
Healing the Broken Hearts: A Glimpse on Next Generation Therapeutics



Abstract
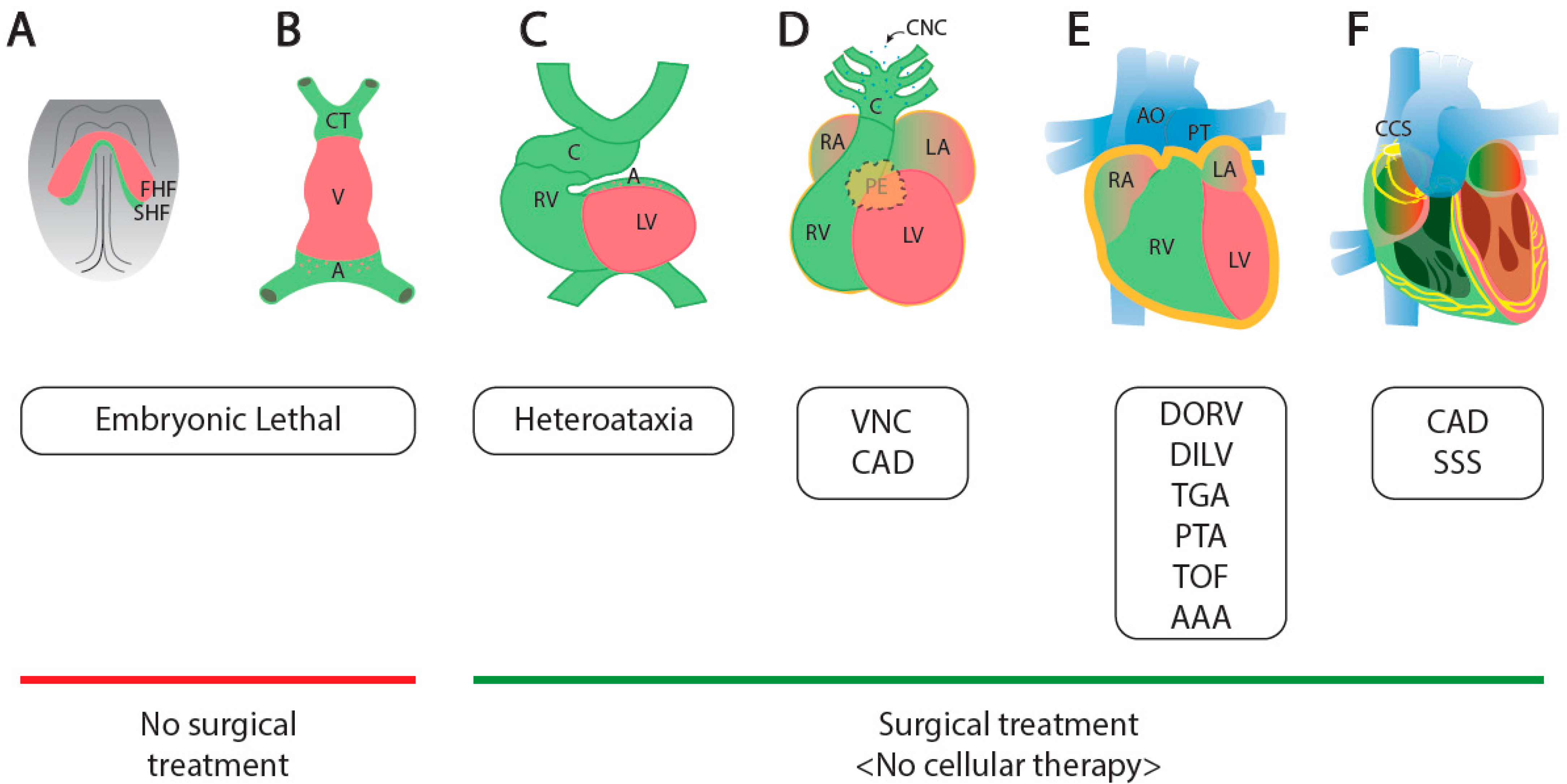
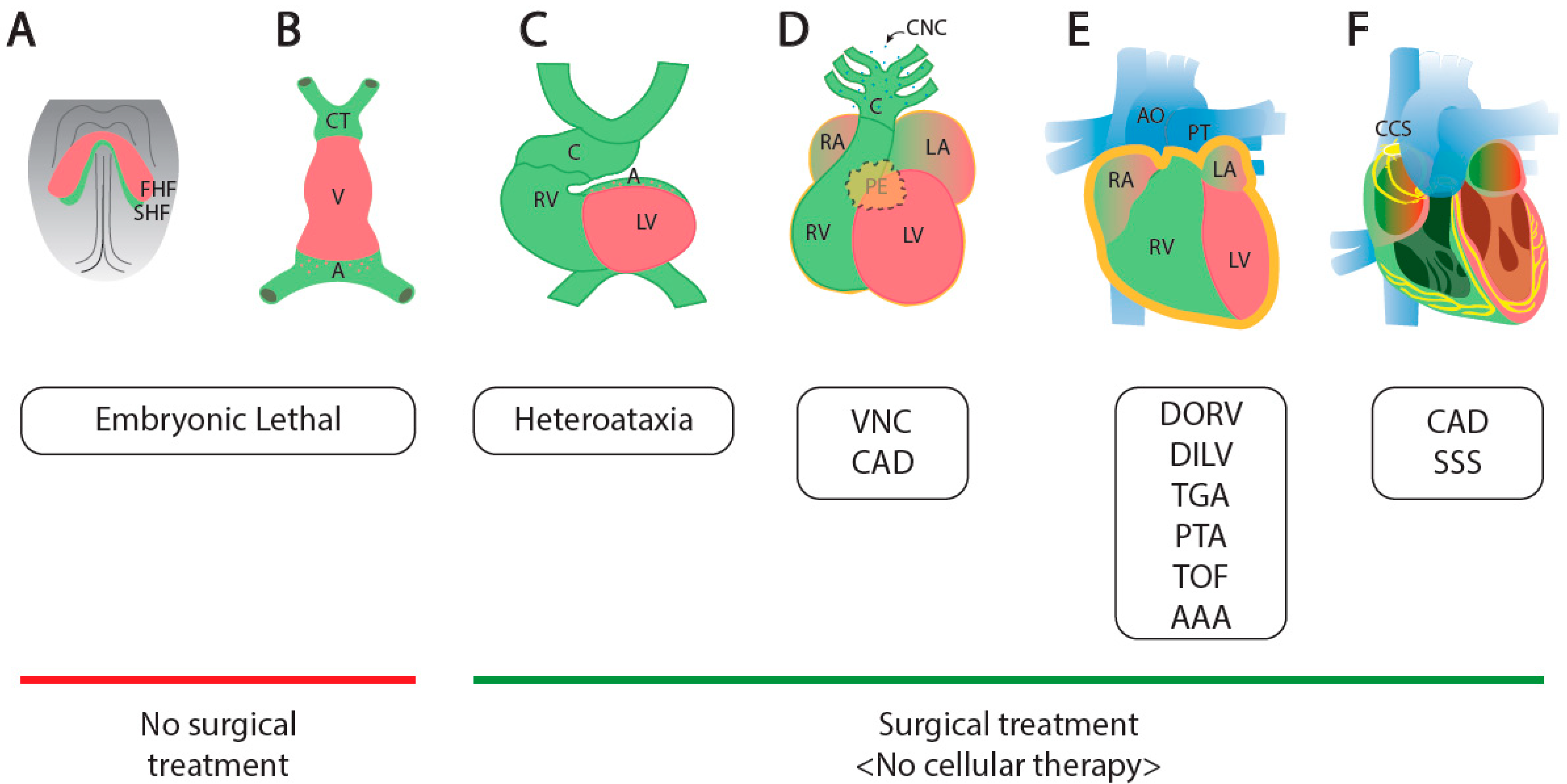
:1. Cardiogenesis and Congenital Heart Diseases
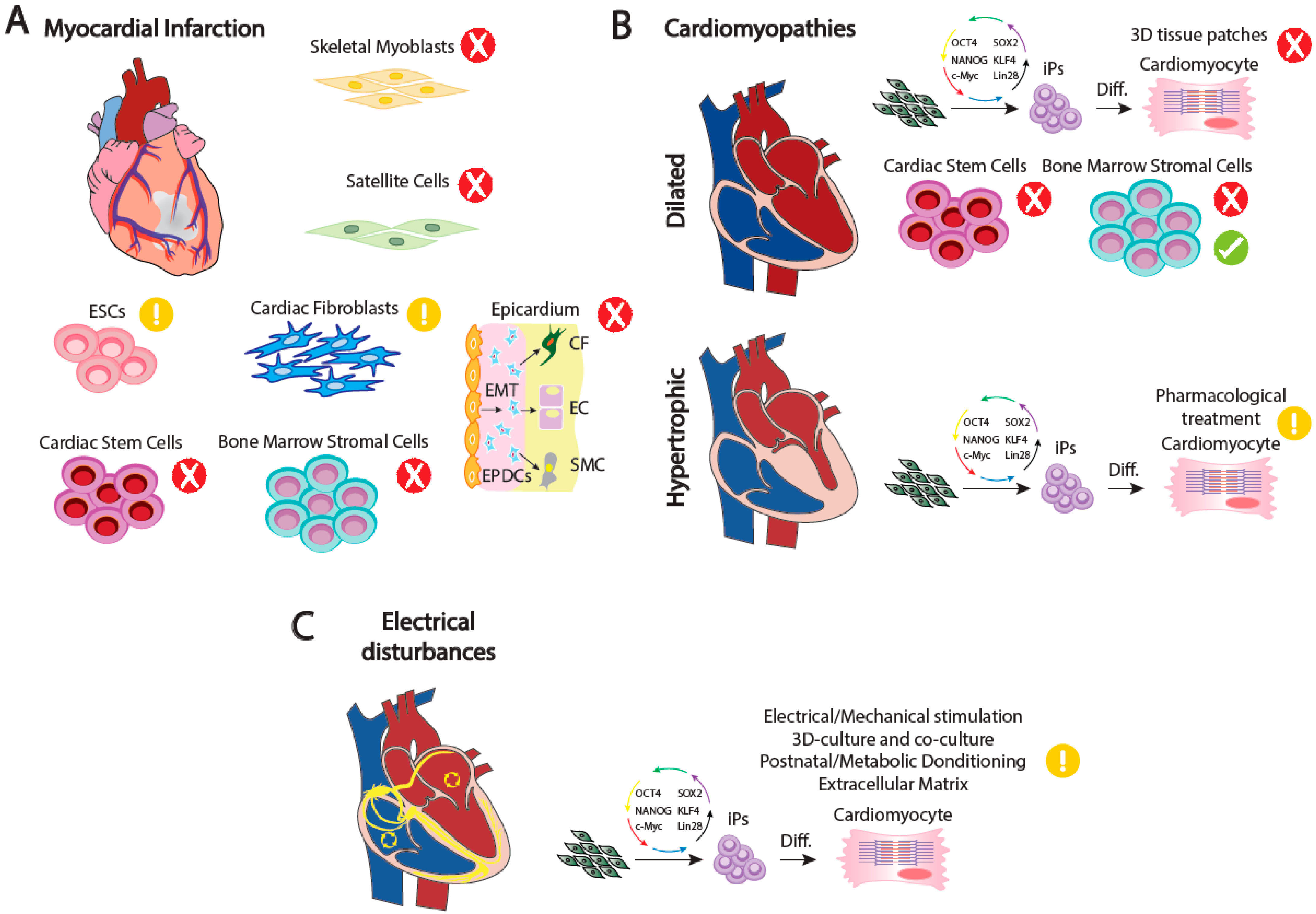
2. Adult Cardiac Performance and the Bases of Malfunction
3. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Meilhac, S.M.; Buckingham, M.E. The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 2018, 15, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Meilhac, S.M.; Lescroart, F.; Blanpain, C.D.; Buckingham, M.E. Cardiac cell lineages that form the heart. Cold Spring Harb. Perspect. Med. 2014, 4, a026344. [Google Scholar] [CrossRef] [PubMed]
- Christoffels, V.; Jensen, B. Cardiac morphogenesis: Specification of the four-chambered heart. Cold Spring Harb. Perspect. Biol. 2020, 12, a037143. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Schwarz, J.; Bucana, C.; Olson, E.N. Control of Mouse Cardiac Morphogenesis and Myogenesis by Transcription Factor MEF2C. Science 2016, 276, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lin, Q.; Duncan, S.A.; Olson, E.N. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997, 11, 1061–1072. [Google Scholar] [CrossRef]
- Campione, M.; Franco, D. Current perspectives in cardiac laterality. J. Cardiovasc. Dev. Dis. 2016, 3, 34. [Google Scholar] [CrossRef]
- Desgrange, A.; Garrec, J.F.L.; Meilhac, S.M. Left-right asymmetry in heart development and disease: Forming the right loop. Development 2018, 145, dev162776. [Google Scholar] [CrossRef]
- Franco, D.; Campione, M. The role of Pitx2 during cardiac development: Linking left-right signaling and congenital heart diseases. Trends Cardiovasc. Med. 2003, 13, 157–163. [Google Scholar] [CrossRef]
- Spoon, J.M. Situs inversus totalis. Neonatal Netw. J. Neonatal Nurs. 2001, 20, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Geddes, G.C.; Samudrala, S.S.; Earing, M.G. Neonatal Assessment of Infants with Heterotaxy. Clin. Perinatol. 2020, 47, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Varghese, R.; Jesudian, V.; Moses, J. The heterotaxy syndrome: Associated congenital heart defects and management. Indian J. Thorac. Cardiovasc. Surg. 2021, 37, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Sempou, E.; Khokha, M.K. Genes and mechanisms of heterotaxy: Patients drive the search. Curr. Opin. Genet. Dev. 2019, 56, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.F.M.; Christoffels, V.M. Cardiac chamber formation: Development, genes, and evolution. Physiol. Rev. 2003, 83, 1223–1267. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G.; Buckingham, M.E.; Moorman, A.F. Heart fields and cardiac morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a015750. [Google Scholar] [CrossRef]
- Kelly, R.G.; Buckingham, M.E. The anterior heart-forming field: Voyage to the arterial pole of the heart. Trends Genet. 2002, 18, 210–216. [Google Scholar] [CrossRef]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Männer, J.; Pérez-Pomares, J.M.; Macías, D.; Muñoz-Chápuli, R. The origin, formation and developmental significance of the epicardium: A review. Cells Tissues Organs 2001, 169, 89–103. [Google Scholar] [CrossRef]
- Carmona, R.; Guadix, J.A.; Cano, E.; Ruiz-Villalba, A.; Portillo-Sánchez, V.; Pérez-Pomares, J.M.; Muñoz-Chápuli, R. The embryonic epicardium: An essential element of cardiac development. J. Cell. Mol. Med. 2010, 14, 2066–2072. [Google Scholar] [CrossRef]
- Filho, D.C.S.; do Rêgo Aquino, P.L.; de Souza Silva, G.; Fabro, C.B. Left Ventricular Noncompaction: New Insights into a Poorly Understood Disease. Curr. Cardiol. Rev. 2020, 17, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Lorts, A.; Jefferies, J.L. Left ventricular non-compaction cardiomyopathy. Lancet 2015, 386, 813–825. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M.; De La Pompa, J.L. Signaling during epicardium and coronary vessel development. Circ. Res. 2011, 109, 1429–1442. [Google Scholar] [CrossRef] [PubMed]
- Cano, E.; Carmona, R.; Ruiz-Villalba, A.; Rojas, A.; Chau, Y.Y.; Wagner, K.D.; Wagner, N.; Hastie, N.D.; Muñoz-Chápuli, R.; and Pérez-Pomares, J.M. Extracardiac septum transversum/proepicardial endothelial cells pattern embryonic coronary arterio-venous connections. Proc. Natl. Acad. Sci. USA 2016, 113, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, G.; Casanova, J.C.; Guadix, J.A.; MacGrogan, D.; Burch, J.B.E.; Pérez-Pomares, J.M.; de la Pompa, J.L. Differential notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ. Res. 2011, 108, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; van den Hoff, M.J.B.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.N.; van Wijk, B.; Pérez-Pomares, J.M.; et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef]
- Männer, J.; Schlueter, J.; Brand, T. Experimental analyses of the function of the proepicardium using a new microsurgical procedure to induce loss-of-proepicardial-function in chick embryos. Dev. Dyn. 2005, 233, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Person, A.D.; Klewer, S.E.; Runyan, R.B. Cell biology of cardiac cushion development. Int. Rev. Cytol. 2005, 243, 287–335. [Google Scholar]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.F.; Lamers, W.H. Molecular Anatomy of the Heart. Trends Cardiovasc. Med. 1994, 4, 257–264. [Google Scholar] [CrossRef]
- Stefanovic, S.; Etchevers, H.C.; Zaffran, S. Outflow tract formation—Embryonic origins of conotruncal congenital heart disease. J. Cardiovasc. Dev. Dis. 2021, 8, 42. [Google Scholar] [CrossRef]
- Briggs, L.E.; Kakarla, J.; Wessels, A. The Pathogenesis of Atrial and Atrioventricular Septal Defects with Special Emphasis on the Role of the Dorsal Mesenchymal Protrusion. Differentiation 2012, 84, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, A.; Franco, D. Regulatory mechanisms of cardiac development and repair. Cardiovasc. Hematol. Disord. Targets 2006, 6, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Katano, W.; Moriyama, Y.; Takeuchi, J.K.; Koshiba-Takeuchi, K. Cardiac septation in heart development and evolution. Dev. Growth Differ. 2019, 61, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Lamers, W.H.; Moorman, A.F.M. Cardiac septation: A late contribution of the embryonic primary myocardium to heart morphogenesis. Circ. Res. 2002, 91, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Geva, T.; Martins, J.D.; Wald, R.M. Atrial septal defects. Lancet 2014, 383, 1921–1932. [Google Scholar] [CrossRef]
- Wang, S.Y.; Welch, T.D.; Elfenbein, A.; Kaplan, A.V. Spontaneous Closure of a Secundum Atrial Septal Defect. Methodist DeBakey Cardiovasc. J. 2018, 14, 60–62. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, M.L.; Levi, D.S. State-of-the-Art Atrial Septal Defect Closure Devices for Congenital Heart. Interv. Cardiol. Clin. 2019, 8, 11–21. [Google Scholar] [CrossRef]
- Shi, D.; Kang, Y.; Zhang, G.; Gao, C.; Lu, W.; Zou, H.; Jianmg, H. Biodegradable atrial septal defect occluders: A current review. Acta Biomater. 2019, 96, 68–80. [Google Scholar] [CrossRef]
- Bissessor, N. Current perspectives in percutaneous atrial septal defect closure devices. Med. Devices Evid. Res. 2015, 8, 297–303. [Google Scholar] [CrossRef]
- Franco, D.; Meilhac, S.M.; Christoffels, V.M.; Kispert, A.; Buckingham, M.; Kelly, R.G. Left and right ventricular contributions to the formation of the interventricular septum in the mouse heart. Dev. Biol. 2006, 294, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.H.; Brown, N.A.; Mohun, T.J. Insights regarding the normal and abnormal formation of the atrial and ventricular septal structures. Clin. Anat. 2016, 29, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Nicolae, M.I.; Summers, K.M.; Radford, D.J. Familial muscular ventricular septal defects and aneurysms of the muscular interventricular septum. Cardiol. Young 2007, 17, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.A.; Jaimes, C.; Abbara, S. Ventricular septal defects: Embryology and imaging findings. J. Thorac. Imaging. 2013, 28, 28–34. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.P.; Leung, P.K.C.; Ho, S.Y. Perimembranous and muscular ventricular septal defects—Morphology revisited in the era of device closure. J. Interv. Cardiol. 2005, 18, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yu, J.; Zhang, Z.; Li, J.; Yu, J. Perimembranous ventricular septal defect closure via ultra-minimal trans intercostal incision in children. J. Card. Surg. 2021, 36, 3131–3137. [Google Scholar] [CrossRef]
- Ho, S.Y.; Mccarthy, K.P.; Rigby, M.L. Morphology of perimembranous ventricular septal fefects: Implications for transcatheter device closure. J. Interv. Cardiol. 2004, 17, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Brewer, S.; Jiang, X.; Donaldson, S.; Williams, T.; Sucov, H.M. Requirement for AP-2α in cardiac outflow tract morphogenesis. Mech. Dev. 2002, 110, 139–149. [Google Scholar] [CrossRef]
- Mesbah, K.; Camus, A.; Babinet, C.; Barra, J. Mutation in the Trap α/Ssr1 Gene, Encoding Translocon-Associated Protein α, Results in Outflow Tract Morphogenetic Defects. Mol. Cell. Biol. 2006, 26, 7760–7771. [Google Scholar] [CrossRef]
- Thompson, L.N.D.; McElhinney, D.B.; Reddy, V.M.; Petrossian, E.; Silverman, N.H.; Hanley, F.L. Neonatal repair of truncus arteriosus: Continuing improvement in outcomes. Ann. Thorac. Surg. 2001, 72, 391–395. [Google Scholar] [CrossRef]
- Goo, H.W. Double Outlet Right Ventricle: In-Depth Anatomic Review Using Three-Dimensional Cardiac CT Data. Korean J. Radiol. 2021, 22, 1894–1908. [Google Scholar] [CrossRef]
- Obler, D.; Juraszek, A.L.; Smoot, L.B.; Natowicz, M.R. Double outlet right ventricle: Aetiologies and associations. J. Med. Genet. 2008, 45, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Lecompte, Y.; Batisse, A.D.C.D. Double-outlet right ventricle: A surgical synthesis. Adv. Card. Surg. 1993, 4, 109–136. [Google Scholar] [PubMed]
- Kirklin, J.K.; Pacifico, A.D.; Kirklin, J.W. Intraventricular Tunnel Repair of Double Outlet Right Ventricle. J. Card. Surg. 1987, 2, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Scholl, A.M.; Kirby, M.L. Signals controlling neural crest contributions to the heart. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 220–227. [Google Scholar] [CrossRef]
- Hutson, M.R.; Kirby, M.L. Neural crest and cardiovascular development: A 20-year perspective. Birth Defects Res. Part C Embryo Today Rev. 2003, 69, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L. Alteration of Cardiogenesis after Neural Crest Ablation. Ann. N. Y. Acad. Sci. 1990, 588, 289–295. [Google Scholar] [CrossRef]
- Bailliard, F.; Anderson, R.H. Tetralogy of Fallot. Orphanet J. Rare Dis. 2009, 4, 2. [Google Scholar] [CrossRef]
- Morgenthau, A.; Frishman, W.H. Genetic Origins of Tetralogy of Fallot. Cardiol. Rev. 2018, 26, 86–92. [Google Scholar] [CrossRef]
- Apitz, C.; Webb, G.D.; Redington, A.N. Tetralogy of Fallot. Lancet 2009, 374, 1462–1471. [Google Scholar] [CrossRef]
- Geerdink, L.M.; Kapusta, L. Dealing with Ebstein’s anomaly. Cardiol. Young 2008, 24, 191–200. [Google Scholar] [CrossRef]
- Yuan, S.M. Ebstein’s Anomaly: Genetics, Clinical Manifestations, and Management. Pediatr. Neonatol. 2017, 58, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Bakker, M.L.; Christoffels, V.M.; Moorman, A.F.M. The cardiac pacemaker and conduction system develops from embryonic myocardium that retains its primitive phenotype. J. Cardiovasc. Pharmacol. 2010, 56, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Christoffels, V.M.; Smits, G.J.; Kispert, A.; Moorman, A.F.M. Development of the pacemaker tissues of the heart. Circ. Res. 2010, 106, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Miquerol, L.; Beyer, S.; Kelly, R.G. Establishment of the mouse ventricular conduction system. Cardiovasc. Res. 2011, 91, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Choquet, C.; Boulgakoff, L.; Kelly, R.G.; Miquerol, L. New insights into the development and morphogenesis of the cardiac purkinje fiber network: Linking architecture and function. J. Cardiovasc. Dev. Dis. 2021, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Gillette, P.C.; Garson, A., Jr. Sudden cardiac death in the pediatric population. Circulation 1992, 85 (Suppl. S1), I64–I69. [Google Scholar]
- Niwa, K.; Warita, N.; Sunami, Y.; Shimura, A.; Tateno, S.; Sugita, K. Prevalence of arrhythmias and conduction disturbances in large population-based samples of children. Cardiol. Young 2004, 14, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, A.; Aranega, A.E.; Franco, D. More than just a simple cardiac envelope; cellular contributions of the epicardium. Front. Cell Dev. Biol. 2017, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Chang, A.; Red-Horse, K. Coronary Artery Development: Progenitor Cells and Differentiation Pathways. Annu. Rev. Physiol. 2017, 79, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pomares, J.M.; De La Pompa, J.L.; Franco, D.; Henderson, D.; Ho, S.Y.; Houyel, L.; Kelly, R.G.; Sedmera, D.; Sheppard, M.; Sperling, S.; et al. Congenital coronary artery anomalies: A bridge from embryology to anatomy and pathophysiology-a position statement of the development, anatomy, and pathology ESC Working Group. Cardiovasc. Res. 2016, 109, 204–216. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.; Beaton, A.Z.; Benjamin, E.J.; Benzinger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Teo, K.K.; Rafiq, T. Cardiovascular Risk Factors and Prevention: A Perspective From Developing Countries. Can. J. Cardiol. 2021, 37, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.S.; Ambrosy, A.P.; Velazquez, E.J. Adverse Remodeling and Reverse Remodeling After Myocardial Infarction. Curr. Cardiol. Rep. 2017, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Lazzara, R.; Scherlag, B.J. Generation of arrhythmias in myocardial ischemia and infarction. Am. J. Cardiol. 1988, 61, A20–A26. [Google Scholar] [CrossRef]
- Osmancik, P.P.; Stros, P.; Herman, D. In-hospital arrhythmias in patients with acute myocardial infarction—The relation to the reperfusion strategy and their prognostic impact. Acute Card. Care 2008, 10, 15–25. [Google Scholar] [CrossRef]
- Crossman, A.W.; D’Agostino, H.J.; Geraci, S.A. Timing of coronary artery bypass graft surgery following acute myocardial infarction: A critical literature review. Clin. Cardiol. 2002, 25, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Pirwitz, M.J.; Hillis, L.D. Emergency coronary artery bypass surgery for acute myocardial infarction. Coron. Artery Dis. 1994, 5, 385–391. [Google Scholar] [CrossRef]
- Perrier, S.; Kindo, M.; Gerelli, S.; Mazzucotelli, J.P. Coronary artery bypass grafting or percutaneous revascularization in acute myocardial infarction? Interact. Cardiovasc. Thorac. Surg. 2013, 17, 1015–1019. [Google Scholar] [CrossRef]
- Rout, A.; Tantry, U.S.; Novakovic, M.; Sukhi, A.; Gurbel, P.A. Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert Opin. Pharmacother. 2020, 21, 1851–1865. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target. Ther. 2022, 7, 78. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar] [CrossRef]
- Horackova, M.; Arora, R.; Chen, R.; Armour, J.A.; Cattini, P.A.; Livingston, R.; Byzcko, Z. Cell transplantation for treatment of acute myocardial infarction: Unique capacity for repair by skeletal muscle satellite cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Smits, P.C.; Van Geuns, R.J.M.; Poldermans, D.; Bountioukos, M.; Onderwater, E.E.M.; Lee, C.H.; Maat, A.P.; Serruys, P.W. Catheter-Based Intramyocardial Injection of Autologous Skeletal Myoblasts as a Primary Treatment of Ischemic Heart Failure Clinical Experience with Six-Month Follow-Up. J. Am. Coll. Cardiol. 2003, 42, 2063–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siminiak, T.; Fiszer, D.; Jerzykowska, O.; Grygielska, B.; Rozwadowska, N.; Kałmucki, P.; Kurpisz, M. Percutaneous trans-coronary-venous transplantation of autologous skeletal myoblasts in the treatment of post-infarction myocardial contractility impairment: The POZNAN trial. Eur. Heart J. 2005, 26, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Menasché, P.; Alfieri, O.; Janssens, S.; McKenna, W.; Reichenspurner, H.; Trinquart, L.; Vilquin, J.T.; Marolleau, J.P.; Seymour, B.; Larghero, J.; et al. The myoblast autologous grafting in ischemic cardiomyopathy (MAGIC) trial: First randomized placebo-controlled study of myoblast transplantation. Circulation 2008, 117, 1189–1200. [Google Scholar] [CrossRef]
- Zhong, H.; Zhu, H.; Zhang, Z. Affects of different access routes on autologous satellite cell implantation stimulating myocardial regeneration. Chin. Med. J. 2002, 115, 1521–1524. [Google Scholar]
- Emmert, M.Y.; Hitchcock, R.W.; Hoerstrup, S.P. Cell therapy, 3D culture systems and tissue engineering for cardiac regeneration. Adv. Drug Deliv. Rev. 2014, 69, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Brand, N.J.; Allen, S.; Khan, M.A.; Farrell, A.O.; Murtuza, B.; El Oakley, R.; Yacoub, M.H. Overexpression of connexin 43 in skeletal myoblasts: Relevance to cell transplantation to the heart. J. Thorac. Cardiovasc. Surg. 2001, 122, 759–766. [Google Scholar] [CrossRef]
- Roell, W.; Lewalter, T.; Sasse, P.; Tallini, Y.N.; Choi, B.R.; Breitbach, M.; Doran, R.; Becher, U.M.; Hwang, S.M.; Bostani, T.; et al. Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature 2007, 450, 819–824. [Google Scholar] [CrossRef]
- Fernandes, S.; van Rijen, H.V.M.; Forest, V.; Evain, S.; Leblond, A.L.; Mérot, J.; Charpentier, F.; de Bakker, J.M.T.; Lemarchand, P. Cardiac cell therapy: Overexpression of connexin43 in skeletal myoblasts and prevention of ventricular arrhythmias. J. Cell. Mol. Med. 2009, 13, 3703–3712. [Google Scholar] [CrossRef]
- García, J.; Tanabe, T.; Beam, K.G. Relationship of calcium transients to calcium currents and charge movements in myotubes expressing skeletal and cardiac dihydropyridine receptors. J. Gen. Physiol. 1994, 103, 125–147. [Google Scholar] [CrossRef]
- Durrani, S.; Konoplyannikov, M.; Ashraf, M.; Haider, K.H. Skeletal myoblasts for cardiac repair. Regen. Med. 2010, 5, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Rohwedel, J.; Hescheler, J.; Wobus, A.M. Embryonic stem cells differentiate in vitro into cardiomyocytes representing sinusnodal, atrial and ventricular cell types. Mech. Dev. 1993, 44, 41–50. [Google Scholar] [CrossRef]
- Thomson, J.A. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai-Brunswick, T.H.; Carvalho, A.B.; de Carvalho, A.C.C. Stem cell therapies in cardiac diseases: Current status and future possibilities. World J. Stem Cells 2021, 13, 1231–1247. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Yang, Y.; Sullivan, M.F.; Ke, Q.; Converso, K.L.; Chen, Y.; Morgan, J.P.; Xiao, Y.F. Long-term improvement of cardiac function in rats after infarction by transplantation of embryonic stem cells. J. Thorac. Cardiovasc. Surg. 2003, 125, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Takahashi, J.; Watanabe, K.; Hayashi, H.; Morizane, A.; Koyanagi, M.; Sasai, Y.; Hashimoto, N. Fluorescence-Activated Cell Sorting-Based Purification of Embryonic Stem Cell-Derived Neural Precursors Averts Tumor Formation after Transplantation. Stem Cells 2006, 24, 763–771. [Google Scholar] [CrossRef]
- Pfannkuche, K.; Liang, H.; Hannes, T.; Xi, J.; Fatima, A.; Nguemo, F.; Matzkies, M.; Wernig, M.; Jaenisch, R.; Pillekamp, F.; et al. Cardiac myocytes derived from murine reprogrammed fibroblasts: Intact hormonal regulation, cardiac ion channel expression and development of contractility. Cell. Physiol. Biochem. 2009, 24, 73–86. [Google Scholar] [CrossRef]
- Glenn, I.; Cohen, J.D.; Eli, Y.; Adashi, M. Human Embryonic Stem-Cell Research under Siege—Battle Won but Not the War. N. Engl. J. Med. 2011, 48, e48. [Google Scholar]
- Chen, W.; Bian, W.; Zhou, Y.; Zhang, J. Cardiac Fibroblasts and Myocardial Regeneration. Front. Bioeng. Biotechnol. 2021, 9, 599928. [Google Scholar] [CrossRef]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex relationship between cardiac fibroblasts and cardiomyocytes in health and disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef]
- Orlic, D.; Kajstura, J.; Chimenti, S.; Limana, F.; Jakoniuk, I.; Quaini, F.; Nadal-Ginard, B.; Bodine, D.M.; Leri, A.; Anversa, P. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc. Natl. Acad. Sci. USA 2001, 98, 10344–10349. [Google Scholar] [CrossRef] [PubMed]
- Orlic, D.; Kajstura, J.; Chimenti, S.; Jakoniuk, I.; Anderson, S.M.; Li, B.; Pickel, J.; McKay, R.; Nadal-Ginard, B.; Bodine, D.M.; et al. Bone marrow cells regenerate infarcted myocardium. Lett. Nat. 2001, 410, 701–705. [Google Scholar] [CrossRef]
- Botleroo, R.A.; Bhandari, R.; Ahmed, R.; Kareem, R.; Gyawali, M.; Venkatesan, N.; Ogeyingbo, O.D.; Elshaikh, A.O. Stem Cell Therapy for the Treatment of Myocardial Infarction: How Far Are We Now? Cureus 2021, 13, e17022. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Y.; Qian, H.Y.; Huang, P.S.; Xu, J.; Xiong, Y.Y.; Jiang, W.Y.; Xu, Y.; Leng, W.X.; Li, X.D.; Chen, G.H.; et al. Transplantation efficacy of autologous bone marrow mesenchymal stem cells combined with atorvastatin for acute myocardial infarction (TEAM-AMI): Rationale and design of a randomized, double-blind, placebo-controlled, multi-center, Phase II TEAM-AMI trial. Regen. Med. 2019, 14, 1077–1087. [Google Scholar] [CrossRef]
- Yamada, Y.; Minatoguchi, S.; Kanamori, H.; Mikami, A.; Okura, H.; Dezawa, M.; Minatoguchi, S. Stem cell therapy for acute myocardial infarction—Focusing on the comparison between Muse cells and mesenchymal stem cells. J. Cardiol. 2021, 80, 80–87. [Google Scholar] [CrossRef]
- He, L.; Nguyen, N.B.; Ardehali, R.; Zhou, B. Heart regeneration by endogenous stem cells and cardiomyocyte proliferation: Controversy, fallacy, and progress. Circulation 2020, 142, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Goichberg, P.; Chang, J.; Liao, R.; Leri, A. Cardiac stem cells: Biology and clinical applications. Antioxid. Redox Signal. 2014, 21, 2002–2017. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Tang, X.L.; Sanganalmath, S.K.; Rimoldi, O.; Mosna, F.; Abdel-Latif, A.; Jneid, H.; Rota, M.; Leri, A.; Kajstura, J. Intracoronary delivery of autologous cardiac stem cells improves cardiac function in a porcine model of chronic ischemic cardiomyopathy. Circulation 2013, 128, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Riley, P. The epicardium as a candidate for heart regeneration. Future Cardiol. 2012, 8, 53–69. [Google Scholar] [CrossRef]
- Smart, N.; Risebro, C.A.; Melville, A.A.D.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin β-4 is essential for coronary vessel development and promotes neovascularization via adult epicardium. Ann. N. Y. Acad. Sci. 2007, 1112, 171–188. [Google Scholar] [CrossRef]
- Redpath, A.N.; Smart, N. Recapturing embryonic potential in the adult epicardium: Prospects for cardiac repair. Stem Cells Transl. Med. 2021, 10, 511–521. [Google Scholar] [CrossRef]
- Limana, F.; Capogrossi, M.C.; Germani, A. The epicardium in cardiac repair: From the stem cell view. Pharmacol. Ther. 2011, 129, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.M.; Dronkers, E.; Goumans, M.J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018, 127, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Quijada, P.; Trembley, M.A.; Small, E.M. The Role of the Epicardium during Heart Development and Repair. Circ. Res. 2020, 126, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Rikhtegar, R.; Pezeshkian, M.; Dolati, S.; Safaie, N.; Afrasiabi Rad, A.; Mahdipour, M.; Nouri, M.; Jodati, A.R.; Yousefi, M. Stem cells as therapy for heart disease: IPSCs, ESCs, CSCs, and skeletal myoblasts. Biomed. Pharmacother. 2019, 109, 304–313. [Google Scholar] [CrossRef]
- Dib, N.; Menasche, P.; Bartunek, J.J.; Zeiher, A.M.; Terzic, A.; Chronos, N.A.; Henry, T.D.; Peters, N.S.; Fernández-Avilés, F.; Yacoub, M.; et al. Recommendations for Successful Training on Methods of Delivery of Biologics for Cardiac Regeneration. A Report of the International Society for Cardiovascular Translational Research. JACC Cardiovasc. Interv. 2010, 3, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Hagège, A.A.; Marolleau, J.P.; Vilquin, J.T.; Alhéritière, A.; Peyrard, S.; Duboc, D.; Abergel, E.; Messas, E.; Mousseaux, E.; Schwartz, K.; et al. Skeletal myoblast transplantation in ischemic heart failure: Long-term follow-up of the first phase I cohort of patients. Circulation 2006, 114 (Suppl. S1), I108. [Google Scholar] [CrossRef] [PubMed]
- Mozid, A.M.; Arnous, S.; Sammut, E.C.; Mathur, A. Stem cell therapy for heart diseases. Br. Med. Bull. 2011, 98, 143–159. [Google Scholar] [CrossRef]
- Sherman, W.; Martens, T.P.; Viles-Gonzalez, J.F.; Siminiak, T. Catheter-based delivery of cells to the heart. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3 (Suppl. S1), 57–64. [Google Scholar] [CrossRef]
- Kim, R.J.; Wu, E.; Rafael, A.; Chen, E.L.; Parker, M.A.; Simonetti, O.; Klocke, F.J.; Bonow, R.O.; Judd, R.M. The Use of Contrast-Enhanced Magnetic Resonance Imaging to Identify Reversible Myocardial Dysfunction. Surv. Anesthesiol. 2001, 343, 1445–1453. [Google Scholar] [CrossRef]
- Halkos, M.E.; Zhao, Z.Q.; Kerendi, F.; Wang, N.P.; Jiang, R.; Schmarkey, L.S.; Martin, B.J.; Quyyumi, A.A.; Few, W.L.; Kin, H.; et al. Intravenous infusion of mesenchymal stem cells enhances regional perfusion and improves ventricular function in a porcine model of myocardial infarction. Basic Res. Cardiol. 2008, 103, 525–536. [Google Scholar] [CrossRef]
- Barbash, I.M.; Chouraqui, P.; Baron, J.; Feinberg, M.S.; Etzion, S.; Tessone, A.; Miller, L.; Guetta, E.; Zipori, D.; Kedes, L.H.; et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation 2003, 108, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Widimsky, P.; Penicka, M.; Lang, O.; Kozak, T.; Motovska, Z.; Jirmar, R.; Aschermann, M. Intracoronary transplantation of bone marrow stem cells: Background, techniques, and limitations. Eur. Heart J. 2006, 8, 16–22. [Google Scholar] [CrossRef]
- Vicario, J.; Piva, J.; Pierini, A.; Ortega, H.H.; Canal, A.; Gerardo, L.; Pfeiffer, H.; Campos, C.; Fendrich, I.; Novero, R.; et al. Transcoronary sinus delivery of autologous bone marrow and angiogenesis in pig models with myocardial injury. Cardiovasc. Radiat. Med. 2002, 3, 91–94. [Google Scholar] [CrossRef]
- Yokoyama, S.I.; Fukuda, N.; Li, Y.; Hagikura, K.; Takayama, T.; Kunimoto, S.; Honye, J.; Saito, S.; Wada, M.; Satomi, A.; et al. A strategy of retrograde injection of bone marrow mononuclear cells into the myocardium for the treatment of ischemic heart disease. J. Mol. Cell. Cardiol. 2006, 40, 24–34. [Google Scholar] [CrossRef]
- Miyahara, Y.; Nagaya, N.; Kataoka, M.; Yanagawa, B.; Tanaka, K.; Hao, H.; Ishino, K.; Ishida, H.; Shimizu, T.; Kangawa, K.; et al. Monolayered mesenchymal stem cells repair scarred myocardium after myocardial infarction. Nat. Med. 2006, 12, 459–465. [Google Scholar] [CrossRef]
- Blum, B.; Benvenisty, N. The Tumorigenicity of Human Embryonic Stem Cells. Adv. Cancer Res. 2008, 100, 133–158. [Google Scholar] [PubMed]
- Yoshihara, M.; Oguchi, A.; Murakawa, Y. Genomic Instability of iPSCs and Challenges in Their Clinical Applications. In Stem Cells Therapeutic Applications; Springer: Cham, Switzerland, 2019; p. 410. [Google Scholar]
- Beohar, N.; Rapp, J.; Pandya, S.; Losordo, D.W. Rebuilding the damaged heart: The potential of cytokines and growth factors in the treatment of ischemic heart disease. J. Am. Coll. Cardiol. 2010, 56, 1287–1297. [Google Scholar] [CrossRef]
- Kapur, N.K.; Rade, J.J. Fibroblast Growth Factor 4 Gene Therapy for Chronic Ischemic Heart Disease. Trends Cardiovasc. Med. 2008, 18, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Banai, S.; Jaklitsch, M.T.; Casscells, W.; Shou, M.; Shrivastav, S.; Correa, R.; Epstein, S.E.; Unger, E.F. Effects of acidic fibroblast growth factor on normal and ischemic myocardium. Circ. Res. 1991, 69, 76–85. [Google Scholar] [CrossRef]
- Kardami, E.; Detillieux, K.; Ma, X.; Jiang, Z.; Santiago, J.J.; Jimenez, S.K.; Cattini, P.A. Fibroblast growth factor-2 and cardioprotection. Heart Fail. Rev. 2007, 12, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Detillieux, K.A.; Sheikh, F.; Kardami, E.; Cattini, P.A. Biological activities of fibroblast growth factor-2 in the adult myocardium. Cardiovasc. Res. 2003, 57, 8–19. [Google Scholar] [CrossRef]
- Matsunaga, T.; Warltier, D.C.; Tessmer, J.; Weihrauch, D.; Simons, M.; Chilian, W.M. Expression of VEGF and angiopoietins-1 and -2 during ischemia-induced coronary angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, 352–358. [Google Scholar] [CrossRef]
- Takeshita, S.; Zheng, L.P.; Brogi, E.; Kearney, M.; Pu, L.Q.; Bunting, S.; Ferrara, N.; Symes, J.F.; Isner, J.M. Therapeutic angiogenesis. A single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. J. Clin. Investig. 1994, 93, 662–670. [Google Scholar] [CrossRef]
- Vandervelde, S.; van Luyn, M.J.A.; Rozenbaum, M.H.; Petersen, A.H.; Tio, R.A.; Harmsen, M.C. Stem cell-related cardiac gene expression early after murine myocardial infarction. Cardiovasc. Res. 2007, 73, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Vöö, S.; Eggermann, J.; Dunaeva, M.; Ramakers-Van Oosterhoud, C.; Waltenberger, J. Enhanced functional response of CD133+ circulating progenitor cells in patients early after acute myocardial infarction. Eur. Heart J. 2008, 29, 241–250. [Google Scholar] [CrossRef]
- Wang, Y.; Gabrielsen, A.; Lawler, P.R.; Paulsson-Berne, G.; Steinbrüchel, D.A.; Hansson, G.K.; Kastrup, J. Myocardial gene expression of angiogenic factors in human chronic ischemic myocardium: Influence of acute ischemia/cardioplegia and reperfusion. Microcirculation 2006, 13, 187–197. [Google Scholar] [CrossRef]
- Katoh, O.; Tauchi, H.; Kawaishi, K.; Kimura, A.; Satow, Y. Expression of the vascular endothelial growth factor (VEGF) receptor gene, KDR, in hematopoietic cells and inhibitory effect of VEGF on apoptotic cell death caused by ionizing radiation. Cancer Res. 1995, 55, 5687–5692. [Google Scholar]
- Kastrup, J.; Jørgensen, E.; Rück, A.; Tägil, K.; Glogar, D.; Ruzyllo, W.; Botker, H.E.; Dudek, D.; Drvota, V.; Hesse, B.; et al. Direct intramyocardial plasmid vascular endothelial growth factor-A 165 gene therapy in patients with stable severe angina pectoris: A randomized double-blind placebo-controlled study: The Euroinject One trial. J. Am. Coll. Cardiol. 2005, 45, 982–988. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor and its inhibitors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, J.P.; Hendy, J.; Takamatsu, Y.; Simmons, P.J.; Bendall, L.J. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by gcsf or cyclophosphamide. J. Clin. Investig. 2003, 111, 187–196. [Google Scholar] [CrossRef]
- Harada, M.; Qin, Y.; Takano, H.; Minamino, T.; Zou, Y.; Toko, H.; Ohtsuka, M.; Matsuura, K.; Sano, M.; Nishi, J.I.; et al. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat. Med. 2005, 11, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Suda, J.; Kajigaya, S.; Nagata, S.; Asano, S.; Saito, M.; Miura, Y. Effects of recombinant murine granulocyte colony-stimulating factor on granulocyte-macrophage and blast colony formation. Exp. Hematol. 1985, 15, 958–965. [Google Scholar]
- Vandervelde, S.; van Luyn, M.J.A.; Tio, R.A.; Harmsen, M.C. Signaling factors in stem cell-mediated repair of infarcted myocardium. J. Mol. Cell. Cardiol. 2005, 39, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Yang, C.; Deng, H.; Yang, A.; Gene, T.; Chen, X.; Ma, A.; Liu, Z. Effects of GM-CSF on the stem cells mobilization and plasma C-reactive protein levels in patients with acute myocardial infarction. Int. J. Cardiol. 2006, 113, 92–96. [Google Scholar] [CrossRef]
- Van Der Meer, P.; Lipsic, E.; Henning, R.H.; Boddeus, K.; Van Der Velden, J.; Voors, A.A.; van Veldhuisen, D.J.; van Gilst, W.H.; Schoemaker, R.G. Erythropoietin induces neovascularization and improves cardiac function in rats with heart failure after myocardial infarction. J. Am. Coll. Cardiol. 2005, 46, 125–133. [Google Scholar] [CrossRef]
- Heeschen, C.; Aicher, A.; Lehmann, R.; Fichtlscherer, S.; Vasa, M.; Urbich, C.; Mildner-Rihm, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood 2003, 102, 1340–1346. [Google Scholar] [CrossRef]
- Nishiya, D.; Omura, T.; Shimada, K.; Matsumoto, R.; Kusuyama, T.; Enomoto, S.; Iwao, H.; Takeuchi, K.; Yoshikawa, J.; Yoshiyama, M. Effects of erythropoietin on cardiac remodeling after myocardial infarction. J. Pharmacol. Sci. 2006, 101, 31–39. [Google Scholar] [CrossRef]
- Hirata, A.; Minamino, T.; Asanuma, H.; Fujita, M.; Wakeno, M.; Myoishi, M.; Tsukamoto, O.; Okada, K.I.; Koyama, H.; Komamura, K.; et al. Erythropoietin Enhances Neovascularization of Ischemic Myocardium and Improves Left Ventricular Dysfunction After Myocardial Infarction in Dogs. J. Am. Coll. Cardiol. 2006, 48, 176–184. [Google Scholar] [CrossRef]
- Fazio, S.; Palmieri, E.A.; Biondi, B.; Cittadini, A.; Saccà, L. The role of the GH-IGF-I axis in the regulation of myocardial growth: From experimental models to human evidence. Eur. J. Endocrinol. 2000, 142, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Duerr, R.L.; Huang, S.; Miraliakbar, H.R.; Clark, R.; Chien, K.R.; Ross, J. Insulin-like growth factor-1 enhances ventricular hypertrophy and function during the onset of experimental cardiac failure. J. Clin. Investig. 1995, 95, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Cameron, D.; Griese, D.P.; Riegger, G.A.J.; Kromer, E.P. Differential effects of growth hormone on cardiomyocyte and extracellular matrix protein remodeling following experimental myocardial infarction. Cardiovasc. Res. 1998, 40, 297–306. [Google Scholar] [CrossRef]
- Fazio, S.; Biondi, B.; Sabatini, D.; Cuocolo, A.; Tommaselli, A.P.; Lombardi, G.; Sacca, L. Long-Term Growth Hormone Deficiency as a Cause of Cardiomyopathy and Its Reversibility with Specific Replacement Therapy. J. Clin. Endocrinol. Metab. 1996, 81, 887–890. [Google Scholar] [PubMed]
- Takahashi, K.; Ito, Y.; Morikawa, M.; Kobune, M.; Huang, J.; Tsukamoto, M.; Sasaki, K.; Nakamura, K.; Dehari, H.; Ikeda, K.; et al. Adenoviral-delivered angiopoietin-1 reduces the infarction and attenuates the progression of cardiac dysfunction in the rat model of acute myocardial infarction. Mol. Ther. 2003, 8, 584–592. [Google Scholar] [CrossRef]
- Roviezzo, F.; Tsigkos, S.; Kotanidou, A.; Bucci, M.; Brancaleone, V.; Cirino, G.; Papapetropoulos, A. Angiopoietin-2 causes inflammation in vivo by promoting vascular leakage. J. Pharmacol. Exp. Ther. 2005, 314, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Kim, H.G.; So, J.N.; Kim, J.H.; Kwak, H.J.; Koh, G.Y. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Circ. Res. 2000, 86, 24–29. [Google Scholar] [CrossRef]
- Lukasz, A.; Beutel, G.; Kümpers, P.; Denecke, A.; Westhoff-Bleck, M.; Schieffer, B.; Bauersachs, J.; Kielstein, J.T.; Tutarel, O. Angiopoietin-2 in Adults with Congenital Heart Disease and Heart Failure. PLoS ONE 2013, 8, e66861. [Google Scholar] [CrossRef]
- Yasuda, S.; Goto, Y.; Baba, T.; Satoh, T.; Sumida, H.; Miyazaki, S.; Nonogi, H. Enhanced secretion of cardiac hepatocyte growth factor from an infarct region is associated with less severe ventricular enlargement and improved cardiac function. J. Am. Coll. Cardiol. 2000, 36, 115–121. [Google Scholar] [CrossRef]
- Bussolino, F.; Di Renzo, M.F.; Ziche, M.; Bocchietto, E.; Olivero, M.; Naldini, L.; Gaudino, G.; Tamgnone, L.; Coffer, A.; Comoglio, P.M. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J. Cell Biol. 1992, 119, 629–641. [Google Scholar] [CrossRef]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisamce, S.; de Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 2003, 9, 936–943. [Google Scholar] [CrossRef]
- Mattei, M.G.; Borg, J.P.; Rosnet, O.; Marmé, D.; Birnbaum, D. Assignment of vascular endothelial growth factor (VEGF) and placenta growth factor (PIGF) genes to human chromosome 6p12-p21 and 14q24-q31 regions, respectively. Genomics 1996, 32, 168–169. [Google Scholar] [CrossRef] [PubMed]
- Autiero, M.; Luttun, A.; Tjwa, M.; Carmeliet, P. Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: Novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J. Thromb. Haemost. 2003, 1, 1356–1370. [Google Scholar] [CrossRef]
- Khurana, R.; Moons, L.; Shafi, S.; Luttun, A.; Collen, D.; Martin, J.F.; Carmeliet, P.; Zachary, C. Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation 2005, 111, 2828–2836. [Google Scholar] [CrossRef]
- Chabot, B.; Stephenson, D.A.; Chapman, V.M.; Besmer, P.; Bernstein, A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature 1988, 335, 88–89. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Hattori, K.; Dias, S.; Friedrich, M.; Ferris, B.; Hackett, N.R.; Crystal, R.G.; Besmer, P.; Lyden, D.; Moore, M.A.S.; et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of Kit-ligand. Cell 2002, 109, 625–637. [Google Scholar] [CrossRef] [Green Version]
- Fazel, S.S.; Chen, L.; Angoulvant, D.; Li, S.; Weisel, R.D.; Keating, A.; Li, R.K. Activation of c-kit is necessary for mobilization of reparative bone marrow progenitor cells in response to cardiac injury. FASEB J. 2008, 22, 930–940. [Google Scholar] [CrossRef]
- Broudy, V.C.; Lin, N.L.; Priestley, G.V.; Nocka, K.; Wolf, N.S. Interaction of stem cell factor and its receptor c-kit mediates lodgment and acute expansion of hematopoietic cells in the murine spleen. Blood 1996, 88, 75–81. [Google Scholar] [CrossRef]
- Kuang, D.; Zhao, X.; Xiao, G.; Ni, J.; Feng, Y.; Wu, R.; Wang, G. Stem cell factor/c-kit signaling mediated cardiac stem cell migration via activation of p38 MAPK. Basic Res. Cardiol. 2008, 103, 265–273. [Google Scholar] [CrossRef]
- Kanellakis, P.; Slater, N.J.; Du, X.J.; Bobik, A.; Curtis, D.J. Granulocyte colony-stimulating factor and stem cell factor improve endogenous repair after myocardial infarction. Cardiovasc. Res. 2006, 70, 117–125. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef]
- Cannon, R.O.; Butany, J.W.; McManus, B.M.; Speir, E.; Kravitz, A.B.; Bolli, R.; Ferrans, V.J. Early degradation of collagen after acute myocardial infarction in the rat. Am. J. Cardiol. 1983, 52, 390–395. [Google Scholar] [CrossRef]
- Whittaker, P.; Boughner, D.R.; Kloner, R.A. Role of collagen in acute myocardial infarct expansion. Circulation 1991, 84, 2123–2134. [Google Scholar] [CrossRef]
- Danielsen, C.C.; Wiggers, H.; Andersen, H.R. Increased amounts of collagenase and gelatinase in porcine myocardium following ischemia and reperfusion. J. Mol. Cell. Cardiol. 1998, 30, 1431–1442. [Google Scholar] [CrossRef]
- Wells, J.M.; Gaggar, A.; Blalock, J.E. MMP generated Matrikines. J. Matrix Biol. 2015, 44, 122–129. [Google Scholar] [CrossRef]
- Senior, R.M.; Griffin, G.L.; Mecham, R.P. Chemotactic activity of elastin-derived peptides. J. Clin. Investig. 1980, 66, 859–862. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Bujak, M.; Zymek, P.; Ren, G.; Entman, M.L.; Frangogiannis, N.G. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006, 324, 475–488. [Google Scholar] [CrossRef]
- Clark, R.A.F. Overview and General Considerations of Wound Repair. In The Molecular and Cellular Biology of Wound Repair; Springer: Boston, MA, USA, 1998; pp. 3–33. [Google Scholar]
- Hudson, M.P.; Armstrong, P.W.; Ruzyllo, W.; Brum, J.; Cusmano, L.; Krzeski, P.; Lyon, R.; Quinones, M.; Theroux, P.; Sydlowski, D.; et al. Effects of Selective Matrix Metalloproteinase Inhibitor (PG-116800) to Prevent Ventricular Remodeling After Myocardial Infarction. Results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) Trial. J. Am. Coll. Cardiol. 2006, 48, 15–20. [Google Scholar] [CrossRef]
- Cerisano, G.; Buonamici, P.; Valenti, R.; Sciagrà, R.; Raspanti, S.; Santini, A.; Carrabba, N.; Dovellini, E.V.; Romito, R.; Pupi, A.; et al. Early short-term doxycycline therapy in patients with acute myocardial infarction and left ventricular dysfunction to prevent the ominous progression to adverse remodelling: The TIPTOP trial. Eur. Heart J. 2014, 35, 184–191. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Yahalom-Ronen, Y.; Rajchman, D.; Sarig, R.; Geiger, B.; Tzahor, E. Reduced matrix rigidity promotes neonatal cardiomyocyte dedifferentiation, proliferation and clonal expansion. eLife 2015, 4, e07455. [Google Scholar] [CrossRef] [PubMed]
- Mercer, S.E.; Odelberg, S.J.; Simon, H.G. A dynamic spatiotemporal extracellular matrix facilitates epicardial-mediated vertebrate heart regeneration. Dev. Biol. 2013, 382, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Baruch Umansky, K.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Bassat, D.R.; et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef]
- Gaetani, R.; Feyen, D.A.M.; Verhage, V.; Slaats, R.; Messina, E.; Christman, K.L.; Giacomello, A.; Doevendans, P.A.F.M.; Sluijter, J.P.G. Epicardial application of cardiac progenitor cells in a 3D-printed gelatin/hyaluronic acid patch preserves cardiac function after myocardial infarction. Biomaterials 2015, 61, 339–348. [Google Scholar] [CrossRef]
- Jang, J.; Park, H.J.; Kim, S.W.; Kim, H.; Park, J.Y.; Na, S.J.; Kim, H.J.; Park, M.N.; Choi, S.H.; Park, S.H.; et al. 3D printed complex tissue construct using stem cell-laden decellularized extracellular matrix bioinks for cardiac repair. Biomaterials 2017, 112, 264–274. [Google Scholar] [CrossRef]
- Serpooshan, V.; Zhao, M.; Metzler, S.A.; Wei, K.; Shah, P.B.; Wang, A.; Mahmoudi, M.; Malkovskiy, A.V.; ar Rajadas, J.; Butte, M.J.; et al. Use of bio-mimetic three-dimensional technology in therapeutics for heart disease. Bioengineered 2014, 5, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Guyette, J.P.; Charest, J.M.; Mills, R.W.; Jank, B.J.; Moser, P.T.; Gilpin, S.E.; Gershlak, J.; Okamoto, T.; Gonzalez, G.; Milan, D.J.; et al. Bioengineering Human Myocardium on Native Extracellular Matrix. Circ. Res. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in regenerative medicine. Adv. Mater. 2009, 21, 3307–3329. [Google Scholar] [CrossRef]
- Sazzad, F.; Kuzemczak, M.; Loh, E.; Wu, W.; Kofidis, T. Targeted myocardial restoration with injectable hydrogels—In search of the holy grail in regenerating damaged heart tissue. Biomedicines 2021, 9, 595. [Google Scholar] [CrossRef] [PubMed]
- Singelyn, J.M.; Sundaramurthy, P.; Johnson, T.D.; Schup-Magoffin, P.J.; Hu, D.P.; Faulk, D.M.; Wang, J.; Mayle, K.M.; Bartel, K.; Salvatore, M.; et al. Catheter-deliverable hydrogel derived from decellularized ventricular extracellular matrix increases endogenous cardiomyocytes and preserves cardiac function post-myocardial infarction. J. Am. Coll. Cardiol. 2012, 59, 751–763. [Google Scholar] [CrossRef]
- Mewhort, H.E.M.; Turnbull, J.D.; Satriano, A.; Chow, K.; Flewitt, J.A.; Andrei, A.C.; Guzzardi, D.G.; Svystonyuk, D.A.; White, J.A.; Fedak, P.W.M. Epicardial infarct repair with bioinductive extracellular matrix promotes vasculogenesis and myocardial recovery. J. Heart Lung Transplant. 2016, 35, 661–670. [Google Scholar] [CrossRef]
- Robinson, K.A.; Li, J.; Mathison, M.; Redkar, A.; Cui, J.; Chronos, N.A.F.; Matheny, R.G.; Badylak, S. Extracellular matrix scaffold for cardiac repair. Circulation 2005, 112 (Suppl. S9), I135. [Google Scholar] [CrossRef]
- Magadum, A.; Kaur, K.; Zangi, L. mRNA-Based Protein Replacement Therapy for the Heart. Mol. Ther. 2019, 27, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Zangi, L. Modified mRNA as a Therapeutic Tool for the Heart. Cardiovasc. Drugs Ther. 2020, 34, 871–880. [Google Scholar] [CrossRef]
- Zangi, L.; Lui, K.O.; Von Gise, A.; Ma, Q.; Ebina, W.; Ptaszek, L.M.; Später, D.; Xu, H.; Tabebordbar, M.; Gorbatov, R.; et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 2013, 31, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Lui, K.O.; Zangi, L.; Silva, E.A.; Bu, L.; Sahara, M.; Li, R.A.; Mooney, D.J.; Chien, K.R. Driving vascular endothelial cell fate of human multipotent Isl1 + heart progenitors with VEGF modified mRNA. Cell Res. 2013, 23, 1172–1186. [Google Scholar] [CrossRef] [Green Version]
- Collén, A.; Bergenhem, N.; Carlsson, L.; Chien, K.R.; Hoge, S.; Gan, L.M.; Fritsche-Danielson, R. VEGFA mRNA for regenerative treatment of heart failure. Nat. Rev. Drug Discov. 2022, 21, 79–80. [Google Scholar] [CrossRef]
- Huang, C.L.; Leblond, A.L.; Turner, E.C.; Kumar, A.H.; Martin, K.; Whelan, D.; O’Sullivan, D.M.; Caplice, N.M. Synthetic chemically modified mRNA-based delivery of cytoprotective factor promotes early cardiomyocyte survival post-acute myocardial infarction. Mol. Pharm. 2015, 12, 991–996. [Google Scholar] [CrossRef]
- Zangi, L.; Oliveira, M.S.; Ye, L.Y.; Ma, Q.; Sultana, N.; Hadas, Y.; Chepurko, E.; Später, D.; Zhou, B.; Chew, W.L.; et al. Cardiovascular. An IGF1R-dependent pathway drives epicardial adipose tissue formation after myocardial injury Lior. Circulation 2017, 135, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Jermendy, A.L.; Merkely, B.; Maurovich-Horvat, P. Clinical importance of epicardial adipose tissue. Arch. Med. Sci. 2017, 13, 864–874. [Google Scholar] [CrossRef]
- Hadas, Y.; Katz, M.G.; Bridges, C.R.; Zangi, L. Modified mRNA as a therapeutic tool to induce cardiac regeneration in ischemic heart disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1367. [Google Scholar] [CrossRef] [PubMed]
- Sabater-Molina, M.; Pérez-Sánchez, I.; Hernández del Rincón, J.P.; Gimeno, J.R. Genetics of hypertrophic cardiomyopathy: A review of current state. Clin. Genet. 2018, 93, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Zhu, W.; Huang, H.C.; Fung, E. Hypertrophic cardiomyopathy: An overview of genetics and management. Biomolecules 2019, 9, 878. [Google Scholar] [CrossRef]
- Chen, S.N.; Mestroni, L.; Taylor, M.R.G. Genetics of dilated cardiomyopathy. Curr. Opin. Cardiol. 2021, 36, 288–294. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Santos Mateo, J.J.; Sabater Molina, M.; Gimeno Blanes, J.R. Hypertrophic cardiomyopathy. Med. Clin. 2018, 150, 434–442. [Google Scholar] [CrossRef]
- Tuohy, C.V.; Kaul, S.; Song, H.K.; Nazer, B.; Heitner, S.B. Hypertrophic cardiomyopathy: The future of treatment. Eur. J. Heart Fail. 2020, 22, 228–240. [Google Scholar] [CrossRef]
- Bottner, P.C.; Fernández, T.C.; Valenzuela, M.L.; Romero, P.C. Dilated cardiomyopathy and severe heart failure. An update for pediatricians. Arch. Argent. Pediatr. 2018, 116, e421–e428. [Google Scholar]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Nat. Rev. Cardiol. 2020, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Günthel, M.; Barnett, P.; Christoffels, V.M. Development, Proliferation, and Growth of the Mammalian Heart. Mol. Ther. 2018, 26, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; Jovinge, S.; et al. Evidence for cardiomyocyte renewal in humans. Natl. Inst. Health 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.H.; Yang, X.; Don, C.W.; Minami, E.; Liu, Y.W.; Weyers, J.J.; Mahoney Jr, W.M.; Van Biber, B.; Palpant, N.J.; Gantz, J.A.; et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 2014, 510, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Santini, M.P.; Forte, E.; Harvey, R.P.; Kovacic, J.C. Developmental origin and lineage plasticity of endogenous cardiac stem cells. Development 2016, 143, 1242–1258. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Bradfute, S.B.; Gallardo, T.D.; Nakamura, T.; Gaussin, V.; Mishina, Y.; Pocius, J.; Michael, L.H.; Behringer, R.R.; Garry, D.J.; et al. Cardiac progenitor cells from adult myocardium: Homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. USA 2003, 100, 12313–12318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amini, H.; Rezaie, J.; Vosoughi, A.; Rahbarghazi, R.; Nouri, M. Cardiac progenitor cells application in cardiovascular disease. J. Cardiovasc. Thorac. Res. 2017, 9, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Keith, M.C.L.; Bolli, R. “String theory” of c-kitpos cardiac cells: A new paradigm regarding the nature of these cells that may reconcile apparently discrepant results. Circ. Res. 2015, 116, 1216–1230. [Google Scholar] [CrossRef]
- Lesizza, P.; Aleksova, A.; Ortis, B.; Beltrami, A.P.; Giacca, M.; Sinagra, G.; Merlo, M.; Pinamonti, B. Regenerative Medicine and Biomarkers for Dilated Cardiomyopathy. In Dilated Cardiomyopathy: From Genetics to Clinical Management; Springer: Cham, Switzerland, 2019; Chapter 11. [Google Scholar]
- Srivastava, D.; DeWitt, N. In Vivo Cellular Reprogramming: The Next Generation. Cell 2016, 166, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Martino, H.; Brofman, P.; Greco, O.; Bueno, R.; Bodanese, L.; Clausell, N.; Arnez Maldonado, J.; Mill, J.; Braile, D.; Moraes Jr, J.; et al. Multicentre, randomized, double-blind trial of intracoronary autologous mononuclear bone marrow cell injection in non-ischaemic dilated cardiomyopathy (the dilated cardiomyopathy arm of the MiHeart study). Eur. Heart J. 2015, 36, 2898–2904. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Rasokat, U.; Assmus, B.; Seeger, F.H.; Honold, J.; Leistner, D.; Fichtlscherer, S.; Schächinger, V.; Tonn, T.; Martin, H.; Dimmeler, S.; et al. A pilot trial to assess potential effects of selective Intracoronary bone marrow-derived progenitor cell infusion in patients with nonischemic dilated cardiomyopathy: Final 1-year results of the transplantation of progenitor cells and functional regenerat. Circ. Heart Fail. 2009, 2, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.; Bhargava, B.; Narang, R.; Ray, R.; Mohanty, S.; Gulati, G.; Kumar, L.; Airan, B.; Venugopal, P. The ABCD (Autologous Bone Marrow Cells in Dilated Cardiomyopathy) Trial. A Long-Term Follow-Up Study. J. Am. Coll. Cardiol. 2010, 55, 1643–1644. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Ousaka, D.; Fukushima, Y.; Kondo, M.; Eitoku, T.; Shigemitsu, Y.; Hara, M.; Baba, K.; Iwasaki, T.; Kasahara, S.; et al. Cardiosphere-derived exosomal microRNAs for myocardial repair in pediatric dilated cardiomyopathy. Sci. Transl. Med. 2020, 12, eabb3336. [Google Scholar] [CrossRef]
- Han, L.; Li, Y.; Tchao, J.; Kaplan, A.D.; Lin, B.; Li, Y.; Mich-Basso, J.; Lis, A.; Hassan, N.; London, B.; et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 2014, 104, 258–269. [Google Scholar] [CrossRef]
- Braunwald, E.; Mann, D.L.; Zipes, D.P.; Libby, P. Braunwald’s Heart Disease, 10th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2015. [Google Scholar]
- Lip, G.Y.H.; Fauchier, L.; Freedman, S.B.; Van Gelder, I.; Natale, A.; Gianni, C.; Nattel, S.; Potpara, T.; Rienstra, M.; Tse, H.F.; et al. Atrial fibrillation. Nat. Rev. Dis. Prim. 2016, 2, 1–26. [Google Scholar] [CrossRef]
- Kornej, J.; Börschel, C.S.; Benjamin, E.J.; Schnabel, R.B. Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circ. Res. 2020, 127, 4–20. [Google Scholar] [CrossRef]
- Li, J.; Gao, M.; Zhang, M.; Liu, D.; Li, Z.; Du, J.; Hou, Y. Treatment of atrial fibrillation: A comprehensive review and practice guide. Cardiovasc. J. Afr. 2020, 31, 153–158. [Google Scholar] [CrossRef]
- January, C.T.; Wann, L.S.; Calkins, H.; Chen, L.Y.; Cigarroa, J.E.; Cleveland, J.C.; Murray, K.T.; Elinor, P.T.; Shea, J.B.; Ezekowitz, M.D.; et al. 2019 AHA/ACC/HRS Focused Update of the 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart R. Circulation 2019, 140, 125–151. [Google Scholar] [CrossRef]
- Lozano-Velasco, E.; Franco, D.; Aranega, A.; Daimi, H. Genetics and epigenetics of atrial fibrillation. Int. J. Mol. Sci. 2020, 21, 5717. [Google Scholar] [CrossRef]
- Kim, J.A.; Chelu, M.G.; Li, N. Genetics of Atrial Fibrillation Jitae. Curr. Opin. Cardiol. 2021, 36, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Wijesurendra, R.S.; Casadei, B. Mechanisms of atrial fibrillation. Heart 2019, 105, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, S.A.; Yi, B.A.; Ellinor, P.T. Genetics of atrial fibrillation. Heart Fail. Clin. 2010, 6, 239–247. [Google Scholar] [CrossRef]
- Ly, O.T.; Brown, G.E.; Han, Y.D.; Darbar, D.; Khetani, S.R. Bioengineering approaches to mature induced pluripotent stem cell-derived atrial cardiomyocytes to model atrial fibrillation. Exp. Biol. Med. 2021, 246, 1816–1828. [Google Scholar] [CrossRef] [PubMed]
- Mora, C.; Serzanti, M.; Giacomelli, A.; Turco, V.; Marchina, E.; Bertini, V.; Piovani, G.; Savio, G.; Refsgaard, L.; Olesen, M.S.; et al. Generation of induced pluripotent stem cells (iPSC) from an atrial fibrillation patient carrying a KCNA5 p.D322H mutation. Stem Cell Res. 2017, 24, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Flinders, D.C.; Roberts, S.D. Ventricular arrhythmias. Prim. Care Clin. Off. Pract. 2000, 27, 709–724. [Google Scholar] [CrossRef]
- Goel, R.; Srivathsan, K.; Mookadam, M. Supraventricular and Ventricular Arrhythmias. Prim. Care Clin. Off. Pract. 2013, 40, 43–71. [Google Scholar] [CrossRef]
- Latif, S.; Dixit, S.; Callans, D.J. Ventricular Arrhythmias in Normal Hearts. Cardiol. Clin. 2008, 26, 367–380. [Google Scholar] [CrossRef]
- Sala, L.; Gnecchi, M.; Schwartz, P.J. Long QT Syndrome modelling with cardiomyocytes derived from human-induced pluripotent stem cells. Arrhythm. Electrophysiol. Rev. 2019, 8, 105–110. [Google Scholar] [CrossRef]
- Sinnecker, D.; Goedel, A.; Dorn, T.; Dirschinger, R.J.; Moretti, A.; Laugwitz, K.L. Modeling long-QT syndromes with iPS cells. J. Cardiovasc. Transl. Res. 2013, 6, 31–36. [Google Scholar] [CrossRef]
- Pan, Z.; Ebert, A.; Liang, P. Human-induced pluripotent stem cells as models for rare cardiovascular diseases: From evidence-based medicine to precision medicine. Pflug. Arch. Eur. J. Physiol. 2021, 473, 1151–1165. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, S.; Malan, D.; Sasse, P. Modeling long QT syndromes using induced pluripotent stem cells: Current progress and future challenges. Trends Cardiovasc. Med. 2013, 23, 91–98. [Google Scholar] [CrossRef] [PubMed]
- De Ponti, R.; Marazzato, J.; Bagliani, G.; Leonelli, F.M.; Padeletti, L. Sick Sinus Syndrome. Card. Electrophysiol. Clin. 2018, 10, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Wellens, H.J.J.; Josephson, M.E. Paroxysmal atrioventricular block. Heart Rhythm 2009, 6, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Bradley, A.; Clark, D.; Eric, N.; Prystowsky, M. Electrocardiography of Atrioventricular Block. Card. Electrophysiol. Clin. 2021, 13, 599–605. [Google Scholar]
- Tan, N.Y.; Witt, C.M.; Oh, J.K.; Cha, Y.-M.M.; Witt, C.M.; Oh, J.K.; Cha, Y.M. Left Bundle Branch Block: Current and Future Perspectives. Circ. Arrhythm. Electrophysiol. 2020, 13, e008239. [Google Scholar] [CrossRef]
- Rowland, E.; Morgado, F. Sino-atrial node dysfunction, atrioventricular block and intraventricular conduction disturbances. Eur. Heart J. 1992, 13, 130–135. [Google Scholar] [CrossRef]
- Mountantonakis, S.E.; Hutchinson, M.D. Indications for implantable cardioverter-defibrillator placement in ischemic cardiomyopathy and after myocardial infarction. Curr. Heart Fail. Rep. 2011, 8, 252–259. [Google Scholar] [CrossRef]
- Franco, E.; Núñez-Gil, I.J.; Vivas, D.; Ruiz Mateos, B.; Ibañez, B.; Gonzalo, N.; Macaya, C.; Fernandez Ortiz, A. Heart failure and non-ST-segment elevation myocardial infarction: A review for a widespread situation. Eur. J. Intern. Med. 2011, 22, 533–540. [Google Scholar] [CrossRef]
- O’Brien, T.M.; Schloss, E.J.; Chung, E.S. Indications for cardiac resynchronization therapy. Cardiol. Clin. 2014, 32, 293–298. [Google Scholar] [CrossRef]
- St. John Sutton, M.; Keane, M.G. Reverse remodelling in heart failure with cardiac resynchronisation therapy. Heart 2007, 93, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, L.M.; Morin, D.P. Cardiac resynchronization therapy: History, present status, and future directions. Ochsner J. 2014, 14, 596–607. [Google Scholar] [PubMed]
- Barbuti, A.; Robinson, R.B. Stem cell-derived nodal-like cardiomyocytes as a novel pharmacologic tool: Insights from sinoatrial node development and function. Pharmacol. Rev. 2015, 67, 368–388. [Google Scholar] [CrossRef] [PubMed]
- Ambesh, P.; Kapoor, A. Biological pacemakers: Concepts and techniques. Natl. Med. J. India 2017, 30, 324–326. [Google Scholar]
- Poon, E.; Kong, C.W.; Li, R.A. Human pluripotent stem cell-based approaches for myocardial repair: From the electrophysiological perspective. Mol. Pharm. 2011, 8, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Pokushalov, E.; Romanov, A.; Steinberg, J.S. Stem Cell Therapy for Electrophysiological Disorders Topical Collection on Invasive Electrophysiology and Pacing. Curr. Cardiol. Rep. 2013, 15, 408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Cell Theraphy | ||
|---|---|---|
| Cell Types | Disease | STATE of the Research |
| Skeletal Myoblasts | Myocardial Infarction | Clinical Trials |
| Satellite Cells | Myocardial Infarction | Basic Research |
| Embryonic Stem Cells | Myocardial Infarction | Basic Research |
| Cardiac Fibroblasts | Myocardial Infarction | Basic Research |
| Bone Marrow Stem Cells | Myocardial Infarction | Clinical Trials |
| Dilated cardiomyopathy | ||
| Cardiac Stem Cells | Myocardial Infarction | Basic Research |
| Dilated cardiomyopathy | ||
| Epicardium | Myocardial Infarction | Basic Research |
| Human iPSCs | Dilated cardiomyopathy | Basic Research |
| Hypertrophic cardiomyopathy | ||
| Electrical distrubances | ||
| Cytokines Treatment | ||
| Cytokin | Function | State of the research |
| FGF1 | Cells mitosis | Basic Research |
| FGF2 | Cell migration | Clinical Trials |
| Angiogenesis | ||
| FGF4 | Cell proliferation | Clinical Trials |
| Angiogenesis Metalloproteinases | ||
| VEGF | Cell proliferation | Clinical Trials |
| Migration | ||
| Neovascularization | ||
| GCSF | Cell mobilization | Clinical Trials |
| GMCSF | Cell mobilization | Basic Research |
| EPO | Angiogenesis | Basic Research |
| Cell proliferation | ||
| Cell mobilization | ||
| GH and IGF-1 | Reduced collagen deposition Reduced fibrosis | Clinical Trials |
| ANG1 | Vessel maturation | Basic Research |
| HGF | Angiogenesis | Basic Research |
| Antiapoptosis | ||
| PIGF | Cell growth | Basic Research |
| Cell survival | ||
| Cell migration | ||
| SCF | Cell mobilization | Basic Research |
| Cardiac Extracellular Matrix Treatment | ||
| cECM protein | Function | State of the research |
| Elastin | Differentiation | Basic Research |
| Proliferation | ||
| Clonal expansion | ||
| Tenascin-C Fibronectin | Cardiac regeneration | Basic Research |
| Agrin | Cardiac regeneration | Basic Research |
| Matrix Patches | Recapitulate embryonic myocardium environment | Basic Research |
| Hydrogels | Cell mobilization | Basic Research |
| Revascularization | ||
| Cardiac marker expression | ||
| Modified RNAs therapy after MI | ||
| modRNA | Function | State of the research |
| VEGFA | Cell proliferation | Clinical Trials |
| Vascularization | ||
| Angiogenesis | ||
| IGF1 | Cardiomyocyte survival rate | Basic Research |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, D.; Lozano-Velasco, E. Healing the Broken Hearts: A Glimpse on Next Generation Therapeutics. Hearts 2022, 3, 96-116. https://doi.org/10.3390/hearts3040013
Franco D, Lozano-Velasco E. Healing the Broken Hearts: A Glimpse on Next Generation Therapeutics. Hearts. 2022; 3(4):96-116. https://doi.org/10.3390/hearts3040013
Chicago/Turabian StyleFranco, Diego, and Estefanía Lozano-Velasco. 2022. "Healing the Broken Hearts: A Glimpse on Next Generation Therapeutics" Hearts 3, no. 4: 96-116. https://doi.org/10.3390/hearts3040013