Transplant Drugs against SARS, MERS and COVID-19




Abstract
:1. Introduction
1.1. Pathophysiology in Coronaviral Infections
1.2. Morbidity and Mortality of Coronaviruses
1.3. Treatment of Coronaviruses
1.4. Scope of This Review
2. Methods
3. Results
4. Discussion
4.1. COVID-19 in Solid Organ Transplant Recipients
4.2. Cytokine Storm Syndrome (CSS)
4.3. Cytokine Storm Syndrome in Other Diseases
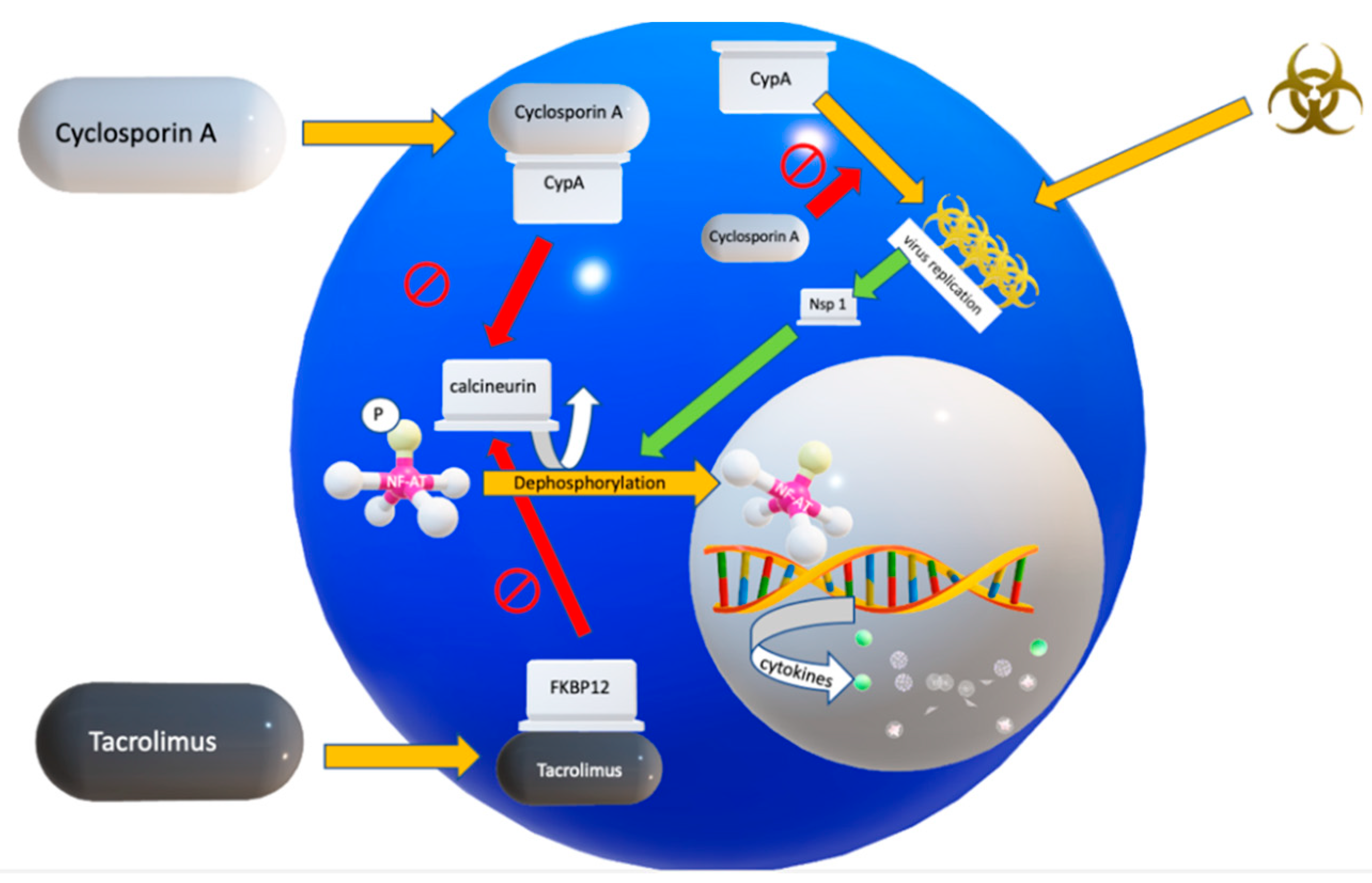
4.4. The Calcineurin/NF-AT Signaling Pathway
4.5. Two Calcineurin Inhibitors in Clinical Use
4.6. Mechanism of Action of Cyclosporin A (CsA)
4.7. Mechanism of Action of Tacrolimus
4.8. Alternative Drugs to Inhibit the Cytokine Storm Syndrome
5. Conclusions
6. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- De Wilde, A.H.; Pham, U.; Posthuma, C.C.; Snijder, E.J. Cyclophilins and cyclophilin inhibitors in nidovirus replication. Virology 2018, 522, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.V. SARS coronavirus: A new challenge for prevention and therapy. J. Clin. Investig. 2003, 111, 1605–1609. [Google Scholar] [CrossRef] [PubMed]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Schönrich, G.; Raftery, M.J.; Samstag, Y. Devilishly radical NETwork in COVID-19: Oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv. Biol. Regul. 2020, 77, 100741. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Antón-Plágaro, C.; Williamson, M.K.; Shoemark, D.K.; Simón-Gracia, L.; Klein, K.; Bauer, M.; Hollandi, R.; Greber, U.F.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. BioRxiv 2020. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ohja, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; Kallio, K.; Kaya, T.; Anastasina, M.; Smura, T.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and provides a possible pathway into the central nervous system. BioRxiv 2020. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 1 August 2020).
- Baharoon, S.; Memish, Z.A. MERS-CoV as an emerging respiratory illness: A review of prevention methods. Travel Med. Infect. Dis. 2019, 32, 101520. [Google Scholar] [CrossRef]
- Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA J. Am. Med. Assoc. 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sato, Y.; Sasaki, T. Suppression of coronavirus replication by cyclophilin inhibitors. Viruses 2013, 5, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- De Wilde, A.H.; Raj, V.S.; Oudshoorn, D.; Bestebroer, T.M.; Van Nieuwkoop, S.; Limpens, R.W.A.L.; Posthuma, C.C.; Van Der Meer, Y.; Bárcena, M.; Haagmans, B.L.; et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-α treatment. J. Gen. Virol. 2013, 94, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, A.H.; Zevenhoven-Dobbe, J.C.; Van Der Meer, Y.; Thiel, V.; Narayanan, K.; Makino, S.; Snijder, E.J.; Van Hemert, M.J. Cyclosporin A inhibits the replication of diverse coronaviruses. J. Gen. Virol. 2011, 92, 2542–2548. [Google Scholar] [CrossRef]
- Loa, C.C.; Lin, T.; Wu, C.C.; Bryan, T.; Hooper, T.; Schrader, D. The effect of immunosuppression on protective immunity of turkey poults against infection with turkey coronavirus. Comp. Immunol. Microbiol. Infect. Dis. 2002, 25, 127–138. Available online: www.elsevier.com/locate/cimid/ (accessed on 1 September 2020). [CrossRef]
- Li, H.; Kuok, D.I.; Cheung, M.C.; Ng, M.M.; Ng, K.; Hui, K.P.; Peiris, J.S.M.; Chan, M.C.W.; Nicholls, J.M. Effect of interferon alpha and cyclosporine treatment separately and in combination on Middle East Respiratory Syndrome Coronavirus (MERS-CoV) replication in a human in-vitro and ex-vivo culture model. Antivir. Res. 2018, 155, 89–96. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, A.H.; Falzarano, D.; Zevenhoven-Dobbe, J.C.; Beugeling, C.; Fett, C.; Martellaro, C.; Posthuma, C.C.; Feldmann, H.; Perlman, S.; Snijder, E.J. Alisporivir inhibits MERS- and SARS-coronavirus replication in cell culture, but not SARS-coronavirus infection in a mouse model. Virus Res. 2017, 228, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfefferle, S.; Schöpf, J.; Kögl, M.; Friedel, C.C.; Müller, M.A.; Carbajo-Lozoya, J.; Stellberger, T.; Von Dall’Armi, E.; Herzog, P.; Kallies, S.; et al. The SARS-Coronavirus-host interactome: Identification of cyclophilins as target for pan-Coronavirus inhibitors. PLoS Pathog. 2011, 7, e1002331. [Google Scholar] [CrossRef] [Green Version]
- Carbajo-Lozoya, J.; Mueller, M.A.; Kallies, S.; Thiel, V.; Drosten, C.; Von Brunn, A. Replication of human coronaviruses SARS-CoV, HCoV-NL63 and HCoV-229E is inhibited by the drug FK506. Virus Res. 2012, 165, 112–117. [Google Scholar] [CrossRef]
- Carbajo-Lozoya, J.; Ma-Lauer, Y.; Malesevic, M.; Theuerkorn, M.; Kahlert, V.; Prell, E.; Von Brunn, B.; Muth, D.; Baumert, T.F.; Drosten, C.; et al. Human coronavirus NL63 replication is cyclophilin A-dependent and inhibited by non-immunosuppressive cyclosporine A-derivatives including Alisporivir. Virus Res. 2014, 184, 44–53. [Google Scholar] [CrossRef]
- Dittmar, M.; Lee, J.S.; Whig, K.; Segrist, E.; Li, M.; Jurado, K.A.; Samby, K.; Ramage, H.; Schultz, D.; Cherry, S. Drug repurposing screens reveal FDA approved drugs active against SARS-Cov-2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Song, Z.; Xu, Y.-F.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Hage, R.; Steinack, C.; Benden, C.; Schuurmans, M.M. COVID-19 in Patients with Solid Organ Transplantation: A Systematic Review. Transplantology 2020, 1, 1–15. [Google Scholar] [CrossRef]
- Steinack, C.; Hage, R.; Benden, C.; Schuurmans, M.M. SARS-CoV-2 and Norovirus Co-Infection after Lung Transplantation. Transplantology 2020, 1, 16–23. [Google Scholar] [CrossRef]
- D’Antiga, L. Coronaviruses and Immunosuppressed Patients: The Facts during the Third Epidemic. Liver Transplant. 2020, 26, 832–834. [Google Scholar] [CrossRef] [PubMed]
- Ritschl, P.V.; Nevermann, N.; Wiering, L.; Wu, H.H.; Morodor, P.; Brandl, A.; Hillebrandt, K.; Tacke, F.; Friedersdorff, F.; Schlomm, T.; et al. Solid organ transplantation programs facing lack of empiric evidence in the COVID-19 pandemic: A By-proxy Society Recommendation Consensus approach. Am. J. Transplant. 2020, 20, 1826–1836. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, M.; Bartoletti, M.; Bussini, L.; Pancaldi, L.; Pascale, R.; Comai, G.; Morelli, M.; Ravaioli, M.; Cescon, M.; Cristini, F.; et al. COVID-19 in solid organ transplant recipients: No difference in survival compared to general population. Transpl. Infect. Dis. 2020, e13421. [Google Scholar] [CrossRef]
- Chaudhry, Z.S.; Williams, J.D.; Vahia, A.; Fadel, R.; Acosta, T.P.; Prashar, R.; Shrivastava, P.; Khoury, N.; Corrales, J.P.; Williams, C.; et al. Clinical characteristics and outcomes of COVID-19 in solid organ transplant recipients: A case-control study. Am. J. Transplant. 2020. [Google Scholar] [CrossRef]
- Chatenoud, L.; Ferran, C.; Reuter, A.; Legendre, C.; Gevaert, Y.; Kreis, H.; Franchimont, P.B.J. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma. N. Engl. J. Med. 1989, 321, 63. [Google Scholar]
- Diamanti, A.P.; Rosado, M.M.; Pioli, C.; Sesti, G.; Laganà, B. Cytokine release syndrome in COVID-19 patients, a new scenario for an old concern: The fragile balance between infections and autoimmunity. Int. J. Mol. Sci. 2020, 21, 3330. [Google Scholar] [CrossRef]
- Recalcati, S.; Invernizzi, P.; Arosio, P.; Cairo, G. New functions for an iron storage protein: The role of ferritin in immunity and autoimmunity. J. Autoimmun. 2008, 30, 84–89. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, G.; Wang, B.; Liu, B. Cytokine storm syndrome in coronavirus disease 2019: A narrative review. J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Boil. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Zhang, H.; Zhan, M.; Jiang, J.; Yin, H.; Dauphars, D.J.; Li, S.-Y.; Li, Y.; He, Y.-W. Preventing Mortality in COVID-19 Patients: Which Cytokine to Target in a Raging Storm? Front. Cell Dev. Biol. 2020, 8, 677. [Google Scholar] [CrossRef]
- Meftahi, G.H.; Jangravi, Z.; Sahraei, H.; Bahari, Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: The contribution of “inflame-aging”. Inflamm. Res. 2020, 69, 825–839. [Google Scholar] [CrossRef] [PubMed]
- England, J.T.; Abdulla, A.; Biggs, C.M.; Lee, A.Y.; Hay, K.A.; Hoiland, R.L.; Wellington, C.L.; Sekhon, M.; Jamal, S.; Shojania, K.; et al. Weathering the COVID-19 storm: Lessons from hematologic cytokine syndromes. Blood Rev. 2020, 100707. [Google Scholar] [CrossRef]
- Lechowicz, K.; Drożdżal, S.; Machaj, F.; Rosik, J.; Szostak, B.; Zegan-Barańska, M.; Biernawska, J.; Dabrowski, W.; Bosiacki, M.; Kotfis, K. COVID-19: The Potential Treatment of Pulmonary Fibrosis Associated with SARS-CoV-2 Infection. J. Clin. Med. 2020, 9, 1917. [Google Scholar] [CrossRef]
- Hage, R.; Steinack, C.; Schuurmans, M.M. Calcineurin inhibitors revisited: A new paradigm for COVID-19? Braz. J. Infect. Dis. 2020, 24, 365–367. [Google Scholar] [CrossRef]
- Willicombe, M.; Thomas, D.; McAdoo, S. COVID-19 and Calcineurin Inhibitors: Should They Get Left Out in the Storm? J. Am. Soc. Nephrol. JASN 2020, 31, 1145–1146. [Google Scholar] [CrossRef] [Green Version]
- Cavagna, L.; Seminari, E.; Zanframundo, G.; Gregorini, M.; Di Matteo, A.; Rampino, T.; Montecucco, C.; Pelenghi, S.; Cattadori, B.; Pattonieri, E.F.; et al. Calcineurin Inhibitor-Based Immunosuppression and COVID-19: Results from a Multidisciplinary Cohort of Patients in Northern Italy. Microorganisms 2020, 8, 977. [Google Scholar] [CrossRef]
- Martínez-Martínez, S.; Redondo, J.M. Inhibitors of the Calcineurin/NFAT Pathway. Curr. Med. Chem. 2004, 11, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Allan, J. Immunosuppression for lung transplantation. Semin. Thorac. Cardiovasc. Surg. 2004, 16, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, M.W.; Galat, A.; Uehling, D.E.; Schreiber, S.L. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 1989, 341, 758–760. [Google Scholar] [CrossRef]
- Satoh, K.; Shimokawa, H.; Berk, B.C. Cyclophilin A: Promising new target in cardiovascular therapy. Circ. J. 2010, 74, 2249–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, B.; Yarlett, N.; Strupp, A.; Cerami, A. Identification of cyclophilin as a proinflammatory secretory product of lipopolysaccharide-activated macrophages (cytokine/inflammation/chemotaxis/neutrophil/endotoxin). Biochemistry 1992, 89, 3511–3515. [Google Scholar]
- Yurchenko, V.; Constant, S.; Eisenmesser, E.; Bukrinsky, M. Cyclophilin-CD147 interactions: A new target for anti-inflammatory therapeutics. Clin. Exp. Immunol. 2010, 160, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.G.; Melaragno, M.G.; Liao, D.-F.; Yan, C.; Haendeler, J.; Suh, Y.-A.; Lambeth, J.D.; Berk, B.C. Cyclophilin A Is a Secreted Growth Factor Induced by Oxidative Stress. Circ. Res. 2000, 87, 789–796. Available online: http://www.circresaha.org (accessed on 1 September 2020). [CrossRef] [Green Version]
- Coppinger, J.A.; Cagney, G.; Toomey, S.; Kislinger, T.; Belton, O.; McRedmond, J.P.; Cahill, D.J.; Emili, A.; Fitzgerald, D.; Maguire, P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004, 103, 2096–2104. [Google Scholar] [CrossRef] [Green Version]
- Ianevski, A.; Zusinaite, E.; Kuivanen, S.; Strand, M.; Lysvand, H.; Teppor, M.; Kakkola, L.; Paavilainen, H.; Laajala, M.; Kallio-Kokko, H.; et al. Novel activities of safe-in-human broad-spectrum antiviral agents. Antivir. Res. 2018, 154, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Frausto, S.D.; Lee, E.; Tang, H. Cyclophilins as modulators of viral replication. Viruses 2013, 5, 1684–1701. [Google Scholar] [CrossRef]
- Flisiak, R.; Jaroszewicz, J.; Flisiak, I.; Łapiński, T. Update on alisporivir in treatment of viral hepatitis C. Expert Opin. Investig. Drugs 2012, 21, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Adegunsoye, A.; Strek, M.E. Therapeutic Approach to Adult Fibrotic Lung Diseases. Chest 2016, 150, 1371–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavagna, L.; Caporali, R.; Abdi-Ali, L.; Dore, R.; Meloni, F.; Montecucco, C. Cyclosporine in anti-Jo1-positive patients with corticosteroid-refractory interstitial lung disease. J. Rheumatol. 2013, 40, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Horita, N.; Akahane, M.; Okada, Y.; Kobayashi, Y.; Arai, T.; Amano, I.; Takezawa, T.; To, M.; To, Y. Tacrolimus and steroid treatment for acute exacerbation of idiopathic pulmonary fibrosis. Intern. Med. 2011, 50, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inase, N.; Sawada, M.; Ohtani, Y.; MlYAKE, S.; Isogai, S.; Sakashita, H.; Miyazaki, Y.; Yoshizawa, Y. Cyclosporin A Followed by the Treatment of Acute Exacerbation of Idiopathic Pulmonary Fibrosis with Corticosteroid. Intern. Med. 2003, 42, 565–570. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, S.; Homma, S.; Miyamoto, A.; Kurosaki, A.; Fujii, T.; Yoshimura, K. Cyclosporin A in the treatment of acute exacerbation of idiopathic pulmonary fibrosis. Intern. Med. 2010, 49, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Kawasumi, H.; Gono, T.; Kawaguchi, Y.; Yamanaka, H. Recent treatment of interstitial lung disease with idiopathic inflammatory myopathies. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9, 9–17. [Google Scholar] [CrossRef]
- Staab-Weijnitz, C.A.; Fernandez, I.E.; Knüppel, L.; Maul, J.; Heinzelmann, K.; Juan-Guardela, B.M.; Hennen, E.; Preissler, G.; Winter, H.; Neurohr, C.; et al. FK506-binding protein 10, a potential novel drug target for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26 and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy 2020. [Google Scholar] [CrossRef]
- Reig, J.A.; Esteve-Valverde, E.; Belizna, C.; Selva-O’Callaghan, A.; Pardos-Gea, J.; Quintana, A.; Mekinian, A.; Anunciacion-Llunell, A.; Miró-Mur, F. Immunomodulatory therapy for the management of severe COVID-19. Beyond the anti-viral therapy: A comprehensive review. Autoimmun. Rev. 2020, 19, 102569. [Google Scholar] [CrossRef]
- Softic, L.; Brillet, R.; Berry, F.; Ahnou, N.; Nevers, Q.; Morin-Dewaele, M.; Softic, L.; Brillet, R.; Berry, F.; Ahnou, N.; et al. Inhibition of SARS-CoV-2 Infection by the Cyclophilin Inhibitor Alisporivir (Debio 025). Antimicrob. Agents Chemother. 2020, 64, e00876-20. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.-M. SARS-CoV-2 pandemic: Time to revive the cyclophilin inhibitor alisporivir. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Alghamdi, M.; Mushtaq, F.; Awn, N.; Shalhoub, S. MERS CoV infection in two renal transplant recipients: Case report. Am. J. Transplant. 2015, 15, 1101–1104. [Google Scholar] [CrossRef] [Green Version]
- Clinical Trial to Evaluate Methylprednisolone Pulses and Tacrolimus in Patients with COVID-19 Lung Injury (TACROVID). Available online: https://clinicaltrials.gov/ct2/show/study/NCT04341038 (accessed on 7 August 2020).
- Trial Cyclosporine in Patients with Moderate COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT044127851/7 (accessed on 1 September 2020).
- Levenfus, I.; Ullmann, E.; Battegay, E.; Schuurmans, M.M. Triage tool for suspected COVID-19 patients in the emergency room: AIFELL score. Braz. J. Infect. Dis. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- The RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Terrazzano, G.; Rubino, V.; Palatucci, A.T.; Giovazzino, A.; Carriero, F.; Ruggiero, G. An Open Question: Is It Rational to Inhibit the mTor-Dependent Pathway as COVID-19 Therapy? Front. Pharmacol. 2020, 11, 856. [Google Scholar] [CrossRef] [PubMed]
- Mirjalili, M.; Shafiekhani, M.; Vazin, A. Coronavirus Disease 2019 (COVID-19) and Transplantation: Pharmacotherapeutic Management of Immunosuppression Regimen. Ther. Clin. Risk Manag. 2020, 16, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Rizk, J.G.; Kalantar-Zadeh, K.; Mehra, M.R.; Lavie, C.J.; Rizk, Y.; Forthal, D.N. Pharmaco-Immunomodulatory Therapy in COVID-19. Drugs 2020, 80, 1267–1292. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Coronaviral Serotype Studies in Humans | CNI | Remarks | Ref. No. |
| MERS-CoV | Tac | renal transplant recipient on tacrolimus survived | [11] |
| MERS-CoV | CsA | inhibition of viral replication | [12] |
| Coronaviral Serotype Studies in Animals | CNI | Remarks | Ref. No. |
| feline CoV | CsA | inhibition of viral replication in dose-dependent manner | [13] |
| turkey CoV | CsA | enhanced virus titers in kidney | [14] |
| Coronaviral Serotype Studies In Vitro | CNI | Remarks | Ref. No. |
| MERS-CoV | CsA + IFN-α | inhibition of viral replication | [15] |
| MERS-CoV, SARS-CoV | ALV | inhibition of viral replication | [16] |
| SARS-CoV, CoV-229E | CsA | inhibition of viral replication SARS-CoV replication impaired, but not fully blocked (1–5% of cells remained SARS-CoV positive, even in high CsA concentrations) | [17] |
| CoV-NL63, CoV-229E, SARS-CoV | CsA | inhibition of viral replication | [18] |
| SARS-CoV, CoV-NL63, CoV-229E | Tac | inhibition of viral replication | [19] |
| CoV-NL63 | CsA-d | inhibition of viral replication by CsA derivatives (Alisporivir, NIM811) | [19] |
| SARS-CoV-2 | CsA | potent antiviral activity in SARS-CoV-2, cyclophillin depedent (and calcineurin independent) | [20] |
| Research Question | Possible Answers in Literature | Refs. |
|---|---|---|
| Which patients with COVID-19 could benefit from the addition of CNI to the standard therapy |
| [65] |
| Does CypA play a role in cardiovascular morbidity in COVID-19 patients? |
| [45] |
| How to screen for patients with a high risk of progression to more severe stages of COVID-19 and thus merit pharmacological interventions |
| [67] |
| Which patients with COVID-19 should be excluded from CNIs? |
| [65] |
| CNI monotherapy or combination therapy with either a corticosteroid, an antimetabolite (Mycophenolate) |
| [68] |
| Alternative immunomodulatory drugs? |
| [69] [70] [71] |
| Alisporivir as non-immunosuppressive cyclophilin inhibitor? |
| [62,63] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hage, R.; Steinack, C.; Gautschi, F.; Schuurmans, M.M. Transplant Drugs against SARS, MERS and COVID-19. Transplantology 2020, 1, 71-84. https://doi.org/10.3390/transplantology1020007
Hage R, Steinack C, Gautschi F, Schuurmans MM. Transplant Drugs against SARS, MERS and COVID-19. Transplantology. 2020; 1(2):71-84. https://doi.org/10.3390/transplantology1020007
Chicago/Turabian StyleHage, René, Carolin Steinack, Fiorenza Gautschi, and Macé M. Schuurmans. 2020. "Transplant Drugs against SARS, MERS and COVID-19" Transplantology 1, no. 2: 71-84. https://doi.org/10.3390/transplantology1020007
APA StyleHage, R., Steinack, C., Gautschi, F., & Schuurmans, M. M. (2020). Transplant Drugs against SARS, MERS and COVID-19. Transplantology, 1(2), 71-84. https://doi.org/10.3390/transplantology1020007