
Multifaceted Role of the Transforming Growth Factor β on Effector T Cells and the Implication for CAR-T Cell Therapy




Abstract
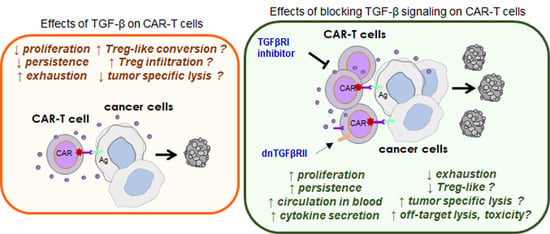
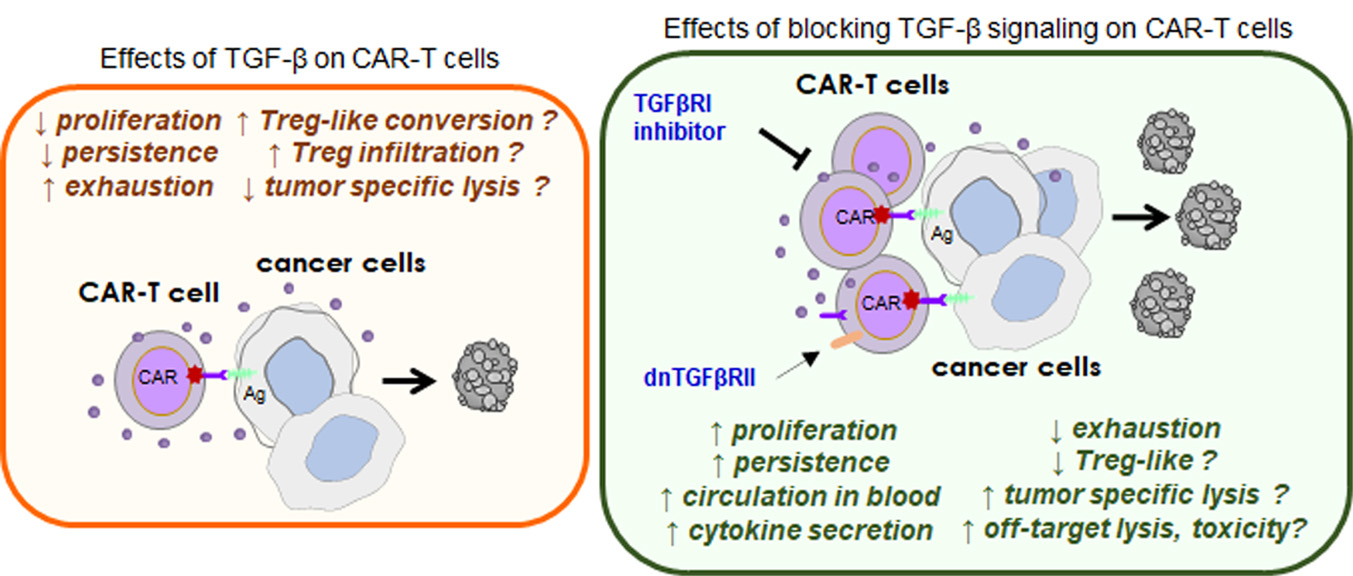
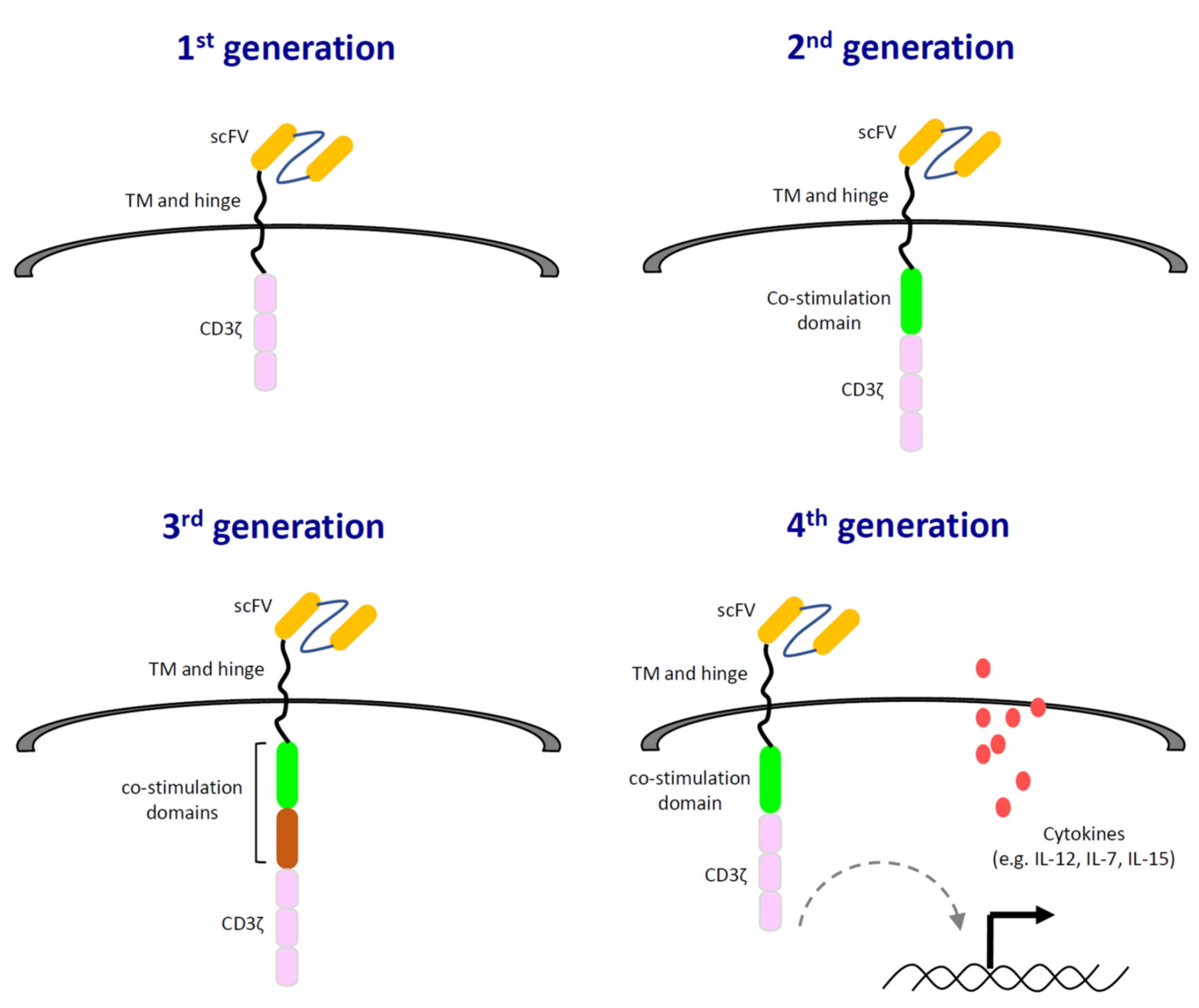
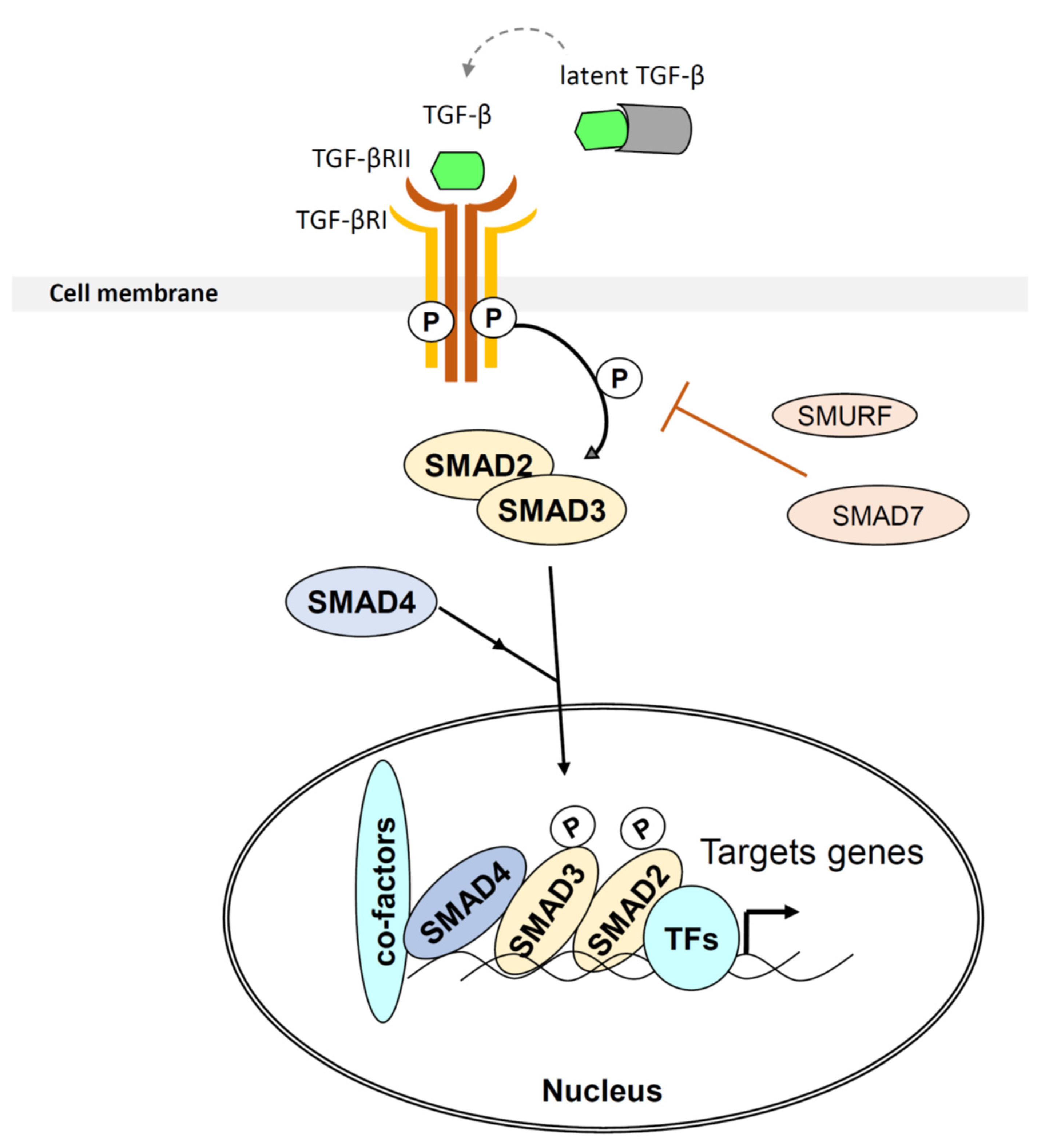
:
{kind=link}
{kind=link}
{kind=link}
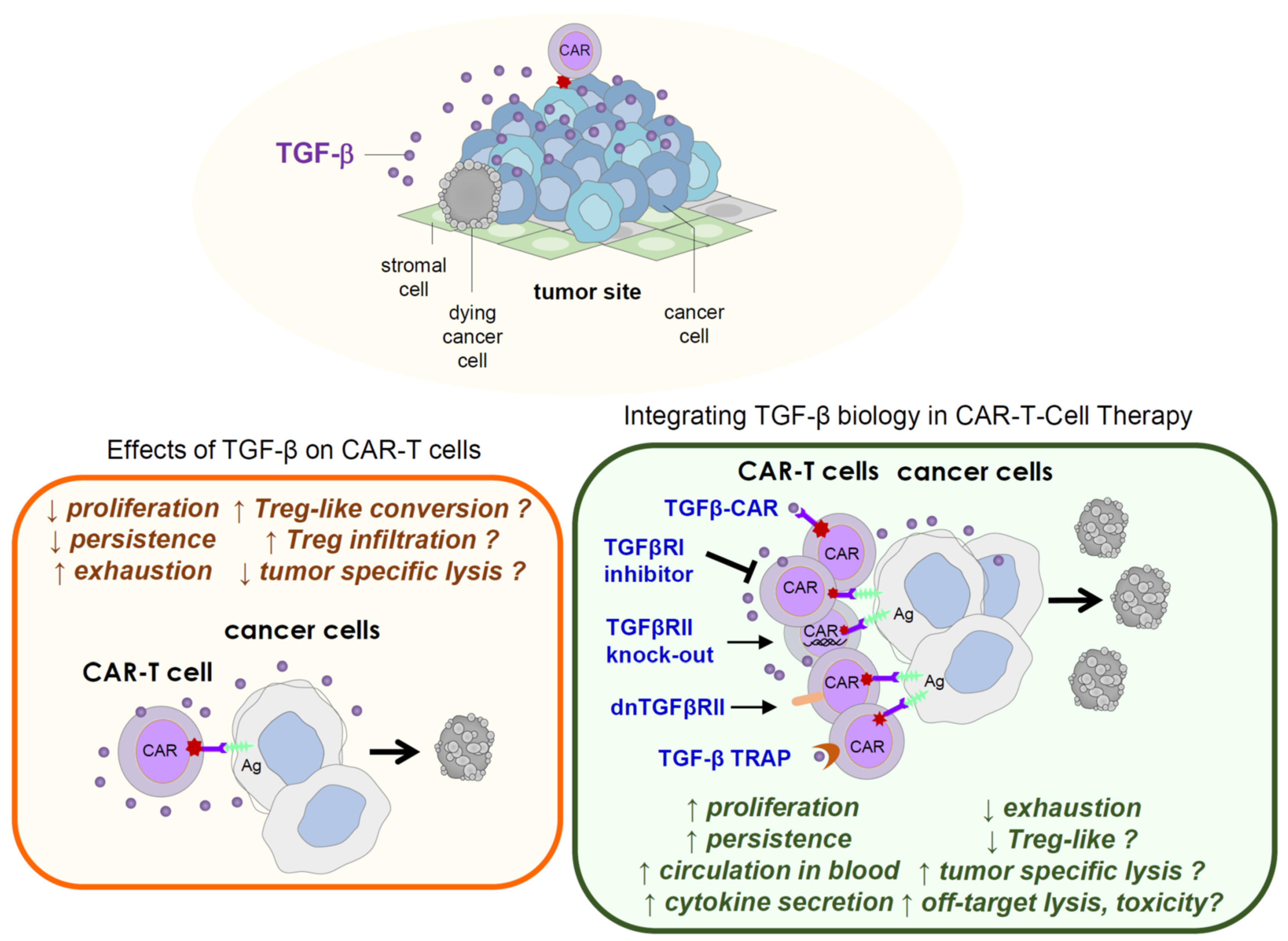{kind=link}
1. Introduction
2. Tumor Microenvironment and Immunosuppression
2.1. TGF-β
2.2. TGF-β, TME Components and Immunosuppression
3. Effects of TGF-β on Effector T Cells and T Regs, and Consequences on Immune Checkpoint Blockade
4. Effects of TGF-β on CAR-T Effector Functions
5. Therapeutic Consequences and Considerations
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Burstein, H.J.; Krilov, L.; Aragon-Ching, J.B.; Baxter, N.N.; Chiorean, E.G.; Chow, W.A.; De Groot, J.F.; Devine, S.M.; DuBois, S.G.; El-Deiry, W.S.; et al. Clinical Cancer Advances 2017: Annual Report on Progress Against Cancer From the American Society of Clinical Oncology. J. Clin. Oncol. 2017, 35, 1341–1367. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; Lopez-Soto, A. The Hallmarks of Successful Anticancer Immunotherapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; Ribas, A. Tumour-Intrinsic Resistance to Immune Checkpoint Blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Boyiadzis, M.M.; Dhodapkar, M.V.; Brentjens, R.J.; Kochenderfer, J.N.; Neelapu, S.S.; Maus, M.V.; Porter, D.L.; Maloney, D.G.; Grupp, S.A.; Mackall, C.L.; et al. Chimeric Antigen Receptor (CAR) T Therapies for the Treatment of Hematologic Malignancies: Clinical Perspective and Significance. J. Immunother. Cancer 2018, 6, 137. [Google Scholar] [CrossRef]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N. The Landscape of CAR-T Cell Clinical Trials against Solid Tumors—A Comprehensive Overview. Cancers 2020, 12, 2567. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.J.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benmebarek, M.-R.; Karches, C.; Cadilha, B.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. IJMS 2019, 20, 1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric Antigen Receptor Signaling: Functional Consequences and Design Implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef] [PubMed]
- Poorebrahim, M.; Melief, J.; Pico de Coaña, Y.; L Wickström, S.; Cid-Arregui, A.; Kiessling, R. Counteracting CAR T Cell Dysfunction. Oncogene 2021, 40, 421–435. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-Cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1–2 Trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of Resistance to CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric Antigen Receptor T Cell (CAR-T) Immunotherapy for Solid Tumors: Lessons Learned and Strategies for Moving Forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ Biology in Cancer Progression and Immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Tu, E.; Chia, P.Z.C.; Chen, W. TGFβ in T Cell Biology and Tumor Immunity: Angel or Devil? Cytokine Growth Factor Rev. 2014, 25, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making Sense of Latent TGFβ Activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.S.; Sheppard, D. Cross Talk among TGF-β Signaling Pathways, Integrins, and the Extracellular Matrix. Cold Spring Harb. Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ren, J.; ten Dijke, P. Targeting TGFβ Signal Transduction for Cancer Therapy. Sig. Transduct. Target. 2021, 6, 8. [Google Scholar] [CrossRef]
- Wakefield, L.M.; Hill, C.S. Beyond TGFβ: Roles of Other TGFβ Superfamily Members in Cancer. Nat. Rev. Cancer 2013, 13, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β Induces M2-like Macrophage Polarization via SNAIL-Mediated Suppression of a pro-Inflammatory Phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-Derived Catabolites Are Responsible for Inhibition of T and Natural Killer Cell Proliferation Induced by Indoleamine 2,3-Dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells through Membrane-Bound TGF-Beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast Heterogeneity in Tumor Micro-Environment: Role in Immunosuppression and New Therapies. Semin. Immunol. 2020, 48, 101417. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The Polarization of Immune Cells in the Tumour Environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratchev, A. TGF-β Signalling in Tumour Associated Macrophages. Immunobiology 2017, 222, 75–81. [Google Scholar] [CrossRef]
- Cinier, J.; Hubert, M.; Besson, L.; Di Roio, A.; Rodriguez, C.; Lombardi, V.; Caux, C.; Ménétrier-Caux, C. Recruitment and Expansion of Tregs Cells in the Tumor Environment—How to Target Them? Cancers 2021, 13, 1850. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kitani, A.; Strober, W. Cell Contact-Dependent Immunosuppression by CD4(+)CD25(+) Regulatory T Cells Is Mediated by Cell Surface-Bound Transforming Growth Factor Beta. J. Exp. Med. 2001, 194, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.C.; Letterio, J.J.; Gavin, M.; Rudensky, A.Y. TGF-Beta1 Maintains Suppressor Function and Foxp3 Expression in CD4+CD25+ Regulatory T Cells. J. Exp. Med. 2005, 201, 1061–1067. [Google Scholar] [CrossRef]
- Donkor, M.K.; Sarkar, A.; Li, M.O. Tgf-Β1 Produced by Activated CD4(+) T Cells Antagonizes T Cell Surveillance of Tumor Development. Oncoimmunology 2012, 1, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-Beta Directly Targets Cytotoxic T Cell Functions during Tumor Evasion of Immune Surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, H.; Chen, G.; Wei, X.; Wang, C.; Zhou, S.; Huang, A.; Zhang, Z.; Zhan, C.; Wu, Y.; et al. Arming Anti-EGFRvIII CAR-T With TGFβ Trap Improves Antitumor Efficacy in Glioma Mouse Models. Front. Oncol. 2020, 10, 1117. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Immune-Mediated Eradication of Tumors through the Blockade of Transforming Growth Factor-Beta Signaling in T Cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, X.; Pins, M.; Javonovic, B.; Kuzel, T.; Kim, S.-J.; Parijs, L.V.; Greenberg, N.M.; Liu, V.; Guo, Y.; et al. Adoptive Transfer of Tumor-Reactive Transforming Growth Factor-Beta-Insensitive CD8+ T Cells: Eradication of Autologous Mouse Prostate Cancer. Cancer Res. 2005, 65, 1761–1769. [Google Scholar] [CrossRef] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel III, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Yap, T.; Araujo, D.; Wood, D.; Denis, J.-F.; Gruosso, T.; Tremblay, G.; O’Connor-McCourt, M.; Ghosh, R.; Sinclair, S.; Nadler, P.; et al. P856 AVID200, First-in-Class TGF-Beta1 and Beta3 Selective Inhibitor: Results of a Phase 1 Monotherapy Dose Escalation Study in Solid Tumors and Evidence of Target Engagement in Patients. J. Immunother. Cancer 2020, 8 (Suppl. 1), A6.2–A7. [Google Scholar] [CrossRef]
- Martin, C.J.; Datta, A.; Littlefield, C.; Kalra, A.; Chapron, C.; Wawersik, S.; Dagbay, K.B.; Brueckner, C.T.; Nikiforov, A.; Danehy, F.T.; et al. Selective Inhibition of TGFβ1 Activation Overcomes Primary Resistance to Checkpoint Blockade Therapy by Altering Tumor Immune Landscape. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Park, B.V.; Freeman, Z.T.; Ghasemzadeh, A.; Chattergoon, M.A.; Rutebemberwa, A.; Steigner, J.; Winter, M.E.; Huynh, T.V.; Sebald, S.M.; Lee, S.-J.; et al. TGFβ1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016, 6, 1366–1381. [Google Scholar] [CrossRef] [Green Version]
- Dodagatta-Marri, E.; Meyer, D.S.; Reeves, M.Q.; Paniagua, R.; To, M.D.; Binnewies, M.; Broz, M.L.; Mori, H.; Wu, D.; Adoumie, M.; et al. α-PD-1 Therapy Elevates Treg/Th Balance and Increases Tumor Cell PSmad3 That Are Both Targeted by α-TGFβ Antibody to Promote Durable Rejection and Immunity in Squamous Cell Carcinomas. J. Immunother. Cancer 2019, 7, 62. [Google Scholar] [CrossRef]
- Horn, L.A.; Riskin, J.; Hempel, H.A.; Fousek, K.; Lind, H.; Hamilton, D.H.; McCampbell, K.K.; Maeda, D.Y.; Zebala, J.A.; Su, Z.; et al. Simultaneous Inhibition of CXCR1/2, TGF-β, and PD-L1 Remodels the Tumor and Its Microenvironment to Drive Antitumor Immunity. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sow, H.S.; Ren, J.; Camps, M.; Ossendorp, F.; Ten Dijke, P. Combined Inhibition of TGF-β Signaling and the PD-L1 Immune Checkpoint Is Differentially Effective in Tumor Models. Cells 2019, 8, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ Pathway with Galunisertib, a TGFβRI Small Molecule Inhibitor, Promotes Anti-Tumor Immunity Leading to Durable, Complete Responses, as Monotherapy and in Combination with Checkpoint Blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef]
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; Gulley, J.L.; Palena, C.; Schlom, J. Dual Targeting of TGF-β and PD-L1 via a Bifunctional Anti-PD-L1/TGF-ΒRII Agent: Status of Preclinical and Clinical Advances. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident Memory T Cells, Critical Components in Tumor Immunology. J. Immunother. Cancer 2018, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; van Gisbergen, K.P.J.M.; Hombrink, P.; van Lier, R.A.W. Tissue-Resident Memory T Cells at the Center of Immunity to Solid Tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.; Gauthier, L.; Leclerc, M.; Gros, G.; de Montpreville, V.; Theret, N.; Donnadieu, E.; Mami-Chouaib, F. TGFbeta Signaling Intersects with CD103 Integrin Signaling to Promote T-Lymphocyte Accumulation and Antitumor Activity in the Lung Tumor Microenvironment. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corgnac, S.; Malenica, I.; Mezquita, L.; Auclin, E.; Voilin, E.; Kacher, J.; Halse, H.; Grynszpan, L.; Signolle, N.; Dayris, T.; et al. CD103+CD8+ TRM Cells Accumulate in Tumors of Anti-PD-1-Responder Lung Cancer Patients and Are Tumor-Reactive Lymphocytes Enriched with Tc17. Cell Rep. Med. 2020, 1, 100127. [Google Scholar] [CrossRef]
- Brown, N.F.; Marshall, J.F. Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment. Cancers 2019, 11, 1221. [Google Scholar] [CrossRef] [Green Version]
- Dodagatta-Marri, E.; Ma, H.-Y.; Liang, B.; Li, J.; Meyer, D.S.; Sun, K.-H.; Ren, X.; Zivak, B.; Rosenblum, M.D.; Headley, M.B.; et al. Integrin Avβ8 on T Cells Is Responsible for Suppression of Anti-Tumor Immunity in Multiple Syngeneic Models and Is a Promising Target for Tumor Immunotherapy. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.05.14.084913v1.full (accessed on 30 March 2021).
- Mokrani, M.; Klibi, J.; Bluteau, D.; Bismuth, G.; Mami-Chouaib, F. Smad and NFAT Pathways Cooperate to Induce CD103 Expression in Human CD8 T Lymphocytes. J. Immunol. 2014, 192, 2471–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Floc’h, A.; Jalil, A.; Vergnon, I.; Le Maux Chansac, B.; Lazar, V.; Bismuth, G.; Chouaib, S.; Mami-Chouaib, F. Alpha E Beta 7 Integrin Interaction with E-Cadherin Promotes Antitumor CTL Activity by Triggering Lytic Granule Polarization and Exocytosis. J. Exp. Med. 2007, 204, 559–570. [Google Scholar] [CrossRef]
- Baas, M.; Besancon, A.; Goncalves, T.; Valette, F.; Yagita, H.; Sawitzki, B.; Volk, H.D.; Waeckel-Enee, E.; Rocha, B.; Chatenoud, L.; et al. TGFbeta-Dependent Expression of PD-1 and PD-L1 Controls CD8(+) T Cell Anergy in Transplant Tolerance. eLife 2016, 5, e08133. [Google Scholar] [CrossRef]
- Abd Hamid, M.; Colin-York, H.; Khalid-Alham, N.; Browne, M.; Cerundolo, L.; Chen, J.-L.; Yao, X.; Rosendo-Machado, S.; Waugh, C.; Maldonado-Perez, D.; et al. Self-Maintaining CD103+ Cancer-Specific T Cells Are Highly Energetic with Rapid Cytotoxic and Effector Responses. Cancer Immunol. Res. 2020, 8, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Dahmani, A.; Janelle, V.; Carli, C.; Richaud, M.; Lamarche, C.; Khalili, M.; Goupil, M.; Bezverbnaya, K.; Bramson, J.L.; Delisle, J.-S. TGFβ Programs Central Memory Differentiation in Ex Vivo-Stimulated Human T Cells. Cancer Immunol. Res. 2019, 7, 1426–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [Green Version]
- Koehler, H.; Kofler, D.; Hombach, A.; Abken, H. CD28 Costimulation Overcomes Transforming Growth Factor-Beta-Mediated Repression of Proliferation of Redirected Human CD4+ and CD8+ T Cells in an Antitumor Cell Attack. Cancer Res. 2007, 67, 2265–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golumba-Nagy, V.; Kuehle, J.; Hombach, A.A.; Abken, H. CD28-ζ CAR T Cells Resist TGF-β Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018, 26, 2218–2230. [Google Scholar] [CrossRef] [Green Version]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Stüber, T.; Monjezi, R.; Wallstabe, L.; Kühnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wöckel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-β-Receptor Signaling Augments the Antitumor Function of ROR1-Specific CAR T-Cells against Triple-Negative Breast Cancer. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β Inhibition via CRISPR Promotes the Long-Term Efficacy of CAR T Cells against Solid Tumors. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Perna, S.K.; Pagliara, D.; Mahendravada, A.; Liu, H.; Brenner, M.K.; Savoldo, B.; Dotti, G. Interleukin-7 Mediates Selective Expansion of Tumor-Redirected Cytotoxic T Lymphocytes (CTLs) without Enhancement of Regulatory T-Cell Inhibition. Clin. Cancer Res. 2014, 20, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Rostamian, H.; Fallah-Mehrjardi, K.; Khakpoor-Koosheh, M.; Pawelek, J.M.; Hadjati, J.; Brown, C.E.; Mirzaei, H.R. A Metabolic Switch to Memory CAR T Cells: Implications for Cancer Treatment. Cancer Lett. 2021, 500, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.J.; Chang, Z.L.; Lorenzini, M.H.; Zah, E.; Chen, Y.Y. TGF-β-Responsive CAR-T Cells Promote Anti-Tumor Immune Function. Bioeng. Transl. Med. 2018, 3, 75–86. [Google Scholar] [CrossRef]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-Cell Responses to Soluble Factors with Chimeric Antigen Receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Recent Advances in CAR T-Cell Toxicity: Mechanisms, Manifestations and Management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.B.; Salama, A.K.S. A Review of Cancer Immunotherapy Toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotolo, R.; Leuci, V.; Donini, C.; Cykowska, A.; Gammaitoni, L.; Medico, G.; Valabrega, G.; Aglietta, M.; Sangiolo, D. CAR-Based Strategies beyond T Lymphocytes: Integrative Opportunities for Cancer Adoptive Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Folmont, A.; Bourhis, J.-H.; Chouaib, S.; Terry, S. Multifaceted Role of the Transforming Growth Factor β on Effector T Cells and the Implication for CAR-T Cell Therapy. Immuno 2021, 1, 160-173. https://doi.org/10.3390/immuno1030010
de Folmont A, Bourhis J-H, Chouaib S, Terry S. Multifaceted Role of the Transforming Growth Factor β on Effector T Cells and the Implication for CAR-T Cell Therapy. Immuno. 2021; 1(3):160-173. https://doi.org/10.3390/immuno1030010
Chicago/Turabian Stylede Folmont, Apolline, Jean-Henri Bourhis, Salem Chouaib, and Stéphane Terry. 2021. "Multifaceted Role of the Transforming Growth Factor β on Effector T Cells and the Implication for CAR-T Cell Therapy" Immuno 1, no. 3: 160-173. https://doi.org/10.3390/immuno1030010
APA Stylede Folmont, A., Bourhis, J.-H., Chouaib, S., & Terry, S. (2021). Multifaceted Role of the Transforming Growth Factor β on Effector T Cells and the Implication for CAR-T Cell Therapy. Immuno, 1(3), 160-173. https://doi.org/10.3390/immuno1030010