
Mimicry of Tumour-Associated Carbohydrates: Is It a Promising Option for Cancer Treatment?

 ,
,
Abstract
:1. Introduction
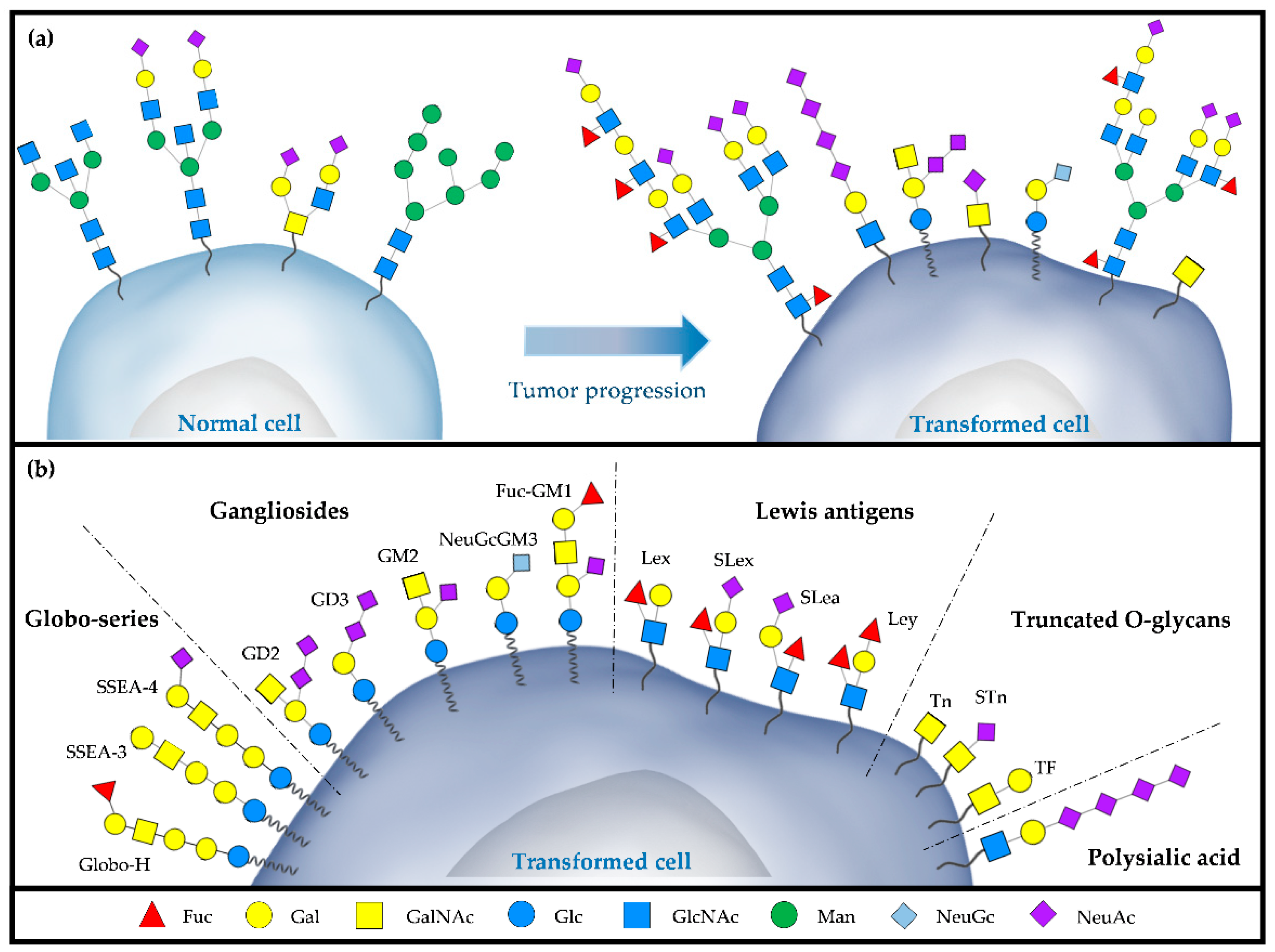
2. TACAs as Cancer Targets
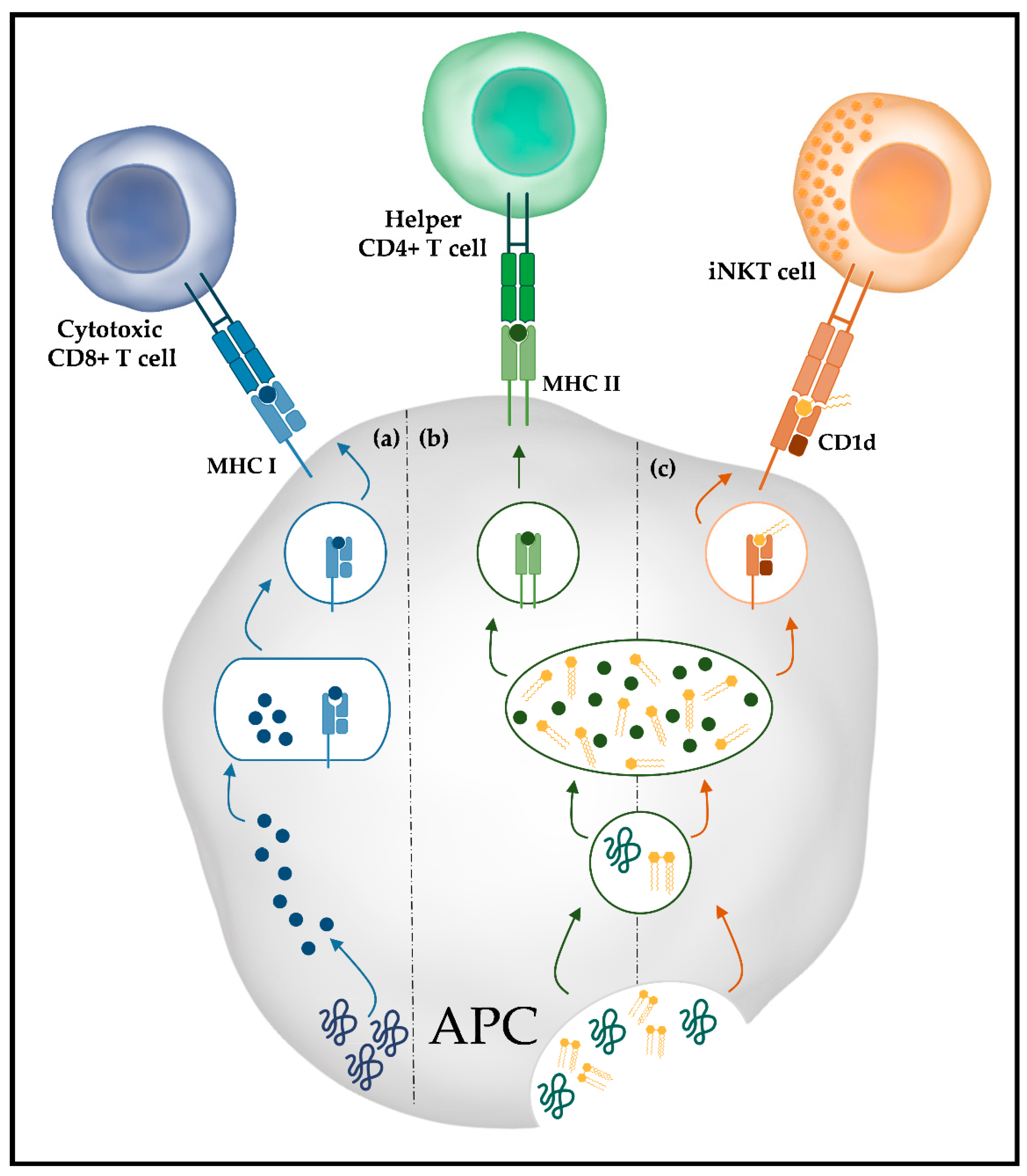
3. Immune Response against Glycans
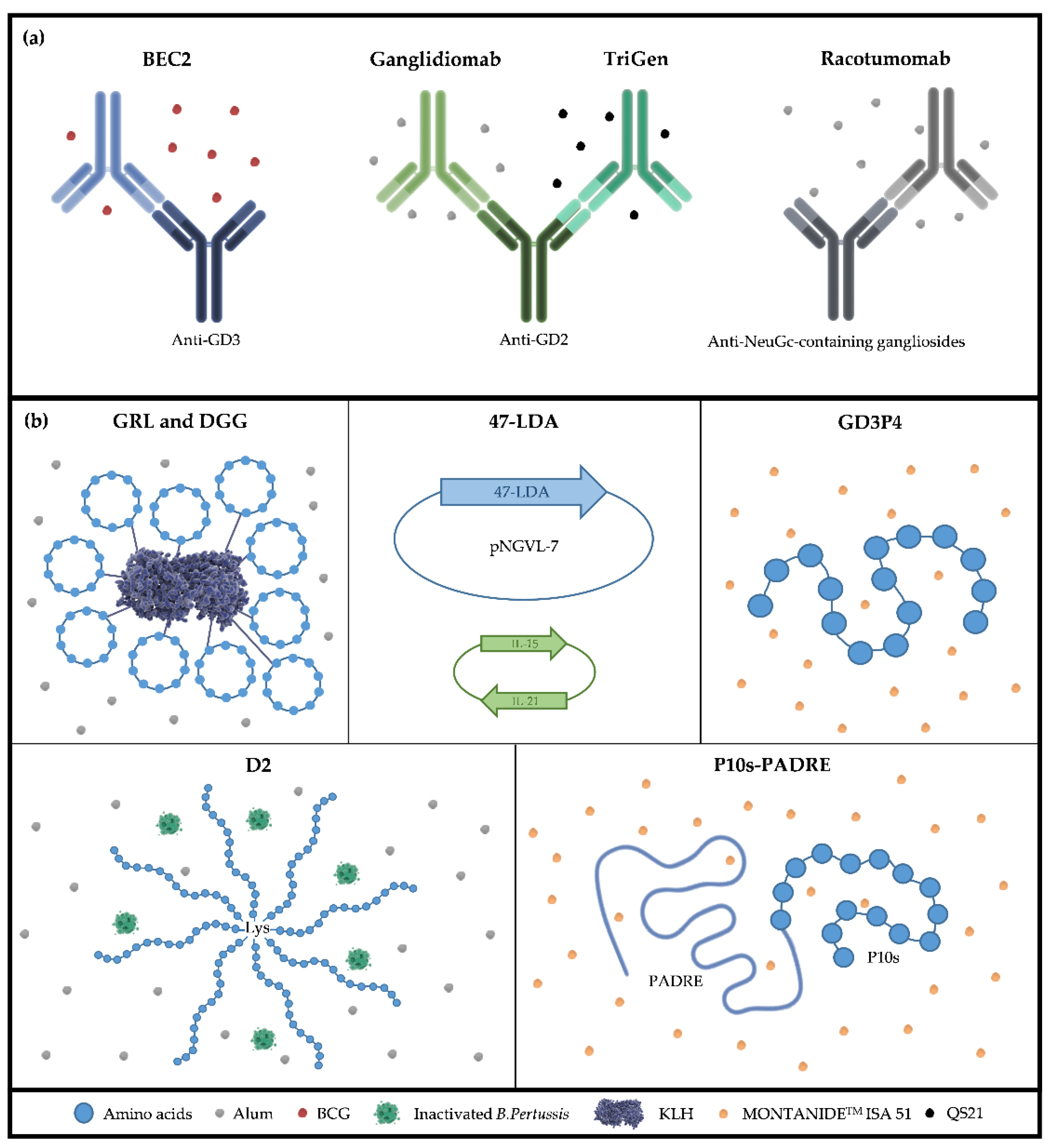
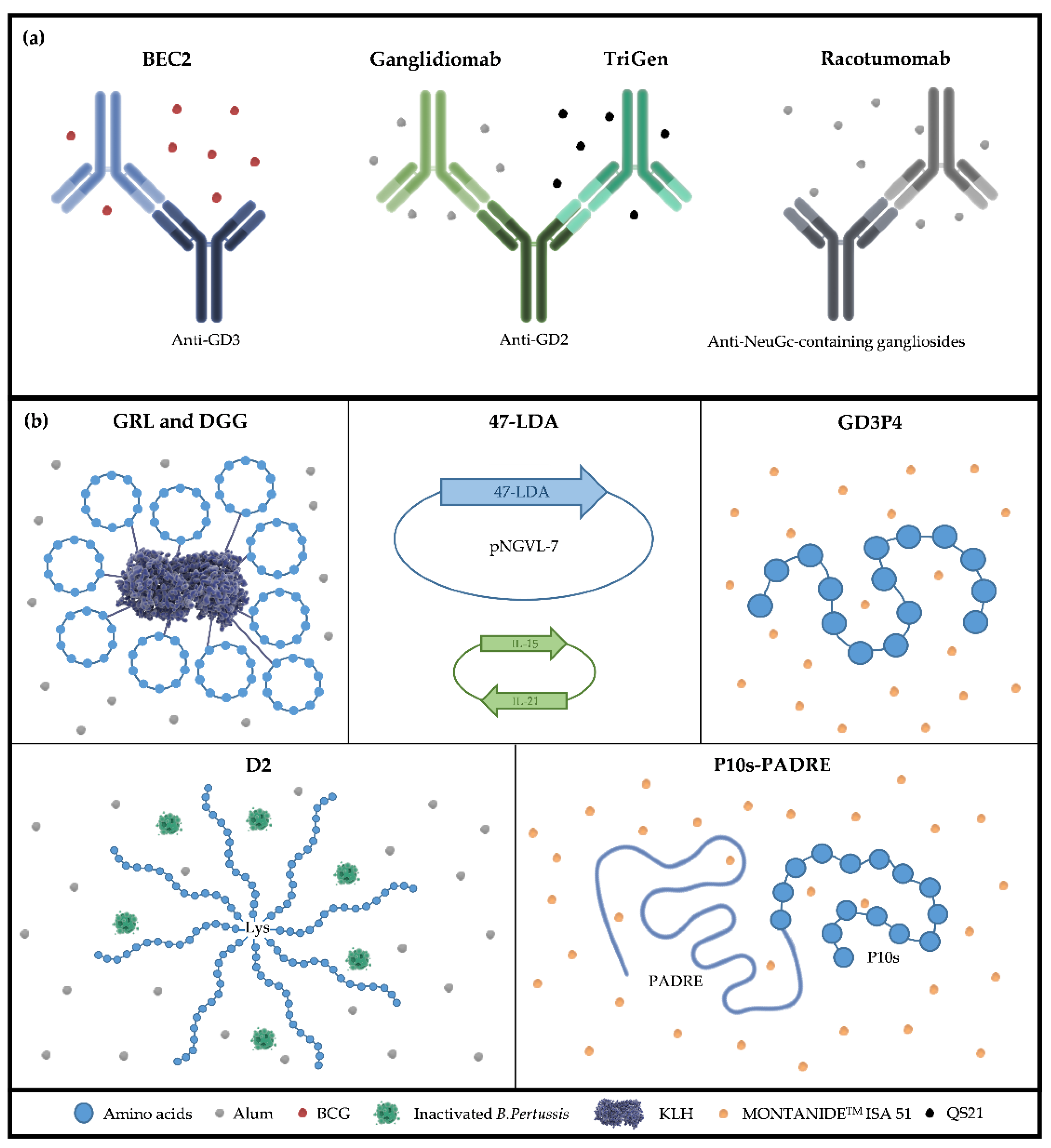
4. TACAs Mimetic Peptides
5. Anti-Idiotype Antibodies as TACA Surrogates
6. Discussion
7. Conclusions
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Peterson, C.; Denlinger, N.; Yang, Y. Recent Advances and Challenges in Cancer Immunotherapy. Cancers 2022, 14, 3972. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Clayton, A.; Colombo, M.P.; Coussens, L.; Dhodapkar, M.V.; Eggermont, A.M.; Fearon, D.T.; Fridman, W.H.; Fučíková, J.; Ghiringhelli, D.I.G.F.; et al. Available online: http://www.impactjournals.com/oncotarget/ (accessed on 30 December 2014).
- Gupta, M.; Wahi, A.; Sharma, P.; Nagpal, R.; Raina, N.; Kaurav, M.; Bhattacharya, J.; Oliveira, S.M.R.; Dolma, K.G.; Paul, A.K.; et al. Recent Advances in Cancer Vaccines: Challenges, Achievements, and Futuristic Prospects. Vaccines 2022, 10, 2011. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: The next immunotherapy frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Baldin, A.V.; Isayev, O.; Werner, J.; Zamyatnin, A.A.; Bazhin, A.V. Cancer vaccines: Antigen selection strategy. Vaccines 2021, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Haen, S.P.; Rammensee, H.G. The repertoire of human tumor-associated epitopes—Identification and selection of antigens and their application in clinical trials. Curr. Opin. Immunol. 2013, 25, 277–283. [Google Scholar] [CrossRef]
- Bright, R.K.; Bright, J.D.; Byrne, J.A. Overexpressed oncogenic tumor-self antigens. Hum. Vaccines Immunother. 2014, 10, 3297–3305. [Google Scholar] [CrossRef] [Green Version]
- Connerotte, T.; Pel, A.V.; Godelaine, D.; Tartour, E.; Schuler-Thurner, B.; Lucas, S.; Thielemans, K.; Schuler, G.; Coulie, P.G. Functions of anti-MAGE T-cells induced in melanoma patients under different vaccination modalities. Cancer Res. 2008, 68, 3931–3940. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Shi, H.; Mu, Y.; Luo, Z.; Zhang, H.; Wan, Y.; Zhang, D.; Lu, L.; Men, K.; Tian, Y.; et al. Effective inhibition of melanoma tumorigenesis and growth via a new complex vaccine based on NY-ESO-1-alum-polysaccharide-HH2. Mol. Cancer 2014, 13, 179. [Google Scholar] [CrossRef] [Green Version]
- Al-Khadairi, G.; Decock, J. Cancer testis antigens and immunotherapy: Where do we stand in the targeting of PRAME? Cancers 2019, 11, 984. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Lutz, E.R.; Laheru, D.A.; Elizabeth, M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2018, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Munkley, J. Aberrant Sialylation in Cancer: Therapeutic Opportunities. Cancers 2022, 14, 4248. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderluh, M.; Berti, F.; Bzducha-Wróbel, A.; Chiodo, F.; Colombo, C.; Compostella, F.; Durlik, K.; Ferhati, X.; Holmdahl, R.; Jovanovic, D.; et al. Recent advances on smart glycoconjugate vaccines in infections and cancer. FEBS J. 2022, 289, 4251–4303. [Google Scholar] [CrossRef]
- Boligan, K.F.; Mesa, C.; Fernandez, L.E.; von Gunten, S. Cancer intelligence acquired (CIA): Tumor glycosylation and sialylation codes dismantling antitumor defense. Cell. Mol. Life Sci. 2015, 72, 1231–1248. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Denys, A.; Delos, M.; Sikora, A.-S.; Carpentier, M.; Julien, S.; Pestel, J.; Allain, F. Macrophage polarization alters the expression and sulfation pattern of glycosaminoglycans. Glycobiology 2015, 25, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Mantuano, N.R.; Natoli, M.; Zippelius, A.; Läubli, H. Tumor-associated carbohydrates and immunomodulatory lectins as targets for cancer immunotherapy. J. ImmunoTherapy Cancer 2020, 8, e001222. [Google Scholar] [CrossRef]
- da Costa Santos, M.A.R.; Dos Reis, J.S.; do Nascimento Santos, C.A.; da Costa, K.M.; Barcelos, P.M.; de Oliveira Francisco, K.Q.; Barbosa, P.A.G.N.; da Silva, E.D.S.; Freire-de-Lima, C.G.; Morrot, A.; et al. Expression of O-glycosylated oncofetal fibronectin in alternatively activated human macrophages. Immunol. Res. 2023, 71, 92–104. [Google Scholar] [CrossRef]
- Stanczak, M.A.; Rodrigues Mantuano, N.; Kirchhammer, N.; Sanin, D.E.; Jacob, F.; Coelho, R.; Everest-Dass, A.V.; Wang, J.; Trefny, M.P.; Monaco, G.; et al. Targeting cancer glycosylation repolarizes tumor-associated macrophages allowing effective immune checkpoint blockade. Sci. Transl. Med. 2022, 14, eabj1270. [Google Scholar] [CrossRef]
- Freeze, H.H.; Chong, J.X.; Bamshad, M.J.; Ng, B.G. Solving glycosylation disorders: Fundamental approaches reveal complicated pathways. Am. J. Hum. Genet. 2014, 94, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astronomo, R.D.; Burton, D.R. Carbohydrate vaccines: Developing sweet solutions to sticky situations? Nat. Rev. Drug Discov. 2010, 9, 308–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Cordon-Cardo, C.; Zhang, H.S.; Reuter, V.E.; Adluri, S.; Hamilton, W.B.; Lloyd, K.O.; Livingston, P.O. Selection of Tumor Antigens as Targets for Immune Attack Using immunohistochemistry: I. focus on Gangliosides. J. Cancer 1997, 73, 42–49. [Google Scholar] [CrossRef]
- Yu, A.L.; Hung, J.T.; Ho, M.Y.; Yu, J. Alterations of Glycosphingolipids in Embryonic Stem Cell Differentiation and Development of Glycan-Targeting Cancer Immunotherapy. Stem Cells Dev. 2016, 25, 1532–1548. [Google Scholar] [CrossRef]
- Ho, M.Y.; Yu, A.L.; Yu, J. Glycosphingolipid dynamics in human embryonic stem cell and cancer: Their characterization and biomedical implications. Glycoconj. J. 2017, 34, 765–777. [Google Scholar] [CrossRef]
- Aloia, A.; Petrova, E.; Tomiuk, S.; Bissels, U.; Déas, O.; Saini, M.; Zickgraf, F.M.; Wagner, S.; Spaich, S.; Sütterlin, M.; et al. The sialyl-glycolipid stage-specific embryonic antigen 4 marks a subpopulation of chemotherapy-resistant breast cancer cells with mesenchymal features. Breast Cancer Res. 2015, 17, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Y.W.; Wang, P.Y.; Yeh, S.C.; Chuang, P.K.; Li, S.T.; Wu, C.Y.; Khoo, K.H.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Stage-specific embryonic antigen-4 as a potential therapeutic target in glioblastoma multiforme and other cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 2482–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Miyata, Y.; Matsuo, T.; Shida, Y.; Hakariya, T.; Ohba, K.; Taima, T.; Ito, A.; Suda, T.; itiroh Hakomori, S.; et al. Stage-specific embryonic antigen-4 is a histological marker reflecting the malignant behavior of prostate cancer. Glycoconj. J. 2019, 36, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Sigal, D.S.; Hermel, D.J.; Hsu, P.; Pearce, T. The role of Globo H and SSEA-4 in the development and progression of cancer, and their potential as therapeutic targets. Future Oncol. 2022, 18, 117–134. [Google Scholar] [CrossRef]
- Chang, W.W.; Chien, H.L.; Lee, P.; Lin, J.; Hsu, C.W.; Hung, J.T.; Lin, J.J.; Yu, J.C.; Shao, L.E.; Yu, J.; et al. Expression of Globo H and SSEA3 in breast cancer stem cells and the involvement of fucosyl transferases 1 and 2 in Globo H synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 11667–11672. [Google Scholar] [CrossRef] [Green Version]
- Chuang, P.K.; Hsiao, M.; Hsu, T.L.; Chang, C.F.; Wu, C.Y.; Chen, B.R.; Huang, H.W.; Liao, K.S.; Chen, C.C.; Chen, C.L.; et al. Signaling pathway of globo-series glycosphingolipids and β1,3-galactosyltransferase V (β3GalT5) in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3518–3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levina, V.; Marrangoni, A.M.; DeMarco, R.; Gorelik, E.; Lokshin, A.E. Drug-Selected Human Lung Cancer Stem Cells: Cytokine Network, Tumorigenic and Metastatic Properties. PLoS ONE 2008, 3, e3077. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Haraguchi, N.; Takahashi, H.; Uemura, M.; Nishimura, J.; Hata, T.; Takemasa, I.; Mizushima, T.; Ishii, H.; Doki, Y.; et al. SSEA-3 as a novel amplifying cancer cell surface marker in colorectal cancers. Int. J. Oncol. 2013, 42, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krengel, U.; Bousquet, P.A. Molecular recognition of gangliosides and their potential for cancer immunotherapies. Front. Immunol. 2014, 5, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides–an overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Ohmi, Y.; Kambe, M.; Ohkawa, Y.; Hamamura, K.; Tajima, O.; Takeuchi, R.; Furukawa, K.; Furukawa, K. Differential roles of gangliosides in malignant properties of melanomas. PLoS ONE 2018, 13, e0206881. [Google Scholar] [CrossRef] [Green Version]
- Portoukalian, J.; David, M.-J.; Richard, M.; Gain, P. Shedding of GD2 ganglioside in patients with retinoblastoma. Int. J. Cancer 1993, 53, 948–951. [Google Scholar] [CrossRef]
- Schnaar, R.L. Gangliosides of the vertebrate nervous system. J. Mol. Biol. 2016, 428, 3325. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Anand, V.; Andreeff, M.; Battula, V.L. Ganglioside GD2: A novel therapeutic target in triple-negative breast cancer. Ann. N. Y. Acad. Sci. 2022, 1508, 35–53. [Google Scholar] [CrossRef]
- Sariola, H.; Terava, H.; Rapola, J.; Saarinen, U.M. Cell-surface ganglioside GD2 in the immunohistochemical detection and differential diagnosis of neuroblastoma. Am. J. Clin. Pathol. 1991, 96, 248–252. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.-L.; Ladisch, S.; Feig, S.; Ulsh, L.; Schwartz, E.; Floutsis, G.; Wiley, F.; Lenarsky, C.; Seeger, R. Shedding of GD2 ganglioside by human neuroblastoma. Int. J. Cancer 1987, 39, 73–76. [Google Scholar] [CrossRef]
- Cavdarli, S.; Groux-Degroote, S.; Delannoy, P. Gangliosides: The Double-Edge Sword of Neuro-Ectodermal Derived Tumors. Biomolecules 2019, 9, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hersey, P.; Jamal, O. Expression of the gangliosides GD3 and GD2 on lymphocytes in tissue sections of melanoma. Pathology 1989, 21, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.I.; Bustos, M.A.; Wu, J.; Jones, P.; Chang, S.C.; Kiyohara, E.; Tran, K.; Zhang, X.; Stern, S.L.; Izraely, S.; et al. Upregulation of cell surface GD3 ganglioside phenotype is associated with human melanoma brain metastasis. Mol. Oncol. 2020, 14, 1760–1778. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. N-glycolylneuraminic acid deficiency in humans. Biochimie 2001, 83, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Mullet, A.; Mazorra, Z.; Vázquez, A.M.; Alfonso, M.; Mesa, C.; Rengifo, E.; Pérez, R.; Fernández, L.E. A Mouse IgG1 Monoclonal Antibody Specific for N-Glycolyl GM3 Ganglioside Recognized Breast and Melanoma Tumors. Hybridoma 2004, 19, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Scursoni, A.M.; Galluzzo, L.; Camarero, S.; Lopez, J.; Lubieniecki, F.; Sampor, C.; Segatori, V.I.; Gabri, M.R.; Alonso, D.F.; Chantada, G.; et al. Detection of N-glycolyl GM3 ganglioside in neuroectodermal tumors by immunohistochemistry: An attractive vaccine target for aggressive pediatric cancer. Clin. Dev. Immunol. 2011, 2011, 245181. [Google Scholar] [CrossRef] [Green Version]
- Torbidoni, A.V.; Scursoni, A.; Camarero, S.; Segatori, V.; Gabri, M.; Alonso, D.; Chantada, G.; Dávila, M.T.G.D. Immunoreactivity of the 14F7 Mab raised against N-Glycolyl GM3 Ganglioside in retinoblastoma tumours. Acta Ophthalmol. 2015, 93, e294–e300. [Google Scholar] [CrossRef]
- Lahera, T.; Calvo, A.; Torres, G.; Rengifo, C.E.; Quintero, S.; Arango, M.d.C.; Danta, D.; Vazquez, J.; Escobar, X.; Carr, A. Prognostic Role of 14F7 Mab Immunoreactivity against N-Glycolyl GM3 Ganglioside in Colon Cancer. J. Oncol. 2014, 2014, 482301. [Google Scholar] [CrossRef] [Green Version]
- Albertó, M.; Cuello, H.A.; Gulino, C.A.; Pifano, M.; Belgorosky, D.; Gabri, M.R.; Eiján, A.N.A.M.; Segatori, V.I. Expression of bladder cancer—Associated glycans in murine tumor cell lines. Oncol. Lett. 2019, 17, 3141–3150. [Google Scholar] [CrossRef] [Green Version]
- Blanco, R.; Domínguez, E.; Morales, O.; Blanco, D.; Martínez, D.; Rengifo, C.E.; Viada, C.; Cedeño, M.; Rengifo, E.; Carr, A. Prognostic Significance of N-Glycolyl GM3 Ganglioside Expression in Non-Small Cell Lung Carcinoma Patients: New Evidences. Patholog. Res. Int. 2015, 2015, 132326. [Google Scholar] [CrossRef] [Green Version]
- Bardor, M.; Nguyen, D.H.; Diaz, S.; Varki, A. Mechanism of uptake and incorporation of the non-human sialic acid N-glycolylneuraminic acid into human cells. J. Biol. Chem. 2005, 280, 4228–4237. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Shewell, L.K.; Day, C.J.; Jennings, M.P. N-glycolylneuraminic acid as a carbohydrate cancer biomarker. Transl. Oncol. 2023, 31, 101643. [Google Scholar] [CrossRef]
- Yin, J.; Hashimoto, A.; Izawa, M.; Miyazaki, K.; Chen, G.; Takematsu, H.; Kozutsumi, Y.; Suzuki, A.; Furuhata, K.; Cheng, F.; et al. Hypoxic Culture Induces Expression of Sialin, a Sialic Acid Transporter, and Cancer-Associated Gangliosides Containing Non-Human Sialic Acid on Human Cancer Cells. Cancer Res. 2006, 66, 2937–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusunoki, S.; Inoue, K.; Iwamori, M.; Nagai, Y.; Mannen, T. Fucosylated glycoconjugates in human dorsal root ganglion cells with unmyelinated axons. Neurosci. Lett. 1991, 126, 159–162. [Google Scholar] [CrossRef]
- Kusunoki, S.; Inoue, K.; Iwamori, M.; Nagai, Y.; Mannen, T.; Kanazawa, I. Developmental changes of fucosylated glycoconjugates in rabbit dorsal root ganglia. Neurosci. Res. 1992, 15, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Brezicka, F.; Olling, S.; Nilsson, O.; Bergh, J.; Holmgren, J.; Sörenson, S.; Yngvason, F.; Lindholm, L. Immunohistological detection of fucosyl-GM1 ganglioside in human lung cancer and normal tissues with monoclonal antibodies. Cancer Res. 1989, 49, 1300–1305. [Google Scholar] [PubMed]
- Livingston, P.O.; Hood, C.; Krug, L.M.; Warren, N.; Kris, M.G.; Brezicka, T.; Ragupathi, G. Selection of GM2, fucosyl GM1, globo H and polysialic acid as targets on small cell lung cancers for antibody mediated immunotherapy. Cancer Immunol. Immunother. 2005, 54, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Blanas, A.; Sahasrabudhe, N.M.; Rodríguez, E.; van Kooyk, Y.; van Vliet, S.J. Fucosylated Antigens in Cancer: An Alliance toward Tumor Progression, Metastasis, and Resistance to Chemotherapy. Front. Oncol. 2018, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Kadota, A.; Masutani, M.; Takei, M.; Horie, T. Evaluation of expression of CD15 and sCD15 in non-small cell lung cancer. Int. J. Oncol. 1999, 15, 1081–1089. [Google Scholar] [CrossRef]
- Koh, Y.W.; Lee, H.J.; Ahn, J.H.; Lee, J.W.; Gong, G. Expression of Lewis X Is Associated With Poor Prognosis in Triple-Negative Breast Cancer. Am. J. Clin. Pathol. 2013, 139, 746–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konety, B.R.; Ballou, B.; Jaffe, R.; Singh, J.; Reiland, J.; Hakala, T.R. Expression of SSEA-1 (Lewisx) on Transitional Cell Carcinoma of the Bladder. Urol. Int. 1997, 58, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Torii, A.; Nakayama, A.; Harada, A.; Nakao, A.; Nonami, T.; Sakamoto, J.; Watanabe, T.; Lto, M.; Takagi, H. Expression of the C D I 5 Antigen in Hepatocellular Carcinoma. Br. J. Cancer 2015, 112, 1911–1920. [Google Scholar] [CrossRef]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Gong, S.; Liao, B.; Pan, J.; Wang, J.; Zou, D.; Zhao, L.; Xiong, S.; Deng, Y.; Yan, Q.; et al. HIF1α/HIF2α induces glioma cell dedifferentiation into cancer stem cells through Sox2 under hypoxic conditions. J. Cancer 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Jin, F.; Wang, F. The physiological and pathological roles and applications of sialyl Lewis x, a common carbohydrate ligand of the three selectins. Glycoconj. J. 2020, 37, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Cuello, H.A.; Ferreira, G.M.; Gulino, C.A.; Toledo, A.G.; Segatori, V.I.; Gabri, M.R. Terminally sialylated and fucosylated complex N-glycans are involved in the malignant behavior of high-grade glioma. Oncotarget 2021, 11, 4822–4835. [Google Scholar] [CrossRef] [PubMed]
- Trinchera, M.; Aronica, A.; Dall’Olio, F. Selectin Ligands Sialyl-Lewis a and Sialyl-Lewis x in Gastrointestinal Cancers. Biology 2017, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, J.M.; Lo, S.K.; Corless, C.; Robertson, M.J.; Lee, N.C.; Barnhill, R.L.; Weinberg, D.S.; Bevilacqua, M.P. Expression of sialyl-Lewis X, an E-selectin ligand, in inflammation, immune processes, and lymphoid tissues. Am. J. Pathol. 1992, 141, 1397. [Google Scholar]
- Baldus, S.E.; Zirbes, T.K.; Mönig, S.P.; Engel, S.; Monaca, E.; Rafiqpoor, K.; Hanisch, F.G.; Hanski, C.; Thiele, J.; Pichlmaier, H.; et al. Histopathological subtypes and prognosis of gastric cancer are correlated with the expression of mucin-associated sialylated antigens: Sialosyl-Lewis(a), Sialosyl-Lewis(x) and sialosyl-Tn. Tumour Biol. 1998, 19, 445–453. [Google Scholar] [CrossRef]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, T.I.S.; Kameyama, M.; Imaoka, S.; Furukawa, H.; Ishikawa, O.; Sasaki, Y.; Kabuto, T.; Iwanaga, T.; Matsushita, Y. Increased expression of sialyl Lewisx antigen correlates with poor survival in patients with colorectal carcinoma: Clinicopathological and immunohistochemical study. Cancer Res. 1993, 53, 3632–3637. Available online: https://pubmed.ncbi.nlm.nih.gov/8101764/ (accessed on 1 August 1993). [PubMed]
- Goonetilleke, K.S.; Siriwardena, A.K. Systematic review of carbohydrate antigen (CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur. J. Surg. Oncol. 2007, 33, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Kolben, T.; Müller, L.; Meister, S.; Keilmann, L.; Buschmann, C.; Trillsch, F.; Burges, A.; Czogalla, B.; Mitter, S.; Schmoeckel, E.; et al. Blood group antigens SLeX, SLeA, and LeY as prognostic markers in endometrial cancer. J. Cancer Res. Clin. Oncol. 2022, 148, 3323–3335. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, J.; Oshima, Y.; Nanami, T.; Suzuki, T.; Yajima, S.; Shiratori, F.; Funahashi, K.; Shimada, H. Prognostic impact of CEA/CA19-9 at the time of recurrence in patients with gastric cancer. Surg. Today 2021, 51, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Wu, H.; Huang, Q.; Yu, Z.; Zhong, Z. Clinical Value of Serum CEA, CA24-2 and CA19-9 in Patients with Colorectal Cancer. Clin. Lab. 2021, 67, 1079–1089. [Google Scholar] [CrossRef]
- Tanaka, F.; Miyahara, R.; Ohtake, Y.; Yanagihara, K.; Fukuse, T.; Hitomi, S.; Wada, H. Lewis Y antigen expression and postoperative survival in non-small cell lung cancer. Ann. Thorac. Surg. 1998, 66, 1745–1750. [Google Scholar] [CrossRef]
- Wakabayashi, M.; Shiro, T.; Seki, T.; Nakagawa, T.; Lmamura, M.; Shiozaki, Y.; Lnoue, K.; Okamura, A. Lewis Y Antigen Expression in Hepatocellular Carcinoma An Immunohistochemical Study. Cancer 1995, 75, 2827–2835. [Google Scholar] [CrossRef]
- Zhuang, H.; Hu, Z.; Tan, M.; Zhu, L.; Liu, J.; Liu, D.; Yan, L.; Lin, B. Overexpression of Lewis y antigen promotes human epididymis protein 4-mediated invasion and metastasis of ovarian cancer cells. Biochimie 2014, 105, 91–98. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, L.; Zhuang, H.; Lin, B. Human Epididymis Protein 4 and Lewis y Enhance Chemotherapeutic Resistance in Epithelial Ovarian Cancer Through the p38 MAPK Pathway. Adv. Ther. 2022, 39, 360–378. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, M.; Qi, Y.; Wang, H.; Liu, M.; Liu, Q.; Lin, B. Lewis(y) antigen-mediated positive feedback loop induces and promotes chemotherapeutic resistance in ovarian cancer. Int. J. Oncol. 2018, 53, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, J.; Zhu, L.; Hu, Z.; Hou, R.; Liu, S.; Tan, M.; Liu, J.; Lin, B. Chemoresistance is associated with MUC1 and Lewis y antigen expression in ovarian epithelial cancers. Int. J. Mol. Sci. 2013, 14, 11024–11033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalziel, M.; Whitehouse, C.; McFarlane, I.; Brockhausen, I.; Gschmeissner, S.; Schwientek, T.; Clausen, H.; Burchell, J.M.; Taylor-Papadimitriou, J. The Relative Activities of the C2GnT1 and ST3Gal-I Glycosyltransferases Determine O -Glycan Structure and Expression of a Tumor-associated Epitope on MUC1. J. Biol. Chem. 2001, 276, 11007–11015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, T.; Aryal, R.P.; Kudelka, M.R.; Wang, Y.; Cummings, R.D. The Cosmc connection to the Tn antigen in cancer. Cancer Biomark. 2014, 14, 63–81. [Google Scholar] [CrossRef] [Green Version]
- Cuello, H.A.; Segatori, V.I.; Albertó, M.; Gulino, C.A.; Aschero, R.; Camarero, S.; Mutti, L.G.; Madauss, K.; Alonso, D.F.; Lubieniecki, F.; et al. Aberrant O-glycosylation modulates aggressiveness in neuroblastoma. Oncotarget 2018, 9, 34176–34188. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.R. Immunoreactive T and Tn antigens in malignancy: Role in carcinoma diagnosis, prognosis, and immunotherapy. Transfus. Med. Rev. 2000, 14, 312–325. [Google Scholar] [CrossRef]
- Springer, G.F. T and Tn, general carcinoma autoantigens. Science 1984, 224, 1198–1206. [Google Scholar] [CrossRef]
- Rømer, T.B.; Aasted, M.K.M.; Dabelsteen, S.; Groen, A.; Schnabel, J.; Tan, E.; Pedersen, J.W.; Haue, A.D.; Wandall, H.H. Mapping of truncated O-glycans in cancers of epithelial and non-epithelial origin. Br. J. Cancer 2021, 125, 1239. [Google Scholar] [CrossRef]
- Festari, M.F.; Da Costa, V.; Rodríguez-Zraquia, S.A.; Costa, M.; Landeira, M.; Lores, P.; Solari-Saquieres, P.; Kramer, M.G.; Freire, T. The tumor-associated Tn antigen fosters lung metastasis and recruitment of regulatory T cells in triple negative breast cancer. Glycobiology 2022, 32, 366–379. [Google Scholar] [CrossRef]
- Hamada, S.-i.; Furumoto, H.; Kamada, M.; Hirao, T.; Aono, T. High expresion rate of Tn antigen in metastatic lesions of uterine cervical cancers. Cancer Lett. 1993, 74, 167–173. [Google Scholar] [CrossRef]
- Konno, A.; Hoshino, Y.; Terashima, S.; Motoki, R.; Kawaguchi, T. Carbohydrate expression profile of colorectal cancer cells is relevant to metastatic pattern and prognosis. Clin. Exp. Metastasis 2002, 19, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.A.; Videira, P.A.; Lima, L.; Pereira, S.; Silva, M.; Carrascal, M.; Severino, P.F.; Fernandes, E.; Almeida, A.; Costa, C.; et al. Overexpression of tumour-ssociated carbohydrate antigen sialyl-Tn in advanced bladder tumours. Mol. Oncol. 2013, 7, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Hashiguchi, Y.; Kasai, M.; Fukuda, T.; Ichimura, T.; Yasui, T.; Sumi, T. Serum Sialyl-Tn (STN) as a Tumor Marker in Patients with Endometrial Cancer. Pathol. Oncol. Res. 2016, 22, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Itzkowitz, S.; Kjeldsen, T.; Friera, A.; Hakomori, S.; Yang, U.S.; Kim, Y.S. Expression of Tn, sialosyl Tn, and T antigens in human pancreas. Gastroenterology 1991, 100, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Videira, P.A.; Delannoy, P. Sialyl-tn in cancer: (How) did we miss the target? Biomolecules 2012, 2, 435–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.; Zhao, H.; Wang, Y.; Cai, H.; Xiao, Y.; Zeng, Y.; Chen, H. Tumor-associated antigens: Tn antigen, sTn antigen, and T antigen. HLA 2016, 88, 275–286. [Google Scholar] [CrossRef]
- Karsten, U.; Goletz, S. What controls the expression of the core-1 (Thomsen-Friedenreich) glycotope on tumor cells? Biochemistry 2015, 80, 801–807. [Google Scholar] [CrossRef]
- Rivinoja, A.; Kokkonen, N.; Kellokumpu, I.; Kellokumpu, S. Elevated Golgi pH in breast and colorectal cancer cells correlates with the expression of oncofetal carbohydrate T-antigen. J. Cell. Physiol. 2006, 208, 167–174. [Google Scholar] [CrossRef]
- Thorens, B.; Vassalli, P. Chloroquine and ammonium chloride prevent terminal glycosylation of immunoglobulins in plasma cells without affecting secretion. Nature 1986, 321, 618–620. [Google Scholar] [CrossRef]
- Monzavi-Karbassi, B.; Pashov, A.; Kieber-Emmons, T. Tumor-Associated Glycans and Immune Surveillance. Vaccines 2013, 1, 174–203. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, Y.; Fukumori, T.; Raz, A. Galectin-3 and metastasis. Glycoconj. J. 2002, 19, 543–549. [Google Scholar] [CrossRef]
- Yu, L.G. The oncofetal Thomsen-Friedenreich carbohydrate antigen in cancer progression. Glycoconj. J. 2007, 24, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Soukhtehzari, S.; Berish, R.B.; Fazli, L.; Watson, P.H.; Williams, K.C. The different prognostic significance of polysialic acid and CD56 expression in tumor cells and lymphocytes identified in breast cancer. NPJ Breast Cancer 2022, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Kapsenberg, M.L. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 2003, 3, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, K. Dendritic Cells. Encycl. Cell Biol. 2016, 3, 741–749. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- Caza, T.; Landas, S. Functional and Phenotypic Plasticity of CD4 (+) T Cell Subsets. Biomed Res. Int. 2015, 2015, 521957. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Kennedy, R.; Celis, E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol. Rev. 2008, 222, 129–144. [Google Scholar] [CrossRef]
- Zimmermann, S.; Lepenies, B. Glycans as vaccine Antigens and Adjuvants: Immunological considerations. Methods Mol. Biol. 2015, 1331, 11–26. [Google Scholar] [CrossRef]
- Yoshida, T.; Mei, H.; Dörner, T.; Hiepe, F.; Radbruch, A.; Fillatreau, S.; Hoyer, B.F. Memory B and memory plasma cells. Immunol. Rev. 2010, 237, 117–139. [Google Scholar] [CrossRef]
- von Bülow, G.-U.; van Deursen, J.M.; Bram, R.J. Regulation of the T-Independent Humoral Response by TACI. Immunity 2001, 14, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Heimburg-Molinaro, J.; Lum, M.; Vijay, G.; Jain, M.; Almogren, A.; Rittenhouse-Olson, K. Cancer Vaccines and Carbohydrate Epitopes. Vaccine 2011, 29, 8802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, T.H.; Boyle, A.L.; Diebolder, C.A.; Kros, A.; Koster, A.J.; Gros, P. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl. Acad. Sci. USA 2019, 116, 11900–11905. [Google Scholar] [CrossRef] [Green Version]
- Bortnick, A.; Chernova, I.; Quinn, W.J.; Mugnier, M.; Cancro, M.P.; Allman, D. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J. Immunol. 2012, 188, 5389–5396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Johnson, T.D.; Nishinaka, Y.; Morton, D.L.; Irie, R.F. IgM Anti-Ganglioside Antibodies Induced by Melanoma Cell Vaccine Correlate with Survival of Melanoma Patients. J. Investig. Dermatol. 1999, 112, 205–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsueh, E.C.; Gupta, R.K.; Qi, K.; Morton, D.L. Correlation of specific immune responses with survival in melanoma patients with distant metastases receiving polyvalent melanoma cell vaccine. J. Clin. Oncol. 1998, 16, 2913–2920. [Google Scholar] [CrossRef]
- Ravindranath, M.H.; Muthugounder, S.; Presser, N.; Ye, X.; Brosman, S.; Morton, D.L. Endogenous immune response to gangliosides in patients with confined prostate cancer. Int. J. Cancer 2005, 116, 368–377. [Google Scholar] [CrossRef]
- Kawashima, A.; Tsugawa, S.; Boku, A.; Kobayashi, M.; Minamoto, T.; Nakanishi, I.; Oda, Y. Expression of alphav integrin family in gastric carcinomas: Increased alphavbeta6 is associated with lymph node metastasis. Pathol. Res. Pract. 2003, 199, 57–64. [Google Scholar] [CrossRef]
- Pashov, A.; Monzavi-Karbassi, B.; Chow, M.; Cannon, M.; Kieber-Emmons, T. Immune surveillance as a rationale for immunotherapy? Hum. Vaccines 2007, 3, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Pashov, A.; Monzavi-Karbassi, B.; Kieber-Emmons, T. Immune surveillance and immunotherapy: Lessons from carbohydrate mimotopes. Vaccine 2009, 27, 3405–3415. [Google Scholar] [CrossRef]
- Zajonc, D.M. The CD1 family: Serving lipid antigens to T cells since the Mesozoic era. Immunogenetics 2016, 68, 561–576. [Google Scholar] [CrossRef] [Green Version]
- Gentilini, M.V.; Pérez, M.E.; Fernández, P.M.; Fainboim, L.; Arana, E. The tumor antigen N-glycolyl-GM3 is a human CD1d ligand capable of mediating B cell and natural killer T cell interaction. Cancer Immunol. Immunother. 2016, 65, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.Y.; Segal, N.H.; Sidobre, S.; Kronenberg, M.; Chapman, P.B. Cross-presentation of disialoganglioside GD3 to natural killer T cells. J. ExMed. 2003, 198, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salio, M.; Silk, J.D.; Jones, E.Y.; Cerundolo, V. Biology of CD1- and MR1-Restricted T Cells. Annu. Rev. Immunol. 2014, 32, 323–366. [Google Scholar] [CrossRef]
- Krovi, S.H.; Gapin, L. Invariant Natural Killer T Cell Subsets—More Than Just Developmental Intermediates. Front. Immunol. 2018, 9, 1393. [Google Scholar] [CrossRef] [Green Version]
- Dellabona, P.; Abrignani, S.; Casorati, G. iNKT-cell help to B cells: A cooperative job between innate and adaptive immune responses. Eur. J. Immunol. 2014, 44, 2230–2237. [Google Scholar] [CrossRef]
- Doherty, D.G.; Melo, A.M.; Moreno-Olivera, A.; Solomos, A.C. Activation and regulation of B cell responses by invariant natural killer T cells. Front. Immunol. 2018, 9, 1360. [Google Scholar] [CrossRef] [Green Version]
- Makhoul, I.; Hutchins, L.; Emanuel, P.D.; Pennisi, A.; Siegel, E.; Jousheghany, F.; Karbassi, B.M.-; Kieber-Emmons, T. Moving a carbohydrate mimetic peptide into the clinic clinical response of a breast cancer patient after mimotope-based immunotherapy. Hum. Vaccines Immunother. 2015, 11, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Buskas, T.; Thompson, P.; Boons, G.-J. Immunotherapy for cancer: Synthetic carbohydrate-based vaccines. Chem. Commun. 2009, 36, 5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettu, R.; Chen, C.Y.; Wu, C.Y. Synthetic carbohydrate-based vaccines: Challenges and opportunities. J. Biomed. Sci. 2020, 27, 9. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S. Dinutuximab: First Global Approval. Drugs 2015, 75, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Suciu, S.; Rutkowski, P.; Marsden, J.; Santinami, M.; Corrie, P.; Aamdal, S.; Ascierto, P.A.; Patel, P.M.; Kruit, W.H.; et al. Adjuvant Ganglioside GM2-KLH/QS-21 Vaccination Versus Observation After Resection of Primary Tumor > 1.5 mm in Patients With Stage II Melanoma: Results of the EORTC 18961 Randomized Phase III Trial. J. Clin. Oncol. 2013, 31, 3831–3837. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-S.; Yu, A.L.; Tseng, L.-M.; Chow, L.W.C.; Hou, M.-F.; Hurvitz, S.A.; Schwab, R.B.; Wong, C.-H.; Murray, J.L.; Chang, H.-K.; et al. Randomized phase II/III trial of active immunotherapy with OPT-822/OPT-821 in patients with metastatic breast cancer. J. Clin. Oncol. 2016, 34, 1003. [Google Scholar] [CrossRef]
- Miles, D.; Roché, H.; Martin, M.; Perren, T.J.; Cameron, D.A.; Glaspy, J.; Dodwell, D.; Parker, J.; Mayordomo, J.; Tres, A.; et al. Phase III Multicenter Clinical Trial of the Sialyl-TN (STn)-Keyhole Limpet Hemocyanin (KLH) Vaccine for Metastatic Breast Cancer. Oncologist 2011, 16, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Hossain, F.; Andreana, P.R. Developments in Carbohydrate-Based Cancer Therapeutics. Pharmaceuticals 2019, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.T.; Lan, H.R.; Chen, X.Y.; Wang, S.B.; Ying, X.J.; Lin, Y.; Mou, X.Z. Recent advances in carbohydrate-based cancer vaccines. Biotechnol. Lett. 2019, 41, 641–650. [Google Scholar] [CrossRef]
- Shivatare, S.S.; Shivatare, V.S.; Wong, C.H. Glycoconjugates: Synthesis, Functional Studies, and Therapeutic Developments. Chem. Rev. 2022, 122, 15603–15671. [Google Scholar] [CrossRef]
- Berois, N.; Pittini, A.; Osinaga, E. Targeting Tumor Glycans for Cancer Therapy: Successes, Limitations, and Perspectives. Cancers 2022, 14, 645. [Google Scholar] [CrossRef]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Cohen, I.R. Antigenic Mimicry, Clonal Selection and Autoimmunity. J. Autoimmun. 2001, 16, 337–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pashov, A.; Perry, M.; Dyar, M.; Chow, M.; Kieber-Emmons, T. Carbohydrate Mimotopes in the Rational Design of Cancer Vaccines. Curr. Top. Med. Chem. 2005, 5, 1171–1185. [Google Scholar] [CrossRef] [PubMed]
- Vyas, N.K.; Vyas, M.N.; Chervenak, M.C.; Bundle, D.R.; Pinto, B.M.; Quiocho, F.A. Structural basis of peptide-carbohydrate mimicry in an antibody-combining site. Proc. Natl. Acad. Sci. USA 2003, 100, 15023–15028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collis, A.V.J.; Brouwer, A.P.; Martin, A.C.R. Analysis of the antigen combining site: Correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J. Mol. Biol. 2003, 325, 337–354. [Google Scholar] [CrossRef]
- Sawada, R.; Sun, S.M.; Wu, X.; Hong, F.; Ragupathi, G.; Livingston, P.O.; Scholz, W.W. Human monoclonal antibodies to sialyl-Lewisa (CA19.9) with potent CDC, ADCC, and antitumor activity. Clin. Cancer Res. 2011, 17, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Canziani, G.; Cunto-Amesty, G.; Kieber-Emmons, T. A molecular basis for functional peptide mimicry of a carbohydrate antigen. J. Biol. Chem. 2000, 275, 16146–16154. [Google Scholar] [CrossRef] [Green Version]
- Horwacik, I.; Golik, P.; Grudnik, P.; Kolinski, M.; Zdzalik, M.; Rokita, H.; Dubin, G. Structural basis of GD2 ganglioside and mimetic peptide recognition by 14G2a antibody. Mol. Cell. Proteom. 2015, 14, 2577–2590. [Google Scholar] [CrossRef] [Green Version]
- Liebert, M.A.; Qiu, J.; Luo, P.; Wasmund, K.; Steplewski, Z.; Kieber-emmons, T. Towards the Development of Peptide Mimotopes of Carbohydrate Antigens as Cancer Vaccines. Hybridoma 1999, 18, 103–112. [Google Scholar]
- Rahbarnia, L.; Farajnia, S.; Babaei, H.; Majidi, J.; Veisi, K.; Ahmadzadeh, V.; Akbari, B. Evolution of phage display technology: From discovery to application. J. Drug Target. 2017, 25, 216–224. [Google Scholar] [CrossRef]
- Jaroszewicz, W.; Morcinek-Orłowska, J.; Pierzynowska, K.; Gaffke, L.; Wȩgrzyn, G. Phage display and other peptide display technologies. FEMS Microbiol. Rev. 2022, 46, fuab052. [Google Scholar] [CrossRef] [PubMed]
- Goracci, M.; Pignochino, Y.; Marchiò, S. Phage display-based nanotechnology applications in cancer immunotherapy. Molecules 2020, 25, 843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemer, A.B.; Förster-Waldl, E.; Brämswig, K.H.; Pollak, A.; Zielinski, C.C.; Pehamberger, H.; Lode, H.N.; Scheiner, O.; Jensen-Jarolim, E. Induction of IgG antibodies against the GD2 carbohydrate tumor antigen by vaccination with peptide mimotopes. Eur. J. Immunol. 2006, 36, 1267–1274. [Google Scholar] [CrossRef]
- Kozbor, D. Cancer vaccine with mimotopes of tumor-associated carbohydrate antigens. Immunol. Res. 2010, 46, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwacik, I.; Czaplicki, D.; Talarek, K.; Kowalczyk, A.; Bolesta, E.; Kozbor, D.; Rokita, H. Selection of novel peptide mimics of the GD2 ganglioside from a constrained phage-displayed peptide library. Int. J. Mol. Med. 2007, 19, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popa, I.; Ishikawa, D.; Tanaka, M.; Ogino, K.; Portoukalian, J.; Taki, T. GD3-replica peptides selected from a phage peptide library induce a GD3 ganglioside antibody response. FEBS Lett. 2006, 580, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Heimburg-Molinaro, J.; Almogren, A.; Morey, S.; Glinskii, O.V.; Roy, R.; Wilding, G.E.; Cheng, R.P.; Glinsky, V.V.; Rittenhouse-Olson, K. Development, characterization, and immunotherapeutic use of peptide mimics of the Thomsen-Friedenreich carbohydrate antigen. Neoplasia 2009, 11, 780–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieber-Emmons, T.; Murali, R.; Greene, M.I. Therapeutic peptides and peptidomimetics. Curr. Opin. Biotechnol. 1997, 8, 435–441. [Google Scholar] [CrossRef]
- Hoffmüller, U.; Knaute, T.; Hahn, M.; Höhne, W.; Schneider-Mergener, J.; Kramer, A. Evolutionary transition pathways for changing peptide ligand specificity and structure. EMBO J. 2000, 19, 4866–4874. [Google Scholar] [CrossRef] [Green Version]
- onzavi-Karbassi, B.; Hennings, L.J.; Artaud, C.; Liu, T.; Jousheghany, F.; Pashov, A.; Murali, R.; Hutchins, L.F.; Kieber-Emmons, T. Preclinical studies of carbohydrate mimetic peptide vaccines for breast cancer and melanoma. Vaccine 2007, 25, 3022–3031. [Google Scholar] [CrossRef]
- Hennings, L.; Artaud, C.; Jousheghany, F.; Monzavi-Karbassi, B.; Pashov, A.; Kieber-Emmons, T. Carbohydrate mimetic peptides augment carbohydrate-reactive immune responses in the absence of immune pathology. Cancers 2011, 3, 4151–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchins, L.F.; Makhoul, I.; Emanuel, P.D.; Pennisi, A.; Siegel, E.R.; Jousheghany, F.; Guo, X.; Pashov, A.D.; Monzavi-Karbassi, B.; Kieber-Emmons, T. Targeting tumor-associated carbohydrate antigens: A phase I study of a carbohydrate mimetic-peptide vaccine in stage IV breast cancer subjects. Oncotarget 2017, 8, 99161–99178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhoul, I.; Ibrahim, S.M.; Abu-Rmaileh, M.; Jousheghany, F.; Siegel, E.R.; Rogers, L.J.; Lee, J.J.; Pina-Oviedo, S.; Post, G.R.; Beck, J.T.; et al. P10s-PADRE vaccine combined with neoadjuvant chemotherapy in ER-positive breast cancer patients induces humoral and cellular immune responses. Oncotarget 2021, 12, 2252–2265. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.; Wierzbicki, A.; Gil, M.; Bambach, B.; Kaneko, Y.; Rokita, H.; Repasky, E.; Fenstermaker, R.; Brecher, M.; Ciesielski, M.; et al. Induction of protective immune responses against NXS2 neuroblastoma challenge in mice by immunotherapy with GD2 mimotope vaccine and IL-15 and IL-21 gene delivery. Cancer Immunol. Immunother. 2007, 56, 1443–1458. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.; Gil, M.; Ciesielski, M.; Fenstermaker, R.A.; Kaneko, Y.; Rokita, H.; Lau, J.T.; Kozbor, D. Immunization with a Mimotope of GD2 Ganglioside Induces CD8 + T Cells That Recognize Cell Adhesion Molecules on Tumor Cells 1. J. Immunol. 2008, 181, 6644–6653. [Google Scholar] [CrossRef] [Green Version]
- Gil, M.; Bieniasz, M.; Wierzbicki, A.; Bambach, B.J.; Rokita, H.; Kozbor, D. Targeting a Mimotope Vaccine to Activating Fcγ Receptors Empowers Dendritic Cells to Prime Specific CD8+ T Cell Responses in Tumor-Bearing Mice. J. Immunol. 2009, 183, 6808–6818. [Google Scholar] [CrossRef] [Green Version]
- Jerne, N.K. Towards a network theory of the immune system. Ann. Immunol. 1974, 125C, 373–389. [Google Scholar]
- Cheung, N.K.; Cheung, I.Y.; Canete, A.; Yeh, S.J.; Kushner, B.; Bonilla, M.A.; Heller, G.; Larson, S.M. Antibody response to murine anti-GD2 monoclonal antibodies: Correlation with patient survival. Cancer Res. 1994, 54, 2228–2233. [Google Scholar]
- Schultes, B.C.; Baum, R.P.; Niesen, A.; Noujaim, A.A.; Madiyalakan, R. Anti-idiotype induction therapy: Anti-CA125 antibodies (Ab3) mediated tumor killing in patients treated with Ovarex mAb B43.13 (Ab1). Cancer Immunol. Immunother. 1998, 46, 201–212. [Google Scholar] [CrossRef]
- Cheung, N.K.; Guo, H.F.; Heller, G.; Cheung, I.Y. Induction of Ab3 and Ab3′ antibody was associated with long-term survival after anti-G(D2) antibody therapy of stage 4 neuroblastoma. Clin. Cancer Res. 2000, 6, 2653–2660. [Google Scholar]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A National Cancer Institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [Green Version]
- Gomez, D.E.; Vázquez, A.M.; Alonso, D.F. Cancer Antigen Prioritization: A Road Map to Work in Defining Vaccines Against Specific Targets. A Point of View. Front. Oncol. 2012, 2, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, P.B.; Houghton, A.N. Induction of IgG antibodies against GD3 ganglioside in rabbits by an anti-idiotypic monoclonal antibody. J. Clin. Investig. 1991, 88, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaffery, M.; Yao, T.J.; Williams, L.; Livingston, P.O.; Houghton, A.N.; Chapman, P.B. Immunization of melanoma patients with BEC2 anti-idiotypic monoclonal antibody that mimics GD3 ganglioside: Enhanced immunogenicity when combined with adjuvant. Clin. Cancer Res. 1996, 2, 679–686. [Google Scholar] [PubMed]
- Yao, T.J.; Meyers, M.; Livingston, P.O.; Houghton, A.N.; Chapman, P.B. Immunization of melanoma patients with BEC2-keyhole limpet hemocyanin plus BCG intradermally followed by intravenous booster immunizations with BEC2 to induce anti-GD3 ganglioside antibodies. Clin. Cancer Res. 1999, 5, 77–81. [Google Scholar]
- Grant, S.C.; Kris, M.G.; Houghton, A.N.; Chapman, P.B. Long survival of patients with small cell lung cancer after adjuvant treatment with the anti-idiotypic antibody BEC2 plus Bacillus Calmette-Guérin. Clin. Cancer Res. 1999, 5, 1319–1323. [Google Scholar]
- Chapman, P.B.; Williams, L.; Salibi, N.; Hwu, W.-J.; Krown, S.E.; Livingston, P.O. A phase II trial comparing five dose levels of BEC2 anti-idiotypic monoclonal antibody vaccine that mimics GD3 ganglioside. Vaccine 2004, 22, 2904–2909. [Google Scholar] [CrossRef]
- Giaccone, G.; Debruyne, C.; Felip, E.; Chapman, P.B.; Grant, S.C.; Millward, M.; Thiberville, L.; D’addario, G.; Coens, C.; Rome, L.S.; et al. Phase III Study of Adjuvant Vaccination With Bec2/Bacille Calmette-Guerin in Responding Patients With Limited-Disease Small-Cell Lung Cancer (European Organisation for Research and Treatment of Cancer 08971-08971B; Silva Study). J. Clin. Oncol. 2005, 23, 6854–6864. [Google Scholar] [CrossRef]
- Sen, G.; Chakraborty, M.; Foon, K.A.; Reisfeld, R.A.; Bhattacharya-Chatterjee, M. Induction of IgG Antibodies by an Anti-Idiotype Antibody Mimicking Disialoganglioside GD2. J. Immunother. 1998, 21, 75–83. [Google Scholar] [CrossRef]
- Sen, G.; Chakraborty, M.; Foon, K.A.; Reisfeld, R.A.; Bhattacharya-Chatterjee, M. Preclinical evaluation in nonhuman primates of murine monoclonal anti-idiotype antibody that mimics the disialoganglioside GD2. Clin. Cancer Res. 1997, 3, 1969–1976. [Google Scholar]
- Foon, K.A.; Sen, G.; Hutchins, L.; Kashala, O.L.; Baral, R.; Banerjee, M.; Chakraborty, M.; Garrison, J.; Reisfeld, R.A.; Bhattacharya-Chatterjee, M. Antibody responses in melanoma patients immunized with an anti-idiotype antibody mimicking disialoganglioside GD2. Clin. Cancer Res. 1998, 4, 1117–1124. [Google Scholar] [PubMed]
- Foon, K.A.; Lutzky, J.; Baral, R.N.; Yannelli, J.R.; Hutchins, L.; Teitelbaum, A.; Kashala, O.L.; Das, R.; Garrison, J.; Reisfeld, R.A.; et al. Clinical and Immune Responses in Advanced Melanoma Patients Immunized With an Anti-Idiotype Antibody Mimicking Disialoganglioside GD2. J. Clin. Oncol. 2000, 18, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Lutzky, J.; Gonzalez-Angulo, A.M.; Orzano, J.A. Antibody-based vaccines for the treatment of melanoma. Semin. Oncol. 2002, 29, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Lode, H.N.; Schmidt, M.; Seidel, D.; Huebener, N.; Brackrock, D.; Bleeke, M.; Reker, D.; Brandt, S.; Mueller, H.-P.; Helm, C.; et al. Vaccination with anti-idiotype antibody ganglidiomab mediates a GD2-specific anti-neuroblastoma immune response. Cancer Immunol. Immunother. 2013, 62, 999–1010. [Google Scholar] [CrossRef]
- Klingel, L.; Siebert, N.; Troschke-Meurer, S.; Zumpe, M.; Ehlert, K.; Huber, S.; Loibner, H.; Mutschlechner, O.; Lode, H.N. Immune Response and Outcome of High-Risk Neuroblastoma Patients Immunized with Anti-Idiotypic Antibody Ganglidiomab: Results from Compassionate-Use Treatments. Cancers 2022, 14, 5802. [Google Scholar] [CrossRef]
- Vázquez, A.M.; Pérez, A.; Hernández, A.M.; Macías, A.; Alfonso, M.; Bombino, G.; Pérez, R. Syngeneic Anti-Idiotypic Monoclonal Antibodies to an Anti-NeuGc-Containing Ganglioside Monoclonal Antibody. Hybridoma 1998, 17, 527–534. [Google Scholar] [CrossRef]
- Hernández, A.M.; Rodríguez, M.; López-Requena, A.; Beausoleil, I.; Pérez, R.; Vázquez, A.M. Generation of anti-Neu-glycolyl-ganglioside antibodies by immunization with an anti-idiotype monoclonal antibody: A self versus non-self-matter. Immunobiology 2005, 210, 11–21. [Google Scholar] [CrossRef]
- Segatori, V.I.; Vazquez, A.M.; Gomez, D.E.; Gabri, M.R.; Alonso, D.F. Preclinical evaluation of racotumomab, an anti-idiotype monoclonal antibody to N-glycolyl-containing gangliosides, with or without chemotherapy in a mouse model of non-small cell lung cancer. Front. Oncol. 2012, 2, 160. [Google Scholar] [CrossRef] [Green Version]
- Guthmann, M.D.; Venier, C.; Toledo, D.; Segatori, V.I.; Alonso, D.F.; Fainboim, L.; Vázquez, A.M.; Ostrowski, H. Anti-ganglioside antibodies induced in chickens by an alum-adsorbed anti-idiotype antibody targeting NeuGcGM3. Front. Immunol. 2012, 3, 422. [Google Scholar] [CrossRef] [Green Version]
- Alfonso, M.; Díaz, A.; Hernández, A.M.; Pérez, A.; Rodríguez, E.; Bitton, R.; Pérez, R.; Vázquez, A.M. An Anti-Idiotype Vaccine Elicits a Specific Response to N -Glycolyl Sialic Acid Residues of Glycoconjugates in Melanoma Patients. J. Immunol. 2002, 168, 2523–2529. [Google Scholar] [CrossRef] [Green Version]
- Díaz, A.; Alfonso, M.; Alonso, R.; Saurez, G.; Troche, M.; Catalá, M.; Díaz, R.M.; Pérez, R.; Vázquez, A.M. Immune responses in breast cancer patients immunized with an anti-idiotype antibody mimicking NeuGc-containing gangliosides. Clin. Immunol. 2003, 107, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.M.; Rodríguez, N.; González, J.E.; Reyes, E.; Rondón, T.; Griñán, T.; Macías, A.; Alfonso, S.; Vázquez, A.M.; Pérez, R. Anti-NeuGcGM3 Antibodies, Actively Elicited by Idiotypic Vaccination in Nonsmall Cell Lung Cancer Patients, Induce Tumor Cell Death by an Oncosis-Like Mechanism. J. Immunol. 2011, 186, 3735–3744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfonso, S.; Valdés-Zayas, A.; Santiesteban, E.R.; Flores, Y.I.; Areces, F.; Hernández, M.; Viada, C.E.; Mendoza, I.C.; Guerra, P.P.; García, E.; et al. A Randomized, Multicenter, Placebo-Controlled Clinical Trial of Racotumomab-Alum Vaccine as Switch Maintenance Therapy in Advanced Non–Small Cell Lung Cancer Patients. Clin. Cancer Res. 2014, 20, 3660–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segatori, V.I.; Cuello, H.A.; Gulino, C.A.; Albertó, M.; Venier, C.; Guthmann, M.D.; Demarco, I.A.; Alonso, D.F.; Gabri, M.R. Antibody-dependent cell-mediated cytotoxicity induced by active immunotherapy based on racotumomab in non-small cell lung cancer patients. Cancer Immunol. Immunother. 2018, 67, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, A.M.; Gabri, M.R.; Hernández, A.M.; Alonso, D.F.; Beausoleil, I.; Gomez, D.E.; Pérez, R. Antitumor properties of an anti-idiotypic monoclonal antibody in relation to N-glycolyl-containing gangliosides. Oncol. Rep. 2000, 7, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Diaz, Y.; Gonzalez, A.; Lopez, A.; Perez, R.; Vazquez, A.M.; Montero, E. Anti-ganglioside anti-idiotypic monoclonal antibody-based cancer vaccine induces apoptosis and antiangiogenic effect in a metastatic lung carcinoma. Cancer Immunol. Immunother. 2009, 58, 1117–1128. [Google Scholar] [CrossRef]
- Fuentes, D.; Avellanet, J.; Garcia, A.; Iglesias, N.; Gabri, M.R.; Alonso, D.F.; Vazquez, A.M.; Perez, R.; Montero, E. Combined therapeutic effect of a monoclonal anti-idiotype tumor vaccine against NeuGc-containing gangliosides with chemotherapy in a breast carcinoma model. Breast Cancer Res. Treat. 2010, 120, 379–389. [Google Scholar] [CrossRef]
- Cacciavillano, W.; Sampor, C.; Venier, C.; Gabri, M.R.; de Dávila, M.T.G.; Galluzzo, M.L.; Guthmann, M.D.; Fainboim, L.; Alonso, D.F.; Chantada, G.L. A Phase I Study of the Anti-Idiotype Vaccine Racotumomab in Neuroblastoma and Other Pediatric Refractory Malignancies. Pediatr. Blood Cancer 2015, 62, 2120–2124. [Google Scholar] [CrossRef]
- Guthmann, M.D.; Bitton, R.J.; Carnero, A.J.L.; Gabri, M.R.; Cinat, G.; Koliren, L.; Lewi, D.; Fernandez, L.E.; Alonso, D.F.; Gómez, D.E.; et al. Active Specific Immunotherapy of Melanoma with a GM3 Ganglioside-Based Vaccine. J. Immunother. 2004, 27, 442–451. [Google Scholar] [CrossRef]
- Neninger, E.; Diaz, R.M.; de la Torre, A.; Rives, R.; Diaz, A.; Saurez, G.; Gabri, M.R.; Alonso, D.F.; Wilkinson, B.; Alfonso, A.M.; et al. Active immunotherapy with 1E10 anti-idiotype vaccine in patients with small cell lung cancer: Report of a phase I trial. Cancer Biol. Ther. 2007, 6, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Gabri, M.R.; Cacciavillano, W.; Chantada, G.L.; Alonso, D.F. Racotumomab for treating lung cancer and pediatric refractory malignancies. Expert Opin. Biol. Ther. 2016, 16, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Bitton, R.J.; Guthmann, M.D.; Gabri, M.R.; Carnero, A.J.L.; Alonso, D.F.; Fainboim, L.; Gomez, D.E. Cancer vaccines: An update with special focus on ganglioside antigens. Oncol. Rep. 2002, 9, 267–276. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11836591 (accessed on 4 April 2002). [CrossRef] [PubMed]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; Mcardle, S.E.B.; Klinman, D. Cancer Vaccines: Adjuvant Potency, Importance of Age, Lifestyle, and Treatments. Front. Immunol. 2021, 11, 3850. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Sondak, V.K.; Smalley, K.S.M.; Kudchadkar, R.; Grippon, S.; Kirkpatrick, P. Ipilimumab. Nat. Rev. Drug Discov. 2011, 10, 411–412. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Mohammad, S.; Kashani, A.; Hosseini-fard, S.R. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Lee, J.B.; Kim, H.R. Immune Checkpoint Inhibitors in 10 Years: Contribution of Basic Research and Clinical Application in Cancer Immunotherapy. Immune Netw. 2022, 22, e2. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; Abaskharoun, M.; Pharm, D.; Hamilton, M.; Keidel, S.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Classification of Glycan | Type of Cancer |
|---|---|---|---|
| Globo H | Fucα1-2Galβ1-3GalNAcβ1-3Galα1-4Galβ1-4Glcβ | Globo Series | Breast, Uterus, Ovary, Prostate, Lung, Liver, Colon |
| SSEA-3 | Galβ1-3GalNAcβ1-3Galα1-4Galβ1 | Globo Series | Breast, NSCLC, Colon |
| SSEA-4 | Neu5Acα1-3Galβ1-3GalNAcβ1-3Galα1-4Galβ1-4Glcβ | Globo Series | Breast, Glioblastoma, Prostate |
| GD2 | GalNAcβ1,4(Neu5Acα2, 8Neu5Acα2,3)Galβ1, 4Glcβ1Cer | Gangliosides | Neuroblastoma, Melanoma, Retinoblastoma, Breast |
| GD3 | Neu5Acα2,8Neu5Acα2,3Galβ1, 4Glcβ1Cer | Gangliosides | Neuroectodermal, Melanoma |
| NeuGcGM3 | Neu5Gcα2-3Galβ1-4GlcβCer | Gangliosides | Melanoma, Neuroblastoma, Retinoblastoma, Colon, Bladder, NSCLC |
| Fuc-GM1 | Fucα1-2Galβ1-3GalNAcβ1-4(Neu5Acα2-3)Galβ1-4GlcβCer | Gangliosides | SCLC |
| Lex | Galβ1-4(Fucα1-3)GlcNAc- | Lewis antigens | Bladder, Hepatic, Breast, Glioblastoma |
| SLex | Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAc | Lewis antigens | Glioma, Gastrointestinal |
| SLea | Neu5Acα2-3Galβ1-3(Fucα1-4)GlcNAc | Lewis antigens | Pancreatic, Gastric, Endometrial, Colon |
| Ley | Fucα1-2Galβ1-4(Fucα1-3)GlcNAc- | Lewis antigens | NSCLC, Hepatocellular, Ovarian |
| Tn | GalNAcαSer/Thr | O-glycans | Cervix, Ovarian, Breast, Prostate, Colon |
| STn | Neu5Acα2-6GalNAcαSer/Thr | O-glycans | Gastric, Endometrial, Bladder |
| TF | Galβ1-3GalNAcαSer/Thr | O-glycans | Ovarian, Prostate, Breast, Colon, Stomach and Bladder |
| Polysialic acid | α2,8-/α2,9 NeuAc | Polysialylated | NSCLC, Breast, Glioblastoma |
| Name | Peptide Size (Feature) | Status Reached | Carbohydrate- Reactive Immune Response | References |
|---|---|---|---|---|
| 47-LDA | 10-mer | Preclinical | GD2 | Kowalczyk, A. et al., 2007 [165] |
| Wierzbicki, A. et al., 2008 [166] Gil, M. et al., 2009 [167] | ||||
| Set of five peptides | 12-mer (internal disulfide bridge) | Preclinical | GD2 | Horwacik, I. et al., 2006 [156] |
| GD3P4 | 15-mer | Preclinical | GD3 | Popa, I et al., 2006 [157] |
| D2 | 15-mer (eight peptides in a single molecule) | Preclinical | TF | Heimburg-Molinaro, J. et al., 2009 [158] |
| P10s | 15-mer | Phase I | GD2 and Ley | Hutchins, L. F. et al., 2017 [163] |
| Makhoul, I et al., 2021 [164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segatori, V.I.; Ferreira, G.M.; Rojo, S.; Nogueira, A.C.; Castillo, J.O.; Gulino, C.A.; Gabri, M.R. Mimicry of Tumour-Associated Carbohydrates: Is It a Promising Option for Cancer Treatment? Immuno 2023, 3, 122-147. https://doi.org/10.3390/immuno3020009
Segatori VI, Ferreira GM, Rojo S, Nogueira AC, Castillo JO, Gulino CA, Gabri MR. Mimicry of Tumour-Associated Carbohydrates: Is It a Promising Option for Cancer Treatment? Immuno. 2023; 3(2):122-147. https://doi.org/10.3390/immuno3020009
Chicago/Turabian StyleSegatori, Valeria Inés, Gretel Magalí Ferreira, Selene Rojo, Aylen Camila Nogueira, Jeremías Omar Castillo, Cynthia Antonella Gulino, and Mariano Rolando Gabri. 2023. "Mimicry of Tumour-Associated Carbohydrates: Is It a Promising Option for Cancer Treatment?" Immuno 3, no. 2: 122-147. https://doi.org/10.3390/immuno3020009
APA StyleSegatori, V. I., Ferreira, G. M., Rojo, S., Nogueira, A. C., Castillo, J. O., Gulino, C. A., & Gabri, M. R. (2023). Mimicry of Tumour-Associated Carbohydrates: Is It a Promising Option for Cancer Treatment? Immuno, 3(2), 122-147. https://doi.org/10.3390/immuno3020009