
A Functional GM-CSF Receptor on Dendritic Cells Is Required for Efficient Protective Anti-Tumor Immunity

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Lines
2.3. Preparation of Bone Marrow-Derived Dendritic Cells
2.4. Animal Immunization and Tumor Challenge
2.5. Antibodies
2.6. Flow Cytometric Analysis
2.7. Peripheral Blood Lymphocyte Analysis
2.8. Statistical Analysis
3. Results
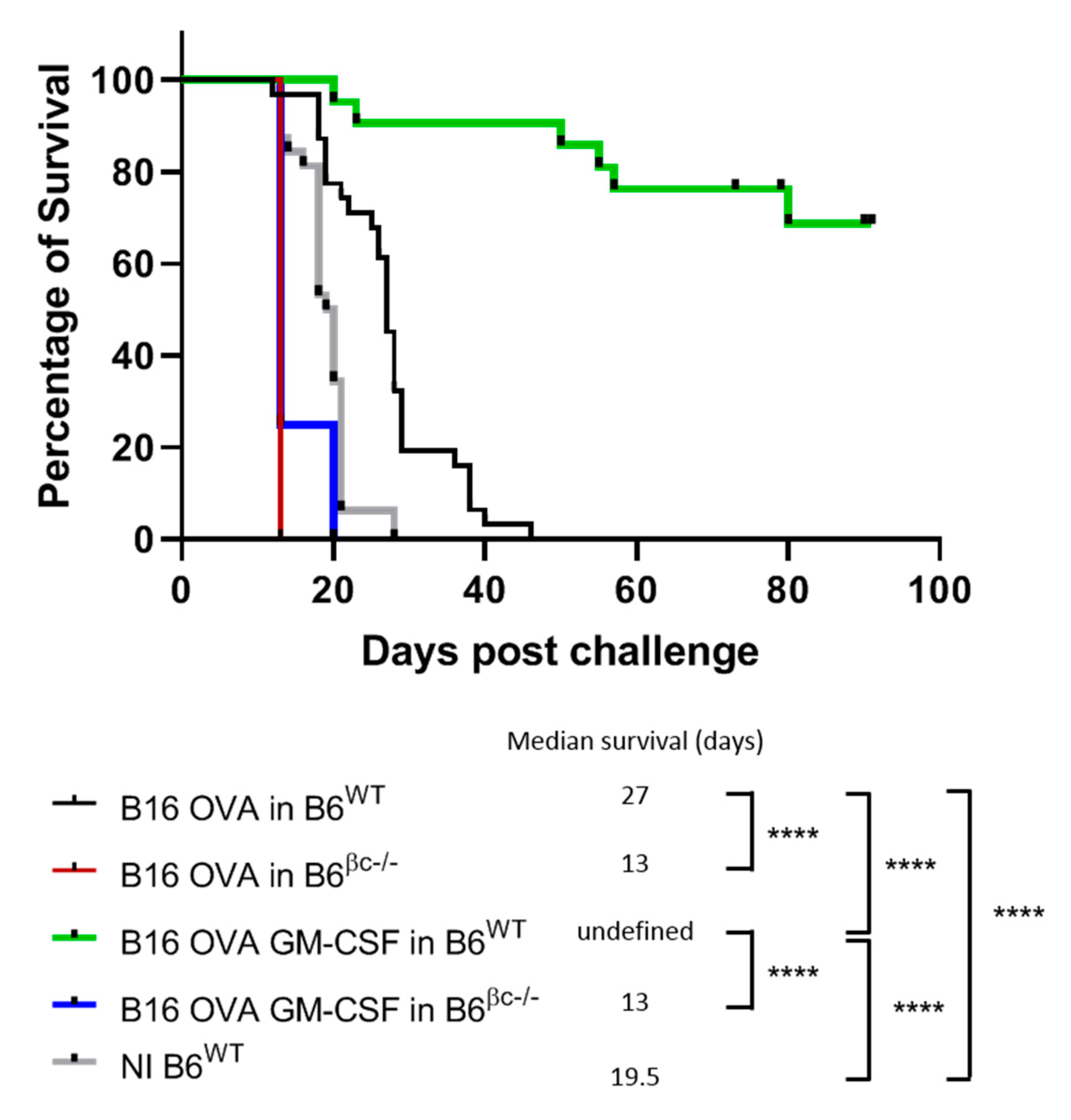
3.1. Mice without a Functional GM-CSF Signaling Do Not Mount Anti-Tumor Immunity
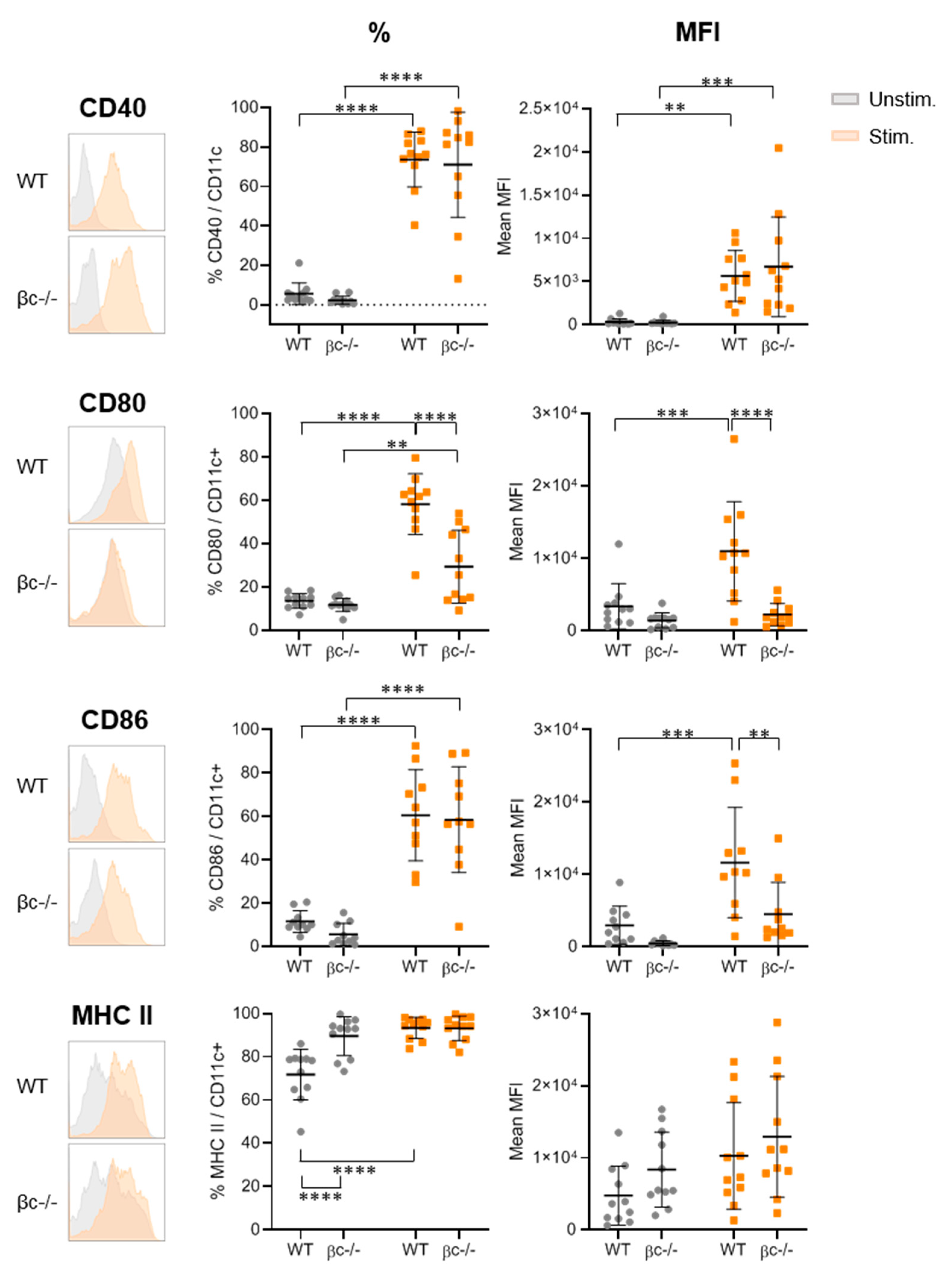
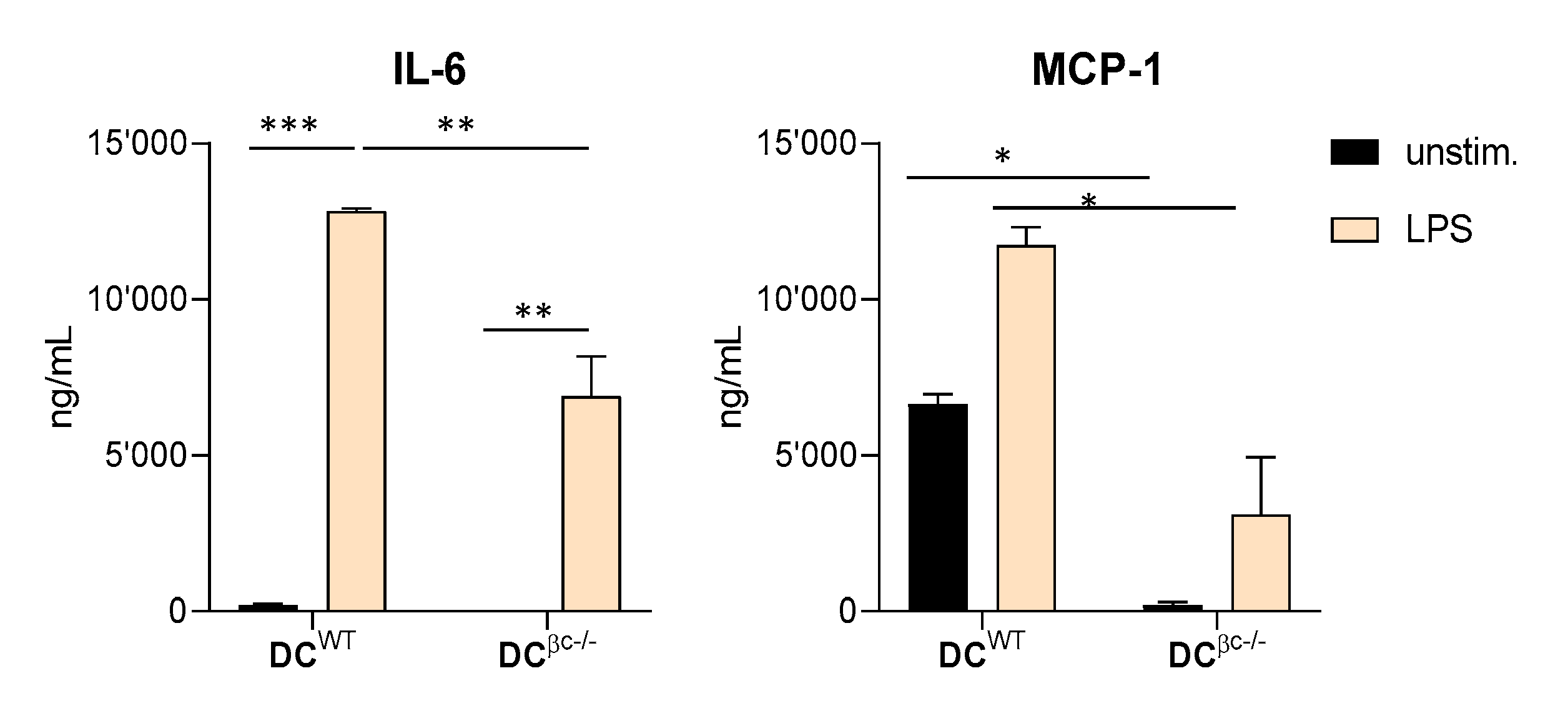
3.2. DCβc-/- Are Not Able to Undergo Full Maturation upon TLR Stimulation
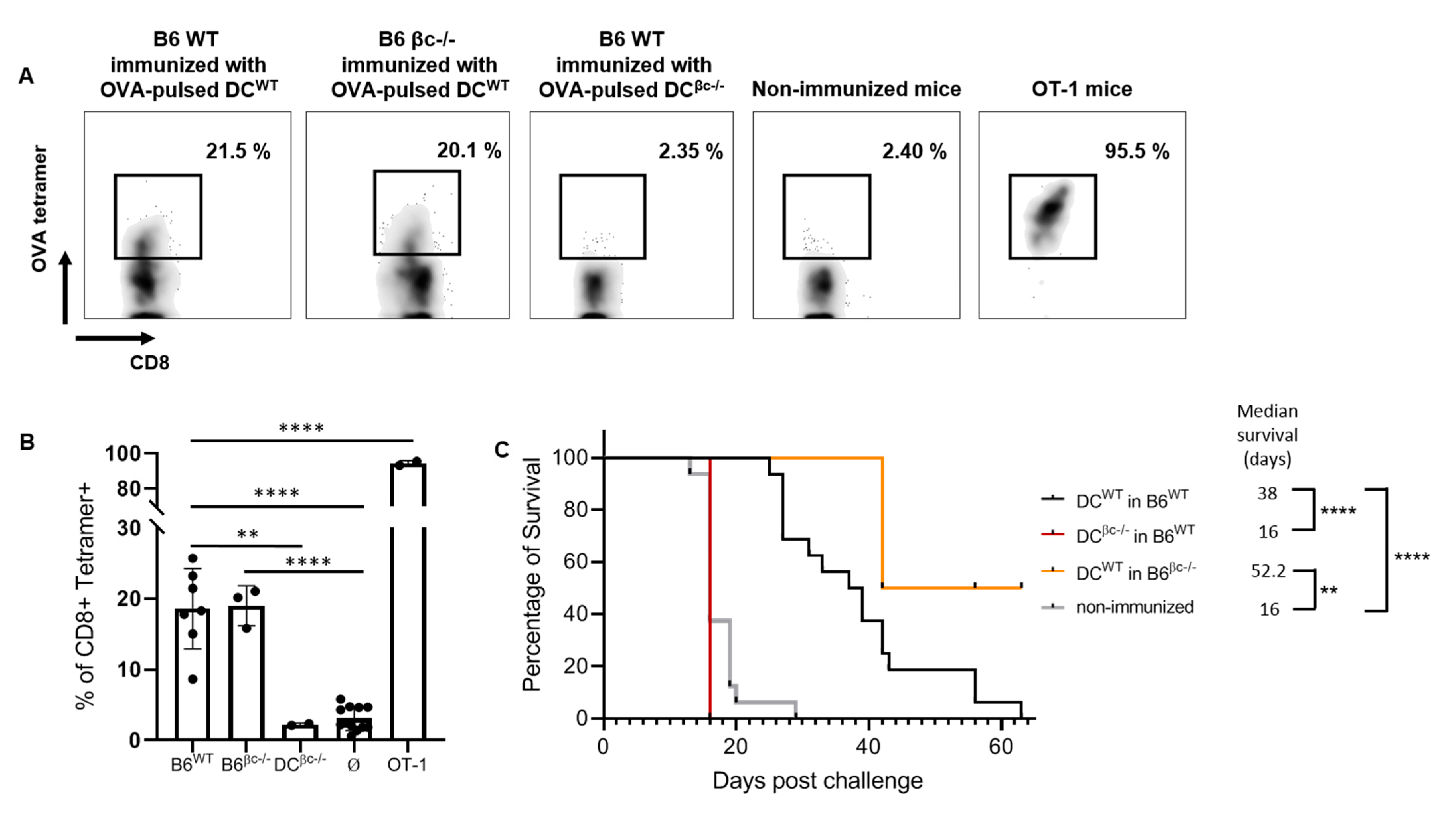
3.3. Immunization with DCβc-/- Is Unable to Mount a Specific Immune Response or to Prevent Tumor Growth
3.4. OVA-Pulsed DCWT Are Able to Rescue the Lack of a Specific Anti-Tumoral Immune Response
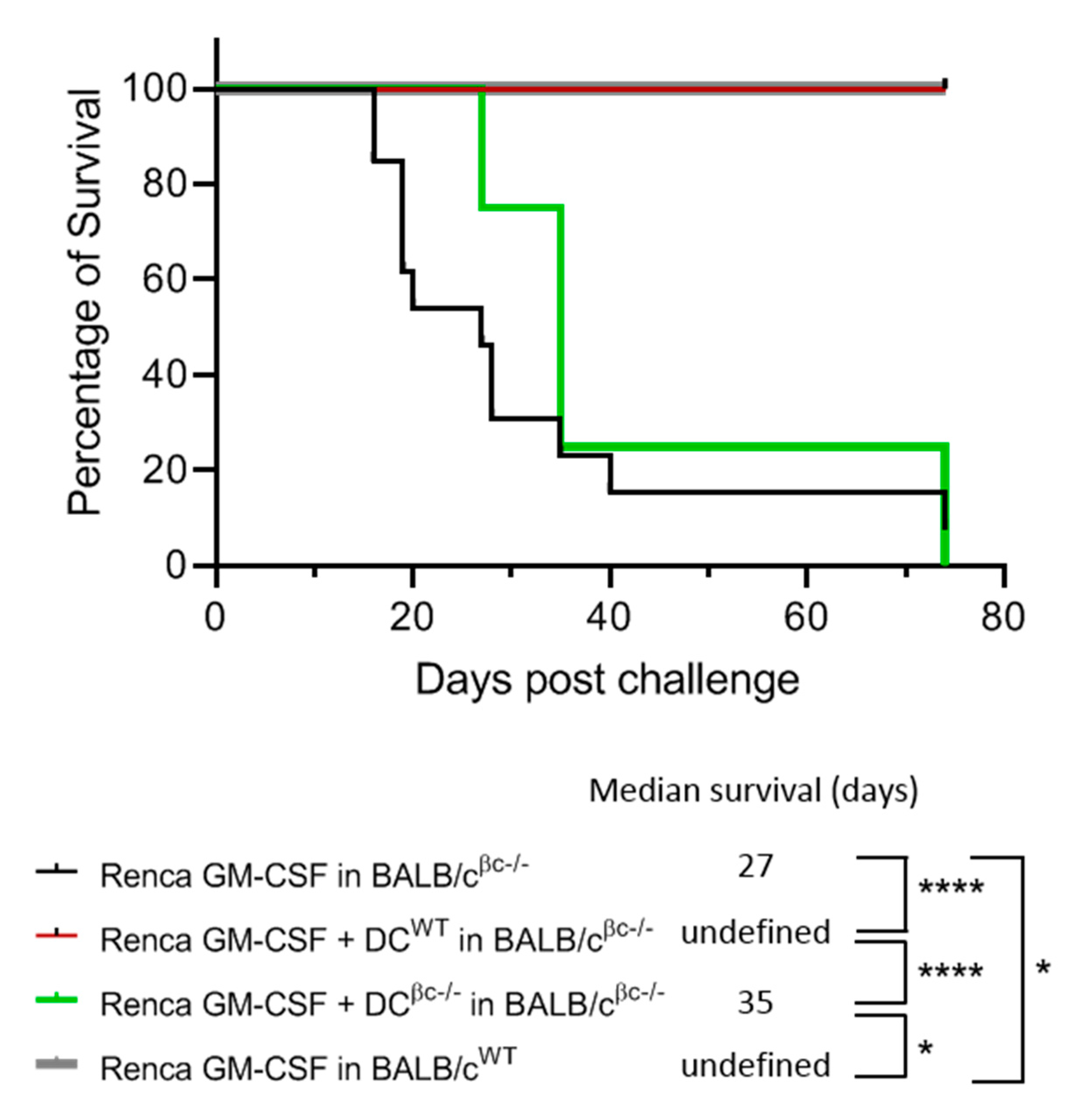
3.5. Immunization of BALB/c βc-/- Mice with a Combination of DCWT and Renca-GM-CSF Cells Restores the Protective Anti-Tumoral Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic Cell-Based Immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Martinek, J.; Wu, T.-C.; Cadena, D.; Banchereau, J.; Palucka, K. Interplay between dendritic cells and cancer cells. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 348, pp. 179–215. ISBN 978-0-12-818351-9. [Google Scholar]
- Lin, C.-C.; Wang, T.-E.; Liu, C.-Y.; Lin, C.-P.; Liu, T.-P.; Chen, M.-J.; Chang, W.-H.; Lin, J.-C.; Chang, K.-M.; Chu, C.-H.; et al. Potentiation of the Immunotherapeutic Effect of Autologous Dendritic Cells by Pretreating Hepatocellular Carcinoma with Low-Dose Radiation. Clin. Investig. Med. 2008, 31, E150–E159. [Google Scholar] [CrossRef]
- Wang, Q.-T.; Nie, Y.; Sun, S.-N.; Lin, T.; Han, R.-J.; Jiang, J.; Li, Z.; Li, J.-Q.; Xiao, Y.-P.; Fan, Y.-Y.; et al. Tumor-Associated Antigen-Based Personalized Dendritic Cell Vaccine in Solid Tumor Patients. Cancer Immunol. Immunother. 2020, 69, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Dhodapkar, K.M.; Palucka, A.K. Interactions of Tumor Cells with Dendritic Cells: Balancing Immunity and Tolerance. Cell Death Differ. 2008, 15, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking Dendritic Cells into Medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Cranmer, L.D.; Trevor, K.T.; Hersh, E.M. Clinical Applications of Dendritic Cell Vaccination in the Treatment of Cancer. Cancer Immunol. Immunother. 2004, 53, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. GM-CSF-Based Cancer Vaccines. Immunol. Rev. 2002, 188, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. GM-CSF-Secreting Melanoma Vaccines. Oncogene 2003, 22, 3188–3192. [Google Scholar] [CrossRef]
- Curry, W.T.; Gorrepati, R.; Piesche, M.; Sasada, T.; Agarwalla, P.; Jones, P.S.; Gerstner, E.R.; Golby, A.J.; Batchelor, T.T.; Wen, P.Y.; et al. Vaccination with Irradiated Autologous Tumor Cells Mixed with Irradiated GM-K562 Cells Stimulates Antitumor Immunity and T Lymphocyte Activation in Patients with Recurrent Malignant Glioma. Clin. Cancer Res. 2016, 22, 2885–2896. [Google Scholar] [CrossRef]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with Irradiated Tumor Cells Engineered to Secrete Murine Granulocyte-Macrophage Colony-Stimulating Factor Stimulates Potent, Specific, and Long-Lasting Anti-Tumor Immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Wei, X.X.; Chan, S.; Kwek, S.; Lewis, J.; Dao, V.; Zhang, L.; Cooperberg, M.R.; Ryan, C.J.; Lin, A.M.; Friedlander, T.W.; et al. Systemic GM-CSF Recruits Effector T Cells into the Tumor Microenvironment in Localized Prostate Cancer. Cancer Immunol. Res. 2016, 4, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Caux, C.; Vanbervliet, B.; Massacrier, C.; Dezutter-Dambuyant, C.; de Saint-Vis, B.; Jacquet, C.; Yoneda, K.; Imamura, S.; Schmitt, D.; Banchereau, J. CD34+ Hematopoietic Progenitors from Human Cord Blood Differentiate along Two Independent Dendritic Cell Pathways in Response to GM-CSF+TNF Alpha. J. Exp. Med. 1996, 184, 695–706. [Google Scholar] [CrossRef]
- Hopewell, E.L.; Cox, C. Manufacturing Dendritic Cells for Immunotherapy: Monocyte Enrichment. Mol. Ther. Methods Clin. Dev. 2020, 16, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Daro, E.; Pulendran, B.; Brasel, K.; Teepe, M.; Pettit, D.; Lynch, D.H.; Vremec, D.; Robb, L.; Shortman, K.; McKenna, H.J.; et al. Polyethylene Glycol-Modified GM-CSF Expands CD11b(High)CD11c(High) but NotCD11b(Low)CD11c(High) Murine Dendritic Cells in Vivo: A Comparative Analysis with Flt3 Ligand. J. Immunol. 2000, 165, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Vremec, D.; Lieschke, G.J.; Dunn, A.R.; Robb, L.; Metcalf, D.; Shortman, K. The Influence of Granulocyte/Macrophage Colony-Stimulating Factor on Dendritic Cell Levels in Mouse Lymphoid Organs. Eur. J. Immunol. 1997, 27, 40–44. [Google Scholar] [CrossRef]
- Shen, Z.; Reznikoff, G.; Dranoff, G.; Rock, K.L. Cloned Dendritic Cells Can Present Exogenous Antigens on Both MHC Class I and Class II Molecules. J. Immunol. 1997, 158, 2723–2730. [Google Scholar]
- Zou, G.M.; Tam, Y.K. Cytokines in the Generation and Maturation of Dendritic Cells: Recent Advances. Eur. Cytokine Netw. 2002, 13, 186–199. [Google Scholar]
- Hong, I.-S. Stimulatory versus Suppressive Effects of GM-CSF on Tumor Progression in Multiple Cancer Types. Exp. Mol. Med. 2016, 48, e242. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Schwenter, F.; Luy, P.; Aurrand-Lions, M.; Morel, P.; Kopf, M.; Dranoff, G.; Mach, N. Role of GM-CSF Signaling in Cell-Based Tumor Immunization. Blood 2009, 113, 6658–6668. [Google Scholar] [CrossRef]
- Robb, L.; Drinkwater, C.C.; Metcalf, D.; Li, R.; Köntgen, F.; Nicola, N.A.; Begley, C.G. Hematopoietic and Lung Abnormalities in Mice with a Null Mutation of the Common Beta Subunit of the Receptors for Granulocyte-Macrophage Colony-Stimulating Factor and Interleukins 3 and 5. Proc. Natl. Acad. Sci. USA 1995, 92, 9565–9569. [Google Scholar] [CrossRef]
- Nicola, N.A.; Robb, L.; Metcalf, D.; Cary, D.; Drinkwater, C.C.; Begley, C.G. Functional Inactivation in Mice of the Gene for the Interleukin-3 (IL- 3)-Specific Receptor Beta-Chain: Implications for IL-3 Function and the Mechanism of Receptor Transmodulation in Hematopoietic Cells. Blood 1996, 87, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Gorski, S.A.; Lawrence, M.G.; Hinkelman, A.; Spano, M.M.; Steinke, J.W.; Borish, L.; Teague, W.G.; Braciale, T.J. Expression of IL-5 Receptor Alpha by Murine and Human Lung Neutrophils. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Gillessen, S.; Wilson, S.B.; Sheehan, C.; Mihm, M.; Dranoff, G. Differences in Dendritic Cells Stimulated in Vivo by Tumors Engineered to Secrete Granulocyte-Macrophage Colony-Stimulating Factor or Flt3-Ligand. Cancer Res. 2000, 60, 3239–3246. [Google Scholar]
- Inaba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of Large Numbers of Dendritic Cells from Mouse Bone Marrow Cultures Supplemented with Granulocyte/Macrophage Colony-Stimulating Factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar] [CrossRef]
- Bogunovic, M.; Ginhoux, F.; Helft, J.; Shang, L.; Hashimoto, D.; Greter, M.; Liu, K.; Jakubzick, C.; Ingersoll, M.A.; Leboeuf, M.; et al. Origin of the Lamina Propria Dendritic Cell Network. Immunity 2009, 31, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Helft, J.; Chow, A.; Hashimoto, D.; Mortha, A.; Agudo-Cantero, J.; Bogunovic, M.; Gautier, E.L.; Miller, J.; Leboeuf, M.; et al. GM-CSF Controls Nonlymphoid Tissue Dendritic Cell Homeostasis but Is Dispensable for the Differentiation of Inflammatory Dendritic Cells. Immunity 2012, 36, 1031–1046. [Google Scholar] [CrossRef]
- King, I.L.; Kroenke, M.A.; Segal, B.M. GM-CSF-Dependent, CD103+ Dermal Dendritic Cells Play a Critical Role in Th Effector Cell Differentiation after Subcutaneous Immunization. J. Exp. Med. 2010, 207, 953–961. [Google Scholar] [CrossRef]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of Dendritic Cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- De Vries, I.J.M.; Krooshoop, D.J.E.B.; Scharenborg, N.M.; Lesterhuis, W.J.; Diepstra, J.H.S.; Van Muijen, G.N.P.; Strijk, S.P.; Ruers, T.J.; Boerman, O.C.; Oyen, W.J.G.; et al. Effective Migration of Antigen-Pulsed Dendritic Cells to Lymph Nodes in Melanoma Patients Is Determined by Their Maturation State. Cancer Res. 2003, 63, 12–17. [Google Scholar] [PubMed]
- Dhodapkar, M.V.; Steinman, R.M.; Krasovsky, J.; Munz, C.; Bhardwaj, N. Antigen-Specific Inhibition of Effector T Cell Function in Humans after Injection of Immature Dendritic Cells. J. Exp. Med. 2001, 193, 233–238. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Steinman, R.M. Antigen-Bearing Immature Dendritic Cells Induce Peptide-Specific CD8(+) Regulatory T Cells in Vivo in Humans. Blood 2002, 100, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor Cells Convert Immature Myeloid Dendritic Cells into TGF-Beta-Secreting Cells Inducing CD4+CD25+ Regulatory T Cell Proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef]
- Kanada, S.; Nishiyama, C.; Nakano, N.; Suzuki, R.; Maeda, K.; Hara, M.; Kitamura, N.; Ogawa, H.; Okumura, K. Critical Role of Transcription Factor PU.1 in the Expression of CD80 and CD86 on Dendritic Cells. Blood 2011, 117, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Hathcock, K.S.; Laszlo, G.; Pucillo, C.; Linsley, P.; Hodes, R.J. Comparative Analysis of B7-1 and B7-2 Costimulatory Ligands: Expression and Function. J. Exp. Med. 1994, 180, 631–640. [Google Scholar] [CrossRef]
- Nabavi, N.; Freeman, G.J.; Gault, A.; Godfrey, D.; Nadler, L.M.; Glimcher, L.H. Signalling through the MHC Class II Cytoplasmic Domain Is Required for Antigen Presentation and Induces B7 Expression. Nature 1992, 360, 266–268. [Google Scholar] [CrossRef]
- Tan, J.K.H.; O’Neill, H.C. Maturation Requirements for Dendritic Cells in T Cell Stimulation Leading to Tolerance versus Immunity. J. Leukoc. Biol. 2005, 78, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Sathe, P.; Pooley, J.; Vremec, D.; Mintern, J.; Jin, J.-O.; Wu, L.; Kwak, J.-Y.; Villadangos, J.A.; Shortman, K. The Acquisition of Antigen Cross-Presentation Function by Newly Formed Dendritic Cells. J. Immunol. 2011, 186, 5184–5192. [Google Scholar] [CrossRef] [PubMed]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 Deficiency Reveals a Critical Role for CD8alpha+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Labeur, M.S.; Roters, B.; Pers, B.; Mehling, A.; Luger, T.A.; Schwarz, T.; Grabbe, S. Generation of Tumor Immunity by Bone Marrow-Derived Dendritic Cells Correlates with Dendritic Cell Maturation Stage. J. Immunol. 1999, 162, 168–175. [Google Scholar]
- Baldin, A.V.; Savvateeva, L.V.; Bazhin, A.V.; Zamyatnin, A.A. Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting. Cancers 2020, 12, 590. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Wu, S.; Ciernik, I.F.; Chen, H.; Nadaf-Rahrov, S.; Gabrilovich, D.; Carbone, D.P. Genetic Immunotherapy of Established Tumors with Adenovirus-Murine Granulocyte-Macrophage Colony-Stimulating Factor. Hum. Gene Ther. 1997, 8, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Hodi, F.S.; Dranoff, G. Enhancing the Clinical Activity of Granulocyte-Macrophage Colony-Stimulating Factor-Secreting Tumor Cell Vaccines. Immunol. Rev. 2008, 222, 287–298. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charrier, E.; Vernet, R.; Schwenter, F.; Luy, P.; Donda, A.; Mach, N. A Functional GM-CSF Receptor on Dendritic Cells Is Required for Efficient Protective Anti-Tumor Immunity. Immuno 2021, 1, 240-252. https://doi.org/10.3390/immuno1030016
Charrier E, Vernet R, Schwenter F, Luy P, Donda A, Mach N. A Functional GM-CSF Receptor on Dendritic Cells Is Required for Efficient Protective Anti-Tumor Immunity. Immuno. 2021; 1(3):240-252. https://doi.org/10.3390/immuno1030016
Chicago/Turabian StyleCharrier, Emily, Rémi Vernet, Frank Schwenter, Patricia Luy, Alena Donda, and Nicolas Mach. 2021. "A Functional GM-CSF Receptor on Dendritic Cells Is Required for Efficient Protective Anti-Tumor Immunity" Immuno 1, no. 3: 240-252. https://doi.org/10.3390/immuno1030016
APA StyleCharrier, E., Vernet, R., Schwenter, F., Luy, P., Donda, A., & Mach, N. (2021). A Functional GM-CSF Receptor on Dendritic Cells Is Required for Efficient Protective Anti-Tumor Immunity. Immuno, 1(3), 240-252. https://doi.org/10.3390/immuno1030016