
Hyperinsulinism

Abstract
:1. Introduction
2. Glucose Homeostasis
3. Hyperinsulinism—General Overview
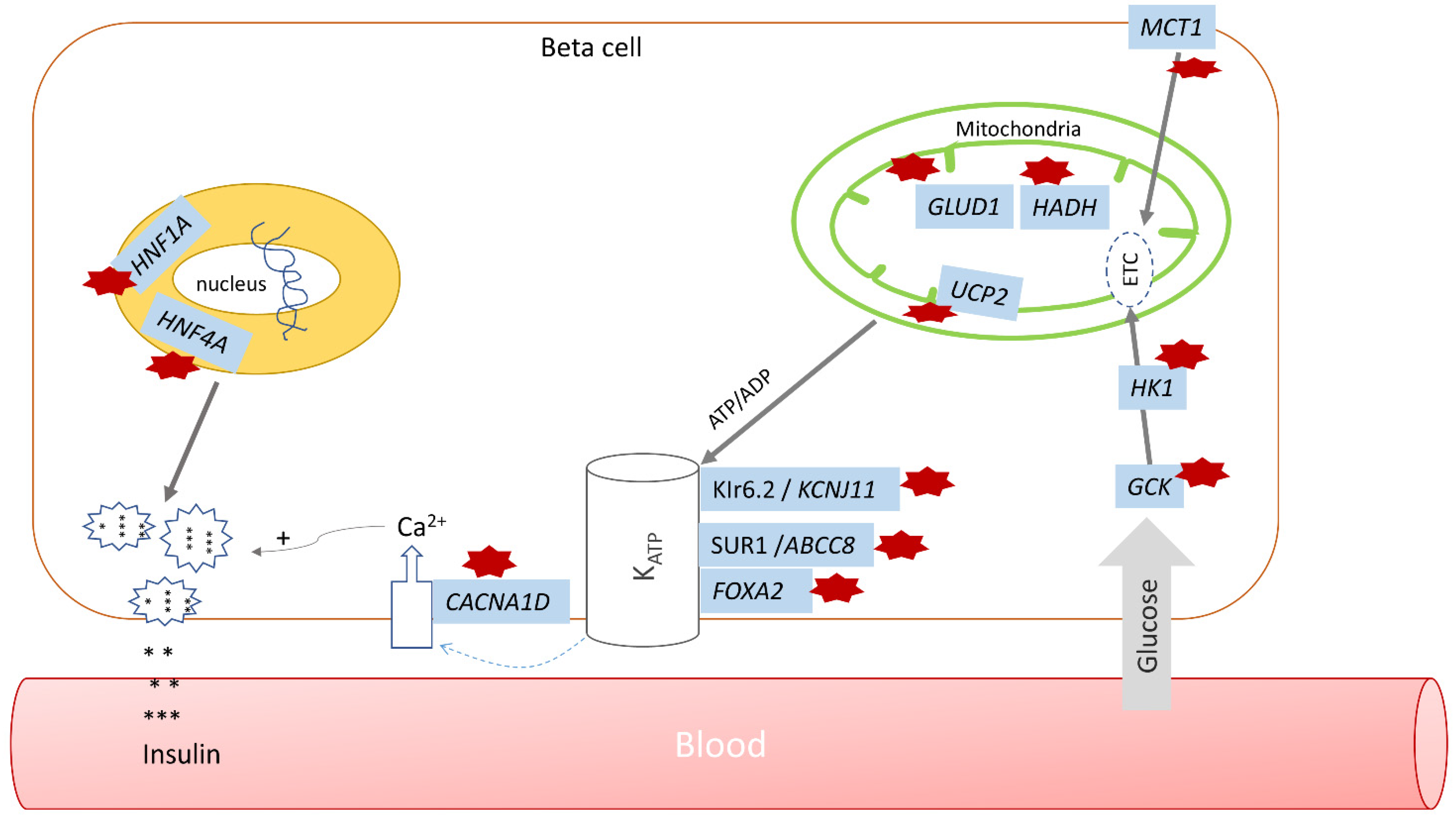
4. Monogenic Hyperinsulinism
4.1. Pancreatic β-Cell KATP Channel Defects
4.2. Glutamate Dehydrogenase (GDH) Hyperinsulinism
4.3. Glucokinase Hyperinsulinism
4.4. SCHAD Hyperinsulinism
4.5. HNF1A and HNF4A Hyperinsulinism
4.6. UCP2 Hyperinsulinism
4.7. MCT1 Hyperinsulinism
4.8. HK-1 Hyperinsulinism
4.9. FOXA2 Hyperinsulinism
4.10. CACNA1D Hyperinsulinism
5. Clinical Presentation, Diagnosis, and General Approach to Management of Hyperinsulinemic Hypoglycemia (HH)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De León, D.D.; Stanley, C.A. Mechanisms of Disease: Advances in diagnosis and treatment of hyperinsulinism in neonates. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 57–68. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, I. Idiopathic spontaneously occurring hypoglycemia in infants; clinical significance of problem and treatment. AMA Am. J. Dis. Child. 1954, 87, 399–428. [Google Scholar] [CrossRef]
- Kapoor, R.R.; Flanagan, S.E.; Arya, V.B.; Shield, P.; Ellard, S.; Hussain, K. Clinical and molecular characterization of 300 patients with congenital hyperinsulinism. Eur. J. Endocrinol. 2013, 168, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Nordlie, R.C.; Foster, J.D.; Lange, A.J. Regulation of glucose production by the liver. Ann. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Han, H.S.; Kim, M.J. Transcriptional regulators of hepatic gluconeogenesis. Arch. Pharm. Res. 2013, 36, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.C.; Ishiyama, N.; Nenquin, M.; Ravier, A.M.; Jonas, J. Signals and pools underlying biphasic insulin secretion. Diabetes 2002, 51 (Suppl. 1), S60–S67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Philippe, J.; Mojsov, S.; Chick, L.W.; Habener, J.F. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc. Natl. Acad. Sci. USA 1987, 84, 3434–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, R.M.; Morgan, L.M.; Tredger, J.A.; Deacon, S.; Wright, J.; Marks, V. Glucagon-like peptide-1 (7-36) amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: Acute post-prandial and 24-h secretion patterns. J. Endocrinol. 1993, 138, 159–166. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, S.; Chen, J.; Su, Z. Unraveling the Regulation of Hepatic Gluconeogenesis. Front. Endocrinol. 2019, 9, 802. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, T.; Donaldson, C.J.; Cáceres, E.; Hunter, E.A.; Cowing-Zitron, C.; Pound, L.D.; Adams, M.W.; Zembrzycki, A.; Grove, K.L.; Huising, M.O. Urocortin3 mediates soomatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 2015, 21, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Morgan, S.A.; Sherlock, M.; Gathercole, L.L.; Lavery, G.G.; Lenaghan, C.; Bujalska, I.J.; Laber, D.; Yu, A.; Convey, G.; Mayers, R.; et al. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 2009, 58, 2506–2515. [Google Scholar] [CrossRef] [Green Version]
- Gathercole, L.L.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W. Glucocorticoid modulation of insulin signaling in human subcutaneous adipose tissue. J. Clin. Endocrinol. Metab. 2007, 92, 4332–4339. [Google Scholar] [CrossRef] [Green Version]
- Sperling, M.A.; DeLamater, P.V.; Phelps, D.; Fiser, H.R.; Oh, W.; Fisher, D.A. Spontaneous and amino acid stimulated glucagon secretion in the immediate postnatal period. Relation to glucose and insulin. J. Clin. Investig. 1974, 53, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Farquhar, J.W. The child of the diabetic woman. Arch. Dis. Child. 1959, 34, 76–96. Available online: https://pubmed.ncbi.nlm.nih.gov/13628237/ (accessed on 5 March 2022). [CrossRef] [Green Version]
- Collins, J.E.; Leonard, J.V. Hyperinsulinism in asphyxiated and small-for-dates infants with hypoglycaemia. Lancet 1984, 324, 311–313. [Google Scholar] [CrossRef]
- Hoe, F.M.; Thornton, P.S.; Wanner, L.A.; Steinkrauss, L.; Simmons, R.A.; Stanley, C.A. Clinical features and insulin regulation in infants with a syndrome of prolonged neonatal hyperinsulinism. J. Pediatr. 2006, 148, 207–212. [Google Scholar] [CrossRef]
- Hussain, K.E.; Shepherd, R.M. Hyperinsulinemic hypoglycemia in Beckwith-Wiedemann syndrome due to defects in the function of pancreatic beta-cell adenosine triphosphate-sensitive potassium channels. J. Clin. Endocrinol. Metab. 2005, 90, 4376–4382. [Google Scholar] [CrossRef]
- Faundes, V.; Goh, S.; Akilapa, R. Clinical delineation, sex differences, and genotype-phenotype correlation in pathogenic KDM6A variants causing X-linked Kabuki syndrome type 2. Genet. Med. 2021, 23, 1202–1210. [Google Scholar] [CrossRef]
- Ben Harouch, S.; Klar, A.; Falik Zaccai, T.C. INSR-Related Severe Syndromic Insulin Resistance. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2018; pp. 1993–2022. [Google Scholar] [PubMed]
- Matsuo, T.; Ihara, K.; Ochiai, M. Hyperinsulinemic hypoglycemia of infancy in Sotos syndrome. Am. J. Med. Genet. A 2013, 161, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.E.; Boodhansingh, K.E.; Li, C. Congenital Hyperinsulinism in Infants with Turner Syndrome: Possible Association with Monosomy X and KDM6A Haploinsufficiency. Horm. Res. Paediatr. 2018, 89, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Moravej, H.; Altassan, R.; Jaeken, J. Hypoglycemia in CDG patients due to PMM2 mutations: Follow up on hyperinsulinemic patients. JIMD Rep. 2019, 51, 76–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tegtmeyer, L.C.; Rust, S.; van Scherpenzeel, M. Multiple phenotypes in phosphoglucomutase 1 deficiency. N. Engl. J. Med. 2014, 370, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, R.; Timothy, K.W.; Golden, A. Update on the Molecular Genetics of Timothy Syndrome. Front. Pediatr. 2021, 9, 668546. [Google Scholar] [CrossRef]
- Giurgea, I.; Bellanné-Chantelot, C.; Ribeiro, M. Molecular mechanisms of neonatal hyperinsulinism. Horm. Res. 2006, 66, 289–296. [Google Scholar] [CrossRef]
- Sajorda, B.J.; Gonzalez-Gandolfi, C.X.; Hathaway, E.R. Simpson-Golabi-Behmel Syndrome Type 1. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2006; pp. 1993–2022. [Google Scholar] [PubMed]
- Thomas, P.; Ye, Y.; Lightner, E. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum. Mol. Genet. 1996, 5, 1809–1812. Available online: https://pubmed.ncbi.nlm.nih.gov/8923010/ (accessed on 5 March 2022). [CrossRef]
- Thomas, P.; Cote, G.; Wohllk, N. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 1995, 268, 426–429. Available online: https://pubmed.ncbi.nlm.nih.gov/7716548/ (accessed on 5 March 2022). [CrossRef]
- Pinney, S.; MacMullen, C.; Becker, S.L. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J. Clin. Investig. 2008, 118, 2877–2886. Available online: https://pubmed.ncbi.nlm.nih.gov/18596924/ (accessed on 5 March 2022). [CrossRef]
- De León, D.D.; Thornton, P.S.; Stanley, C.A. Hypoglycemia in the newborn and infant. In Pediatric Endocrinology, 4th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2014; pp. 157–185. [Google Scholar]
- De Franco, E.; Saint-Martin, C.; Brusgaard, K. Update of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum. Mutat. 2020, 41, 884–905. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, E.; Ganguly, A.; De Leon, D. Congenital hyperinsulinism disorders: Genetic and clinical characteristics. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 682–692. Available online: https://pubmed.ncbi.nlm.nih.gov/31414570/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Adzick, N.S.; Leon, D.D.D.; States, L.J. Surgical Treatment of Congenital Hyperinsulinism: Results from 500 Pancreatectomies in Neonates and Children. J. Pediatr. Surg. 2019, 54, 27. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6339589/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Kelly, A.; Ng, D.; Ferry, R. Acute insulin responses to leucine in children with the hyperinsulinism/hyperammonemia syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 3724–3728. Available online: https://pubmed.ncbi.nlm.nih.gov/11502802/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Hsu, B.Y.L.; Kelly, A.; Thornton, P.S. Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. J. Pediatr. 2001, 138, 383–389. [Google Scholar] [CrossRef]
- Palladino, A.A.; Stanley, C.A. The hyperinsulinism/hyperammonemia syndrome. Rev. Endocr. Metab. Disord. 2010, 11, 171–178. Available online: https://pubmed.ncbi.nlm.nih.gov/20936362/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Bahi-Buisson, N.; Roze, E.; Dionisi, C. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev. Med. Child. Neurol. 2008, 50, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Snider, K.E.; Becker, S.; Boyajian, L. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J. Clin. Endocrinol. Metab. 2013, 98, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Sayed, S.; Matchinsky, F.M.; Stanley, C.A. Hyperinsulinism due to activating mutations of glucokinase. In Monogenic Hyperinsulinism Hypoglycemia Disorders; Stanley, C.A., De León, D.D., Eds.; Karger: Basel, Switzerland, 2012; pp. 146–157. [Google Scholar]
- Wabitsch, M.; Lahr, G.; Van de Bunt, M. Heterogeneity in disease severity in a family with a novel G68V GCK activating mutation causing persistent hyperinsulinaemic hypoglycaemia of infancy. Diabet. Med. 2007, 24, 1393–1399. Available online: https://pubmed.ncbi.nlm.nih.gov/17976205/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Clayton, P.; Eaton, S.; Aynsley-Green, A. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J. Clin. Investig. 2001, 108, 457–465. Available online: https://pubmed.ncbi.nlm.nih.gov/11489939/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Hussain, K.; Clayton, P.T.; Krywawych, S. Hyperinsulinism of infancy associated with a novel splice site mutation in the SCHAD gene. J. Pediatr. 2005, 146, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.; Boj, S.; Steele, A. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007, 4, 760–769. Available online: https://pubmed.ncbi.nlm.nih.gov/17407387/ (accessed on 5 March 2022). [CrossRef] [PubMed] [Green Version]
- Fajans, S.S.; Bell, G.I. Macrosomia and neonatal hypoglycaemia in RW pedigree subjects with a mutation (Q268X) in the gene encoding hepatocyte nuclear factor 4α (HNF4A). Diabetologia 2007, 50, 2600–2601. [Google Scholar] [CrossRef] [PubMed]
- Arya, V.B.; Rahman, S.; Senniappan, S. HNF4A mutation: Switch from hyperinsulinaemic hypoglycaemia to maturity-onset diabetes of the young, and incretin response. Diabet. Med. 2014, 31, e11–e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, C.T.; Boodhansingh, K.E.; Paradies, E. Novel hypoglycemia phenotype in congenital hyperinsulinism due to dominant mutations of uncoupling protein 2. J. Clin. Endocrinol. Metab. 2017, 102, 942–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Barroso, M.M.; Giurgea, I.; Bouillaud, F. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS ONE. 2008, 3, e3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullen, T.; Sylow, L.; Sun, G. Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic β-cells leads to relative hyperinsulinism during exercise. Diabetes 2012, 61, 1719–1725. Available online: https://pubmed.ncbi.nlm.nih.gov/22522610/ (accessed on 5 March 2022). [CrossRef] [PubMed] [Green Version]
- Otonkoski, T.; Jiao, H.; Kaminen-Ahola, N. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic β cells. Am. J. Hum. Genet. 2007, 81, 467–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinney, S.; Ganapathy, K.; Bradfield, J. Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm. Res. Paediatr. 2013, 80, 18–27. Available online: https://pubmed.ncbi.nlm.nih.gov/23859901/ (accessed on 5 March 2022). [CrossRef] [PubMed] [Green Version]
- Henquin, J.; Sempoux, C.; Marchandise, J.; Godecharles, S.; Guiot, Y.; Nenquin, M.; Rahier, J. Congenital hyperinsulinism caused by hexokinase I expression or glucokinase-activating mutation in a subset of β-cells. Diabetes 2013, 62, 1689–1696. Available online: https://pubmed.ncbi.nlm.nih.gov/23274908/ (accessed on 5 March 2022). [CrossRef] [PubMed] [Green Version]
- Giri, D.; Vignola, M.L.; Gualtieri, A. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Hum. Mol. Genet. 2017, 26, 4315–4326. [Google Scholar] [CrossRef]
- Vajravelu, M.E.; Chai, J.; Krock, B. Congenital Hyperinsulinism and Hypopituitarism Attributable to a Mutation in FOXA2. J. Clin. Endocrinol. Metab. 2018, 103, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; Vairo, F.; Johnson, M.B. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr. Diabetes 2017, 18, 320–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, P.; Stanley, C.; De Leon, D. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J. Pediatr. 2015, 167, 238–245. Available online: https://pubmed.ncbi.nlm.nih.gov/25957977/ (accessed on 5 March 2022). [CrossRef] [PubMed] [Green Version]
- Menni, F.; de Lonlay, P.; Sevin, C. Neurologic outcomes of 90 neonates and infants with persistent hyperinsulinemic hypoglycem ia. Pediatrics 2001, 107, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Erstad, B. Octreotide, a new somatostatin analogue. Clin. Pharm. 1989, 8, 255–273. Available online: https://pubmed.ncbi.nlm.nih.gov/2653711/ (accessed on 5 March 2022). [PubMed]
- Hosokawa, Y.; Kawakita, R.; Yokoya, S. Efficacy and safety of octreotide for the treatment of congenital hyperinsulinism: A prospective, open-label clinical trial and an observational study in Japan using a nationwide registry. Endocr. J. 2017, 64, 867–880. Available online: https://pubmed.ncbi.nlm.nih.gov/28701683/ (accessed on 5 March 2022). [CrossRef] [PubMed] [Green Version]
- Van Der Steen, I.; Van Albada, M.E.; Mohnike, K. A Multicenter Experience with Long-Acting Somatostatin Analogues in Patients with Congenital Hyperinsulinism. Horm Res. Paediatr. 2018, 89, 82–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corda, H.; Kummer, S.; Welters, A. Treatment with long-acting lanreotide autogel in early infancy in patients with severe neonatal hyperinsulinism. Orphanet. J. Rare Dis. 2017, 12, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yorifuji, T.; Kawakita, R.; Hosokawa, Y. Efficacy and safety of long-term, continuous subcutaneous octreotide infusion for patients with different subtypes of KATP-channel hyperinsulinism. Clin. Endocrinol. 2013, 78, 891–897. [Google Scholar] [CrossRef]
- Alexandrescu, S.; Tatevian, N.; Olutoye, O. Persistent hyperinsulinemic hypoglycemia of infancy: Constitutive activation of the mTOR pathway with associated exocrine-islet transdifferentiation and therapeutic implications. Int. J. Clin. Exp. Pathol. 2010, 3, 691–705. [Google Scholar]
- Yang, S.B.; Lee, H.Y.; Young, D.M. Rapamycin induces glucose intolerance in mice by reducing islet mass, insulin content, and insulin sensitivity. J. Mol. Med. 2012, 90, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Senniappan, S.; Alexandrescu, S.; Tatevian, N. Sirolimus therapy in infants with severe hyperinsulinemic hypoglycemia. N. Engl. J. Med. 2014, 370, 1131–1137. [Google Scholar] [CrossRef]
- Abraham, M.B.; Shetty, V.B.; Price, G. Efficacy and safety of sirolimus in a neonate with persistent hypoglycaemia following near-total pancreatectomy for hyperinsulinaemic hypoglycaemia. J. Pediatr. Endocrinol. Metab. 2015, 28, 1391–1398. [Google Scholar] [CrossRef]
- Minute, M.; Patti, G.; Tornese, G. Sirolimus Therapy in Congenital Hyperinsulinism: A Successful Experience Beyond Infancy. Pediatrics 2015, 136, e1373–e1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korula, S.; Chapla, A.; Priyambada, L. Sirolimus therapy for congenital hyperinsulinism in an infant with a novel homozygous KCNJ11 mutation. J. Pediatr. Endocrinol. Metab. 2018, 31, 87–89. [Google Scholar] [CrossRef]
- De León, D.D.; Li, C.; Delson, M.I. Exendin-(9–39) corrects fasting hypoglycemia in SUR-1−/− mice by lowering cAMP in pancreatic beta-cells and inhibiting insulin secretion. J. Biol. Chem. 2008, 283, 25786–25793. [Google Scholar] [CrossRef] [Green Version]
- Calabria, A.C.; Li, C.; Gallagher, P.R. GLP-1 receptor antagonist exendin-(9–39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP-sensitive K+ channel. Diabetes 2012, 61, 2585–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, P.S. Recent updates in the management of infants and children with hyperinsulinism. Curr. Opin. Pediatr. 2021, 33, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Avatapalle, B.; Padidela, R. Integrating genetic and imaging investigations into the clinical management of congenital hyperinsulinism. Clin. Endocrinol. 2013, 78, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, M.J.; Boddaert, N.; Delzescaux, T. Functional imaging of the pancreas: The role of [18F]fluoro-L-DOPA PET in the diagnosis of hyperinsulinism of infancy. Endocr. Dev. 2007, 12, 55–66. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene Involved | Protein Affected | Pattern of Inheritance | Clinical Features | Response to Diazoxide | Other Treatments |
|---|---|---|---|---|---|
| ABCC8 KCNJ11 | Sulfonylurea receptor 1 (SUR1) Kir6.2 | Autosomal-recessive Autosomal-dominant | Recessive inactivating mutations cause the most severe HI Milder form; varying degrees of HI, from asymptomatic to persistent | − +/− | |
| GLUD1 | Glutamate Dehydrogenase (GDH) | Autosomal-dominant | Hyperinsulinism-hyperammonemia syndrome (HI/HA) Mild fasting hypoglycemia More severe protein-induced hypoglycemia | + | Dietary modifications—carbohydrate preloading and avoidance of protein |
| GCK | Glucokinase (GCK) | Autosomal-dominant | Delayed presentation until late infancy or early childhood History of LGA | +/− | Pancreatectomy Long-acting somatostatin |
| HADH | 3-hydroxyacyl-CoA dehydrogenase (HADH) | Autosomal-recessive | Fasting and protein-induced hypoglycemia No hyperammonemia | + | Dietary modifications—carbohydrate preloading and avoidance of protein |
| HNF1A | HNF1α | Autosomal-dominant | Varying degrees of hyperinsulinism; from transient to severe persistent Older age of presentation | + | |
| HNF4A | HNF4α | Autosomal-dominant | Varying degrees of hyperinsulinism; from transient to severe persistent Older age of presentation | + | |
| UCP2 | Uncoupling protein 2 (UPC2) | Varying degrees of hyperinsulinism; from transient to persistent forms | + | ||
| SLC16A1 | Monocarboxylase transporter 1 (MCT1) | Autosomal-dominant | Exercise-induced hypoglycemia | +/− | Increased carb intake during intake |
| HK-1 | Hexokinase 1 (HK1) | Autosomal-dominant | + | ||
| FOXA2 | Forkhead box protein A1 (FOXA2) | Sporadic | Hypopituitarism Craniofacial dysmorphisms Endoderm-derived organ abnormalities | + | Replacement of pituitary hormone deficiencies |
| CACNA1D | Calcium-voltage-gated channel subunit α1 D (CACNA1D) | Sporadic | Developmental delays Cardiac defects hypotonia | + | Possibly Ca channel blockers |
| Genes Involved | Syndrome | Clinical Manifestations |
|---|---|---|
| IGF2 H19 CDKN1C | Beckwith–Wiedemann Syndrome [19] | Most common syndrome associated with HI presenting with neonatal hypoglycemia Macrosomia/LGA, macroglossia, hemi-hypertrophy, omphalocele |
| KDM6A KMT2D | Kabuki Syndrome [20] | Persistent HI during neonatal period Characteristic facial dysmorphic features Cranial/skeletal abnormalities Dermatoglyphic abnormalities Short stature and/or failure to thrive |
| INSR | Donohue Syndrome [21] | Fasting hypoglycemia/postprandial hyperglycemia Severe intrauterine growth restriction/postnatal growth failure Developmental delays Hypotonia Organomegaly |
| NSD1 | Sotos Syndrome [22] | Transient HI during neonatal period Prenatal and postnatal overgrowth Characteristic craniofacial features, dolichocephalic head Developmental delays; learning disabilities |
| KDM6A | Turner Syndrome [23] | HI presenting in neonatal/early infancy period with +/− response to diazoxide Short stature, ovarian insufficiency/delayed puberty development Multisystemic involvement with varying degrees of severity (mosaic forms typically with milder phenotypes) |
| PMM2 | Congenital disorder of glycosylation (CDG) Type Ia [24] | Hyperinsulinemic hypoglycemia in the first year of life, responsive to diazoxide Macrosomia at birth Broad and variable symptomatology depending on stage of life |
| PGM1 | Congenital disorder of glycosylation (CDG) Type Ib [25] | Postprandial hyperinsulinemic hypoglycemia Fasting hyperketotic hypoglycemia |
| CACNA1C | Timothy Syndrome [26] | Intermittent hypoglycemia Facial dysmorphism, syndactyly Prolonged QT interval, Congenital heart defects Seizures, autism spectrum disorder, learning disabilities |
| DIS3L2 | Perlman Syndrome [27] | Macrosomia Facial dysmorphisms Increased risk of renal hamartomas |
| GPC3 | Simpson–Golabi–Behmel Syndrome [28] | Macrosomia Hypertelorism, macrostomia, macroglossia Higher risk for embryonal tumors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemente, E.G.; Kanungo, S.; Schmitt, C.; Maajali, D. Hyperinsulinism. Endocrines 2022, 3, 115-126. https://doi.org/10.3390/endocrines3010011
Clemente EG, Kanungo S, Schmitt C, Maajali D. Hyperinsulinism. Endocrines. 2022; 3(1):115-126. https://doi.org/10.3390/endocrines3010011
Chicago/Turabian StyleClemente, Ethel Gonzales, Shibani Kanungo, Christine Schmitt, and Dana Maajali. 2022. "Hyperinsulinism" Endocrines 3, no. 1: 115-126. https://doi.org/10.3390/endocrines3010011