
The “6B” Strategy: Build Back a Better Blood–Brain Barrier


{kind=link}
Abstract
:1. Introduction
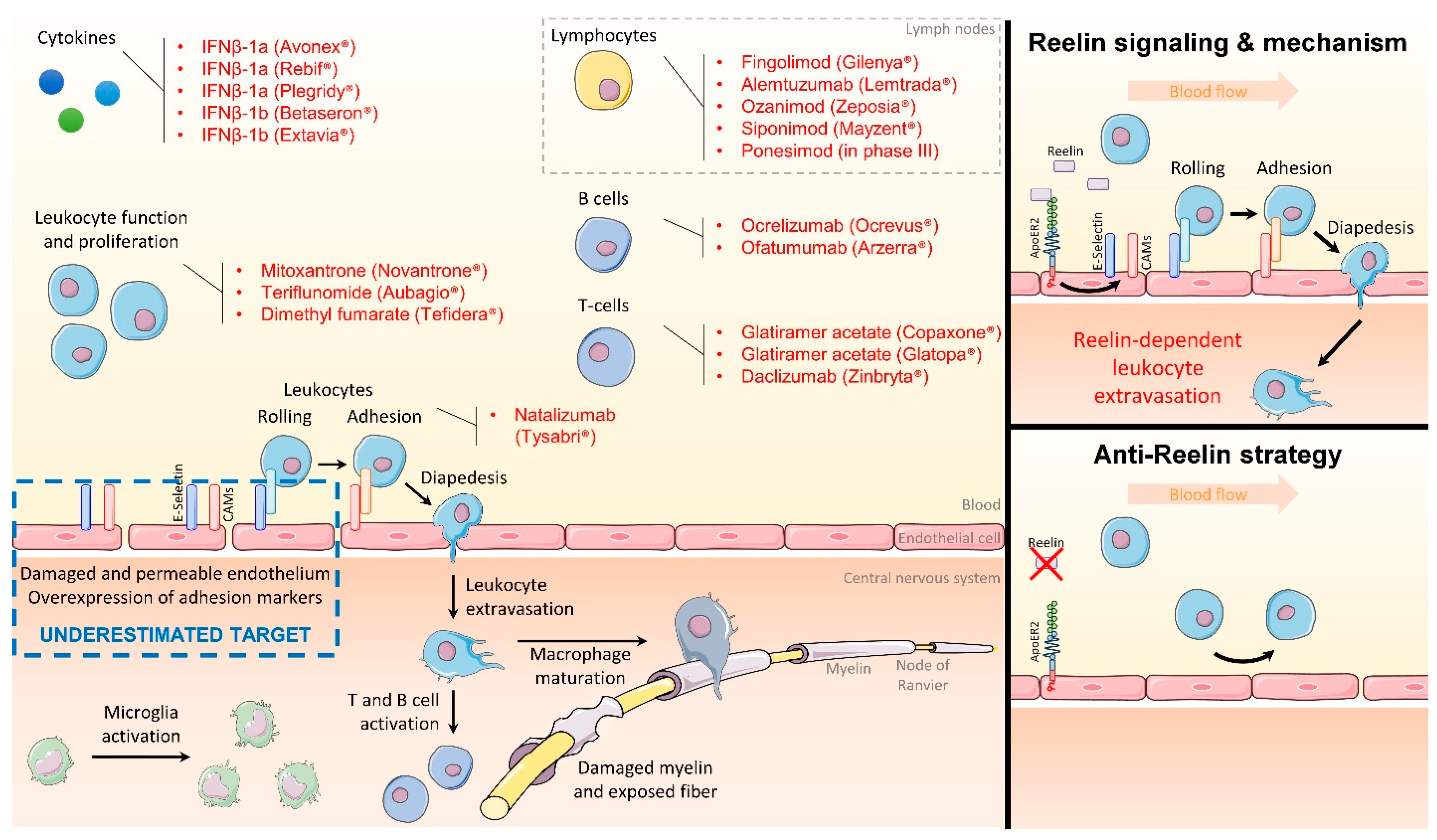
2. Targeting the Endothelial Function to Block Inflammation
3. Reelin Inhibition Protects the Endothelial Function and Presents Clinical Potential
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M.V. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, E.N. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J. Mol. Med. 1997, 75, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Sminia, T.; Wouterlood, F.G.; Dijkstra, C.D. Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J. Neurosci. Res. 1994, 38, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Manouchehri, N.; Hussain, R.Z.; Cravens, P.D.; Esaulova, E.; Artyomov, M.N.; Edelson, B.T.; Wu, G.F.; Cross, A.H.; Doelger, R.; Loof, N.; et al. CD11c+CD88+CD317+ myeloid cells are critical mediators of persistent CNS autoimmunity. Proc. Natl. Acad. Sci. USA 2021, 118, e2014492118. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Hafler, D.A.; Lucchinetti, C.F. Multiple sclerosis-a quiet revolution. Nat. Rev. Neurol. 2015, 11, 134–142. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Atarashi, K.; Hirata, T.; Matsumoto, M.; Kanemitsu, N.; Miyasaka, M. Rolling of Th1 cells via P-selectin glycoprotein ligand-1 stimulates LFA-1-mediated cell binding to ICAM-1. J. Immunol. 2005, 174, 1424–1432. [Google Scholar] [CrossRef]
- Feinstein, A.; Freeman, J.; Lo, A.C. Treatment of progressive multiple sclerosis: What works, what does not, and what is needed. Lancet Neurol. 2015, 14, 194–207. [Google Scholar] [CrossRef]
- Ontaneda, D.; Fox, R.J. Progressive multiple sclerosis. Curr. Opin. Neurol. 2015, 28, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Rommer, P.S.; Milo, R.; Han, M.H.; Satyanarayan, S.; Sellner, J.; Hauer, L.; Illes, Z.; Warnke, C.; Laurent, S.; Weber, M.S.; et al. Immunological Aspects of Approved MS Therapeutics. Front. Immunol. 2019, 10, 1564. [Google Scholar] [CrossRef] [PubMed]
- Callegari, I.; Derfuss, T.; Galli, E. Update on treatment in multiple sclerosis. La Presse Médicale 2021, 50, 104068. [Google Scholar] [CrossRef] [PubMed]
- Stüve, O.; Marra, C.M.; Jerome, K.R.; Cook, L.; Cravens, P.D.; Cepok, S.; Frohman, E.M.; Theodore Phillips, J.; Arendt, G.; Hemmer, B.; et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann. Neurol. 2006, 59, 743–747. [Google Scholar] [CrossRef]
- Stüve, O.; Marra, C.M.; Bar-Or, A.; Niino, M.; Cravens, P.D.; Cepok, S.; Frohman, E.M.; Theodore Phillips, J.; Arendt, G.; Jerome, K.R.; et al. Altered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch. Neurol. 2006, 63, 1383–1387. [Google Scholar] [CrossRef]
- Cutter, G.R.; Stüve, O. Does risk stratification decrease the risk of natalizumab-associated PML? Where is the evidence? Mult. Scler. 2014, 20, 1304–1305. [Google Scholar] [CrossRef]
- Thompson, A.J. A much-needed focus on progression in multiple sclerosis. Lancet Neurol. 2015, 14, 133–135. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef]
- Spencer, J.I.; Bell, J.S.; DeLuca, G.C. Vascular pathology in multiple sclerosis: Reframing pathogenesis around the blood-brain barrier. J. Neurol. Neurosurg. Psychiatry 2018, 89, 42–52. [Google Scholar] [CrossRef]
- Schnoor, M.; Alcaide, P.; Voisin, M.-B.; van Buul, J.D. Crossing the Vascular Wall: Common and Unique Mechanisms Exploited by Different Leukocyte Subsets during Extravasation. Mediat. Inflamm. 2015, 2015, 946509. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Y.; Shen, Q.; Rigor, R.R.; Wu, M.H. Neutrophil transmigration, focal adhesion kinase and endothelial barrier function. Microvasc. Res. 2012, 83, 82–88. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; Nakajima, K.; Miyata, T.; Ogawa, M.; Mikoshiba, K.; Curran, T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J. Neurosci. 1997, 17, 23–31. [Google Scholar] [CrossRef]
- Herz, J.; Chen, Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 2006, 7, 850–859. [Google Scholar] [CrossRef]
- Tissir, F.; Goffinet, A.M. Reelin and brain development. Nat. Rev. Neurosci. 2003, 4, 496–505. [Google Scholar] [CrossRef]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; Hammer, R.E.; Richardson, J.A.; Herz, J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef]
- Marckx, A.T.; Fritschle, K.E.; Calvier, L.; Herz, J. Reelin changes hippocampal learning in aging and Alzheimer’s disease. Behav. Brain Res. 2021, 414, 113482. [Google Scholar] [CrossRef]
- Madonna, R.; De Caterina, R. Relevance of new drug discovery to reduce NF-κB activation in cardiovascular disease. Vascul. Pharmacol. 2012, 57, 41–47. [Google Scholar] [CrossRef]
- Ding, Y.; Huang, L.; Xian, X.; Yuhanna, I.S.; Wasser, C.R.; Frotscher, M.; Mineo, C.; Shaul, P.W.; Herz, J. Loss of Reelin protects against atherosclerosis by reducing leukocyte-endothelial cell adhesion and lesion macrophage accumulation. Sci. Signal. 2016, 9, ra29. [Google Scholar] [CrossRef] [Green Version]
- Calvier, L.; Xian, X.; Lee, R.G.; Sacharidou, A.; Mineo, C.; Shaul, P.W.; Kounnas, M.Z.; Tsai, S.; Herz, J. Reelin Depletion Protects Against Atherosclerosis by Decreasing Vascular Adhesion of Leukocytes. Arter. Thromb. Vasc. Biol. 2021, 41, 1309–1318. [Google Scholar] [CrossRef]
- Calvier, L.; Demuth, G.; Manouchehri, N.; Wong, C.; Sacharidou, A.; Mineo, C.; Shaul, P.W.; Monson, N.L.; Kounnas, M.Z.; Stüve, O.; et al. Reelin depletion protects against autoimmune encephalomyelitis by decreasing vascular adhesion of leukocytes. Sci. Transl. Med. 2020, 12, eaay7675. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Herz, J.; Hansmann, G. Interplay of Low-Density Lipoprotein Receptors, LRPs, and Lipoproteins in Pulmonary Hypertension. JACC Basic Transl. Sci. 2022, 7, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Manouchehri, N.; Sacharidou, A.; Mineo, C.; Shaul, P.W.; Hui, D.Y.; Kounnas, M.Z.; Stüve, O.; Herz, J. Apolipoprotein E receptor 2 deficiency decreases endothelial adhesion of monocytes and protects against autoimmune encephalomyelitis. Sci. Immunol. 2021, 6, eabd0931. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Genome-wide association study of severity in multiple sclerosis. Genes Immun. 2011, 12, 615–625. [Google Scholar] [CrossRef]
- Baranzini, S.E.; Wang, J.; Gibson, R.A.; Galwey, N.; Naegelin, Y.; Barkhof, F.; Radue, E.-W.; Lindberg, R.L.; Uitdehaag, B.M.; Johnson, M.R.; et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet. 2009, 18, 767–778. [Google Scholar] [CrossRef]
- Carotti, S.; Perrone, G.; Amato, M.; Gentilucci, U.V.; Righi, D.; Francesconi, M.; Pellegrini, C.; Zalfa, F.; Zingariello, M.; Picardi, A.; et al. Reelin expression in human liver of patients with chronic hepatitis C infection. Eur. J. Histochem. 2017, 61, 2745. [Google Scholar] [CrossRef]
- Smalheiser, N.R.; Costa, E.; Guidotti, A.; Impagnatiello, F.; Auta, J.; Lacor, P.; Kriho, V.; Pappas, G.D. Expression of reelin in adult mammalian blood, liver, pituitary pars intermedia, and adrenal chromaffin cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1281–1286. [Google Scholar] [CrossRef]
- Samama, B.; Boehm, N. Reelin immunoreactivity in lymphatics and liver during development and adult life. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2005, 285, 595–599. [Google Scholar] [CrossRef]
- Botella-López, A.; De-Madaria, E.; Jover, R.; Bataller, R.; Sancho-Bru, P.; Candela, A.; Compañ, A.; Pérez-Mateo, M.; Martinez, S.; Sáez-Valero, J. Reelin is overexpressed in the liver and plasma of bile duct ligated rats and its levels and glycosylation are altered in plasma of humans with cirrhosis. Int. J. Biochem. Cell Biol. 2008, 40, 766–775. [Google Scholar] [CrossRef]
- Lutter, S.; Xie, S.; Tatin, F.; Makinen, T. Smooth muscle-endothelial cell communication activates Reelin signaling and regulates lymphatic vessel formation. J. Cell Biol. 2012, 197, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Boucher, P.; Herz, J.; Hansmann, G. LRP1 Deficiency in Vascular SMC Leads to Pulmonary Arterial Hypertension That Is Reversed by PPARγ Activation. Circ. Res. 2019, 124, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Storck, S.E.; Kurtyka, M.; Pietrzik, C.U. Brain endothelial LRP1 maintains blood–brain barrier integrity. Fluids Barriers CNS 2021, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sagheddu, C.; Melis, M.; Muntoni, A.L.; Pistis, M. Repurposing Peroxisome Proliferator-Activated Receptor Agonists in Neurological and Psychiatric Disorders. Pharmaceuticals 2021, 14, 1025. [Google Scholar] [CrossRef]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hansmann, G. Transforming Growth Factor β1– and Bone Morphogenetic Protein 2/PPARγ–regulated MicroRNAs in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2017, 196, 1227–1228. [Google Scholar] [CrossRef]
- Kökény, G.; Calvier, L.; Legchenko, E.; Chouvarine, P.; Mózes, M.M.; Hansmann, G. PPARγ is a gatekeeper for extracellular matrix and vascular cell homeostasis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 171–179. [Google Scholar] [CrossRef]
- Hansmann, G.; Calvier, L.; Risbano, M.G.; Chan, S.Y. Activation of the Metabolic Master Regulator PPARγ: A Potential PIOneering Therapy for Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 62, 143–156. [Google Scholar] [CrossRef]
- Németh, Á.; Mózes, M.M.; Calvier, L.; Hansmann, G.; Kökény, G. The PPARγ agonist pioglitazone prevents TGF-β induced renal fibrosis by repressing EGR-1 and STAT3. BMC Nephrol. 2019, 20, 1–9. [Google Scholar] [CrossRef]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hoffmann, N.; Geldner, J.; Borchert, P.; Jonigk, D.; Mozes, M.M.; Hansmann, G. PPARγ Links BMP2 and TGFβ1 Pathways in Vascular Smooth Muscle Cells, Regulating Cell Proliferation and Glucose Metabolism. Cell Metab. 2017, 25, 1118–1134.e7. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvier, L.; Alexander, A.E.; Herz, J. The “6B” Strategy: Build Back a Better Blood–Brain Barrier. Immuno 2022, 2, 506-511. https://doi.org/10.3390/immuno2030032
Calvier L, Alexander AE, Herz J. The “6B” Strategy: Build Back a Better Blood–Brain Barrier. Immuno. 2022; 2(3):506-511. https://doi.org/10.3390/immuno2030032
Chicago/Turabian StyleCalvier, Laurent, Anna E. Alexander, and Joachim Herz. 2022. "The “6B” Strategy: Build Back a Better Blood–Brain Barrier" Immuno 2, no. 3: 506-511. https://doi.org/10.3390/immuno2030032
APA StyleCalvier, L., Alexander, A. E., & Herz, J. (2022). The “6B” Strategy: Build Back a Better Blood–Brain Barrier. Immuno, 2(3), 506-511. https://doi.org/10.3390/immuno2030032