021; 2(3):307-330. https://doi.org/10.3390/hearts2030026
Chicago/Turabian StyleLozano-Velasco, Estefania, Amelia Aranega, and Diego Franco. 2021. "Non-Coding RNAs in the Cardiac Action Potential and Their Impact on Arrhythmogenic Cardiac Diseases" Hearts 2, no. 3: 307-330. https://doi.org/10.3390/hearts2030026
APA StyleLozano-Velasco, E., Aranega, A., & Franco, D. (2021). Non-Coding RNAs in the Cardiac Action Potential and Their Impact on Arrhythmogenic Cardiac Diseases. Hearts, 2(3), 307-330. https://doi.org/10.3390/hearts2030026