
Activated Carbon from Corncobs Doped with RuO2 as Biobased Electrode Material

 , , ,
, , ,  ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Activated Carbon (AC)
2.1.2. Composite Production
2.1.3. Electrode Preparation
2.2. Physico-Chemical Characterization
2.2.1. Specific Surface Area (SSA) and Pore Size Distribution (PSD)
2.2.2. Immersion Calorimetry (IC)
2.2.3. Zeta-Potential
2.2.4. X-ray Diffraction (XRD)
2.2.5. X-ray Photoelectron Spectroscopy (XPS)
2.2.6. Scanning Electron Micrsocopy (SEM)
2.3. Electrochemical Properties
2.3.1. Electric Conductivity (EC) Measurements
2.3.2. Cyclic Voltammetry (CV)
3. Results and Discussion
3.1. Physico-Chemical Properties of the Activated Carbon
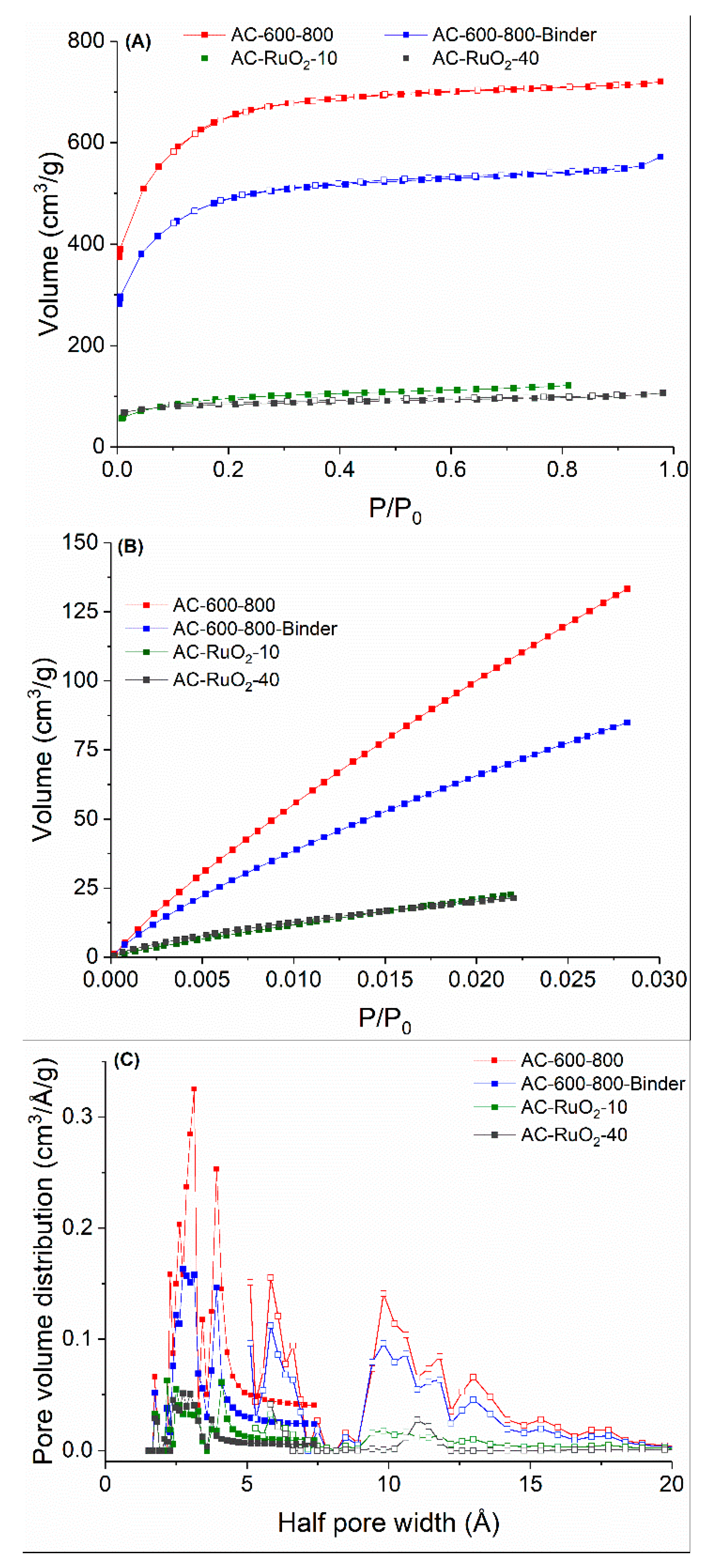
3.1.1. SSA and PSD
3.1.2. Immersion Calorimetry
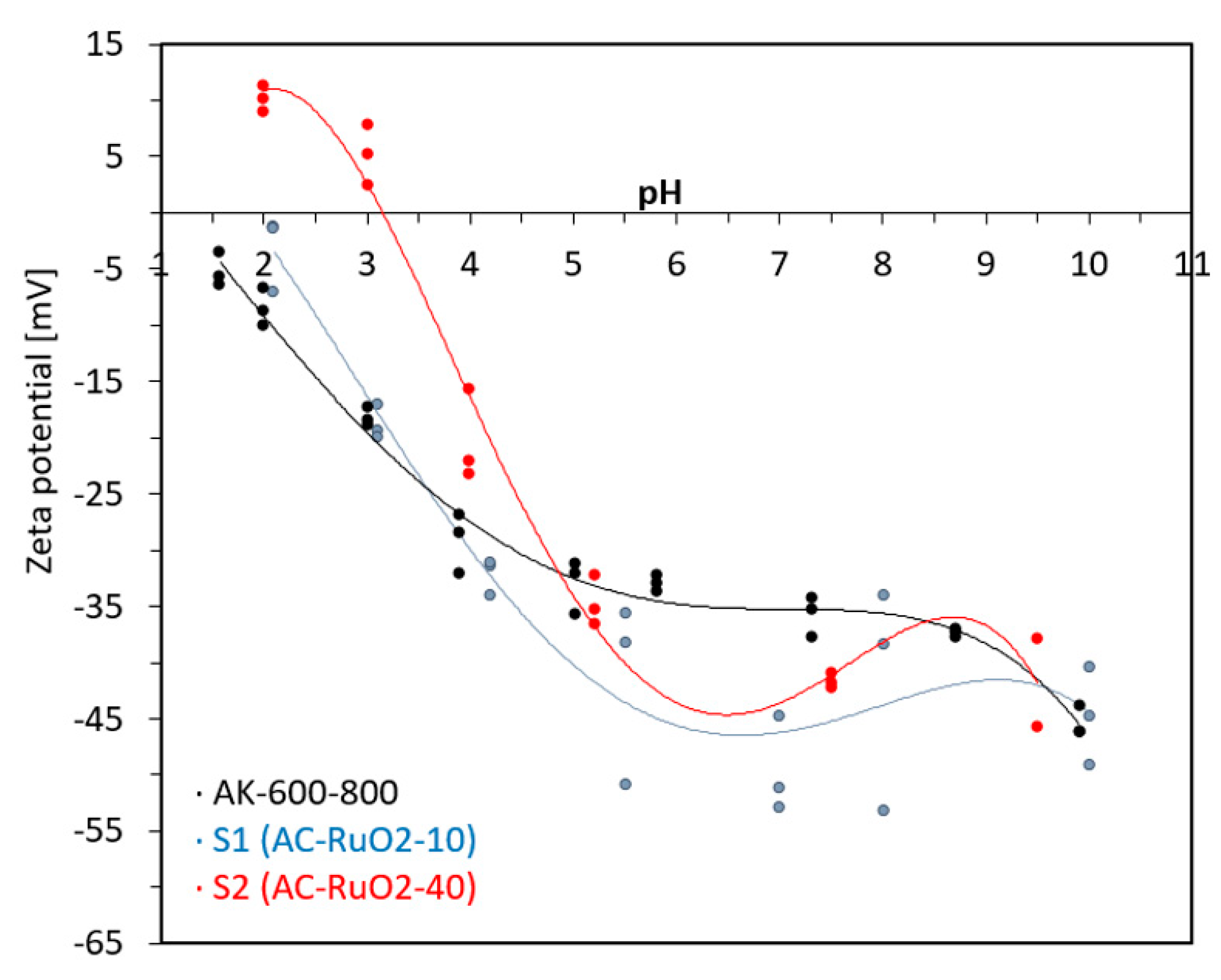
3.1.3. Zeta Potential
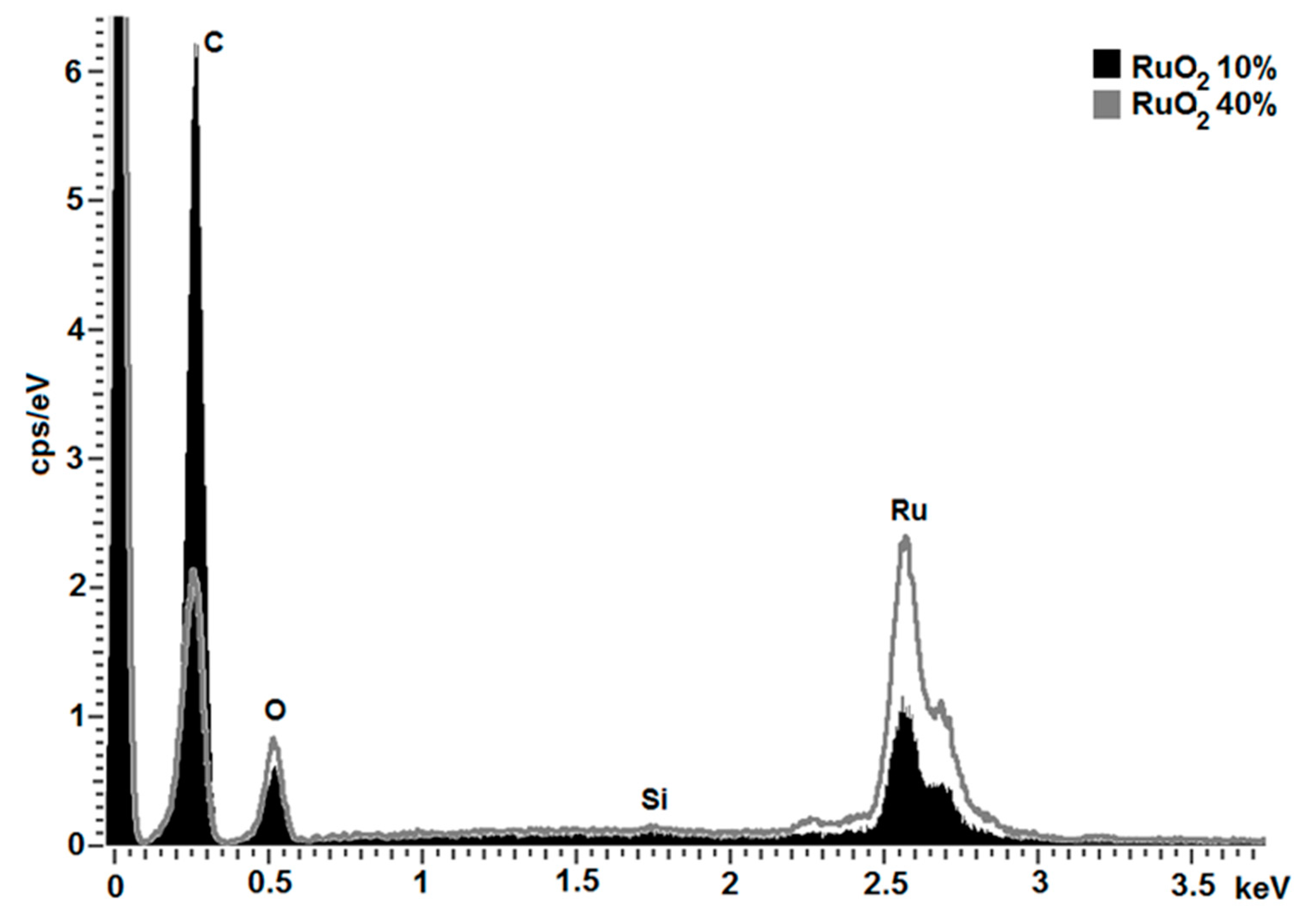
3.1.4. Composition
- (1)
- The roughness of the sample surface, since the evaluated Lα spectral line of Ru has the highest energy (25,586 keV) and is overestimated in shadowed areas towards the Kα lines of C and O.
- (2)
- The superficial coverage of the carbon particles, since the depth of X-ray generation is conceivably not as big as the particles are. This means that the inner part of the particles is neglected by the measurement.
- (3)
- The inhomogeneity of the sample leads to statistical errors, since only a small area of approximately 1 mm2 was analyzed.
3.1.5. X-ray Diffraction (XRD)
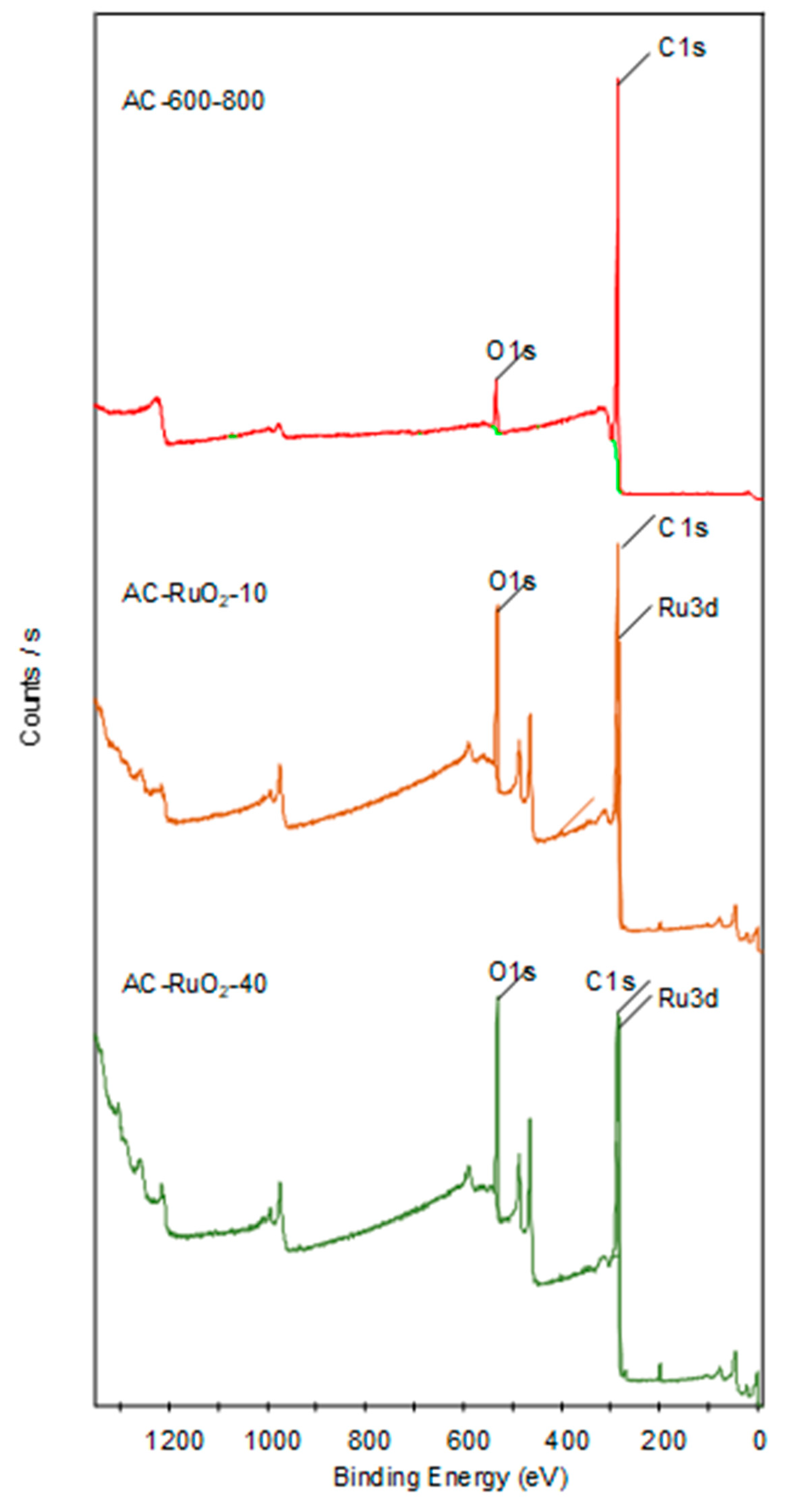
3.1.6. XPS
3.2. Electrochemical Performance
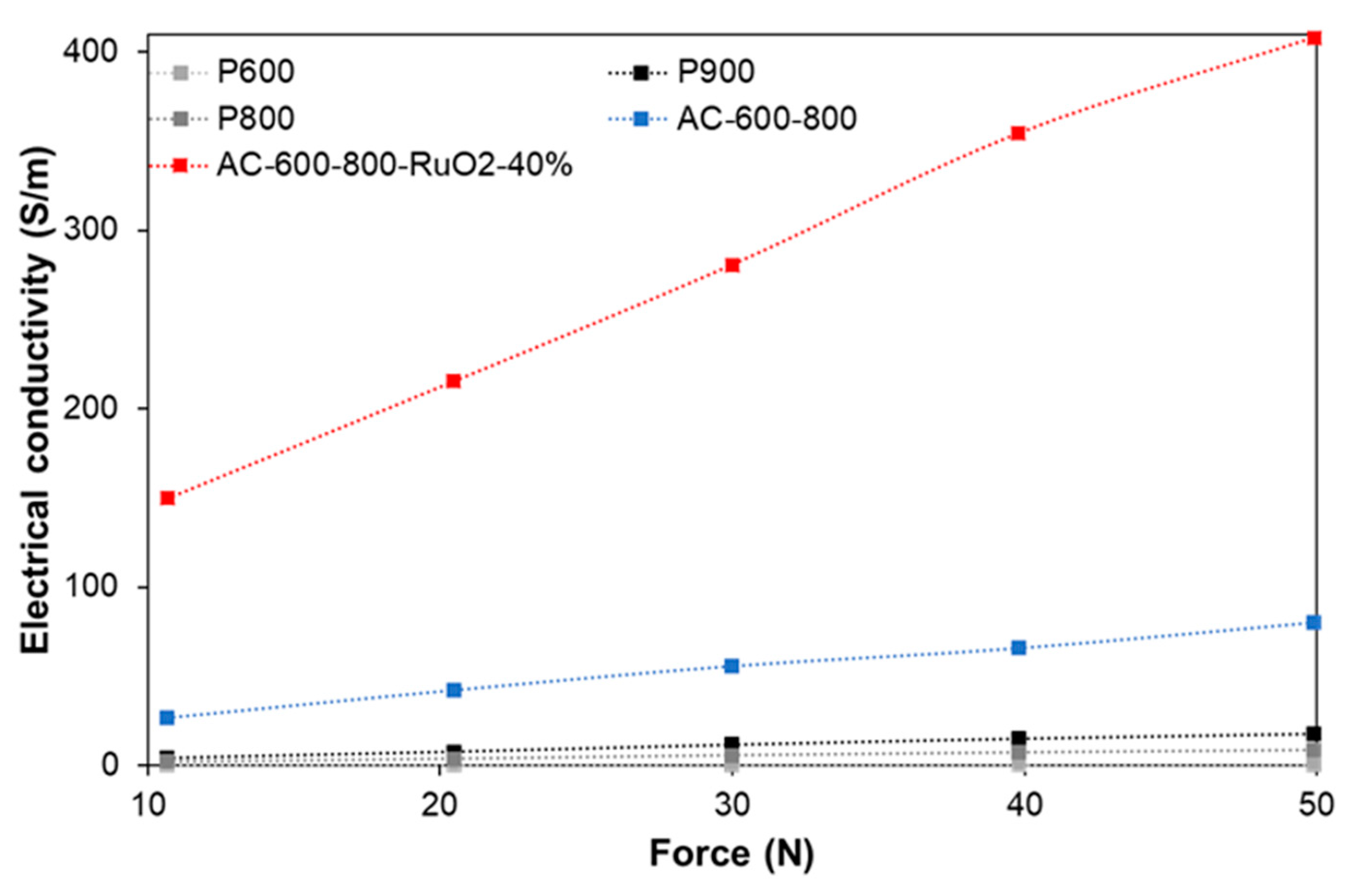
3.2.1. Electric Conductivity (EC)
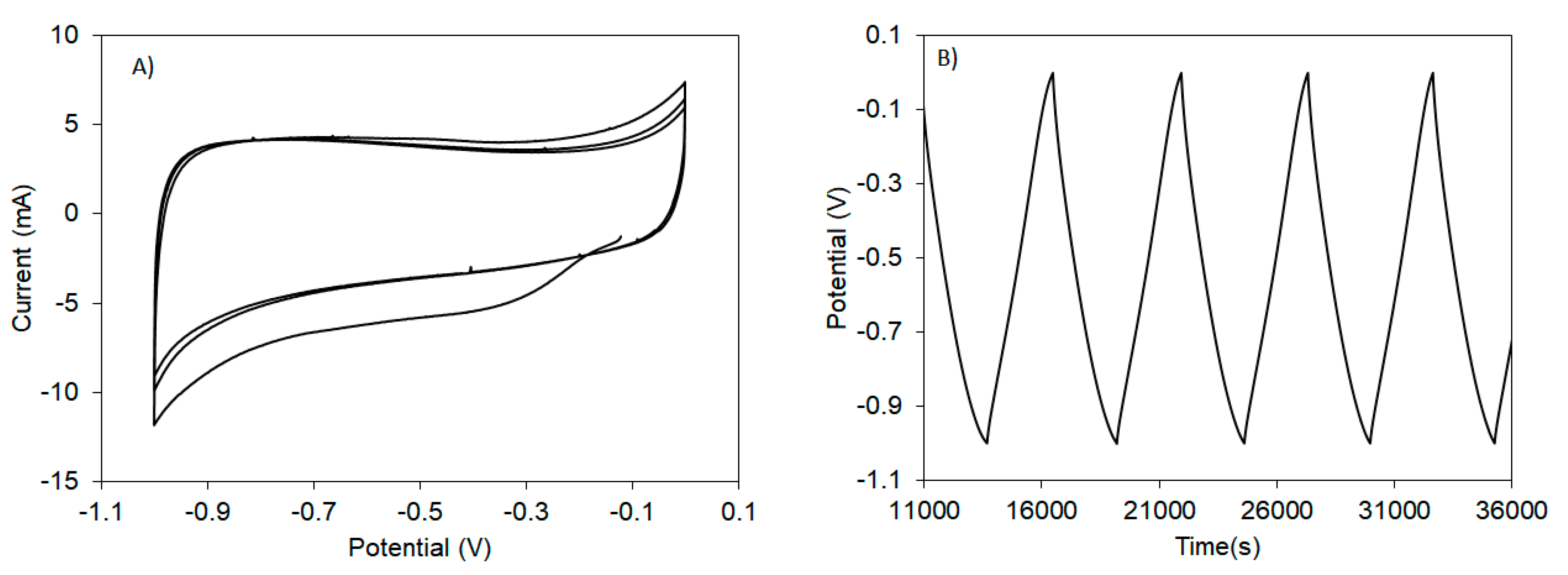
3.2.2. Capacitive Behavior
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- BMBF. Energiewende und Nachhaltiges Wirtschaften Bioökonomie—Neue Konzepte zur Nutzung Natürlicher Ressourcen. Energiewende und Nachhaltiges Wirtschaften. 2017. Available online: https://www.bmbf.de/de/biooekonomie-neue-konzepte-zur-nutzung-natuerlicher-ressourcen-726.html (accessed on 8 March 2017).
- Miller, J.M. Ultracapacitor Applications; IET Power and Energy Series; The Institution of Engineering and Technology: London, UK, 2011. [Google Scholar]
- Frackowiak, E.; Béguin, F. Carbon materials for the electrochemical storage of energy in capacitors. Carbon 2001, 39, 937–950. [Google Scholar] [CrossRef]
- Titirici, M.-M.; White, R.; Falco, C.; Sevilla, M. Black perspectives for a green future: Hydrothermal carbons for environment protection and energy storage. Energy Environ. Sci. 2012, 5, 6796–6822. [Google Scholar] [CrossRef]
- Li, W.; Chen, D.; Li, Z.; Shi, Y.; Wan, Y.; Wang, G.; Jiang, Z.; Zhao, D. Nitrogen-containing carbon spheres with very large uniform mesopores: The superior electrode materials for EDLC in organic electrolyte. Carbon 2007, 45, 1757–1763. [Google Scholar] [CrossRef]
- Su, Y.; Wu, F.; Bao, L.; Yang, Z. RuO2/activated carbon composites as a positive electrode in an alkaline electrochemical capacitor. New Carbon Mater. 2007, 22, 4–9. [Google Scholar] [CrossRef]
- Titirici, M.-M.; Antonietti, M. Chemistry and materials options of sustainable carbon materials made by hydrothermal carbonization. Chem. Soc. Rev. 2010, 39, 103–116. [Google Scholar] [CrossRef]
- Rodriguez Correa, C.; Otto, T.; Kruse, A. Influence of the biomass components on the pore formation of activated carbon. Biomass Bioenergy 2016, 97, 53–64. [Google Scholar] [CrossRef]
- Rodríguez Correa, C.; Stollovsky, M.; Hehr, T.; Rauscher, Y.; Rolli, B.; Kruse, A. Influence of the Carbonization Process on Activated Carbon Properties from Lignin and Lignin-Rich Biomasses. ACS Sustain. Chem. Eng. 2017. [Google Scholar] [CrossRef]
- Zhu, Z.; Tang, S.; Yuan, J.; Qin, X.; Deng, Y.; Qu, R. Effects of Various Binders on Supercapacitor Performances. Int. J. Electroche 2016, 11, 8270–8279. [Google Scholar] [CrossRef]
- Abbas, Q.; Pajak, D.; Frąckowiak, E.; Béguin, F.; Frackowiak, E.; Beguin, F. Effect of binder on the performance of carbon / carbon symmetric capacitors in salt aqueous electrolyte. Electrochim. Acta 2014, 140, 132–138. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Rouquerol, F.; Rouquerol, J.; Sing, K.S.W. Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: Cambridge, MA, USA, 1999. [Google Scholar]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. Computations from nitrogen isotherms. Vol. area Distrib. Porous Subst. 1951, 73, 373–380. [Google Scholar]
- Lastoskie, C.; Gubbins, K.E.; Quirke, N. Pore size distribution analysis of microporous carbons: A density functional theory approach. J. Phys. Chem. 1993, 97, 4786–4796. [Google Scholar] [CrossRef]
- Ravikovitch, P.I.; O’Domhnaill, S.C.; Neimark, A.V.; Schiith, F.; Unger, K.K. Capillary Hysteresis in Nanopores: Theoretical and Experimental Studies of Nitrogen Adsorption on MCM-41. Langmuir 1995, 11, 4765–4772. [Google Scholar] [CrossRef]
- Madani, S.H.; Silvestre-Albero, A.; Biggs, M.J.; Rodríguez-Reinoso, F.; Pendleton, P. Immersion Calorimetry: Molecular Packing Effects in Micropores. ChemPhysChem 2015, 16, 3984–3991. [Google Scholar] [CrossRef] [PubMed]
- Villar-Rodil, S.; Denoyel, R.; Rouquerol, J.; Martínez-Alonso, A.; Tascón, J.M.D. The use of microcalorimetry to assess the size exclusion properties of carbon molecular sieves. Thermochim. Acta 2004, 420, 141–144. [Google Scholar] [CrossRef]
- Echeverría, J.; Estella, J.; Barbería, V.; Musgo, J.; Garrido, J. Synthesis and characterization of ultramicroporous silica xerogels. J. Non-Cryst. Solids 2010, 356, 378–382. [Google Scholar] [CrossRef]
- Hoffmann, V.; Rodriguez Correa, C.; Sautter, D.; Maringolo, E.; Kruse, A. Study of the electrical conductivity of biobased carbonaceous powder materials under moderate pressure for the application as electrode materials in energy storage technologies. GCB Bioenergy 2018, 11, 230–248. [Google Scholar] [CrossRef]
- Mrozowski, S. Studies of carbon powders under compression. In Proceedings of the 3rd Carbon Conference, Buffalo, NY, USA, 17–21 June 1957; p. 495. [Google Scholar]
- Celzard, A.; Marêché, J.F.; Payot, F.; Furdin, G. Electrical conductivity of carbonaceous powders. Carbon 2002, 40, 2801–2815. [Google Scholar] [CrossRef]
- Zoski, C.G. Handbook of Electrochemistry, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Garrido, J.; Linares-Solano, A.; Martin-Martinez, J.M.; Molina-Sabio, M.; Rodriguez-Reinoso, F.; Torregrosa, R. Use of N2 vs. CO2 in the Characterization of Activated Carbons. Langmuir 1987, 3, 76–81. [Google Scholar] [CrossRef]
- Jirkovsky, J.; Makarova, M.; Krtil, P. Particle size dependence of oxygen evolution reaction on nanocrystalline. Electrochem. Commun. 2006, 8, 1417–1422. [Google Scholar] [CrossRef]
- Vargas, D.P.; Giraldo, L.; Moreno-Piraján, J.C. Calorimetric study of activated carbons impregnated with CaCl2. Open Chem. 2015, 13, 683–688. [Google Scholar] [CrossRef]
- Madani, S.H.; Hu, C.; Silvestre-Albero, A.; Biggs, M.J.; Rodríguez-Reinoso, F.; Pendleton, P. Pore size distributions derived from adsorption isotherms, immersion calorimetry, and isosteric heats: A comparative study. Carbon 2016, 96, 1106–1113. [Google Scholar] [CrossRef]
- Barroso-Bogeat, A.; Alexandre-Franco, M.; Fernández-González, C.; Gómez-Serrano, V. Physico-chemical characterization of activated carbon–metal oxide photocatalysts by immersion calorimetry in benzene and water. J. Therm. Anal. Calorim. 2016, 125, 65–74. [Google Scholar] [CrossRef]
- Luxton, T.P.; Eick, M.J.; Scheckel, K.G. Characterization and dissolution properties of ruthenium oxides. J. Colloid Interface Sci. 2011, 359, 30–39. [Google Scholar] [CrossRef]
- Msagati, T.A.M.; Mamba, B.B.; Sivasankar, V.; Omine, K. Surface restructuring of lignite by bio-char of Cuminum cyminum - Exploring the prospects in defluoridation followed by fuel applications. Appl. Surf. Sci. 2014, 301, 235–243. [Google Scholar] [CrossRef]
- Takagi, H.; Maruyama, K.; Yoshizawa, N.; Yamada, Y.; Sato, Y. XRD analysis of carbon stacking structure in coal during heat treatment. Fuel 2004, 83, 2427–2433. [Google Scholar] [CrossRef]
- Sugimoto, W.; Iwata, H.; Yokoshima, K.; Murakami, Y.; Takasu, Y. Proton and electron conductivity in hydrous ruthenium oxides evaluated by electrochemical impedance spectroscopy: The origin of large capacitance. J. Phys. Chem. B 2005, 109, 7330–7338. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.P.; Cygan, P.J.; Jow, T.R. Hydrous Ruthenium Oxide as an Electrode Material for Electrochemical Capacitors. J. Electrochem. Soc. 1995, 142, 2699–2703. [Google Scholar] [CrossRef]
- Foelske, A.; Barbieri, O.; Hahn, M.; Kötz, R. An X-ray Photoelectron Spectroscopy Study of Hydrous Ruthenium Oxide Powders with Various Water Contents for Supercapacitors. Electrochem. Solid-State Lett. 2006, 9, A268. [Google Scholar] [CrossRef]
- Hilgendorff, M.; Diesner, K.; Schulenburg, H.; Bogdanoff, P.; Bron, M.; Fiechter, S. Preparation Strategies towards Selective Ru-Based Oxygen Reduction Catalysts for Direct Methanol Fuel Cells. J. New Mater. Electrochem. Syst. 2002, 5, 71–81. [Google Scholar]
- Eblagon, K.M.; Tsang, S.C.E. Structure-reactivity relationship in catalytic hydrogenation of heterocyclic compounds over ruthenium black-Part A: Effect of substitution of pyrrole ring and side chain in N-heterocycles. Appl. Catal. B Environ. 2014, 160–161, 22–34. [Google Scholar] [CrossRef]
- González-Huerta, R.G.; González-Cruz, R.; Citalán-Cigarroa, S.; Montero-Ocampo, C.; Chavez-Carvayar, J.; Solorza-Feria, O. Development and electrochemical studies of ruthenium nanoparticles as cathode in a PEMFC. J. New Mater. Electrochem. Syst. 2005, 8, 15–23. [Google Scholar]
- Zhang, R.H.; Zhao, T.S.; Tan, P.; Wu, M.C.; Jiang, H.R. Ruthenium dioxide-decorated carbonized tubular polypyrrole as a bifunctional catalyst for non-aqueous lithium-oxygen batteries. Electrochim. Acta 2017, 257, 281–289. [Google Scholar] [CrossRef]
- Gui, Z.; Duay, J.; Hu, J.; Lee, S.B. Redox-exchange induced heterogeneous RuO2-conductive polymer nanowires. Phys. Chem. Chem. Phys. 2014, 16, 12332–12340. [Google Scholar] [CrossRef] [PubMed]
- Kwak, K.H.; Kim, D.W.; Kang, Y.; Suk, J. Hierarchical Ru- and RuO2-foams as high performance electrocatalysts for rechargeable lithium-oxygen batteries. J. Mater. Chem. A 2016, 4, 16356–16367. [Google Scholar] [CrossRef]
- Holm, R. Electric Contacts: Theory and Applications, 4th ed.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2000; p. 221. [Google Scholar]
- Hoffmann, V.; Jung, D.; Zimmermann, J.; Rodriguez Correa, C.; Elleuch, A.; Halouani, K.; Kruse, A. Conductive Carbon Materials from the Hydrothermal Carbonization of Vineyard Residues for the Application in Electrochemical Double-Layer Capacitors (EDLCs) and Direct Carbon Fuel Cells (DCFCs). Materials 2019, 12, 1703. [Google Scholar] [CrossRef]
- Kötz, R.; Carlen, M. Principles and applications of electrochemical capacitors. Electrochim. Acta 2000, 45, 2483–2498. [Google Scholar] [CrossRef]
- Bernstein, H. Bauelemente der Elektronik; Walter de Gruyter GmbH: Berlin, Germany; München, Germany; Boston, MA, USA, 2015; p. 273. [Google Scholar]
- Ma, X.; Chen, Y.A.; Zhou, K.; Wu, P.C.; Hou, C.H. Enhanced desalination performance via mixed capacitive-Faradaic ion storage using RuO2-activated carbon composite electrodes. Electrochim. Acta 2019, 295, 769–777. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Corncobs | Activated Carbon (AC-600-800) | |
|---|---|---|
| CDAF (wt.%) | 47.9 | 95.9 |
| HDAF (wt.%) | 6.5 | 0.1 |
| NDAF (wt.%) | 2.9 | 0.3 |
| SDAF (wt.%) | 0.1 | 0.0 |
| ODAF (wt.%) | 42.6 | 3.7 |
| Sample | SBET,N2 (m2/g) | SBET,CO2 (m2/g) | Smicro (cm2/g) | Vmicro (cm3/g) |
|---|---|---|---|---|
| AC-600-800 | 2334 | 3145 | 2037 | 0.89 |
| AC-600-800-binder | 1759 | 1452 | 1516 | 0.67 |
| AC-RuO2-10 | 344 | 384 | 248 | 0.11 |
| AC-RuO2-40 | 317 | 299 | 264 | 0.11 |
| Sample | ΔHImmC6H6(a) (J/g) | ΔHInmH2O (b) (J/g) | |
|---|---|---|---|
| AC-600-800 | −218.0 | −173.1 | 0.79 |
| AC-RuO2-10 | −71.71 | −132.0 | 1.84 |
| AC-RuO2-40 | −204.4 | −21.24 | 0.10 |
| C (at%) | O (at%) | Ru (at%) | |
|---|---|---|---|
| AC-600-800 | 93.2 | 6.8 | 0 |
| AC-RuO2-10 | 74.4 | 20.6 | 5.0 |
| AC-RuO2-40 | 80.6 | 15.3 | 4.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, V.; Rodriguez Correa, C.; Sachs, S.; del Pilar Sandoval-Rojas, A.; Qiao, M.; Brown, A.B.; Zimmermann, M.; Rodriguez Estupiñan, J.P.; Cortes, M.T.; Moreno-Piraján, J.C.; et al. Activated Carbon from Corncobs Doped with RuO2 as Biobased Electrode Material. Electron. Mater. 2021, 2, 324-343. https://doi.org/10.3390/electronicmat2030023
Hoffmann V, Rodriguez Correa C, Sachs S, del Pilar Sandoval-Rojas A, Qiao M, Brown AB, Zimmermann M, Rodriguez Estupiñan JP, Cortes MT, Moreno-Piraján JC, et al. Activated Carbon from Corncobs Doped with RuO2 as Biobased Electrode Material. Electronic Materials. 2021; 2(3):324-343. https://doi.org/10.3390/electronicmat2030023
Chicago/Turabian StyleHoffmann, Viola, Catalina Rodriguez Correa, Saskia Sachs, Andrea del Pilar Sandoval-Rojas, Mo Qiao, Avery B. Brown, Michael Zimmermann, Jenny Paola Rodriguez Estupiñan, Maria Teresa Cortes, Juan Carlos Moreno-Piraján, and et al. 2021. "Activated Carbon from Corncobs Doped with RuO2 as Biobased Electrode Material" Electronic Materials 2, no. 3: 324-343. https://doi.org/10.3390/electronicmat2030023
APA StyleHoffmann, V., Rodriguez Correa, C., Sachs, S., del Pilar Sandoval-Rojas, A., Qiao, M., Brown, A. B., Zimmermann, M., Rodriguez Estupiñan, J. P., Cortes, M. T., Moreno-Piraján, J. C., Titirici, M.-M., & Kruse, A. (2021). Activated Carbon from Corncobs Doped with RuO2 as Biobased Electrode Material. Electronic Materials, 2(3), 324-343. https://doi.org/10.3390/electronicmat2030023