Abstract
Framework silicates form about 70% of the Earth’s crust, mainly feldspars ~50–60% and quartz ~10–15%. Less-abundant feldspathoids include nepheline-, leucite-, and sodalite-group minerals, rich in structurally challenging properties. This review paper deals with anhydrous feldspar-, nepheline-, and leucite/pollucite groups, emphasising the importance of parallel studies on natural and synthetic samples. Four topics are covered. For decades, petrologists have analysed nephelines and recalculated their compositions as endmember molecules but, by not following rules of stuffed-tridymite crystal chemistry, have not estimated reliably the excess SiO2 present in solid solution. Some materials scientists make similar mistakes, and a new approach is described here. Synthesis studies of analogue feldspars, nephelines, and leucite/pollucites led to collaborative studies, mainly using laboratory and synchrotron X-ray powder diffraction methods at room and elevated temperatures, to study thermal expansion and displacive phase transitions. Such work was recently expanded to address the spontaneous strain relations. Topics covered here include work on nepheline/kalsilite analogues in the system SrAl2O4—BaAl2O4; thermal expansion of (K,Na)Al-, RbAl-, RbGa-, and SrAl-feldspars; and thermal expansion and phase transitions in analogue leucites KGaSi2O6 (tetragonal to cubic) and K2MgSi5O12 (monoclinic to orthorhombic). Results are reviewed in the context of research published in mineralogical and more-widely in physical sciences journals.
1. Introduction
Aluminosilicate minerals make up more than 50% of the Earth’s crust but are essentially absent in the deeper zones. Feldspars make up the vast proportion of this and mainly consist of alkali feldspar solid solutions between the compositional “endmembers” albite (NaAlSi3O8) and orthoclase (KAlSi3O8) and plagioclase solid solutions between albite and anorthite (CaAl2Si2O8). Both mineral groups show high-temperature and low-temperature structural varieties which provide crucial information for the elucidation of the conditions of rock formation, particularly for igneous conditions (both magmatic and post-magmatic in nature). Much less abundant are feldspar varieties celsian (BaAl2Si2O8), slawsonite (SrAl2Si2O8), rubicline (RbAlSi3O8), reedmergnerite (NaBSi3O8), and buddingtonite (NH4AlSi3O8); all of these species form during late-stage magmatic or hydrothermal processes or are associated with low temperature sedimentary rock alteration. However, a very interesting occurrence of an arsenic-rich volcanic sublimate feldspar (named filatovite) is known which has variable compositions in the solid solution series between high-temperature KAlSi3O8 (sanidine) and a hypothetical phase of formula K(Al,Zn)2(As,Si)2O8 (see below). Much less abundant aluminosilicate framework minerals are anhydrous feldspathoids: nepheline (Na3KAl4Si4O16), kalsilite (KAlSiO4), leucite/pollucite (KAlSi2O6/CsAlSi2O6), and sodalite (Na8Al6Si6O24·Cl2) mineral groups (which occur mainly in late-stage igneous rocks and metasomatic equivalents) and zeolites (hydrous minerals with many compositional varieties occurring as alteration products of igneous felsic minerals and volcanic glasses), in low-temperature metamorphic rocks, and in altered sediments. Feldspar-, nepheline-, and leucite/pollucite-groups of minerals all show chemical and structural varieties which include natural mineral and synthetic analogues; these are reflected in very interesting physical properties including compositional-, thermal-, and high pressure- phase transitions. Indeed, physical and chemical properties have led to very widespread industrial and environmental applications for feldspars and feldspathoid minerals and bulk rocks to semiconductors, glass making/ceramics, building and construction, and functional fillers in paints and composites [1]. Specific examples include nepheline and leucite in glass ceramics [2,3,4]; feldspar and nepheline in pottery, porcelain, and sanitary ware [5,6,7]; ionic and thermal conductivity [8,9,10]; catalysis and molecular sieving [11,12,13]; radioactive waste disposal [14,15,16]; dental ceramics [17,18,19]; and geophysical surveying [20,21,22].
Over the years, together with colleagues and collaborators, I have studied the stability and structural relationships of natural and synthetic framework alumino-silicates depending on composition and thermal expansion including samples belonging to feldspar, nepheline, leucite, and sodalite mineral groups. Such framework materials are characterized by having fully polymerized structures where all oxygens of tetrahedrally coordinated cations are bridging oxygens joining adjacent tetrahedra to build up the 3-dimensional framework. Minerals with only SiO4 tetrahedra as the “primary building unit” have essentially pure-SiO2 bulk compositions (e.g., quartz, tridymite, and cristobalite) and contain no interframework (cavity) cations occupying the “holes” in these structures. By contrast, compounds with Al replacing Si in some tetrahedral sites have cations entering the cavities to restore electrostatic neutrality; each mineral group tends to have distinctive linkages of the tetrahedral units to build up different types of “secondary building units” (SBU’s). This type of research is aided by interdisciplinary studies which results in publications in journals covering science areas ranging solid state chemistry, condensed matter physics, materials science, and mineral chemistry and physics. The new journal should cover all of these areas and in this invited paper I will deal with representative problems involved in studying the chemistry and structure of the anhydrous silicate mineral/materials groups feldspar, nepheline, and leucite and will try to keep “narrow” terminology to a minimum.
2. Research Topic 1: Natural Nepheline Chemistry and Structural Formulae
Context. The nepheline family of crystalline materials contains both natural minerals and synthetic compounds. All of these are variants (defined as hettotypes) of an ideal structure (defined as the aristotype) based on the hexagonal high-tridymite tetrahedral framework of the SiO2 polymorph. The aristotype has equal sized hexagonal interframework cavity sites formed by six-membered rings of SiO4 tetrahedra. The natural mineral kalsilite (KAlSiO4, note that its name is based on the formula cation symbols) is a hettotype of this ideal structure with essentially equal-sized cavity sites although distortions of the rings and tetrahedral tilting may reduce their size. The nepheline framework is collapsed around the cavity sites occupied by the smaller Na cations, so that distortions lead to the development of two different sizes and shapes of one of the sites (see later). Within these six-rings, Si and Al are ordered into adjacent tetrahedra which also point “up” and “down” alternately; these hexagonal rings are stacked along the c axis via linked SiO4 and AlO4 tetrahedra (cf. Al-avoidance). Compounds with these types of framework geometries are described as having stuffed-tridymite structures [23]. In this nepheline-based topic, the recalculation of wt.% mineral analyses into endmember molecules will be considered first followed by discussion of phase relations and thermal expansion properties and phase transitions of synthetic analogues in the next major section (research topic 2).
The chemical compositions of natural nephelines are conventionally reported as wt.% oxides, and most authors now calculate atomic proportions of cations on a 32 oxygen basis equivalent to the unit cell composition which contains 8 formula units of the fundamental nepheline endmember NaAlSiO4 (denoted Ne here). However, over the years, some authors have reported the proportions of chosen endmember mineral molecules on a wt.% basis while others have used mole % values for such components, but often the reported method used is not specified. Experimentally determined phase diagrams are conventionally used to display liquidus isotherms, crystal–silicate liquid field boundaries, and equilibrium coexisting mineral tie lines; such phase diagrams are always determined and discussed as wt.% systems, and this is perhaps why many igneous petrologists reported phase analyses using wt.% endmember mineral proportions. In particular, much attention was paid to determining the magmatic and sub-solidus phase stabilities of silica minerals, feldspars and feldspathoids in silica-saturated and silica-undersaturated bulk compositions with the stability relations in the quartz (SiO2 denoted Qz)—nepheline (Ne)—kalsilite (denoted Ks) system displayed in the so-called Petrogeny’s Residua System (PRS, Schairer and Bowen, 1935) [24].
Electron microprobe studies of coexisting feldspars and feldspathoids in magmatic rocks provided new data, and authors continued to report compositions as wt.% proportions of endmember minerals and plotted these in the wt.% Qz—Ne—Ks (PRS) phase diagram. Reporting the wt.% of endmember mineral molecules is an unambiguous procedure, but differences emerge when recalculating mole % proportions of these molecules, which results in different values being obtained by different authors for similar composition nephelines. Although different sets of data are all on a mole % basis, it is clear that different calculation procedures have been used whereby one method might have followed the strict rules of nepheline stoichiometry and structure while others did not. In this first section of the paper, I will go back to first principles in an attempt to decipher the different calculation procedures that have been used and to develop a standardized, rigorous and logical way of dealing with this problem.
2.1. Background to a New Approach on Reporting Nepheline Compositional Relationships
Early chemical analyses of nepheline date back to the early 19th century, and by the early 20th century, many analyses, obtained by classical, gravimetric “wet” methods on mineral separates from coarse grained rocks had been published. In a key paper, Bannister and Hey (1931) [25] gave new analyses, 6 from the volcanic region of Mt Somma, Vesuvius, which involved use of micro-analytical techniques with as little as 64.1 mg for one sample. This was before the crystal structure had been determined using single-crystal X-ray diffraction techniques, but they combined their chemical analyses, densities, optical properties, and unit cell dimensions to deduce that the unit cell atomic formulae should be calculated to 32 oxygen atoms. On that basis, they found that Si + Al averaged 15.995, consistent with 16 framework cations per unit cell. They also calculated 32 oxygen atomic formulae for about 100 nepheline analyses published over the previous 130 years and found that the sum of the atoms of Na, K, and Ca was closely balanced by the amount of Al [i.e., (2Ca + Na + K) − Al] averaged ~0.02 per 32 oxygens [25]. This relationship points to each divalent Ca being complexed with 2 Al atoms, compared with 1 Al for each univalent cavity cation. The available cavity cation sites remain the same implying that entry of a Ca atom must be accompanied by a linked cavity cation site vacancy. For all of the analyses considered by Bannister and Hey [25], the Si/Al averaged 1.10 and is more siliceous than the (Na,K)AlSiO4 composition with about 10 atom % of excess Si. This reflects the atomic substitution Si → Na + Al, and if one extra Si replaces one Al in the framework, one less Na is present in the cavity (per 32 oxygens) pointing to formation of a linked vacancy in the cavity site (see below). Many researchers had pointed out that natural nephelines had Si > Al in the tetrahedral framework (i.e., the T sites) and the eminent, pioneer experimental petrologist/mineralogist N.L. Bowen (1912a) [26] reported a mean formula of (Na,K)8Al8Si9O34 ([i.e., (Na,K)8Al8Si8O32 + SiO2; note that at that time, he didn’t realise that the ideal formula should be reported to 32 O], which is equivalent to the presence of 10 mol % excess silica. Bowen was the first researcher to attribute this to the solid solution of albite feldspar (NaAlSi3O8) in nepheline. It was subsequently determined that the limit of solid solution of albite in Na-nepheline at 1 bar pressure dry is ~33 wt. % (Ne67Ab33, wt.%) [27], equivalent to Ne85Qz15 (wt.) and Ne83Qz17 (mol.).
The literature survey of nepheline compositions in [25] showed a significant Ca component, and Bowen (1912b) [28] had earlier experimentally determined at 1 bar that the maximum amount of solid solution of anorthite in nepheline is ~35 wt.% (Ne65An35 and ~7 wt.% CaO) equivalent to Ne65.5An34.5 (mol.) Experimental studies for the system NaAlSiO4—CaO·Al2O3 at 1 bar showed that nepheline could form solid solutions with at least 60 wt.% of CaAl2O4 [29], but there is little evidence that natural nephelines could have Ca present as that component.
The first single crystal X-ray diffraction study of a natural nepheline (from Mt Somma, Vesuvius) was published in 1955 (Hahn and Buerger) [30]; no analysis was given, and they assumed the ideal composition Na3K[Al4Si4O16] (where square brackets denote an 8 T-site tetrahedral framework composition). The structure was hexagonal (P63) with Si and Al ordered into separate sites, and two distinct cavity cation sites were reported, consisting of a single larger pseudo-hexagonal site (occupied by K) and three smaller more distorted sites (occupied by Na). They were not specific on which site might be occupied by Ca [30]. Buerger (1954) [23] had already suggested that the framework consisted of linked, staggered six-rings of tetrahedra based on the structure of high-tridymite with the Na, K, Ca and associated vacancies occupying the cavity cation sites which are defined by the holes in the six-ring, polyhedral framework; this confirmed Bannister and Hey’s [25] earlier prediction. Buerger (1954) [23] described the structures of nepheline and kalsilite using the term “stuffed-tridymite” and also described carnegieite (high-temperature NaAlSiO4) as a “stuffed cristobalite”.
Donnay et al. (1959) [31] later used powder X-ray diffraction methods to assess changes in hexagonal unit cell parameters in 4 series of synthetic nepheline-structured solid solution series: NaAlSiO4 (Ne)—KAlSiO4 (Ks), Ne—NaAlSi3O8 (albite, denoted Ab), Ne—CaAl2Si2O8 (anorthite, denoted An), and Ne—CaAl2O4; both the Ne—Ab and the Ne—An series would have vacancies present in the cavity sites. They also suggested that any Fe3+ present substitutes for Si and Al in tetrahedral coordination, while the small amounts of Mg, Mn, and Ti that might be present would substitute for Na, K, or Ca (but see below). Hamilton and MacKenzie (1960) [32] reported on the results for the experimental determination of nepheline compositions in the system NaAlSiO4—KAlSiO4—SiO2 (Qz) at 1 kbar water pressure (PH2O); this is the Ne—Ks—Qz (PRS) system [24], which has been used for decades to consider the melting and crystallization relations of igneous rocks rich in silica minerals, alkali feldspars [NaAlSi3O8 (Ab)—KAlSi3O8 (orthoclase, denoted Or)], nepheline group minerals, and leucite (KAlSi2O6) (i.e., “salic” magmatic rocks). Hamilton and MacKenzie [32] plotted a mole % version of the ternary Ne—Ks—Qz system in terms of the compositions Na8Al8Si8O32 (8Ne)—K8Al8Si8O32 (8Ks)—8SiO2 (8Qz). In that diagram (their Figure 6), they drew the theoretical nepheline solid solution join from Na6K2Al8Si8O32 (ideal naturally occurring nepheline) to a point at 40% of the Qz component, which is labelled Na6□2Al8Si10O32 on the Na8Al8Si8O32 (8Ne)—8SiO2 join; note that the Qz corner has 8 Si atoms matching the Si content of the Na and K-component corners. The number of oxygens of the Qz corner is half of that defined for an ideal 32 oxygen stuffed tridymite unit cell, and thus, the excess Qz solid solutions do not match what is defined here as strict nepheline stoichiometry. If they had used a Qz component with 32 oxygens (16SiO2), the Ne-Q endmember would have been correctly plotted at Ne75Q25 rather than the Ne60Q40 that they show (see [33]).
Barth (1963) [34] tackled the need to define different nepheline endmember components with the same numbers of oxygens per formula unit (pfu) and to take account of vacancies in the cavity sites. Thus, he suggested that the nepheline structure contains four mineral molecules plus “holes” (vacant interframework cavity sites). The tetrahedral T cations and cavity cation sites plus holes total 24 sites per 32 oxygens as follows:
| 24 Ne (nepheline, NaAlSiO4) | Na2 | Na6 | Al8 | Si8 | O32 |
| 24 Ks (kalsilite, KAlSiO4) | K2 | K6 | Al8 | Si8 | O32 |
| 24 An′ (20 An anorthite, CaAl2Si2O8 + 4 □ ) | □4 | Ca4 | Al8 | Si8 | O32 |
| 24 Q’ (16 Q tridymite, SiO2 + 8 □) | □2 | □6 | Si8 | Si8 | O32 |
Two cavity cation sites represent two distinct interframework cations in natural nepheline [30]. Barth concluded that the “holes” are potential sites for cavity cations and that these “will be reckoned as cations and included in the equivalent molecular percentages” but he did not always follow that “rule”. Barth ([34]; his Figure 2) also used the quaternary system Ne—Ks—Q’—An’ to define a triangular “compositional plane for natural nephelines” with corners defined by the components Na6K2Al8Si8O32, Na6□2Al6Si10O32, and Na4Ca2□2Al8Si8O32; that figure correctly shows the second and third components at Ne75Q’25 and Ne50An’50, respectively, consistent with their vacancy contents.
The presence of other chemical components in the nepheline structure should also be considered. Ferric iron (Fe3+) substituting for Al in the tetrahedral sites is commonly found in chemical and microprobe analyses of nepheline. However, older wet chemical analyses of nepheline separates from rocks also show very small amounts of Mg, Fe2+, Mn and Ti, but these components might result from grain impurities and/or alteration products [35]. However, it is well known that synthetic leucite analogues occur with a variety of divalent cations (e.g., Mg, Zn, Co, Fe2+, Cd) in the polymerized framework of general formulae K2M2+Si4+5O12 [36,37,38,39]). Such leucite analogues show the coupled substitution 2Al3+ → M2+ + Si4+. In particular, note that the synthesis of hexagonal nepheline-like phases of composition K2MgSi3O8 [36] and K2Fe2+Si3O8 showing the same type of tetrahedral cation substitution [40] have been reported; also see Buerger [23]. Other synthetic silicate phases have stuffed-tridymite nepheline structures with linked 6-rings of tetrahedra containing divalent cations and Si in separate sites, e.g., BaMSiO4 with M = Co, Zn, Mg [41]. Grain impurities can be avoided using the electron microprobe [35], and if found using high-quality microprobe analysis, it is likely that small contents of Mg, Mn, and Ti occupy the nepheline framework and that all Ca replaces Na in the cavity sites. Note that complete solid solution in the system SrAl2O4—BaAl2O4 shows that Sr and Ba occupy cavity sites in kalsilite-like structures which have stuffed tridymite topology (Henderson and Taylor, 1982) [42]. Thus, it will be assumed here that smaller divalent cations (e.g., Mg, Mn, and Fe2+ if some of the Fe is known to be in this reduced form) and Ti will replace Si, Al (and Fe3+) in the T sites, whereas larger divalent cations (Ca, Sr, Ba) and Rb [43] will replace Na and K in the cavity sites. In this paper, the calcic endmember molecule will be referred to as CaNe (calcium-Ne, formula □Ca0.5Ca0.5AlSiO4) rather than An’ [34] and total excess silica as Q’ (□SiSi2O4).
2.2. Calculation of Nepheline Endmember Molecules
Based on a stuffed-tridymite unit cell with 32 oxygens, the unit cell of nepheline contains a total of 24 cations, 16 of which are in the tetrahedral framework, and 8 cavity cations or vacancies occupy the interframework sites; these proportions define strict nepheline stoichiometry. The sodic endmember is defined to have the unit cell formula Na8Al8Si8O32, but the simplest nepheline formula is still written as NaAlSiO4, with 8 formula units per unit cell. The atomic occupancies in the intermediate compositions all follow the stuffed-tridymite structure.
2.2.1. Ne—Qz Solid Solution Series
The data for the full series are given in Table 1. The atomic occupancies in the intermediate compositions all follow the stuffed-tridymite structure. Thus, if a “solid solution” contains 90 mol % Ne, it will have 7.2 atoms each of Na, Al, and Si. The Si + Al atoms in the T site total 14.4 atoms pfu leaving 1.6 excess Si atoms (Si’) to make up the T sites to 16.0. The cavity cation site will contain 7.2 Na and to make this site up to 8 atoms pfu, there must be 0.8 vacant sites pfu. The first column gives the compositions as mol % proportions of Ne and Qz at 10% intervals together with intermediate compositions with integral numbers of vacancies. The next columns are the number of vacancies associated with Qz (□Si), then numbers of atoms of Na, Al, and Si in Ne, followed by the atoms of “excess” Si (denoted Si’ here). Note that the vacancy number is exactly Si’/2 and the sum of Na, Al, Si, Si’, and vacancies is exactly 24 for stoichiometric nepheline solid solutions. The next 4 columns give formulae for calculating the proportions of Ne and excess silica (denoted Q’ here) based on the cavity occupancies. Molecular proportions can also be calculated as follows:

Table 1.
Stoichiometric nepheline compositions in the system Ne—Qz.
Mol % Ne = [(atoms Na) × 100]/(atoms Na + □Si ) or
= (atoms Na) × 100/8
and considering the complete 24 atom unit cell as follows:
Mol % Ne = [(atoms Na + Al + Si)Ne × 100]/[(atoms Na + Al + Si)Ne + Si’ + □Si ]
or for Na = Al = Si in stoichiometric NaAlSiO4:
= (atoms 3Na)Ne × 100/(atoms 3Na + 1.5Si’) or
= (atoms 3Na)Ne × 100/24
and
Mol % Q’ = [(atoms Si’ + □Si) × 100)]/[(atoms Na + Al + Si)Ne + Si’ + □Si ] or
= (atoms 1.5Si’) × 100)/(atoms 3Na + 1.5Si’) or
= (atoms 1.5Si’) × 100)/24
Because endmember Ne has numbers of Al and Si fixed to the Na (or 2Ca and K) content the excess Si can be simply defined as the excess of Si over the Al content; both of these make up the 16-atom framework, and one more equation can be defined to calculate the mol % Q’. Thus:
Mol % Q’ = (Sitotal – Al) × 100/16
The next columns show the masses for each of the oxide components recast as wt.% of Na2O, Al2O3, and the sum of SiO2 in Ne and Q’; these values are the input for the method to check that the calculations of wt % analysed oxides in nephelines provide the correct answers which match the defined atomic numbers for each sample in Table 1. Note that the formula Na6□2Al6Si10O32 occurs for the bulk composition Ne75Q’25 mol. % (see above).
2.2.2. Ne—An’ (CaNe) Solid Solution Series
A similar approach is used to assess the Ne—CaNe system and Table 2 shows the atomic compositions for this system at 10 % intervals for 32 oxygens. The endmembers have 8 Na and 4 Ca; all members have 8 Al and 8 Si, and the cavity sites are filled to total 8 by the presence of 4 vacancies in the Ca endmember. Thus, for strict nepheline stoichiometry, the number of Ca atoms is matched by the same number of vacancies. As stressed by Barth (1963), calculation of molecular endmembers requires combining vacancy numbers and related cation numbers. Thus, the molecular composition can be calculated for the cavity occupants (total 8) from atom numbers in column 8, Table 2 as follows:
Table 2.
Stoichiometric nepheline compositions for the system Ne—An’.
% Ne = (atoms Na × 100)/(atoms Na + Ca + vacancies); or (atoms Na × 100)/(atoms Na + 2Ca); or (atoms Na × 100)/8
and in column 9:
% CaNe = [(atoms Ca + □Ca) × 100]/(atoms Na + Ca + □Ca); or = [(atoms 2 × Ca) × 100] /(atoms Na + 2Ca); or = [(atoms 2 × Ca) × 100] /8).
Based on the full 24-cation plus vacancies data in column 12:
% Ne = [(atoms 3 × Na)Ne × 100]/[(3 × Na)Ne + (5 × Ca)An + □Ca]; or [(atoms 3 × Na)Ne × 100]/[(3 × Na)Ne + (6 × Ca)CaNe]; or [(atoms 3 × Na)Ne × 100]/24;
and in column 11:
% CaNe = [(atoms 5 × Ca + □Ca) × 100[/(atoms 3 × Na + 5 × Ca + □Ca); or = [(atoms 6 × Ca) × 100]/(atoms 3 × Na + 6 × Ca); or = (atoms 6 × Ca) × 100/24
As mentioned above, Barth [34] stressed that the number of vacancies associated with both Q’ and the Ca component (which he treated as anorthite; note that I use the name CaNe) had to be included as equivalent to cations to calculate nepheline formulae. However, he calculated the Ca as An using the terms 5 × Ca (representing Ca + 2Al + 2Si) but using that method to calculate the Ca component leads to an endmember containing only 83.33% of the chosen component (see column 10, Table 2). In other words, the vacancy associated with Ca had not been included. Thus, the Barth method [34] gives a proportion of CaNe, which is 1/6 too small and the vacancy number associated with excess Q’ is 1/6 Ca too high.
In the next 4 columns wt.% data for Na2O, CaO, Al2O3, and SiO2 are given.
2.2.3. Ne—An’ (CaNe)—Q Solid Solutions
Table 3 gives atom and vacancy numbers for representative ternary compositions in the system Ne—Q—CaNe based on a 32-oxygen unit cell (Z = 8). The columns in order are vacancies associated with entry of Ca and excess Si, then atoms of Na and Ca, followed by Al and Si in Ne and CaNe, and finally “excess” Si (i.e., Si’). The next columns give calculated mol. % values for Ne, An’ (CaNe), and excess silica (Q’) which are most conveniently calculated on a 24 atom basis as follows.
Table 3.
Stoichiometry for nepheline compositions in system Ne-An-Qz.
% Ne = (3 × Na) × 100/24; % An’ = (6 × Ca) × 100/24; % Q’ = (24 − 3Na − 6 Ca) × 100/24
The last columns give wt.% Na2O, CaO, total Al2O3 (in Ne and An’), and total SiO2 (in Ne, An, and Q’)
2.2.4. Natural Nephelines
For potassium bearing samples, it is simple to add a term for the Ks component to the equations developed above. Thus, based on [34], the key points to remember here are that, for strict nepheline stoichiometry, the vacancies derived from the CaNe component can be set to the Ca atomic content (which is known from the analyses of natural minerals recalculated on a 32 O basis) while those associated with the Q content are represented by half of the atoms of excess Si’ calculated from the analyses as follows: excess Si (Si’) = total Si atoms − 3 × Na − 3 × K − 6 × Ca. In considering the occupancy of the cavity cation site, the vacancies associated with the entry of Ca and with the excess Si’ content must both be considered as “virtual” cavity cations. However, other researchers have used different oxygen bases and/or endmember components. Peterson [44] used an 8-oxygen basis to define the endmember components Ne (Na2Al2Si2O8), Ks (K2Al2Si2O8), Nf (Na2Fe3+2Si2O8) (i.e., iron nepheline), An (CaAl2Si2O8), Qz (Si4O8), and Cn (Al16/3O8); the last component (in effect a theoretical corundum [Al2O3] content) was introduced in an attempt to account for analyses so rich in Na and K that deficiencies were reported for both Al and Si. Rossi et al. [45] used a 32 O basis and endmembers K2Na6Al8Si8O32, Na2Na6Al8Si8O32 and Ca□CaNa6Al8Si8O32, and more recently, Blancher et al. [33] reported nepheline endmembers based on 4-oxygen cells as NaAlSiO4, KAlSiO4, □SiSi2O4, and □Ca0.5Ca0.5AlSiO4. Hamada et al. [46] also reported nepheline endmembers based on 4-oxygen cells as NaAlSiO4, KAlSiO4, □SiSi2O4, and □Ca0.5(Ca,Mg)0.5AlSiO4; for the last component, it was assumed that Mg occupied cavity sites, but this approach is not followed here (see above).
The main nepheline endmembers chosen here are based on the 32 O unit cell and are defined as NaAlSiO4 (Ne), KAlSiO4 (Ks), □Ca0.5Ca0.5AlSiO4 (CaNe), and □SiSi2O4 (Q’); the equations developed for calculating these components are summarised in Table 4. This table also gives the equation for a minor component defined by its content of divalent tetrahedral cations (e.g., Mg, Mn, and Fe2+); this potential endmember would have the formula K8M2+4Si12O32 with M = Mg + Fe2+ + Mn (see above) where Mg tends to be the most concentrated divalent component reported in natural nephelines. Based on the simplest formula for this component of KM2+0.5Si1.5O4 (8 per equivalent nepheline unit cell), the proportion of the KsM molecule would be based on 0.5 atoms of M2+.
Table 4.
Formulae for calculations based on 32 oxygen nepheline structure unit cell.
Equations (1) and (2) (Table 4) give the excess Si (Si’a and Si’b), and Equations (3)–(6) the Qxs, Ne, Ks, and CaNe values taking account of essential vacancies in the cavity cation sites. The divalent tetrahedral cation endmember KsM is calculated with Equation (7). Equation (8) provides another estimate of total excess silica (QSi) which is based on a 16 T-atom framework; the actual % of Si in the framework is given by Si’ = (Sitotal − Na − K − 2Ca) × 100/24 and the amount of vacancy associated with the excess Si component □Si = (8 − Na − K − 2Ca) × 100/24. These Si parameter proportions are 3:2:1, which are fixed by the ideal nepheline stoichiometry and structural symmetry. Equation (9) provides a fourth estimate for the total excess silica (Q(Si-Al)) for a 16 T-atom framework; this parameter does not depend on the reliability of analyses for the cavity cations.
Endmember proportions can also be described from the occupancy of the cavity cation sites alone (Equations (10) and (12)–(14)), which give the same values as Equations (3)–(6). Equation (11) provides another estimate of the total excess silica proportion (Qcav2) based on the total cations. For ideal stoichiometry, all calculated total excess silica values should be identical; different values will depend on the scale of non-stoichiometry and/or the presence of analytical errors. Note that the second value defined for Qcav would be influenced by any analytical error for Al (Equation (11)).
Departures from stoichiometry based on variations of the ratio (Al + Fe3−)/(Na + K + 2Ca) from zero should also be considered. Thus the endmember molecules calculated for natural nephelines by Peterson [44] provide values for both excess and deficient Si and Al molecules which, based on the 32 O cell used here, have the ideal formulae □8Si16O32 and □2.666Al21.333O32; the percentage values for these are simply calculated as [(total Si − Na − K − 2Ca) × 100]/16 and [(Al − Na − K − 2Ca) × 100]/21.333, respectively. In considering an excess or deficient content of Al (+ Fe3+) compared with the Na, K, and Ca content, analytical error must be considered; thus, an excess of Al could be related to alkali “loss” in the electron microprobe if a defocussed beam or raster scan is not used [47,48]; a lower-than-normal beam current is also essential. If the analysis conditions are appropriate, an excess of alumina (Al2O3) implies that the framework contains both tetrahedral and octahedral Al sites, perhaps similar to an anhydrous, hexagonal kappa- or chi- alumina phase [49,50]). A small amount of such a molecule in solid solution might be possible, but the occurrence of significant interstitial octahedral Al cations is less likely. Although some CaAl2O4 in solid solution might be possible [28,31], this would not explain a deficiency in the delta Al parameter [atoms (Al + Fe3+) − (2Ca + Na + K)]. Another possibility is that a small amount of an alkali-rich and Al and Si poor alteration phase is present in such natural nephelines. The most probable alteration mineral would be cancrinite, which has a hexagonal structure with six-rings of tetrahedra similar to those of nepheline, although the linkages of these units are different in the two mineral groups. The general formula for the cancrinite group is (Na,K)6Ca2[(Al,Si)12O24](CO3, SO4, Cl, OH)2. nH2O which on a 32 O basis would be (Na,K)8Ca2.66[(Al,Si)16O32](CO3, SO4, Cl, OH)2.66. nH2O. This is chemically similar to nepheline but is structurally different with the extra Na, Ca and large anions occupying “cancrinite cages” and large continuous channels (Deer et al. 2004, their Figure 245) [51]; also see [52]). However, in that case, any extra Ca—large anion complex would NOT be associated with entry of an equivalent vacancy in a cavity site. Some cancrinite species are stable at high temperatures [53], and experimental studies have shown the presence of a triple point with nepheline, cancrinite, and silicate melt coexisting at ~950 °C, 2 kbar [54].
2.3. Assessment of Igneous Rock Nepheline Compositions
Based on recalculation of nepheline analyses to a 32-oxygen nepheline unit cell, Bannister and Hey [25] pointed out that nepheline compositions should have a valence sum for cavity cations closely balanced by the number of tetrahedral trivalent cations; the samples studied showed that 2Ca + Na + K was in excess of the total Al + Fe3+ by an average of about +0.02. If there is an excess of cavity cation charges over the total trivalent tetrahedral cation sum, the positive charge from the excess Na+ could be balanced by Si-O components of the framework, but that would imply disruption of some Si—O—Si linkages. Any Na-O-Si linkages could be similar to those occurring in beta sodium disilicate (β-Na2Si2O5); this compound exists as the natural mineral natrosilite, which is a monoclinic, pseudo-hexagonal sheet-like compound with the sheets containing six-rings of SiO44− alternating with layers of 5- or 6-coordinated Na-O polyhedra [55,56]. However, it is more likely that the charge on any excess alkali content could be neutralised by the presence of large anions (e.g., CO32−, Cl) as in cancrinites (see later).
In a key later paper Dollase and Thomas [35] assessed the Bannister and Hey criteria and concluded that the difference between 2Ca + Na + K and Al + Fe3+ (or the difference between total valence sum for tetrahedral cations and the “ideal” value for 16 × mean tetrahedral cation valence) should fall within ± 0.25 atoms per 32 oxygens for acceptable analyses. To assess these chemical controls in more detail, two parameters were provided in Henderson (in press) [57] calculated from cation numbers per 32 O formula units as follows:
∆Alcc = [tetrahedral (Al + Fe3+) − cavity cation (Na + K + 2Ca)]
∆Tcharge = [{(Al + Fe3+) × 3 + (Si + Ti) × 4} − {16 × mean tetrahedral charge for the sample}].
For strict nepheline stoichiometry (unit cell 32 oxygens, 16 T atoms, 8 cavity sites, Si = Al) ∆Alcc = 0 and ∆Tcharge = 0. Henderson [57] showed that for departures from these “ideal” values, delta parameters would be coupled with either positive or negative values, but both should have the same sign and have similar values in the absence of significant analytical errors. The fact that natural nephelines mostly have Si/Al ratios >1.0 has been accounted for by the presence of a feldspar-like component (e.g, NaAlSi3O8) and the same could be said for an anhydrous analcime component (NaAlSi2O6); such solid solutions would of course have stuffed tridymite frameworks (see above). Using “nepheline” 32 oxygen unit cell stoichiometry, ∆Alcc and ∆Tcharge parameters for ideal NaAlSiO4, NaAlSi2O6 and NaAlSi3O8 should have zero values for both delta parameters in such endmembers where the different Si/Al ratios are 1, 2, and 3, respectively. By allowing a very small degree of non-stoichiometry for the Si, Al, or Na components, the ∆Alcc/∆Tcharge ratios appear to have values close to 1.14, 1.09, and 1.07, respectively; the size of the delta values relative to zero reflects their departure from ideal nepheline stoichiometry. Henderson [57] showed that many high-quality nepheline analyses have ∆Alcc/∆Tcharge values of ~1.134, and he considered the significance of that ratio for nepheline solid solutions, and how it is affected by wider limits of solid solution, in particular the variation with excess silica content.
2.3.1. Understanding the Dependence of ∆Alcc/∆Tcharge on Composition
Henderson [57] showed a derivation of the relationship in collaboration with Dr Kevin Knight where N, K, C, Al, S represent the numbers of atoms of Na, K, Ca, Al (or Al + Fe3+), and Si, respectively, per 32 O unit cell (ideal Na-endmember Na8Al8Si8O32). Thus, for strict stuffed-tridymite stoichiometry, the total T-site atoms equal 16, cavity sites equal 8, and the total of cation sites is 24. For 32 oxygens, charge balance requires the constraint N + K + 2C + 3Al + 4S = 64.
It was shown that ΔAlcc = Al − 2C − N − K and that from the charge balance constraint ΔAl = −64 + 4S + 4Al, and this definition is used here as it contains the same variables as ΔTcharge. ΔTcharge is defined as the difference in total charge calculated from the ideal charge [(4S + 3Al)/16] and the total charge per T-site [(4S + 3Al)/(S + Al)]. Thus, ΔTcharge = 16((4S + 3Al)/16 − (4S + 3Al)/(S + Al)). By combining these expressions for ΔAlcc and ΔTcharge, multiplying through and simplifying, the following key equation was defined [57]:
(ΔAlcc/ΔTcharge ) = (64S + 64Al − 4S2 − 8SAl − 4Al2)/(64S + 48Al − 4S2 − 7SAl − 3Al2)
This expression can be recast in terms of S and the difference between S and Al (Al = S − δ) leading to another key equation [57]:
ΔAlcc/ΔTcharge = (128S − 16S2 + 16Sδ − 64δ − 4δ2)/(112S − 14S2 + 13Sδ − 48δ − 3δ2)
For the relationships δ is small, S ~ Al, S ~ 8, 128S ~ 16S2, and 112 ~ 14S2, δ2 is small enough to be ignored and δ can be cancelled in the numerator and denominator:
Thus ΔAlcc/ΔTcharge ~ (128 − 64)/(104 − 48) = (64/56) = 8/7 = 1.1429
Even though the individual ΔAlcc and ΔTcharge parameters are each effectively zero, the strict nepheline stoichiometry still delivers a ΔAlcc/ΔTcharge ratio of 1.1429 for endmember NaAlSiO4!
In the context of a strict stuffed-tridymite stoichiometry defining the nepheline unit cell, the integers 64 and 8 can be rationalised as referring to the ideal tridymite cell, which has a total charge of 64 for 16 quadrivalent Si atoms (total S = 16) in the 32 O unit cell. The unit cell has 8 □SiSi2O4 “nepheline” molecules, and thus 64/8 = 8 per unit molecule. The integers 56 and 7 refer to the Na8Al8Si8O4 species (8 NaAlSiO4 per unit cell) where the T site total is 56 (8 × 4 + 8 × 3) and 56/8 = 7, and 8/7 = 1.1429; the CaNe (and An, CaAl2Si2O8) framework would also have that ratio. Following the same logic for nepheline solid solutions containing excess silica (Si/Al > 1), an albite molecule expressed as having a 32 O nepheline cell would have the formula Na4Al4Si12O32 (total T charge (12 × 4 + 4 × 3) = 60) and 60/8 = 7.5; an anhydrous analcime (NaAlSi2O6) would have the formula Na5.333Al5.333Si10.667O32 ((total T charge (10.667 × 4 + 5.333 × 3) = 58.667) and 58.667/8 = 7.3334 [57]. These values lead to ΔAcc/ΔTcharge ratios of 8/7.5 (1.0667) and 8/7.333 (1.0909), respectively; see the values given earlier. The three sodic endmembers all have ideal stoichiometries and would have zero values for each delta parameter, so the presence of excess silica in nephelines is not a sign of non-stoichiometry, and departures from such ideal nepheline stoichiometry would allow delta ratios to deviate from these “standard” values as would the presence of analytical errors.
2.3.2. Compositions of Naturally Occurring Nephelines and Some Ideal “Nepheline” Analogues
Henderson [57] recently assembled a database of 310 analyses (mainly electron microprobe data) and tried to deduce how different authors had calculated parameters such as Ne, Ks, Qxs, together with a Ca component which was usually reported as An. Most authors did not specify the calculation method used, and different groups of researchers sometimes obtained different results for nepheline analyses which are broadly similar. Henderson used that database to assess the criteria for acceptability of analytical data. Departures from stoichiometry were assessed using the parameters ∆Alcc and ∆Tcharge. The values for these parameters are closely coupled for reliable analyses, and acceptable analyses were defined as (i) having values within ±0.6 for both ∆Alcc and ∆Tcharge parameters (much larger than the ±0.25 originally recommended [35]) and (ii) lying within the range 1.0 to 1.25 for the coupled ∆Alcc/∆Tcharge ratio. Many high-quality nepheline analyses have ∆Alcc/∆Tcharge values of ~1.134. Other criteria used to select reliable analyses were that the CaO concentrations should be <3.5 wt.%; that 32 O cell formulae should have tetrahedral atom totals within the range 15.9–16.1; and the different approaches to calculating the excess silica-excess parameter (Q’) should show only limited differences between the different ways of defining excess silica Q’ (see above in this paper and [57]).
Figure 1a shows the ∆Alcc vs. ∆Tcharge relationship for the full original database of natural nepheline compositions [57] with all of the analyses shown with the same small blue diamond symbol. Most analyses plot close to the main linear trend, but those less reliable are over-plotted with large open symbols with the data sources defined in the inset box. For the main database samples Figure 1b shows how the ratio ∆Alcc/∆Tcharge varies as a function of the Si/Al ratio; it is clear that some analyses plot well away from the general trend which is defined by the small dark blue diamond symbols (labelled “All samples”). Based on the criteria for acceptability defined in [57], “reliable” analyses plot between ∆Alcc/∆Tcharge values from 1.00 to 1.25 and the vast majority (small blue symbols) define a trend of ∆Alcc/∆Tcharge decreasing with increasing Si/Al; the most silica-rich compositions on this trend are from synthetic samples.
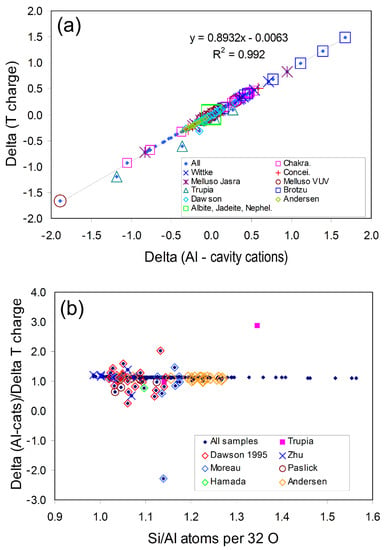
Figure 1.
(a) Plot of delta parameters ∆Alcc vs. ∆Tcharge; all the original database of 310 nepheline compositions [57] are plotted with the same symbol (small blue diamond). These points define a linear trend with the statistics of the fit displayed. Most analyses lie close to the line over the range ±0.5 for both ∆Alcc and ∆Tcharge, showing that these parameters are closely coupled for reliable analyses. Analyses falling outside that range and falling further away from the linear trend are also marked with larger symbols; the sources for these points are identified in the inset box. (b) Plot of atomic Si/Al vs. ∆Alcc/∆Tcharge. All of the database analyses [57] are plotted with the same symbol, and most analyses are seen to fall on a slightly decreasing delta ratio over the range Si/Al from 1.0 to 1.55. As in Figure 1a, points falling away from the main trend are identified with different symbols. Only analyses with delta rations from 1.0 to 1.25 were considered to be reliable. After [57].
Table 5 illustrates some compositional information using “acceptable” compositions from the original database and also includes analyses that were published after that database was compiled and are discussed here for the first time, together with some that had not been identified earlier. The first three columns give recalculated data for three theoretical, stoichiometric nephelines, one from each of Table 1, Table 2 and Table 3. Cations per 32 O are given for each along with calculated values for ∆Alcc and ∆Tcharge, together with mole % values for endmember nepheline molecules described above including a sodic iron nepheline (Nf, NaFe3+SiO4), a magnesian kalsilite (KsM, e.g., K2MgSi3O8), excess Al2O3 (corundum Cn), and four different excess silica endmembers; all of these are calculated using the equations described above (Table 4). The mole % Ne, An, and Qxs values all match the expected values. Because these are stoichiometric compositions the calculated ∆Alcc and ∆Tcharge ratios are zero and all four different Q’ parameters give the same endmember compositions. The remaining columns give data for published wt.% oxide analyses for natural nephelines together with the mole % values for endmembers reported by the original authors; mostly gave Ne—Ks—Qz ternary compositions, but some also included Ca-nepheline (An) values. The remaining rows all give values recalculated to 32 oxygens following the methods described above.
Table 5.
Recalculation of nepheline atomic and molecular formulae for published analyses based on ideal nepheline stoichiometry.
The data in columns 4 to 8 [35,58,59,60] were all originally calculated following the approach of Dollase and Thomas [35], and the values reported by the original authors all have Q’ values (row 13) significantly higher than those reported here (row 40), which inevitably leads to lower Ne and Ks values than reported here. The Dollase and Thomas method is explained in Henderson [57]; note also that the data in column 5 [58] shows that the CaNe values calculated here are about double those delivered with the Dollase and Thomas method. The difference is due to the latter method not following strict stuffed-tridymite stoichiometry so that the vacancies in the cavity cations associated with the presence of excess Si and Ca were not considered in the recalculation protocol.
The cation numbers given by Vulic (column 9) [61] are also reported as an atomic formula, but endmembers were not calculated. However, using their Ca occupancies quoted for the cavity site would only give half of the total CaNe molecule while the vacancy content of the cavity site would be too high by including vacancies related to both the Si’ content and that associated with Ca. The composition in the “Vruble” column for analysis 10 [62] appear to be reliable and the cation numbers per 32 O look sensible but all the endmember component values published are unreliable; the Ne/Ks ratio quoted (0.251) is an atomic ratio (not identified as such), but the K/Na values quoted do not match that number and indeed appear to have been converted to wt.% values which appear to be unrelated to the oxide wt.% values in their table. For the analyses in columns 11 [33], 12 [63] and 13 [64], the data given here are close to the values quoted in the original papers. The analysis in column 14 [65] had correctly calculated Ne and Ks values but the Q values are too high, and this seems to be accounted for by that value not having been corrected for the vacancy proportion reflecting the presence of a Ca component; all of the analyses in that paper show the same problem. The Berkesi analysis given in column 14 [65] had neither accompanying atomic cell formulae nor information on molecular endmember proportions, so it is not possible to comment on how such a calculation might have been made by the author. These analyses at first sight appear to be reliable but see immediately below. Column 15 [66] has an analysis of a very sodic nepheline from a volcanic sublimate; this is a good analysis and is a very useful addition to the nepheline database. Finally, an analysis of a glassy phase from a xenolith in the brecciated Murchison (CM) chondrite is given in column 16 [67]; this phase has a bulk composition with many similarities to a sodic nepheline although its very low ∆Tcharge (−0.006) does not match its ∆Alcc value (0.417) (see Table 5) This analysis also has a fairly high content of Ni (NiO 1.05 wt.%) and Fe is reported as FeO. However, it seems likely that most of the Fe should be oxidised, and the recalculation was done on that basis; the Ni was assumed to enter the KsM2+ molecule. It is probable that this phase formed from an original crystalline nepheline phase by shock metamorphism akin to the formation of glassy maskelynite from crystalline plagioclase feldspar.
It seems from this survey of recent analyses that many authors do not always carry out calculations that provide reliable and useful data for endmember nepheline components. Henderson [57] has attempted to make the case for rectifying this problem and provided a spreadsheet to make it easier for researchers to obtain correct and standardised molecular nepheline components for this phase which is a key mineral in alkaline rocks.
All of the published data that have newly been added to the nepheline database (59 microprobe analyses) have been plotted on Figure 2a together with all the reliable analyses extracted from the original database samples. The “acceptable” original analyses are all plotted with the same symbol, and the new data are plotted with a different symbol for each literature source. All of the new data plot on the trend for acceptable analyses. Moreover, plotted on Figure 2a are the compositions for a sodic and a calcic cancrinite; Henderson [57] suggested that the “nepheline” analyses in the original database with the most negative ∆Alcc and ∆Tcharge values were probably samples contaminated with cancrinite as an alteration phase and the plotted points for the cancrinites shown in Figure 2b are consistent with this possibility. Figure 2b shows the same data illustrating the relationship between ∆Alcc/∆Tcharge and Si/Al. All of the new data fall very close to the most reliable analyses except for most of the analyses from Berkesi [65], which show significant departures from acceptable nepheline analyses. This is ironic because that paper provides excellent data for the existence of a very Na- and F-rich aqueous fluid in the active, peralkaline Olodinyo Lengai volcano, Tanzania. In that paper, analyses are given for quenched immiscible natro-carbonatite and highly peralkaline silicate melts that occur as melt inclusions in nepheline phenocrysts from peralkaline nephelinites. Many of the analyses were obtained using EDS techniques with a very low beam current (0.2 nA) to minimise Na loss. The nepheline microprobe analyses given in that paper were obtained by WDS electron microprobe, with a 7 to 10 µm spot and a beam current of 2 nA. These conditions should have delivered reliable Na analyses, but many of them have bulk compositions suggesting loss of Na. Note that the term peralkaline is usually defined as the molecular ratio ([Na2O + K2O]/Al2O3]) or atomic ratio ([Na + K]/Al) ratio having values greater than unity and a peralkaline index used by many researchers for the study of ultra-alkaline rock types including Oldoinyo-Lengai nephelinite and carbonatite samples uses this formulation. However, such rock types often contain quenched silicate glasses and nephelines that tend to have very low Al leading to very high peralkalinity indices (i.e., up to 13 in silicate glass inclusions [64]). The nephelines in these rocks tend to have high iron contents where the Fe3+ undoubtedly proxies for Al; it would therefore be sensible to use a redefined peralkalinity index as (Na + K)/(Al + Fe3+), which would provide a more reliable indicator of alkali enrichment.
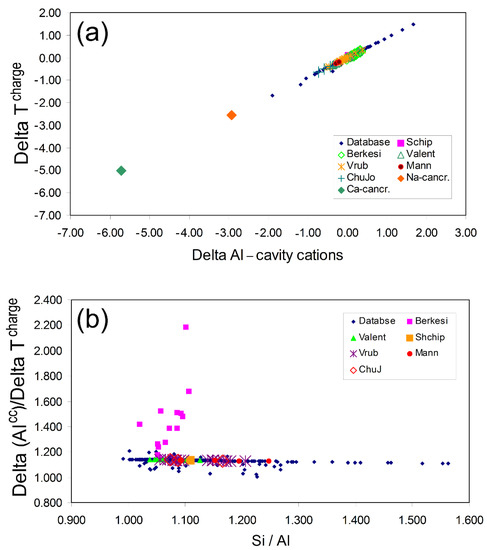
Figure 2.
(a) Plots of delta parameters ∆Alcc vs. ∆Tcharge for the most reliable analyses from the original database [57] are shown as small blue diamonds. The nepheline analyses highlighted in the present paper are shown with different symbols for the different source analyses, and all lie on or close to the main trend. Most analyses lie close to the line over the range ±0.5 for both ∆Alcc and ∆Tcharge; nepheline analyses with high + ve ratios have probably involved loss of Na during the EMP analyses. (b) Plot of ∆Alcc/∆Tcharge vs. Si/Al; only the most reliable analyses from the original database are shown, while all the analyses for the newly added samples are included. All lie on the main trend except for those from the Berkesi paper [65], which are not reliable; see text. After [57].
Henderson [57] showed that many microprobe analyses of nephelines from Oldoinyo Lengai nephelinites have very low or even negative amounts of a Q’ component, and this was attributed to the nepheline having excess Na contents, possibly consistent either with the presence of cancrinite as an alteration phase or even the possibility of some cancrinite molecule occurring in solid solution in primary nepheline. With the Berkesi paper [65] discussed here, there is now the possibility that nepheline analyses could also include components from “hidden” alkali carbonate or peralkaline silicate melt phases or perhaps even a component of the Na-F-rich aqueous fluid. Henderson suggested a variation for dealing with the possibility of excess Na in nephelines by calculating an excess QSi content using Equation (9) in Table 4 (see above) and then applying this to calculate the amount of Na present in a stoichiometric Ne component. (NaNe) This Na value is then subtracted from the total calculated Na atoms to define the amount of excess Na (Naxs). Note that none of this excess Na (and its associated O) is considered to be part of the bulk nepheline, and it is necessary to recalculate the data to a new 32 O total. Of course, peraluminous (i.e., (Na + K)/Al < 1.0) compositions would return negative Naxs values, but as seen earlier, such compositions would have positive Cn values. The Naxs could then be used to estimate the amount of a Na-cancrinite component present in the nepheline analysis (% cancrinite = Naxs × 100/10.666), and this value is given in the bottom rows of Table 5 just below the mole % data for nepheline analyses corrected for the presence of excess Na. Note that the endmember component calculations of peralkaline nephelines are much improved by making this correction with the Oldoinyo Lengai nepheline in Table 5 column 13 [64] now returning an excess Q value of 3.19% compared to the original best estimate of 0.65%.
2.3.3. Possible Significance of a Cancrinite Compon ent Existing in Nepheline
The cancrinite group mineral vishnevite is characterised by having lower Si and Al than nepheline with significantly higher Na contents in sodic varieties, and comparable Na contents and higher Ca occurs in cancrinite itself [68,69,70]. These papers show that Na and H2O are located within three-fold symmetry cancrinite “cages” defined by top and bottom 6-rings of ordered SiO4 and AlO4 tetrahedra, while Na, Ca, OH−, Cl−, CO32− (or SO42−) occur in large, continuous pseudo-hexagonal channels bounded by puckered 12-rings of tetrahedra; the two sites occupied by Na are structurally distinct. Figure 2a shows the effect that the presence of cancrinite would have on the composition of nephelines.
It has been suggested here that Na-rich nephelines might contain a sodic cancrinite impurity phase, perhaps concentrated in cleavage cracks or occurring as inconspicuous fine alteration products. However, the absence of any features associated with such an alteration process might suggest a small degree of solid solution of an alkali-rich molecule with cancrinite-like characteristics. Thus Henderson [57] speculated that some embryo cage-like modifications might occur in the large hexagonal channels within the six-ring nepheline structure, which might contain the excess alkalis in some nephelines and considered the following evidence. Many natural nephelines show features such as satellite diffraction peaks indicative of an incommensurate structure [71,72,73] and Friese et al. [74] re-determined the modulated structure of one of the original nephelines studied by McConnell [71] using superspace crystallography, which was developed to study periodic structures that show long-range order but which lack translational symmetry. They found that all atoms are displacively modulated with amplitudes <0.1 Å and that Na fills the smaller (oval) channels; K, Na and Ca and vacancies are disordered within the larger (hexagonal) channels. Framework oxygens show split-atom modulations which are coupled to the occupational modulations of the cations (and vacancies) in the large channels. Perhaps this disordered local geometry could allow access of “additional” small cations (Na and Ca) into the large nepheline channels to extend the effects of non-stoichiometry in natural nephelines. Henderson [57] considered the implications of how a cancrinite-like molecule occurring in nepheline would lead to a content of large anions and hydrous species associated with the extra Na. A review of infrared and NMR spectroscopic results for nepheline, cancrinite, and hydrous silicate glasses led him to suggest that there might be some prima facie evidence for a small amount of a cancrinite-like Na-H2O complex in nepheline. Thus, the high Na-content of nephelines from peralkaline rocks could suggest the presence of up to about 5% of a cancrinite-like phase.
Comments. Reliable calculations of nepheline endmember molecular proportions for sodic (Ne), potassic (Ks), calcic (CaNe), and excess-silica (Q) require the proportions of both filled (Ca) and vacant (□Ca) sites to be included to calculate CaNe and of all the □Si sites to calculate Q’ values.
The reliability of nepheline analyses can be assessed using the parameters ∆Alcc and ∆Tcharge. Acceptable analyses are defined as having values within ±0.6 for both ∆Alcc and ∆T-charge parameters and within the range 1.0 to 1.25 for the coupled ∆Alcc/∆Tcharge ratio. Many high-quality nepheline analyses have ∆Alcc/∆Tcharge values of ~1.134. CaO concentrations should be <3.5 wt.%; that 32 O cell formulae should have tetrahedral atom totals within the range 15.9–16.1 and also should show only limited differences between the different ways of calculating the excess silica parameters Qxs, QSi, Qcavity, and Q(Si-Al).
Good analyses of nepheline phenocrysts from nephelinites and peralkaline igneous rocks associated with Na-rich carbonatites often show Al deficiencies reflected in negative ∆Alcc parameters, the presence of excess Na contents (Naxs), and anomalously low or negative excess Q’ parameters. For such nephelines, the presence of excess cavity cations over (Al + Fe3+) suggests that the Q(Si-Al) calculation provides the most reliable excess silica estimate.
3. Research Topic 2: Nepheline and Structural Analogues Including Sr-Ba Aluminates
Context. Henderson and Roux [75] studied the thermal expansion and displacive phase transitions in synthetic sub-potassic nephelines and found that samples with <2.5 mol. % Ks showed variable features having lower symmetry structures (orthorhombic supercell) at room temperature, which transformed reversibly to the Hahn and Buerger structure at ~ 473 K. This paper was referred to in recent work by Deshkar et al. [3] on the occurrence of nepheline and carnegieite phases in glass-ceramic systems; it was suggested that the sub-potassic, type-B nepheline [75] might be a mixture of nepheline and carnegieite phases. These samples do appear to have two phases, but the extra peaks disappear reversibly at ~473 K implying that the two phases must homogenise and separate out on a very rapid timescale. This is unlikely as nepheline and carnegieite are polymorphs based, respectively, on stuffed-tridymite and stuffed-cristobalite structures which would involve major structural reorganization in a phase transition that undoubtedly would have a reconstructive mechanism. Other aspects covered in the study of a NaAlSiO4−based, glass-ceramic system by Deshkar et al. [3] should be considered. Detailed results for XRD powder diffraction and advanced MAS NMR and multi-quantum NMR characterisation of the glass—part-crystalline samples provide interesting results for two systems: a “meta-aluminous” system with variable Na2O/CaO molar ratios having a constant AlSiO4 stuffed-SiO2 framework (NC series); and a “peralkaline” system with variable CaO/SiO2 molar ratios and constant Na2O and Al2O3 proportions (SC series). The second series is a curious choice as varying two components that do not act structurally in the same way takes the Ca-rich, Si-poor compositions out of a nepheline—stuffed-SiO2 compositional field. In addition, the atomic 4-oxygen formulae for the different samples provided in Deshkar’s [3] Table 3 do not match the molar oxide proportions given in their Table 1.
For the NC series, the proportion for Ca is double its correct amount leading to a mismatch of the cation valence sums from the 8 negative charges provided by the four oxygens. Taking full account of the vacant sites and following strict stuffed-tridymite stoichiometry rules (see above) the correct atomic formula for NC-17.5 is Na0.3Ca0.35AlSiO4 and this is equivalent to a molar composition of Ne30CaNe70. For the SC series the Ca values given are again twice the correct values, and the Si values are too small; the correct formula should be Na1.053Ca0.210Al1.053Si0.842O4; note that atoms Na = atoms Al as intended. This is equivalent to a molar composition Ne0.842(NaAlO2)0.211(CaO)0.210 with balanced +ve and –ve charges, which seems to be far removed from a model nepheline-type composition. Henderson [57] showed that many petrological papers give incorrectly calculated nepheline compositions because the vacancies associated with substitution of Ca2+ for Na+ in the cavity sites and for the presence of excess Si (Sixs) in the framework have not been accounted for. It seems that in Deshkar et al. [3] a similar problem has been encountered so that the Ca value given is actually the sum of the Ca content plus the associated same value for a Ca-vacancy; the reason for incorrect, low-Si value for the SC series in [3] is unclear. Note also that there seems to be some confusion regarding reconstructive vs. displacive phase transitions in carnegieites; the transition from high-carnegieite (β-form) to low-carnegieite (α-form) at ~940 K, is definitely a reversible, displacive transition (not reconstructive) (see [3]). Moreover, the transition from nepheline to carnegieite at high temperature has many more properties of a reconstructive than a displacive transition despite the suggestion that the presence of a coexisting melt phase might play some mediating role [76].
3.1. The Nepheline/Kalsilite-Analogue SrAl2O4—BaAl2O4 System
In a related project on nepheline analogue systems, Henderson and Taylor [42] studied the synthetic system SrAl2O4—BaAl2O4 to investigate how a compound with a single framework cation (Al is the only T cation) might vary from those with both Al and Si; this is treated below in the second research topic.
In the last four decades the electronic, luminescence, ferroelastic, and ferroelectric properties of these aluminates have led to many studies using different techniques [77,78]. The endmember BaAl2O4 is hexagonal at room temperature (P63) but SrAl2O4 is monoclinic (P21); structural data are complicated by axis choice and by the presence of satellite reflections in some samples [42,79,80]. Henderson and Taylor [42] synthesized samples across the solid solution series and studied the thermal expansion and monoclinic to hexagonal (P63) phase transitions shown within this sample series. Two-phase regions were reported, where the monoclinic and hexagonal phases coexist depending on both composition and temperature. SrAl2O4 was found to transform over the range 938 to 978 K with a volume contraction of 0.2 to 0.3 %, while the two-phase region occurred at room temperature over the compositional range ~0.3 to 0.45 mol fraction BaAl2O4. Powder XRD was used to study the thermal expansion of all the samples with the monoclinic phases indexed in space group P21 (with the b parameter equal to √3a) and the hexagonal phases indexed as P63 [42]. Rodehorst et al. [79] made further studies of the phase transitions in these samples (together with newly synthesized samples), using high-temperature IR methods. A key conclusion in [42] is that, based on the discontinuity in volume and the presence of the two-phases within the so-called “region of coexistence”, the transition is a first-order, athermal, displacive transition with additional characteristics of a strain-related, martensitic transition mechanism. To quote Henderson and Taylor, “Thus the transformation takes place with a volume discontinuity and a marked anisotropy of shape change which together generate the elastic strain and strain energy associated with martensitic transformations. At high temperatures the transformation is athermal; that is, once the temperature for the start of the transformation is reached, it only takes place when the temperature is changing. If the temperature is held constant part way through the transformation, then an equilibrium proportion of the two forms is achieved virtually instantaneously.” These conclusions were based on in situ studies of transitions in ZrO2 [81,82], BaTiO3 [83,84], and cristobalite [85] This type of phase transition has also been reported to occur in synthetic KGaSi2O6 leucite and in synthetic and natural K-Al leucites (Bell and Henderson, 2020) [86] and a Supporting file attached to that paper summarises characteristics of the phase transitions shown by SiO2, ZrO2 and BaTiO3. In the present paper I will use the unit cell thermal expansion data from [42] to analyse the spontaneous strain relationships for the monoclinic to hexagonal phase transition for the samples of composition Sr100, Sr90, Sr80, and Sr70.
3.1.1. Thermal Expansion and Phase Transition Data for the SrAl2O4—BaAl2O4 System
The unit cell parameter data versus temperature from Henderson and Taylor [42] are plotted here in Figure 3. Note that for the monoclinic phases the b axis points refer to the values bm/√3 and the volume to Vm/2. For compositions Sr100, Sr 90, Sr 80, and Sr 70, the a and bm/√3 values (where in all cases a > bm/√3) show parallel increases initially with increasing temperature. However, the Sr 100 a parameter shows an initial increase from 5.148 Å at room temperature, to ~5.175 Ǻ at ~673 K before decreasing to 5.163 Ǻ at 933 K, just before the transition to a hexagonal cell with lower values a. Rodehorst et al. [79] described this trend as “anomalous”. Ishii et al. [80] have recently published on Sr-rich solid solutions in the series SrAl2O4—BaAl2O4; solid solutions of compositions Sr90, Sr80 and Sr70 have very similar room temperature cell parameters to the same compositions described here (see [42]). However, their Sr100 sample has a significantly larger room temperature c parameter than our cell (equivalent to our a) of ~5.17 Å; their a (equivalent to our c), b, β and V are all similar to our values. For another SrAl2O4 Avdeev et al. [87] report room temperature cell parameters of a (our c) = 8.44365, b = 8.8898, c (our a) 5.15964 Å; β = 93.411°; their c is significantly higher than our a for Sr100 while the other parameters are in good agreement with our data. The discrepancy for the a parameter in our sample is unclear but might suggest the presence of a different structural variety for this particular sample at low temperature; although the cell parameters are displayed in Figure 3, the more recent data of Avdeev [87] are considered to be definitive for the Sr endmember, and those values will be used here in the treatment of the thermal expansion characteristics of the Sr-Ba solid solution series.
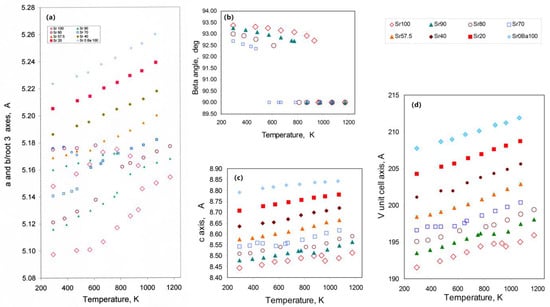
Figure 3.
Thermal expansion trends for the (Sr, Ba) solid-solution series determined by Henderson and Taylor [42]; different symbols are used for each composition. Figure 3a shows variations for the a and b/√3 parameters, the same symbol is used for both of these; note that for the monoclinic polymorphs a > b/√3. These parameters become identical when the phase transition from monoclinic to hexagonal takes place. Compositions more Ba-rich than the Sr70 are all hexagonal at room temperature. Figure 3b–d shows data for the c, V/2 and β cell parameters. All the parameters show clear discontinuities at the phase transitions; the most significant changes are shown by the c and V parameters pointing to a small negative ∆V.
The Sr 100, 90, 80, and 70 samples all show clear transitions to hexagonal forms with distinct discontinuities in the trends for a and b/√3 before they reach the hexagonal a expansion trend; as expected, the transition temperature decreases with increasing Ba content as was also found by Ishii [80]. With increasing temperature, the c axis trend for the monoclinic phases is slightly shallower than that extrapolated to lower temperatures for the hexagonal polymorphs, and there is a clear negative ∆c. The c axis defines the perpendicular to the “sheet” of linked six-rings of tetrahedra and Andeev et al. [87] have concluded that the ∆c results from the tilting of AlO4 in that plane. Note that in considering the structural changes occurring in framework materials as a result of compositional or thermal expansion, it is conventional to consider the “change in shape” of the entire framework in terms of cooperative tetrahedral rotations or tilting rather than the separate expansion or contraction of particular interatomic bond-distances or angles. In any case, it is more straightforward to interpret trends of overall framework expansion or collapse using such cell parameter variaition data. Note that if the high-temperature polymorph has a larger cell volume than the low-temperature form, the ∆V is defined here to be positive, while a negative ∆V would be associated with the high-temperature volume being smaller than that for the low-temperature phase. The trends for volume and the β angle show significant steps to lower values for each of the Sr-rich samples pointing to the phase transition having a small negative ∆V (−0.2 to −0.3%) and being a first-order transition. This has been confirmed by Ishii [80] for samples of composition Sr100, 90, 80, and 70 and for Sr 100 by Avdeev et al. [87] who also reported a ∆V of −0.25% associated with a phase transition at 953 K, in excellent agreement with the work discussed here. Henderson and Taylor [42] described two-phase regions at the phase transitions for SrAl2O4 and for the solid solutions with low BaAl2O4 contents which they assumed represented mixtures of the monoclinic and hexagonal phases, and this was confirmed by Ishii [80] and Avdeev [87]. However, those workers distinguished between lower temperature P21 and higher temperature P2122 polymorphs with satellites for the latter phase appearing alongside the peaks for the monoclinic phases [80,87]. These effects are accounted for by the occurrence of a first order phase transition with Henderson and Taylor [42], Bell and Henderson [86] and Avdeed [87] suggesting martensitic, strain-related characteristics, whereas Ishii [80] mentions “latent instabilities”.
3.1.2. Spontaneous Strain Analyses for the System BaAl2O4—SrAl2O4.
Carpenter et al. [88] reviewed how the thermal expansion and deformation of the three-dimensional framework structure of minerals and inorganic compounds can be dealt with in terms of a strain ellipsoid. The strain tensor would have six independent components for triclinic symmetry materials (three axial and three involving angles relating the orientation of the strain ellipsoid axes to the crystallographic axes); higher symmetry materials have strain tensors with fewer independent components. Henderson and Taylor [42] assumed a direct transition from hexagonal to monoclinic P21 symmetry in SrAl2O4 (and in Sr-rich solid solutions with BaAl2O4), but Rodehorst et al. [79] suggested that there are two discrete transitions in the sequence P6322 → C2 (monoclinic) → P21. In a later study using high-temperature X-ray and neutron powder diffraction techniques, Avdeev [87] did not detect a monoclinic intermediate phase and reported the sequence P6322 → P63 → P21. The temperature range used by Henderson and Taylor for SrAl2O4 would not have reached a second phase transition so it is assumed that the transition is simply P63 → P21 (i.e., point groups 6 to 2) which should allow a single transition (also see [87]).
For the Sr-Ba aluminates, the spontaneous strains for the unit cell data for the monoclinic to hexagonal phase transition were calculated here using the approach of Rodehorst [79] with the six components e1 to e6 as follows:
note for this monoclinic to hexagonal transition e4 = e6 = 0 so the strain tensor only has four independent components. In these equations a0, c0, and V0 are the high temperature parameters for the hexagonal phase extrapolated into the lower temperature, monoclinic phase field, with monoclinic cell parameters defined as a, b/√3, c, and V/2 to enable direct comparison with the unit cell data for the hexagonal polymorph. For a hexagonal to monoclinic transition, where the high temperature hexagonal phase is considered to have b = √3a, a symmetry-breaking strain can be defined by e1 – e2 [88]. Thus, the approach of Rodehorst is followed to define both (e1 – e2) and e5 = ccosβ/c0 as symmetry breaking strains with (e1 + e2), e3, and Vss being non-symmetry-breaking strains. The shear strain e5 behaves as the symmetry adapted strain, determined by the behaviour of the monoclinic β angle, which decreases to zero at the transition and can therefore be considered to be the primary ferroelastic order parameter Q1 (cf. Redfern and Henderson [89]); (e1 – e2) can be considered as the secondary elastic order parameter Q2.
e1 = (a − a0)/a0; e2 = (b/√3 − a0)/a0; e3 = (c − c0)/c0; Vss = (V − V0)/V0; and e5 = (c/c0) cosβ;
The spontaneous strain parameters are calculated for the thermal expansion data of Henderson and Taylor for Sr100, Sr90, Sr80, and Sr70 in an Excel spreadsheet, giving the same spontaneous strain values to those reported in [79]. I have also calculated strain parameters for the SrAl2O4 data extracted from Figure 3 of Avdeev [87], which provides a more reliable dataset than ours for SrAl2O4. The starting point is to use the cell parameters for the high temperature hexagonal polymorphs for each of the monoclinic samples from [42]; a linear extrapolation into the monoclinic fields provides the necessary values for a0, b/√3 = a0, c0, and V0. Figure 4 illustrates the procedure for the Sr80 and Sr100 samples.
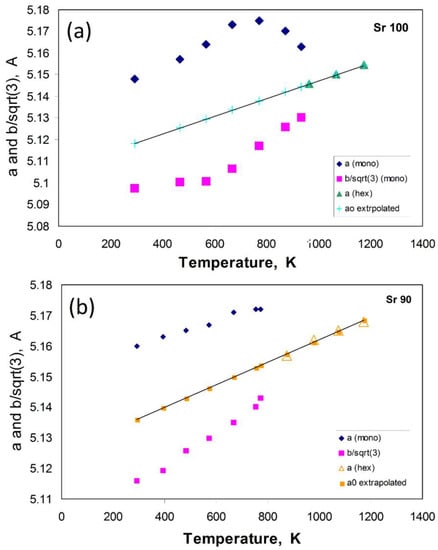
Figure 4.
Dependence of the cell parameters a and b/√3 with temperature for Sr100 (Figure 4a) and Sr90 (Figure 4b); also shown are the linear fits to the hexagonal polymorph a parameters, which are projected into the monoclinic stability fields to obtain the appropriate a0 values. Figure 4b shows steady trends for both parameters in Sr90, which will provide reliable data for calculating the strains as a function of temperature. For Sr100, the anomalous values for a will lead to anomalous trends for the (a — a0) strain estimates; see text.
The Sr90 sample shows easily understood trends, with both a and b/√3 showing steady increases with increasing temperature, and the extrapolated ao trend lies consistently further away from the a trend. The separation between the a and b/√3 values shows a steady decrease with increasing temperature and this will reflect a steadily decreasing degree of strain between these two cell parameters with increasing temperature. The plots for Sr80 and Sr70 show similarly clear trends. However, our Sr100 sample shows a more complicated relationship for the a and b/√3 parameters which as explained above is due to the anomalous values recorded for the a parameter; the separation between the values for a and b/√3 increases with increasing temperature implying increasing strain within the cell, and this would be reflected in unreliable (e1 – e2) strain parameters (see later). By contrast, the temperature dependence of the a and b/√3 parameters of the Avdeev Sr100 sample is entirely normal, and in the following discussion, those data will be used as the most reliable dataset for the Sr100 composition.
Figure 5 shows the variation of the main axis strains e1, e2, and e3 depending on temperature for all four monoclinic samples; the e1 and e3 strains are always positive and decrease with increasing temperature, and e2 is always negative and shows little change. The strain dataset for Sr100 is the most reliable and shows e3 > e1 with e3 decreasing faster than e1 with the values merging below the transition temperature. For the other three samples, e1 is larger than e3 with values for Sr80 and Sr70 tending to merge at higher temperature while for Sr90 the strains increase which presumably indicates larger errors in the dataset for this sample. In each case, it is clear that the strains show clear discontinuities at the respective phase transitions.
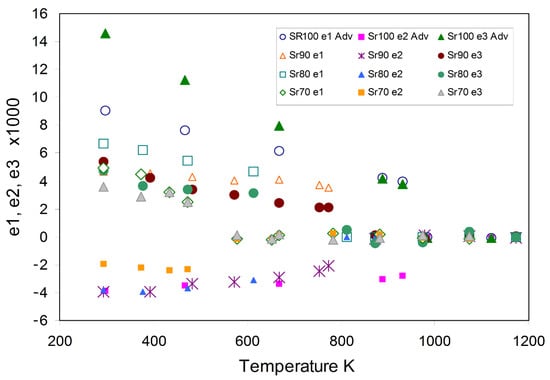
Figure 5.
Temperature dependences of the calculated spontaneous strains for e1, e2, and e3 for the monoclinic samples Sr90, Sr80, and Sr70 (this work); the data for Sr100 are based on the work of Avdeev et al. [87]. See text for explanation of trends.
Figure 6 shows the temperature dependences of the (e1 – e2) and e52 strain parameters; the curves are guides to the eye and are extrapolated towards the discontinuities marking the phase transitions; the discontinuities are clear for each composition consistent with the first-order nature of the transition. Note that it is conventional to plot the values for a secondary order parameter (e1 – e2) versus the square of a primary order parameter values (e52) so that the plotted points show a linear trend consistent with the linear/quadratic relationship expected for a ferroelastic phase transition (see below).
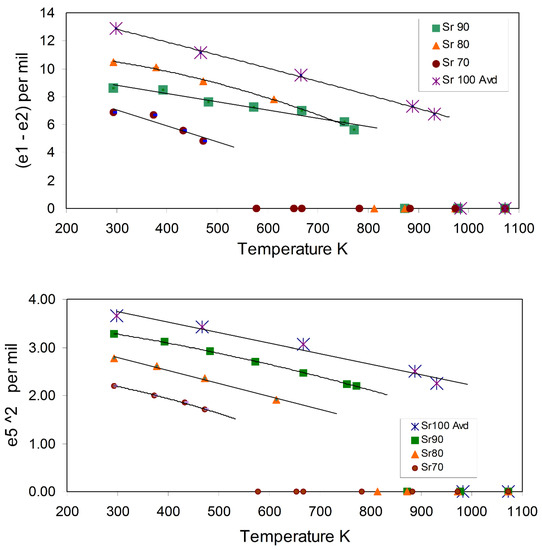
Figure 6.
Temperature dependences of the calculated spontaneous strains for the primary ferroelastic, symmetry-adapted strain e52 and for the secondary strain (e1 – e2) for the monoclinic samples. The lines are “guides to the eye” which terminate at the approximate temperatures of the phase transitions; of course, above these temperatures, the hexagonal polymorphs show zero strains. See text for further discussion.
Figure 7a shows that the plots of (e1 – e2) (Q2) vs. e52 (Q1) for the monoclinic, low-temperature phases are close to linear and trend toward the common origin defined by the hexagonal polymorphs. Thus, the secondary order parameter Q2 behaves as the square of the primary order parameter (Q1); this is a general situation for ferroelastic phase transitions and points to linear-quadratic relationships [88,89,90] in the operation of the ferroelastic, displacive, first-order phase transitions in these Sr-Ba aluminate solid solutions. Figure 7b shows that the plots for the volume strains (Vss) vs. e52 for samples Sr100, Sr90, and Sr80 all show linear segments implying linear-quadratic relationships, but these tend to have steeper segments for the highest strain regions (i.e., lower temperatures) with the lower-sloped points trending towards the hexagonal values at the common origin. For Sr100 and Sr90, the changes in slope occur at e52 ~3 × 10−3 and for Sr80 at ~2.5 × 10−3. The equivalent temperatures for these effects are Sr100 ~670K and for Sr90 and Sr80 a lower value of ~400 K; for Sr90 and Sr80, these values are in line with the observations, based on cell parameter and high-temperature infared measurements observations, by Rodehorst [79] that a second transition point occurred in the region of ~400–500 K. However, the data for Sr70 are not good enough to identify this effect.
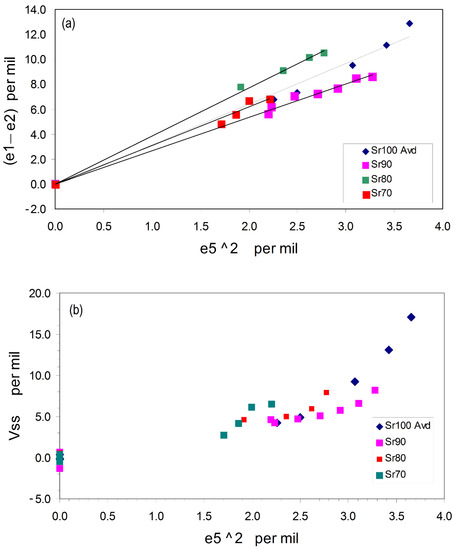
Figure 7.
Trends shown for the variations of the (e1 – e2) (Figure 7a) and Vss versus the e52 value (Figure 7b) for the monoclinic samples. The linear trends for Figure 7a confirm the coupled linear-quadratic relationship of the Q1 and Q2 order parameters and Figure 7b shows that the higher temperature regions (lowest strains) for Sr100, Sr90, and Sr80 appear to have linear trends projecting towards the zero-strain origin. See text for further discussion.
Comments. While the original data obtained by Henderson and Taylor [42] for their Sr100 sample are anomalous and indeed the high-temperature diffraction data for their other samples do not have the quality of those available from recent studies (Ishii [80], Avdeev et al. [87]), however, their overall conclusions have stood the “test of time”, and it is pleasing that Avdeev et al. [87] reported that “aside from the issue of space group assignment all other features of the monoclinic—hexagonal phase transition for SrAl2O4 we observed are in agreement with the results reported before”.
4. Research Topic 3: Thermal Expansion and Phase Transitions in the Feldspar Group
Context. Most naturally occurring feldspars belong to two complete solid-solution series between endmembers KAlSi3O8 (orthoclase, denoted Or here) and NaAlSi3O8 (albite, Ab) (the alkali feldspar series) and between Ab and CaAl2Si2O8 (anorthite, An) (the plagioclase series). The (Si,Al)O4 tetrahedra are linked by bridging oxygens to form 4-rings of tetrahedra that in turn are linked to build up a continuous three-dimensional polymerized framework. This framework can be considered as being composed of linked “crankshaft” chains which are aligned along the a crystallographic axis (Megaw (1974) [91]). Cavities in the framework contain Na and K in alkali feldspars and Na and Ca in plagioclases; both series crystallize in either monoclinic or triclinic symmetries depending on composition and “thermal state”. “High-temperature” feldspars tend to have disordered tetrahedral cations and “low-temperature” forms have Si and Al ordered into separate framework sites but an AlO4 tetrahedron never has another AlO4 tetrahedron as its nearest neighbour (c.f the Al-avoidance principle). See Deer et al. (1992) [52] for further information.
Henderson [92] published laboratory-based powder XRD studies on the thermal expansion of hydrothermally-synthesised disordered alkali feldspars in the system KAlSi3O8 (Or)—NaAlSi3O8 (Ab) having the compositions Or100, Or38, and Or19. Endmember sanidine (Or100) and the solid solution Or38 are monoclinic (C2/m) at room-T and do not show any phase transitions at elevated temperature but Or19 is triclinic at room-T and transforms reversibly to monoclinic symmetry at 773 ± 10 K. Mean linear and volume thermal expansion coefficients and thermal expansion ellipsoid data were given over the ranges 293 to 773 K and 773 to 1273 K. Similar studies on the synthetic disordered feldspar analogues RbAlSi3O8 and RbGaSi3O8 followed and preliminary data were reported (Henderson, 1978) [93] in the U.K. Natural Environment Research Council, Publication Series D “Progress in Experimental Petrology” reports, which were circulated to libraries and internationally to leading researchers. Similar studies were also carried out (Henderson, 1984) [94] on a synthetic, triclinic, ordered microcline—albite solid solution (Or62.4) and on a synthetic analogue plagioclase (SrAl2Si2O8). However, the experimental data were never published in established journals, although they have been referred to over the years by various international authors, and the journal series was cancelled many years ago, and they are no longer available. This invited paper gives the opportunity to rectify this and to provide some interpretation with reference to other relevant published work on related samples.
Synthetic Feldspar Analogues
A Rb-sanidine (RbAlSi3O8) sample (HF 348) was prepared hydrothermally from a glass starting material at 773 K, 1 kbar water vapour pressure for 7 days in a cold-seal pressure vessel. Monoclinic (C2/m) unit cell parameters were determined by powder XRD and are given in Table 6 with typical refinement errors on a, b and c of (2), (3) and (1) in the third decimal place and for β (1) in the second decimal place. Cell parameters at room-T are within error of those reported by Hovis et al. [95] for his sample ScOc; they are slightly different to those of Bruno and Pentinghaus [96] (a 8.839, b 13.034, c 7.182 Å, β 116.35°) and Kyono and Kimata [97] (a 8.839, b 13.035, c 7.175 Å, β 116.11), but note that this latter sample was hydrothermally crystallized in a sealed silver tube which was half-full of water and it is said to have excess Si and a deficiency in Rb. There is no reason to believe that sample HF348 is off composition as it was synthesized in a sealed gold tube with only 5% water to catalyse the crystallization of the amorphous starting material. Table 6 shows that as temperature was increased all the cell parameters show smooth changes with a and V increasing and b and β decreasing, and c showing only a small decrease. The sample started breaking down at 1273 K with the formation of a leucite-type structure [98]. The mean linear thermal expansion coefficients are shown in Table 7, and the orientation of the principal expansion ellipsoid axes are given in Table 8. These data will be discussed below together with similar data for other feldspars studied. The strain ellipsoid data were calculated using the STRAIN computer programme [99].
Table 6.
Feldspar thermal expansion unit cell parameters.
Table 7.
Mean linear and volume thermal expansion coefficients for feldspars for a defined difference in temperature: αx = (1/x) × ∆x/∆T where x is a unit cell parameter (linear or volume), and T is temperature (units: per degree × 106).
Table 8.
Strain ellipsoid magnitudes and orientations for natural and synthetic feldspars.
A sample of RbGa-sanidine (RbGaSi3O8) was hydrothermally synthesized (823 K, 1 kbar, 140 h) and was provided by Dr H. Pentinghaus. The monoclinic (C2/m) unit cell parameters given in Table 6 have slightly smaller errors than those indicated for Rb-sanidine. Cell parameters after the experiments were within error of the starting values showing that the sample had not changed during the heating. Cell parameters at room-T are within error of those reported by Bruno and Pentinghaus [96], i.e., a 8.916, b 13.089, c 7.251 Å, β 116.43°. With increasing temperatures, cell parameters show smooth variations with a and c increasing and b and β decreasing. Linear and volume thermal expansion coefficients are given in Table 7 and thermal ellipsoid orientation values in Table 8 (see below).
A monoclinic (I2/c), ordered “Sr-plagioclase” (SrAl2Si2O8) sample was also provided by Dr Pentinghaus, and its room-T cell parameters are close to those in [96] and to those in reference [100]. Cell parameters a, b, and c up to 1273 K all show regular increases with increasing temperature and a small decrease in β (Table 6) with similar trends to those reported up to 943 K for a single crystal sample [100]. Cell parameters after the experiments were within error of the starting values. Expansion coefficients and data for the orientation of the strain ellipsoid are given in Table 7 and Table 8, respectively (see below).
A sample of an ordered maximum microcline, triclinic (C-1), alkali feldspar solid solution (Or65.4Ab44.6) was provided by Prof W.L. Brown; it was prepared by Waldbaum and Robie (1971) [101] from a natural Amelia albite starting material with intermediate stages involving heating, ion-exchange and then mixing and homogenizing samples with different Na and K contents to get the chosen bulk composition with a homogeneous, low-albite ordered framework. The room-T cell parameters are close to those reported by Waldbaum and Robie [101]. High temperature cell parameters (Table 6) mainly show increasing a, almost no change in b, a small increase in c, a small decrease in β and essentially no change in the triclinic α and γ angles. Linear and volume expansion coefficients for the samples discussed here are given in Table 7, and the strain ellipse orientation data in Table 8. Published data for related feldspar samples are also provided in these tables.
The top halves of both Table 7 and Table 8 show thermal expansion data for both natural and synthetic feldspars and include samples with T-site disordered and ordered members of the Na—K alkali feldspar system. The top four sets of data are all hydrothermally synthesized with fully disordered Al and Si atoms in the three-dimensional polymerized framework and are thus members of the high-albite–high-sanidine solid solution series. The Na-rich Or19 sample is triclinic at room temperature (space group C-1), and it shows a displacive, non-quenchable, phase transition to a monoclinic polymorph (C2/m) at 833 K [92]. This transition is equivalent to that shown by endmember NaAlSi3O8 [102] which, for a fully disordered polymerized (Si,Al)O4 framework has been named analbite, and the high-temperature form is named monalbite. Kroll et al. [103] found that a hydrothermally crystallized Ab100 sample showed the analbite—monalbite transition occurred at 968 ± 8 °C. Note that analbite and sanidine have topochemically monoclinic structures with fully disordered Al-Si distributions. For the Or19 sample, the mean linear expansion coefficients for the triclinic cell show that the maximum expansion rate occurs along the a axis; this value is twice as large as for αb and about three times larger than for αc. The published high albite single crystal data [102] for endmember Ab100 samples show very similar expansion coefficient values to those for the Or19 sample with αa>> αb > αc; the mean volume expansion coefficient αV values for these samples are very similar and lie in the range 28.1 to 31.4 × 10−6 per degree. Above the phase transition in Or19, the rate of increase along the a axis is much larger (24.1 × 10−6) than that below (14.4 × 10−6 per degree), the αb and αc are now much smaller, and the αV is essentially unchanged at 32.8 × 10−6 per degree. For the more potassic disordered sanidine samples Or38 and Or100, the αa values are around an order of magnitude larger than those for αb and αc, and the αV is very large but shows decreasing expansion rates with increasing potassium content. The published data given in Table 6 for two other sanidine samples (Or86 and Or58) confirm each of these relationships. The same unit cell parameter thermal expansion relations are also shown by potassium-rich, ordered triclinic feldspars and microcline samples Or62.4 (this work) and Or100 (Openshaw et al. [104]) confirm these trends. Hovis et al. [95] have exploited the fact that both ordered and disordered K-rich alkali feldspars show the same relationships between increasing albite content and increasing volume thermal expansion rate to predict the expansion rate of alkali feldspars from their volumes measured at room temperature. The monoclinic disordered Rb- and RbGa-sanidine analogues (this work, Table 6) show very similar mean linear and volume expansion rates to those for the other sanidines described above, and this again indicates that the directional thermal expansion characteristics of the feldspar unit cells is more dependent on the framework symmetry than its composition and degree of tetrahedral cation order.
The other data given in Table 6 are for natural plagioclase feldspars (Ab—An solid solutions) and for Sr, Ba, and Pb analogue feldspars having an Al2Si2O8 anorthite-plagioclase composition framework. Note that the calcic endmember plagioclase feldspar has a fully ordered, triclinic (C-1) structure which also occurs as a natural Ba-rich feldspar species (celsian, BaAl2Si2O8); both of these have unit cells with doubled c axes. Thermal expansion data for varying composition natural plagioclases first became available with the Grundy and Brown paper in 1974 [105]. Much later, Tribaudino et al. [106] and Hovis et al. [107] provided better data. Table 6 gives the complete dataset from [104] with other representative data from the Hovis [107] and Grundy and Brown [105] work. It is clear that the most sodic plagioclases have mean expansion coefficients close to those for low albite [108]. With increasing An content all linear expansion coefficient decrease but show the same αa > αb > αc relationship. The mean volume expansion coefficient αV shows a steady decrease from 31 × 10−6 degree−1 for low albite to ~9 × 10−6 degree−1 for fully ordered anorthite; these data provide a reliable picture of the expansion geometry for the plagioclase feldspars. High-temperature structural studies on SrAl2Si2O8 and PbAl2Si2O8 have also provided information on the expansion characteristics of the plagioclase framework. Thus, two samples of synthetic SrAl2Si2O8 and one sample of PbAl2Si2O8 all show similar thermal expansion geometries with αa > αc > αb (Table 7); the αV for the Pb-plagioclase is slightly smaller than those for the Sr-feldspar samples.
It is clear that for all feldspars, the main expansion is recorded along the a crystallographic axis which is the direction of the framework “crankshaft” (Megaw, 1974, [91]) but analysis of the strain ellipsoid using the STRAIN computer programme (e.g., Ohashi and Finger [109]) shows that this is not the full story. Table 8 gives the strain ellipsoid information for many of the samples referred to above. The ellipsoid axes are labelled α1, α2, and α3 and for monoclinic cells α2 is always coincident with the b crystallographic axis with α1 and α3 lying in the a–c plane; the orientations of the axis relative to the crystallographic axes are given in degrees in the table. The αP coefficient is the isobaric volume expansion term; this has the same value as the volume expansion coefficient given in Table 7. In all cases, α1 is larger, sometimes very much larger, than α2 and α3. For the triclinic, disordered, alkali feldspar Or19 the principal axis α1 has a very high value (53.7 × 10−6 degree−1) and that is oriented at 80° to the a axis; note that the negative α3 coefficient marks a shrinkage along the defined orientation axis. Above 560 °C this sample is monoclinic with a smaller α1 which is oriented at 24° to the a axis and lies in the a–c plane. For the monoclinic Or38 and Or100 samples the α1 coefficient has the same orientation to that for Or19 above the phase transition except that its magnitude decreases with increasing K-content as discussed above. The RbAl and RbGa disordered feldspars both show similar expansion ellipsoids with α1 having a similar orientation to the Or100 sample but with a slightly larger angle relative to a. The ordered K-rich microcline feldspars (6434 and 71134) both have very similar α1 values for their orientations and magnitudes to the α1 values for disordered Or100 confirming the earlier conclusion that the main expansion characteristics of alkali feldspars are not dependent on the T-site order in the feldspar framework. The triclinic feldspars with divalent cavity cations Ca, Sr, and Pb all have the same ordered Al2Si2O8 frameworks with α1 values, which are smaller than those for the alkali feldspars but which have very similar orientations to the a axis.
Henderson [92] discussed the orientation and magnitude of the α1 coefficient for the monoclinic cells for Or19, Or38, and Or100 and pointed out that the angle it makes with a is 22°, which was only a few degrees from the orientation of the reciprocal cell a* axis which for most feldspars is aligned at 26° to a (i.e., angle from a to a* = β minus 90). This confirmed the result of Ohashi and Finger [109] for the Laacher See sanidine where they suggested that a major factor controlling its expansion was the elongation of the K—OA1 and K—OA2 bonds, which resulted in the elongation of the expansion polyhedron along a*. Benna et al. [110] and Benna and Bruno [100] made the same conclusion to explain the thermal expansion of triclinic PbAl2Si2O8 and SrAl2Si2O8 using an [010] crystallographic projection of the structure. The angle from the a* direction to the a axis for both of these feldspar axis is 26° but for the Pb feldspar the actual angle is found to be 19° and for the Sr-analogue is 32°. For the strain ellipsoid data reported here (Table 7) the a ^ a* angles for the disordered RbAlSi3O8-sanidine, RbGaSi3O8-sanidines and the powder data for SrAl2Si2O8 (this work) are 16.9 ± 0.5, 16.6 ± 0.3, and 30 ± 3° all significantly displaced from the a* direction. It is possible that the overall expansion of the framework is affected by tetrahedral tilting strains as well as expansion of the cavity cation – oxygen polyhedra along a* with the result that the main expansion direction is displaced from being orientated exactly along the a* axis.
Brown et al. [111] used the results from such studies to develop an integrated explanation of how the effects of composition, temperature and pressure could be combined to understand the controls on the expansion behaviour of the alkali feldspar structure. Information from natural minerals and synthetic samples with cavity cations Na, K, and Rb, and framework cations Si, Al, Fe3+, Ga, and Ge for endmembers and intermediate solid solutions were incorporated. The conventionally-used b–c diagram, which has been so useful for displaying the parallel but separate trends for order and disordered alkali feldspar Si-Al frameworks, was used to define three expansion trends: trend I is defined by substitution of Na for K or by cooling Na-K feldspars and shows a coupled b–c relationship; trend II shows an uncoupled relationship for K—Rb feldspars with c increasing and b decreasing; and trend III is shown by some microclines which appear to have reached a limit for compositional expansion but on heating show little change in c and a decrease in b. A significant conclusion was that the expansion is dominated by the framework and not by the expansion of short cavity-cation—O bonds.
Thus, it seems that the 1970s and 80s was a busy period for experimental studies on feldspar crystal chemistry with investigation on both natural minerals and synthetic samples, including rarer element analogues at both room- and elevated temperatures. This intensity fell away to some extent, but Hovis and his collaborators extended these studies on endmembers together with studies on solid-solution series between them, with samples prepared by direct synthesis and ion-exchange of cavity cations in a chosen crystalline sample so that series with the same framework composition could be investigated. Thus Hovis et al. [95,107] described relationships between room-T cell volumes and mean volume thermal expansion coefficients for alkali- and plagioclase- feldspar series and defined model equations that can be used to predict thermal expansion rates for almost any feldspar based on type and cell volume at ambient conditions. Hovis et al. [107] also pointed out that feldspars with Al2Si2O8 frameworks with Ca, Sr, Pb, or Ba present as the cavity cation had essentially the same thermal expansion characteristics even though they have very different room-T cell volumes; however, this was not the case for alkali feldspars with AlSi3O8 framework compositions and monovalent cavity cations (Na, K, Rb). It was suggested that the latter showed differences because the local bonding relationships between the monovalent cavity cations and the associated T—O—T linkage oxygens are weaker and allow much more vibrational freedom within the AlSi3O8 alkali framework. By contrast, the local bonding for the divalent cation in the Al2Si2O8 framework is considered to be stronger with short, covalent M2+–O bonds leading to much “stiffer” frameworks; but why do Ca, Sr, Pb, and Ba feldspars all show similar thermal expansion coefficients? They have very different ionic radii: (e.g., radii for 7-coordination are 1.06, 1.21, 1.23, 1.38 Ǻ (Shannon, [112]); and different electronegativities of 1.0, 0.95, 2.3, and 0.89 for Ca, Sr, Pb, and Ba, respectively. The intertetrahedral T—O—T angle tends to increase as tetrahedral frameworks expand, yet the mean T—O—T angles at room temperature for these Ca-, Sr-, Pb- and Ba-feldspars are all very similar at room temperature (within 1% of each other) even though their room-T unit cell volumes are very different. In all cases, the available crystallographic data show that the mean M2+—O bond lengths have much larger % thermal expansion changes than the related mean T—O—T angles. Thus M—O polyhedral changes must play a key role in the mechanism of the framework thermal expansion. The fact that all of the M2+ feldspar frameworks have very similar levels of framework “collapse” at room temperature means that they are all at the same stage when the temperature starts to rise, and thus, similar rates of thermal expansion might be expected for each divalent cation feldspar.
Comments. Over the last 20 years, research has continued including single crystal structure determination of synthetic Ga-feldspar analogues (e.g., Fleet, [113]); synchrotron powder X-ray diffraction studies of natural plagioclases where thermal expansion measurements provide a basis for investigating thermoelastic and thermodynamic properties (e.g., Tribaudino et al., [114]); and single crystal X-ray diffraction at controlled high pressure to determine the elastic properties of plagioclase feldspars (Brown et al., [115]). High-resolution neutron powder diffraction experiments at controlled T or P, in particular for synthetic solid-solution series involving analogue endmembers, would provide better data for oxygen positions than X-ray methods. In addition, it would be interesting to explore the solid solutions compositions which might exist within the volcanic sublimate system between KAlSi3O8 (sanidine) and a hypothetical phase of formula K[(Al,Zn)2(As,Si)2O8] (Shchipalkina et al., [66]).
5. Research Topic 4: The Leucite/Pollucite Group of Materials and Their Variable Stoichiometries and Structures
Context. Naturally occurring, anhydrous members of the tetrahedral framework ANA structure [116] have relatively large univalent (A) cations located in the large interframework cavity cation site (W site) with the smaller S site being vacant (general formula A+[C3+Si2O6]); square brackets enclose tetrahedral framework stoichiometry. These phases are represented by the aluminosilicate minerals leucite (K[AlSi2O6] [117]), pollucite (ideally Cs[AlSi2O6] [118,119]), and ammonioleucite (NH4[AlSi2O6] [120]), and by the borosilicate mineral kirchhoffite (Cs[BSi2O6] [121]). Analogous, hydrous ANA phases include analcime (Na[AlSi2O6].H2O; [122]), the ANA prototype, and wairakite (Ca2+[Al2Si4O12]. 2H2O [123]) which contain smaller Na (or Ca) in the S site and H2O in W. The ANA framework is conventionally considered to be made up of two types of secondary building units (SBUs), i.e., 4-membered rings and 6-membered rings of tetrahedra, but some authors also draw attention to a third SBU formed by distorted 8-membered rings (see below). This framework has maximum symmetry Ia-3d and is characterised by two types of channels, one formed by the distorted 8-membered rings and more regular channels formed by 6-membered rings; the latter are oriented along the crystallographic [111] direction of the cubic structure and contain the W-site cations. These channels are the “so-called” percolation channels, which are believed to influence the properties of diffusion, ion conductivity, and molecular sieving in zeolite-type materials and glasses [124]. Thus, while these minerals are conventionally classified as feldspathoids [51], they are also considered to be members of the zeolite structure type [125]. As mentioned above, analcime is generally considered to have a cubic Ia-3d structure (a = 13.725 Å, V = 2585.3 Å3 [51]); other samples have been refined with lower symmetries including an orthorhombic Ibca structure having cell parameters a = 13.733, b = 13.729, c = 13.712 Å, V = 2585.26 Å3 [51] with a mean “pseudocubic” cell edge also of 13.725 Å. By contrast, at room temperature, KAlSi2O6 is tetragonal I41/a with cell parameters a = 13.09, c = 13.75 Å, V = 2356.04 Å3 [117] with a pseudocubic cell edge of 13.306 Å.
Anhydrous phases characterized by the presence of large cavity cations (mainly K and Cs) in W sites are denoted here as having the “leucite/pollucite” structure-type, and over the years, samples have been synthesized with cations forming the tetrahedral oxygen-based framework varying from univalent (Li+) to pentavalent (P5+), and with tetrahedral cation radii varying from 0.11 (B3+) to 0.78 Å (Cd2+). In addition, synthetic analogues are known with Ga3+ or Fe3+ replacing Al3+, Ge4+ or Ti4+ replacing Si4+, and with Rb+ or Tl+ replacing K+ or Cs+. General formulae can also be written as follows: A+[U+0.333T4+2.666O6] (e.g., U = Li, T = Si); A+[D2+0.5T4+2.5O6] (e.g., D = Mg, Cu, T = Si); A+[C3+2E5+O6] (e.g., C = Al, E = P). See [37,38,39] and [98] for a summary of samples having these types of stoichiometry. Such leucites with tetrahedral framework M2+ cations have also been used as model compounds for characterising the short-range order environments of tetrahedral cations in quenched silicate glasses using EXAFS and XANES X-ray absorption techniques [126]. Synthesis of a cubic Ia-3d pollucite phase with Ti4+ replacing Al3+ shows another type of non-stoichiometry with extra oxygen being present to preserve electrostatic neutrality (i.e., CsTiSi2O6.5 [127]). Silica-rich alumino- and boro-silicates are also known with feldspar composition frameworks (e.g., Cs[AlSi3O8] [128,129,130,131], which on the basis of a leucite/pollucite stoichiometry would have the general formula A0.75□0.25[C0.75Si2.25O6]; note the vacancies (□) in the cavity cation site.
The flexible chemistry of the leucite/pollucite phases is also reflected in variable structures with the Space Groups observed including tetragonal I41/a, I41/acd; cubic Ia˗3d, I-43d, Pa˗3; orthorhombic Pbca; and monoclinic P21/c. However, all are isotropy subgroups of the aristotype [98] Ia-3d with either ordered or disordered tetrahedral, multiple symmetry-controlled sites, which would have the same type of atoms in different symmetry sites for some of the different Space Groups. Nevertheless, all of the leucite/pollucite structures can be considered as being related to a cubic Ia-3d “pseudocell”. This can be rationalized, for example, as polymorphic transitions between tetragonal I41/a and cubic Ia-3d, between cubic I-43d and Ia-3d, and between orthorhombic P21/c and monoclinic Pbca are all permitted to be continuous transformations [132], but this situation is clearly sample dependent. Thus, any structural differences may decrease continuously with either increasing temperature or increasing size of the alkali cation reflecting their identical topologies.
The structural relations for the basic framework are shown in Figure 8 using the synthetic leucite analogue, orthorhombic Pbca Cs2CuSi5O12 which was studied by high-resolution neutron powder diffraction to obtain good oxygen data to help to resolve the correct space group for this material [133]. Figure 8 shows the framework tetrahedral linkages for this sample and displays the three different secondary building units and the interframework cavity sites occupied in this case by Cs [133]. The T site Si and Cu atoms are fully ordered, and each divalent Cu is separated from another Cu by two Si atoms; this is equivalent to the “Al avoidance principal” shown by ordered Si-Al frameworks. The 4-membered ring SBUs are shown in Figure 8a,b and the 6-membered rings in Figure 8d. This type of framework is characterised by two types of channels, one formed by the distorted 8-membered rings (Figure 8c) and more regular channels formed by 6-membered rings; the latter are oriented along the crystallographic [111] direction of the cubic structure and contain the W-site cations (Figure 8d). The Cs2CuSi5O12 leucite sample has cell parameters a = 13.589, b = 13.574, c = 13.523 Å, and V = 2512.85 Å3, with a pseudocubic cell edge of 13.595 Å. Note that the tetrahedral species are labelled taking account of the nearest neighbour types; thus, a Q4Si(3Si,1Cu) tetrahedron is defined to have a central Si atom, which is linked by bridging oxygens to four tetrahedra three of which are SiO4 species and one is a CuO4 tetrahedron (see Figure 8 and caption).
Figure 8.
Structure of Pbca crystal structure of hydrothermal Cs2CuSi5O12 [133]. Cs+ cations are shown by light blue circles, CuO4 by brown tetrahedra, Q4Si(4Si) by green tetrahedra, and Q4Si(3Si,1Cu) by grey tetrahedra: (a) projection on (100); note that 4-rings are composed only of Q4Si(3Si,1Cu) secondary building units, and 6-rings are adjacent to the 4-rings; (b) on (001), 4-rings have CuO4 and Q4Si(4Si) opposite each other, and Q4Si(3Si,1Cu) lie between them. (010) shows the same type of 4-rings; (c) view to show the 8-ring of tetrahedra, numbered for clarity; (d) view down the crystallographic [111] direction showing Cs cations in the channel; the 6-rings are fairly symmetrical, reflecting that this orthorhombic leucite shows only a small departure from cubic symmetry. Crystal structure plots provided by Dr Tony Bell.
In this review of leucite/pollucite crystal chemistry, the displacive phase transitions shown by leucite (KAlSi2O6) and its KGaSi2O6 analogue will be discussed together with that for the monoclinic P21/c to othorhombic Pbca K2MgSi5O12 transition. The conclusions from a multivariant regression analysis for the pseudocubic Ia-3d structure type will also be used to show how the pseudocubic unit cell edge (V1/3) can be predicted from the mean ionic radii of both the tetrahedral and the cavity cations and how the mean cavity size can be predicted from the pseudocell edge V1/3 and the mean tetrahedral cation ionic radius.
Leucite Structure Phase Transitions
The natural and synthetic phases belonging to the leucite/pollucite structure type show several types of crystallographic phase transitions which have been studied over the years by mineralogists and materials scientists as a function of size of the cation occupying the W site and temperature. For example, the tetragonal (I41/a) to cubic (Ia-3d) phase transition in natural K-leucite and in synthetic analogues with K replaced by Rb and Cs (e.g., [134,135]), or with Al replaced by Fe3+ [135,136] are well characterized. The compositional and temperature controls of transitions between polymorphs in the cubic space groups I-43d and Ia-3d are known for phases in the system ABSi2O6 (A = Rb, Cs) [131] while Redfern and Henderson [89] have shown that monoclinic (P21/c) K2[MgSi5O12], with ordered Mg and Si T cations, shows a first-order ferroelastic transition to the ordered Pbca structure at elevated temperature.
Phase transitions from I41/a (point group 4/m) tetragonal to (point group m3m) cubic structures [134,135] have been observed as the temperature increases in natural leucite and leucite analogues. Note that an intermediate tetragonal phase in this transition (I41/acd, point group 4/mmm) has been reported [135,137,138]. This phase transition in KAlSi2O6 leucite is displacive, rapid, reversible second order, and continuous [134,135,138]. It has been shown [86] that at ambient temperature, KGaSi2O6 is isostructural with KAlSi2O6 and KFe3+Si2O6; all have the T-site cation disordered I41/a tetragonal structure. The tetragonal structure for KGaSi2O6 is retained on heating to 573 K; however, at 673 K, a phase transition begins, and a high-temperature cubic polymorph (isostructural with pollucite CsAlSi2O6) appears. Between 673 and 943 K, a two-phase region occurs. Thus, at about 673 K, the cubic (004)c peak appears between the tetragonal (400)t and (004)t peaks, and on further heating, this (400)c peak gets stronger, and the two tetragonal peaks move together and get weaker; the tetragonal peaks are no longer detectable at 973 K. The end of the phase transition is placed at 970 ± 10 K. After cooling to ambient temperature, the structure had reverted to the I41/a tetragonal polymorph consistent with it being a reversible displacive transition [134,135].
With increasing temperature, the a axis for the tetragonal polymorph of KGaSi3O8 shows a marked increase, and the c axis a much smaller decrease [86]; these are the expected changes for the leucite structure as the trend towards a more isometric phase takes place steadily (Figure 9a). The a lattice parameter for the newly formed cubic polymorph shows a steady increase for the cubic component of the mixed phase region (680 to 880 K), which continues without any discontinuity into the cubic single-phase region. This type of two-phase assemblage has been referred to as the “region of coexistence” [42]. As expected, the c/a ratio for the I41/a tetragonal structure decreases progressively on heating as the temperature approaches the phase transition temperature. The cubic phase has a larger volume than the coexisting I41/a tetragonal phase in the two-phase region and, for example, at 773 K shows a positive ∆V of ~1.0%. This clear ∆V and the presence of the two phases over a small temperature range, together with it being an unquenchable transition, point to the transition being a first-order displacive transition with a martensitic, strain mediated mechanism [86].
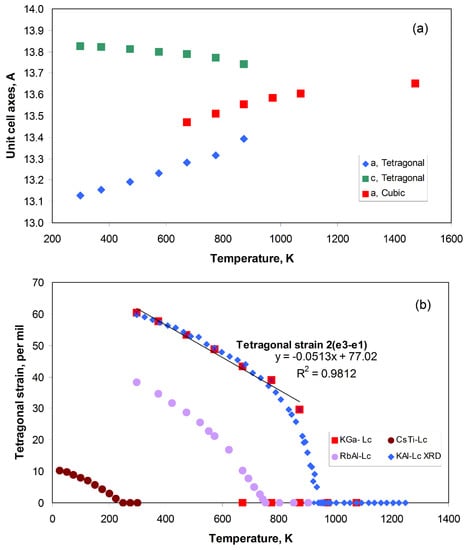
Figure 9.
(a) Temperature dependence of a and c cell parameters for the tetragonal I41/a KGaSi2O6 leucite analogue as a function of temperature (data in [86]). The c/a ratio decreases with increasing temperature trending towards the phase transition to the cubic Ia-3d polymorph. A 2-phase region characterises the smeared transition. The a cell parameters for the cubic polymorphs over the temperature range from 673 to 1073 K define a linear trend which is extrapolated into the tetragonal field to define values for a0 to calculate the strains on a and c. (b) shows the temperature dependence of the calculated tetragonal strain for KGaSi2O6 and compares its features with those for the same symmetry change in natural KAlSi2O6 [138] and synthetic CsAlSi2O6 [135] and CsTiSi2O6.5 [139]. The first order nature of the transition in KGaSi2O6 is clear. See text for further details.
The lattice parameters and phase proportions of the coexisting tetragonal and cubic phases for KGaSi2O6 over the temperature range defining the region of coexistence (673–873 K) show important differences to the characteristics shown by the phase transition for natural KAlSi2O6 leucite [134,135,138]. The continuous nature of the transition for natural leucite with no volume discontinuity suggests that it is a 2nd order transition while those for KGaSi2O6 show that it is a 1st order transition, and it is interesting to consider how such differences might affect the spontaneous strain associated with this type of phase transformation.
The thermal expansion and deformation of the three-dimensional framework structure of natural leucite has been dealt with in terms of a strain ellipsoid [88,138,139]; the strain associated with displacive phase transitions in the leucite structure can be estimated by comparing the measured a and c parameters at particular temperatures below the phase transition to those for the cubic polymorphs extrapolated to these lower temperatures (ao values) [86,138]; see Figure 9a for KGaSi3O8. Thus, e11 = e22 = (a − a0)/a0 and e33 = (c − a0)/a0. The tetragonal strain is defined as et = (1/√3)(2e33 – 2e11) [88], and the measured volume strain (Vs) at a given temperature in the tetragonal leucite field is given as Vss = (VT − VoT)/VoT.
The tetragonal strain parameters calculated using the above equations for the tetragonal polymorphs of KGaSi2O6 are shown in Figure 9b using per mil units; all the cubic polymorphs plot on the zero et axis. It is clear that the tetragonal strain versus temperature data at up to 773 K define an excellent linear trend and even the point at 873 K falls close to this trend (et = −0.0513 × T + 77.02). The tetragonal phases defining the top three temperature points all fall within the region of coexistence, and the matching cubic phase data points are also shown for these temperatures; this serves to show the discontinuity between the tetragonal and cubic structures for KGaSi2O6. Figure 9b also shows calculated data for the symmetry-breaking ferroelastic tetragonal strain using the published unit cell parameters for KAlSi2O6 [135,138], CsAlSi2O6 leucite [135] and Cs2Ti0.1Al0.9Si2O6.05 [139]. The et vs. temperature data for the CsAlSi2O6 and Cs2Ti0.1Al0.9Si2O6.05 analogues have well-defined flat curves which shows a clear intersection with the zero-strain axis; this provides well defined transition temperatures to cubic symmetry (Tc). Figure 9b shows a smoothly curved spontaneous strain trends for KAlSi2O6 which at the highest temperatures is very steep but does not intersect the zero axis; nevertheless, a clearly defined Tc intercept is indicated. It is clear that at temperatures up to about ~770 K the KGaSi2O6 and KAlSi2O6 and leucites both fall within the same spontaneous strain “band” but diverge significantly at higher temperatures. These features are consistent with natural leucite showing a continuous, 2nd order transition and the KGaSi2O6 synthetic analogue leucite a first-order transition.
A phase transition from monoclinic P21/c to orthorhombic Pbca is also known in analogue leucites with compositions based on K2M2+Si5O12. The Space Group P21/c is an isotropy sub-group of Pbca which is permitted to show a continuous transition [132], but as mentioned above, that is sample dependent. The hydrothermally crystallized K2MgSi5O12 leucite analogue was shown by integrated synchrotron powder XRD, electron diffraction and 29Si MAS NMR to have P21/c symmetry [140], and the same techniques showed that hydrothermal Cs2CdSi5O12 is orthorhombic Pbca [141]; many other leucite analogues have now been synthesized with that space group [e.g., 133]. Guinier camera, high-temperature powder XRD techniques show the presence of a transition for K2MgSi5O12 leucite from P21/c to Pbca at ~622 K [89]. This transition showed a small + ve ∆V (+ 1.6%) and both monoclinic and cubic phases coexist over a temperature range of ~ 25 K. A Landau/Lifshitz analysis showed that the symmetry operated strain e13 = [(c − c0)/c0]cos β acts as the symmetry-adapted strain with this shear strain determined by the behaviour of the monoclinic angle β [89]. The transition is an unquenchable, displacive, ferroelastic transition whose first-order character is attributed to a large excess volume, which is related to the collapse of the framework about the cavity alkali cation site. In this paper, a strain analysis is shown to characterize another type of displacive transition shown by a leucite group compound.
The components of the spontaneous strain tensor for a monoclinic–orthorhombic transition, where a0, b0, c0, and V0 are orthorhombic parameters extrapolated into the lower temperature monoclinic field (to each measured temperature), can be calculated as follows [88]:
e11 = a/a0 − 1 (Equation (49) in [88]); e22 = b/b0 − 1 (Equation (50) in [88]); e33 = ((c/c0) sin β) − 1 (Equation (51) in [88]); e13 = (0.5((b/b0)cos β) − 1 (Equation (53)); e12 = e23 = 0 (Equations (52) and (54)).
The volume strain is defined as Vss = (V − V0)/V0 (Equation (69) in [88]).
The K2MgSi5O12 leucite high-temperature cell parameters from (Table 1 [89]) are plotted here in Figure 10. For the monoclinic phases, a and c show smooth increases with increasing temperature; over the same temperature range but the b parameter shows little change. At the phase transition a, b and c, all show increases over a temperature range of 10 K and then a step up to larger cell edges for the cubic phase. The unit cell volumes show the same upturn at the phase transition and then a distinct ∆V. The β angle decreases steadily from 91.7 to 91.1° and then a distinct step down to 90° at the transition.
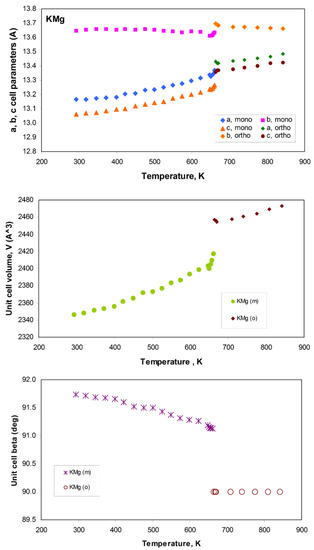
Figure 10.
Unit cell parameters for P21/c K2MgSi5O12 leucite analogue depending on temperature; see [89] for data. All the parameters show distinct discontinuities at the phase transition to Pbca with a clear negative ∆V.
The spontaneous strains for K2MgSi5O12 leucite were calculated from the cell parameters using an Excel spreadsheet and are displayed in Figure 11. The e11, e33, Vss spontaneous strains and the symmetry adapted e13 strain all have negative values for the monoclinic polymorphs and all become less negative with increasing temperature as the strains lessen. The e22 strain also has a negative value, which shows a very slight change initially, and then, all the strain parameters show a small upturn at the transition. The e13 strain has a value very similar value to the e33 strain but lacks the first upturn. All of the strain parameters show clear discontinuities confirming the first-order nature of the transition.
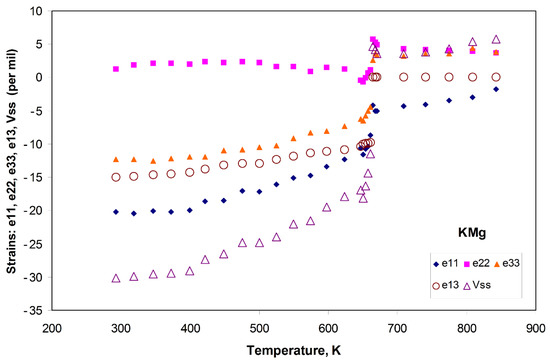
Figure 11.
Temperature dependence of the strain parameters e11, e22, e33, e13, and the volume strain Vss for the monoclinic P21/c to orthorhombic Pbca K2MgSi5O12 phase transition; strains calculated using the data from [89]. The discontinuity at 663 K is clear. See text for further interpretations.
Figure 12 shows a good linear relationship between e132 vs. unit cell volume V that extrapolates towards the values for the orthorhombic structure; this trend is interpreted as a linear-quadratic relationship between cos(β*)2 (effectively e132, proportional to Q12) and V (proportional to Q2) [89].
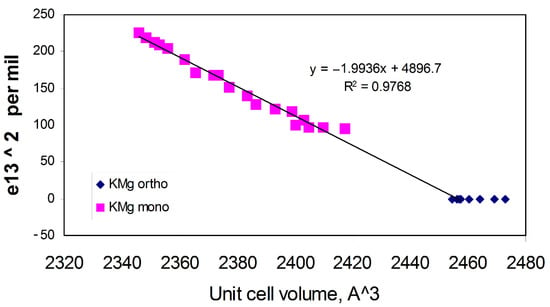
Figure 12.
The plot of shear strain monitored from e132 (effectively cos(β)2) for the monoclinic polymorphs shows a good linear relationship with the cell volume. This trend extrapolates to zero (β = 90°) at the phase transition. The volume discontinuity at the transition is clear and this together with the existence of a 2-phase field points to a first-order phase transition. See text for further interpretation.
It has been pointed out that all of the space groups which have been used describe the different leucite analogue structures are isotropy sub-groups of the cubic Ia-3d aristotype. Henderson et al. (2017) [98] established a database for natural and synthetic members of this group. In that paper, multivariant least-squares regression analyses, using a series of independent variables, established that the unit cell edge (V1/3) can be estimated, for any chosen leucite stoichiometry, from the mean T-site cation ionic radius and the mean cavity-cation ionic radius as follows:
Pseudo-cubic cell edge (Ǻ) = 9.06 (intercept constant) + 6.20 × (mean T ionic radius) + 1.41 × (mean A ionic radius)
(R2 = 0.973; S.E. = 0.00667; F = 981.9; Null significance parameter < 0.000)
Note that the equivalent equation given in [98] has values for the coefficients that would give a cell edge that is 10 times too small. The equation given here gives a pseudocubic cell edge for leucite analogue Cs2CuSi5O12 of 13.61 Å, very close to the value of 13.595 Å reported above.
For all the leucite database samples, a value for the mean A–O dependent variable (i.e., the mean distance for the first 6 oxygens in the cavity cation coordination polyhedron) can be estimated as follows:
A–O distance (Ǻ) = −5.357 + 7.325 × (cell size V1/3 pseudocubic cell size, nm) − 4.421 × (mean tetrahedral cation radius, Ǻ) + 0.98 (temperature/1000, K).
The A–O distance is a measure of the width of the large channel in phases having the leucite/pollucite structure, and as technical applications are associated with functions depending on that macroscopic properties, the mean T-site and A-site sizes can be balanced by choosing the appropriate cavity and mean T-site cations that would deliver a channel of the required size. Time will tell how useful this approach might become.
6. Final Comment
Compounds having framework structures form different types of mineral groups and also find many industrial and environmental applications as summarized above. They are widely studied by different types of scientific specialists but terminology and approach differences sometimes complicate cross-disciplinary understanding. The contributions made by investigating samples of both natural minerals and the diverse compositions represented by synthetic analogues have been emphasized in this review. I hope that other physical scientists and material scientists, broadly defined, will have found this approach useful.
Funding
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Acknowledgments
Jack Zussman advised on nepheline endmember molecule calculations and read an early version of Research Topic 1. Kevin Knight gave much advice and kindly provided the expressions to understand the fundamental significance of the ∆Alcc/∆Tcharge ratios. Tony Bell provided the figure for the Pbca structure shown in Figure 8. The enforced closure of the University and the voluntary lockdown at home for 8 months during the coronavirus epidemic provided the time to embark on this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zharadyik, J.; Jirásek, J.; Starý, J.; Sivek, M. Production, reserves, and processing of feldspar and feldspathoid rocks in the Czech Republic from 2005 to 2019–An overview. Minerals 2020, 10, 722. [Google Scholar] [CrossRef]
- Hogan, M.A.; Risbud, S.H. Gel-derived amorphous cesium-aluminosilicate powders for formation of pollucite glass-ceramics. J. Mater. Sci. 1991, 6, 217–219. [Google Scholar] [CrossRef]
- Deshkar, A.; Marcial, J.; Southern, S.A.; Kobera, L.; Bryce, D.L.; McCloy, J.S.; Goel, A. Understanding the structural origin of crystalline phase transformations in nepheline (NaAlSiO4)-based glass-ceramics. J. Amer. Ceram. Soc. 2017, 100, 2859–2878. [Google Scholar] [CrossRef]
- Maeda, K.; Hirose, M.; Kobayashi, T. High performance transparent glass-ceramics for optical components. Ceram. Soc. Jpn. 2015, 123, 949–954. [Google Scholar] [CrossRef][Green Version]
- Koenig, C.J. Use of nepheline syenite in sanitary porcelain. J. Am. Ceram. Soc. 2006, 22, 38–46. [Google Scholar] [CrossRef]
- Diella, V.; Adamo, H.; Pagliari, L.; Francescon, F. Effect of particle size and starting composition in Na-feldspar/kaolinite system at high temperature. J. Eur. Ceram. Soc. 2015, 35, 1327–1335. [Google Scholar] [CrossRef]
- Bernasconi, A.; Marinoni, N.; Pavese, A.; Francescon, F. Feldspar and firing cycle effects on the evolution of sanitary-ware body. Ceram. Int. 2014, 40, 6389–6398. [Google Scholar] [CrossRef]
- Roth, G.; Bȍhm, H. Ionic conductivity of sodium-nepheline single crystals. Solid State Ion. 1986, 18–19, 553–556. [Google Scholar] [CrossRef]
- Palmer, D.C.; Salje, E.K.H. Phase transitions in leucite: Dielectric properties and transition mechanism. Phys. Chem. Miner. 1990, 17, 444–452. [Google Scholar] [CrossRef]
- Jones, R.J.; Thrall, M.; Henderson, C.M.B. Complex impedance spectroscopy and transport properties of a natural leucite as a function of temperature and pressure. Miner. Mag. 2010, 74, 507–519. [Google Scholar] [CrossRef]
- Kumar, R.; Rakiewicz, E.F.; Rajagopalan, K. Preparation and characterization of of fluid cracking catalysts containing pollucite. J. Catal. 1993, 143, 304–307. [Google Scholar] [CrossRef]
- Nishioka, N.; Yanigisawa, K.; Yamasaki, N. Solidification of sludge ash by hydrothermal hot-pressing. Res. J. Water Pollut. Fed. 1990, 62, 926–932. [Google Scholar]
- Ng, E.-P.; Aleid Ghadah, M.S.; Rigolet, S.; Daou, T.J.; Mintova, S.; Chuan Ling, T. Micro- and macroscopic observations of the nucleation process and crystal growth of nanosized Cs-pollucite in an organotemplate-free hydrosol. New J. Chem. 2019, 43, 17433–17440. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Nishioka, M.; Yamasaki, N. Immobilization of cesium into pollucite structure by hydrothermal hot-pressing. J. Nucl. Sci. Tech. 1987, 24, 51–60. [Google Scholar] [CrossRef]
- Gatta, G.S.; Rinaldi, R.; Mclntyre, G.J.; Nenert, G.; Bellatreccia, F.; Guastoni, A.; Della Ventura, G. On the crystal structure and crystal chemistry of pollucite, (Cs,Na)16Al16Si32O96.H2O. Am. Miner. 2009, 94, 1560–1568. [Google Scholar] [CrossRef]
- Orlova, A.I.; Ojovan, M.I. Ceramic mineral waste-forms for nuclear waste immobilization. Materials 2019, 12, 2638. [Google Scholar] [CrossRef] [PubMed]
- Cesar, P.F.; Yoshimura, H.N.; Miranda, C.Y.; Okada, C.Y. Correlation between fracture toughness and leucite content in dental porcelains. J. Dent. 2005, 33, 721–729. [Google Scholar] [CrossRef]
- Nakashima, J.; Taira, T.; Sawasa, T. In vitro wear of four ceramic materials and human enamel on enamel antagonist. Eur. J. Oral Sci. 2016, 124, 219–300. [Google Scholar] [CrossRef]
- Cattell, M.J.; Patzig, C.; Bissasu, S.; Tsoutsos, A. Nucleation efficacy and flexural strength of novel leucite glass ceramics. Dent. Mater. 2020, 36, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Linvill, M.L.; Vandersande, J.W.; Pohl, R.O. Thermal conductivity of feldspars. Bull. Min. 1984, 107, 521–527. [Google Scholar] [CrossRef]
- Wu, X.; Zeng, Y.-F. Compensation effect for electrical conductivity and its application to estimate oxygen diffusivity in minerals. J. Geophys. Res. Solid Earth 2003, 108, 12. [Google Scholar] [CrossRef]
- Hu, H.; Dai, L.; Li, H.; Jiang, J. Electrical conductivity of K-feldspar at high-temperature and pressure. Min. Pet. 2014, 108, 609–618. [Google Scholar] [CrossRef]
- Buerger, M.J. The stuffed derivatives of the silica structures. Am. Min. 1954, 39, 600–614. [Google Scholar]
- Schairer, J.F.; Bowen, N.L. Preliminary report on equilibrium relations between feldspathoids, alkali feldspars and silica. Am. Geophys. Union Trans. 1935, 16, 325–328. [Google Scholar] [CrossRef]
- Bannister, F.A.; Hey, M.H. A chemical, optical and X-ray study of nepheline and kaliophilite. Min. Mag. 1931, 22, 569–608. [Google Scholar] [CrossRef]
- Bowen, N.L. The composition of nephelite. Am. J. Sci. 1912, 33, 49–54. [Google Scholar] [CrossRef]
- Greig, J.W.; Barth, T.F.W. The system Na2O.Al2O3.2SiO2 (nephelite, carnegieite)–Na2O.Al2O3.6SiO2 (albite). Am. J. Sci. 1938, 35A, 93–112. [Google Scholar]
- Bowen, N.L. The binary system: Na2Al¬2Si2O8 (nephelite, carnegieite)–CaAl2Si2O8 (anorthite). Am. J. Sci. 1912, 33, 551–573. [Google Scholar] [CrossRef]
- Goldsmith, J.R. Some aspects of the system NaAlSiO4-CaO.Al2O3. Am. Miner. 1949, 34, 471–493. [Google Scholar]
- Hahn, T.; Buerger, M.J. The detailed structure of nepheline, KNa3Al4Si4O16. Z. Krist. 1955, 106, 308–338. [Google Scholar] [CrossRef]
- Donnay, G.; Schairer, J.F.; Donnay, J.D.H. Nepheline solid solutions. Miner. Mag. 1959, 32, 93–109. [Google Scholar] [CrossRef]
- Hamilton, D.L.; MacKenzie, W.S. Nepheline solid solution in the system NaAlSiO4–KAlSiO4–SiO2. J. Pet. 1960, 1, 56–72. [Google Scholar] [CrossRef]
- Blancher, S.; D’Arco, P.; Fonteilles, M.; Pascal, M.L. Evolution of nepheline from mafic to highly differentiated members of the alkaline series: The Messum complex, Namibia. Min. Mag. 2010, 74, 413–432. [Google Scholar] [CrossRef]
- Barth, T.F.W. The composition of nepheline. Schweiz. Miner. Pet. Mitt. 1963, 43, 153–164. [Google Scholar]
- Dollase, W.A.; Thomas, W.M. The crystal chemistry of silica-rich, alkali-deficient nepheline. Contrib. Min. Pet. 1978, 66, 311–318. [Google Scholar] [CrossRef]
- Roedder, E.W. The system K2O-MgO-SiO2. Am. J. Sci. 1951, 249, 81–130, 224–248. [Google Scholar] [CrossRef]
- Torres-Martinez, L.M.; West, A.R. Pollucite- and leucite-related phases: A2BX5O12 and ACX2O6 (A = K,Rb,Cs; B = Be,Mg,Fe,Co,Ni,Zn,Cd; C = B, Al, Ga,Fe,Cr; X = Si, Ge). Zeitsschrift. Anorg. Allg. Chem. 1989, 573, 223–230. [Google Scholar] [CrossRef]
- Torres-Martinex, L.M.; Gard, J.A.; Howie, R.A.; West, A.R. Synthesis of Cs2BeSi5O12 with a pollucite structure. J. Solid State Chem. 1984, 51, 100–103. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Bell, A.M.T.; Kohn, S.C.; Page, C.S. Leucite-pollucite structure-type variability and the structure of a synthetic end-member wairakite (CaAl2Si4O12H2O). Miner. Mag. 1998, 62, 165178. [Google Scholar] [CrossRef]
- Roedder, E. A reconnaissance of the liquidus relationships in the system K2O.2SiO2–FeO–SiO2. Am. J. Sci. Bowen Vol. 1952, 250, 435–456. [Google Scholar]
- Liu, B.; Barbier, J. Structures of the stuffed-tridymite derivatives, BaMSiO4 (M = Co, Zn, Mg). J. Solid State Chem. 1993, 102, 115–125. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Taylor, D. The structural behaviour of the nepheline family: 1. Sr and Ba aluminates (MAl2O4), Miner. Mag. 1982, 45, 111–127. [Google Scholar] [CrossRef]
- Roux, J. Etude des solutions solides des néphélines (Na,K)AlSiO4 et (Na,Rb)AlSiO4). Geochim. Cosmochim. Acta 1974, 38, 1213–1224. [Google Scholar] [CrossRef]
- Peterson, T.D. Peralkaline nephelinites. 1. Comparative petrology of Shombole and Oldoinyo Lengai, East Africa. Contrib. Miner. Pet. 1989, 101, 458–478. [Google Scholar] [CrossRef]
- Rossi, G.; Oberti, R.; Smith, D.C. The crystal structure of a K-poor, Ca-rich silicate with the nepheline framework, and crystal-chemical relationships in the compositional space (K,Na,Ca,□)8(Al,Si)16O32. Eur. J. Miner. 1989, 1, 59–70. [Google Scholar] [CrossRef]
- Hamada, M.; Akasaka, M.; Ohfuji, H. Crystal chemistry of K-rich nepheline in nephelinite from Hamada Shimane Province. Miner. Mag. 2019, 83, 239–247. [Google Scholar] [CrossRef]
- Morgan, G.B., VI; London, D. Effect of current density on the electron microprobe analysis of alkali aluminosilicate glasses. Am. Miner. 2005, 90, 1131–1138. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Pierozynski, W.J. An experimental study of Sr, Ba and Rb partitioning between alkali feldspar and silicate liquid in the system nepheline- kalsilite-quartz at 0.1 GPa P(H2O). Miner. Mag. 2012, 76, 157–190. [Google Scholar] [CrossRef]
- Okumiya, M.; Yamaguchi, G. The crystal structure of κ’-Al2O3, the new intermediate phase. Bull. Chem. Soc. Jpn. 1971, 44, 1567–1570. [Google Scholar] [CrossRef]
- Levin, I.; Brandon, D. Metastable alumina polymorphs: Crystal structures and transition sequences. J. Am. Ceram. Soc. 1998, 81, 1995–2012. [Google Scholar] [CrossRef]
- Deer, W.A.; Howie, R.A.; Zussman, J. Rock-Forming Minerals, Framework Silicates: Silica Minerals, Feldspathoids and the Zeolites; The Geological Society: London, UK, 2004; Volume 4B, 982p. [Google Scholar]
- Deer, W.A.; Howie, R.A.; Zussman, J. An Introduction to the Rock-Forming Minerals, 2nd ed.; Longman Scientific & Technical: Harlow, UK, 1992; 696p. [Google Scholar]
- Edgar, A.D. Studies on cancrinite: II–Stability fields and cell dimensions of calcium and potassium-rich cancrinites. Can. Miner. 1964, 8, 53–67. [Google Scholar]
- Sirbescu, M.; Jenkins, D.M. Experiments on the stability of cancrinite in the system Na2O-CaO-Al2O3-SiO2-CO2-H2O. Am. Miner. 1999, 84, 1850–1860. [Google Scholar] [CrossRef]
- Pant, A.K. A reconsideration of the crystal structure of β-Na2Si2O5. Acta Cryst. 1968, B24, 1077–1083. [Google Scholar] [CrossRef]
- Kahlenberg, V. Structural chemistry of anhydrous sodium silicate—A review. Chimia 2010, 64, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.M.B. Nepheline solid solution compositions: Stoichiometry revisited, reviewed, clarified and rationalised. Mineral. Mag. 2020. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Gibb, F.G.F. Felsic mineral crystallization trends in differentiating alkaline basic magmas. Contrib. Miner. Pet. 1983, 84, 355–364. [Google Scholar] [CrossRef]
- Wilkinson, J.F.G.; Hensel, H.D. Nephelines and analcimes in some alkaline igneous rocks. Contrib. Miner. Pet. 1994, 118, 79–91. [Google Scholar] [CrossRef]
- Mann, U.; Marks, M.; Markl, G. Influence of oxygen fugacity on mineral compositions in peralkaline melts. The Katzenbuckel volcano, Southwest Germany. Lithos 2006, 91, 262–285. [Google Scholar] [CrossRef]
- Vulic, P.; Balić-Žunić, T.; Belmonte, L.J.; Kahlenberg, V. Crystal chemistry of nephelines from ijolites and nepheline-rich pegmatites: Influence of composition and genesis on the crystal structure investigated by X-ray diffraction. Miner. Pet. 2011, 101, 185–194. [Google Scholar] [CrossRef]
- Vrublevskii, V.V.; Nikiforov, A.V.; Sugorakova, A.M.; Kozulina, T.V. Petrogenesis and tectonic setting of the Cambrian Kharly alkaline-carbonatite complex (Sangilen Plateau, Southern Siberia): Implications for the Early Paleozoic evolution of magmatism in the western Central Asian Orogenic Belt. J. Asian Earth Sci. 2020, 188, 104163. [Google Scholar] [CrossRef]
- Valentin, E.; Botelho, N.F.; Dantas, E.L. Monte Santo suite, an example of Ediacaran-Cambrian deformed alakline rocks in the Aragaia Belt Central Brazil. Implications for Western Gondwana evolution. Lithos 2020, 365–367, 105552. [Google Scholar] [CrossRef]
- Mitchell, R.H.; Dawson, J.B. The 24th September ash eruption of the carbonatite volcano Oldoinyo Lengai, Tanzania: Mineralogy of the ash and implications for formation of a new magma type. Miner. Mag. 2007, 71, 483–492. [Google Scholar] [CrossRef]
- Berkesi, M.; Bali, E.; Bodnar, R.J.; Szabó, A.; Guzmics, T. Carbonatite and highly peralkaline nephelinite melts from Oldoinyo Lengai volcano, Tanzania. The role of natrite-normative fluid degassing. Gondwana Res. 2020, 85, 76–83. [Google Scholar] [CrossRef]
- Shchipalkina, N.V.; Pekov, I.V.; Koshiykova, N.N.; Britvin, S.N.; Zubkova, N.V.; Varlamov, D.A.; Sidorov, E.G. Unusual mineralization in fumarolic sublimates of the Tolbachik volcano, Kamchatka, Russia–Part 2: Tectosilicates. Eur. J. Miner. 2020, 32, 121–136. [Google Scholar] [CrossRef]
- Kerraouch, I.; Ebert, S.; Patzek, M.; Bischoff, A.; Zolensky, M.E.; Pack, A.; Schmitt-Kopplin, P.; Belhai, D.; Bendaoud, A.; Le, L. A light, chondritic xenolith in the Murchison (CM) chondrite–Formation by fluid-assisted percolation during metasomatism. Geochemistry 2019, 79, 125518. [Google Scholar] [CrossRef]
- Grundy, H.D.; Hassan, I. The crystal structure of a carbonate-rich cancrinite. Can. Miner. 1982, 20, 239–251. [Google Scholar]
- Hassan, I.; Grundy, H.D. The character of the cancrinite-vishnevite solid-solution series. Can. Miner. 1984, 22, 333–340. [Google Scholar]
- Hassan, I.; Grundy, H.D. The crystal structure of basic cancrinite, ideally Na8[Al6Si6O24](OH)2.3H2O. Can. Miner. 1991, 29, 377–383. [Google Scholar]
- McConnell, J.D.C. Electron diffraction study of subsidiary maxima of scattered intensity in nepheline. Miner. Mag. 1962, 33, 114–124. [Google Scholar] [CrossRef][Green Version]
- McConnell, J.D.C. Time-temperature study of the intensity of satellite reflections in nepheline. Am. Miner. 1981, 66, 990–996. [Google Scholar]
- Antao, S.M.; Hassan, I. Nepheline: Structure of three samples from the Bancroft area, Ontario, obtained using synchrotron high-resolution powder X-ray diffraction. Can. Miner. 2010, 48, 69–80. [Google Scholar] [CrossRef]
- Friese, K.; Grzechnik, A.; Petřīč, V.; Schönleber, A.; van Smaalen, S.; Morgenroth, W. Modulated structure of nepheline. Acta Cryst. 2011, B67, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.M.B.; Roux, J. Inversions in sub-potassic nephelines. Contrib. Miner. Petrol. 1977, 61, 279–298. [Google Scholar] [CrossRef]
- Schneider, O.W.; Stoeck, H. The NaAlSiO4 nepheline-carnegieite solid-state transformation. Zeits. Krist. 1994, 209, 113–117. [Google Scholar] [CrossRef]
- Tanaka, E.; Ishii, Y.; Tsukasaki, H.; Tanigucha, H.; Mori, S. Structural changes and microstructures in stuffed-tridymite compounds Ba1-xSrxAl2O4. Jpn. J. Appl. Phys. 2014, 53, 09PB01. [Google Scholar] [CrossRef]
- Shivaruma, N.J.; Coetsee, E.; Roos, W.D.; Nagabhushana, K.R.; Swart, H.C. Charge carrier trapping processes in un-doped and BaAl2O4:Eu3+ nanophosphor for thermoluminescent dosimeter applications. J. Phys. D Appl. Phys. 2020, 53, 475305. [Google Scholar] [CrossRef]
- Rodehorst, U.; Carpenter, M.A.; Marion, S.; Henderson, C.M.B. Structural phase transitions and mixing behaviour of the Ba-aluminate (BaAl2O4)–Sr-aluminate (SrAl2O4) solid solution. Miner. Mag. 2003, 67, 989–1013. [Google Scholar] [CrossRef]
- Ishii, Y.; Tsukasaki, H.; Kawaguchi, S.; Ouchi, Y.; Mori, S. Structural investigation of the SrAl2O4–BaAl2O4 solid solution system with unstable domain walls. J. Solid State Chem. 2017, 249, 149–153. [Google Scholar] [CrossRef]
- Wolten, G.M. Diffusionless phase transformations in zirconia and hafnia. J. Amer. Ceram. Soc. 1963, 46, 418–422. [Google Scholar] [CrossRef]
- Garvie, R.C. The occurrence of metastable tetragonal zirconia as a crystallite size effect. J. Phys. Chem. 1965, 69, 1238–1243. [Google Scholar] [CrossRef]
- Megaw, H.D. Temperature changes in the crystal structure of barium titanium oxide. Proc. R. Soc. A 1947, 189, 261–283. [Google Scholar]
- Parker, T.J.; Burfoot, J.C. Cubic-tetragonal phase transitions in high resistivity BaTiO3 single crystals. Br. J. Appl. Phys. (J. Phys. D) 1969, 2, 1168–1170. [Google Scholar] [CrossRef]
- Leadbetter, A.J.; Wright, A.F. The α-β transition in cristobalite phases of SiO2 and AlPO4 I. X-ray studies. Phil. Mag. 1976, 33105–33112. [Google Scholar] [CrossRef]
- Bell, A.M.T.; Henderson, C.M.B. Tetragonal-cubic phase transition in KGaSi2O6 synthetic leucite analogue and its probable mechansim. J. Solid State Chem. 2020, 284, 121142. [Google Scholar] [CrossRef]
- Avdeev, M.; Yakovlev, S.; Yaramchenko, A.; Kharton, V. Transitions between P21, P63(√3A), and P6322 modifications of SrAl2O4 by in situ high-temperature X-ray and neutron diffraction. J. Solid State Chem. 2007, 180, 3535–3544. [Google Scholar] [CrossRef]
- Carpenter, M.A.; Salje, E.K.H.; Graeme-Barber, A. Spontaneous strain as a determinant of thermodynamic properties for phase transitions in minerals. Eur. J. Miner. 1998, 10, 621–691. [Google Scholar] [CrossRef]
- Redfern, S.A.T.; Henderson, C.M.B. Monoclinic-orthorhombic phase transition in the K2MgSi5O12 leucite analog. Am. Miner. 1996, 81, 369–374. [Google Scholar] [CrossRef]
- Ardit, M.; Martucci, A.; Cruciani, G. Monoclinic-orthorhombic phase transition in ZSM-5 zeolite: Spontaneous strain variation and thermodynamic properties. J. Phys. Chem. C 2015, 119, 7351–7359. [Google Scholar] [CrossRef]
- Megaw, H.D. The architecture of feldspars. In The Feldsparsi; MacKenzie, W.S., Zussman, J., Eds.; NATO Advanced Study Institute, 1972; Manchester University Press: Manchester, UK, 1974; pp. 2–24. [Google Scholar]
- Henderson, C.M.B. An elevated temperature X-ray study of synthetic disordered Na-K alkali feldspars. Contrib. Miner. Petrol. 1979, 79, 71–79. [Google Scholar] [CrossRef]
- Henderson, C.M.B. Thermal expansion of alkali feldspars II. Rb-sanidine and maximum microcline. In Progress in Experimental Petrology; NERC: Swindon, UK, 1978; pp. 51–54. [Google Scholar]
- Henderson, C.M.B. Thermal expansion of feldspars III. RbGaSi3O8-sanidine, Sr-feldspar and an ordered microcline-albite solid solutions (Or62.4, mole %). In Progress in Experimental Petrology; NERC Publication Series D, Sixth Progress Report; NERC Publication: Swindon, Wiltshire, UK, 1984; pp. 78–83. [Google Scholar]
- Hovis, G.L.; Morabito, J.R.; Spooner, A.; Mott, A.; Person, E.L.; Henderson, C.M.B.; Roux, J.; Harlov, D. A simple predictive model for the thermal expansion of Al,Si3 feldspars. Am. Miner. 2008, 93, 1568–1573. [Google Scholar] [CrossRef]
- Bruno, E.; Pentinghaus, H. Substitutions of cations in natural and synthetic feldspars. In The Feldspars; MacKenzie, W., Zussman, J., Eds.; NATO Advance Study Institute, 1972; Manchester University Press: Manchester, UK, 1974; pp. 574–614. [Google Scholar]
- Kyono, A.; Kimata, M. Refinement of the crystal structure of a synthetic non-stoichiometric Rb-feldspar. Miner. Mag. 2001, 65, 523–531. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Bell, A.M.T.; Knight, K.S. Variable stoichiometry in tectosilicates having the leucite/pollucite-type structure with particular emphasis on modeling the interframework cavity cation environment. J. Solid State Chem. 2017, 251, 90–104. [Google Scholar] [CrossRef]
- Ohashi, Y. A program to calculate the strain tensor from two sets of unit cell parameters. In Comparative Crystal Chemistry; Hazen, R.M., Finger, L.W., Eds.; Wiley: Chichester, UK; New York, NY, USA, 1982; pp. 92–102. [Google Scholar]
- Benna, P.; Bruno, E. Single crystal in situ high-temperature investigation on strontium feldspar. Am. Miner. 2001, 86, 690–696. [Google Scholar] [CrossRef]
- Waldbaum, D.R.; Robie, R.A. Calorimetric investigations of Na-K mixing and polymorphism in the alkali feldspars. Z. Krist. 1971, 134, 381–420. [Google Scholar]
- Winter, J.K.; Okamura, F.P.; Ghose, S. A high-temperature structural study of high-albite, monalbite and the analbite-monalbite phase transition. Am. Miner. 1979, 64, 409–423. [Google Scholar]
- Kroll, H.; Bambauer, H.-U.; Schirmer, U. The high-albite-monalbite andanalbite-monalbite transitions. Am. Miner. 1980, 65, 1192–1211. [Google Scholar]
- Openshaw, R.E.; Henderson, C.M.B.; Brown, W.L. A room-temperature phase transition in maximum microcline. Phys. Chem. Miner. 1979, 5, 95–104. [Google Scholar] [CrossRef]
- Grundy, H.D.; Brown, W.L. A high-temperature X-ray study of low and high plagiclase feldspars. In The Feldsparsi; MacKenzie, W.S., Zussman, J., Eds.; NATO Advance Study Institute, 1972; Manchester University Press: Manchester, UK, 1974; pp. 162–173. [Google Scholar]
- Tribaudino, M.; Angel, R.J.; Camara, F.; Nestola, F.; Pasqual, D.; Margiolaki, I. Thermal expansion of plagioclase feldspars. Contrib. Miner. Petrol. 2010, 160, 899–908. [Google Scholar] [CrossRef]
- Hovis, G.L.; Medford, A.; Conlon, M.; Tether, A.; Romanoski, A. Principles of thermal expansion in the feldspars. Am. Miner. 2010, 95, 1060–1068. [Google Scholar] [CrossRef]
- Winter, J.K.; Ghose, S.; Okamura, F.P. A high-temperature study of the thermal expansion and the anisotropy of the sodium atom in low albite. Am. Miner. 1977, 62, 921–931. [Google Scholar]
- Ohashi, Y.; Finger, L.W. An effect of temperature on the feldspar structure: Crystal structure of sanidine at 800 °C. Carnegie Inst. Wash. Yearb. 1975, 74, 569–572. [Google Scholar]
- Benna, P.; Tribaudino, M.; Bruno, E. High-temperature in situ structural investigation on lead feldspar. Am. Miner. 1999, 84, 120–129. [Google Scholar] [CrossRef]
- Brown, W.L.; Openshaw, R.E.; McMillan, P.F.; Henderson, C.M.B. A review of the expansion behaviour of alkali feldspars: Coupled variations in cell parameters and possible phase transitions. Am. Miner. 1984, 69, 1058–1071. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of intratomic distances in halides and chalcogenides. Acta Cryst. 1976, A32, 733–767. [Google Scholar]
- Fleet, M.E. Tetrahedral-site occupancies in sodium aluminum-gallium feldspar solid solutions [Na(Al1-xGax)Si3O8]. J. Solid State Chem. 1991, 92, 295–300. [Google Scholar] [CrossRef]
- Tribaudino, M.; Bruno, M.; Nestola, F.; Pasqual, D.; Angel, R.J. Thermoelastic and thermodynamic properties of plagioclase feldspars from thermal expansion measurements. Am. Miner. 2011, 96, 992–1002. [Google Scholar] [CrossRef]
- Brown, J.M.; Angel, R.J.; Ross, N.L. Elasticity of plagioclase feldspars. J. Geophys. Res. 2016, 121, 663–675. [Google Scholar] [CrossRef]
- Baerlocher, C.; Meier, W.M.; Olson, D.H. Atlas of zeolite framework types. In Structure Commission of the International Zeolite Association, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2001; 308p. [Google Scholar]
- Mazzi, F.; Galli, E.; Gottardi, G. The crystal structure of tetragonal leucite. Am. Miner. 1976, 61, 108–115. [Google Scholar]
- Beger, R.M. The crystal structure and chemical composition of pollucite. Zeits. Krist. 1969, 129, 280–302. [Google Scholar] [CrossRef]
- Teerstra, D.K.; Cȇrny, P. First natural occurrence of end-member pollucite: A product of low-temperature re-equilibration. Eur. J. Miner. 1995, 7, 1137–1148. [Google Scholar] [CrossRef]
- Hori, H.; Nagashima, K.; Yamada, M.; Miyawaki, R.; Marabashi, T. Ammonioleucite, a product of low temperature equilibrium. Am. Miner. 1986, 71, 1022–1027. [Google Scholar]
- Agakhanov, A.A.; Pautov, L.A.; Karpenko, V.Y.; Sokolova, E.; Hawthorne, F.C. Kirchhoffite, CsBSi2O6, a new mineral species from the Darai-Pioz alkaline massif, Tajikistan: Description and crystal structure. Can. Miner. 2012, 50, 523–529. [Google Scholar] [CrossRef]
- Ferraris, G.; Jones, D.W.; Yerken, J. A neutron diffraction study of the crystal structure of analcime, NaAlSi2O6.H2O. Zeits Krist. 1972, 135, 240–252. [Google Scholar] [CrossRef]
- Takéuchi, Y.; Mazzi, F.; Haga, N.; Galli, E. The crystal structure of wairakite. Am. Miner. 1979, 64, 993–1001. [Google Scholar]
- Holakovsky, J.; Kratochvilova, I.; Kocirik, M. Assessment of percolation thresholds for channel systems in MFI zeolite based on the MFI and diamond lattice. Microporous Mesoporous Mater. 2006, 91, 170–171. [Google Scholar] [CrossRef]
- Coombs, D.S. Recommended nomenclature for zeolite minerals: Report of the Subcommittee on Zeolites of International Mineralogical Association. Committee on new minerals and mineral names. Can. Miner. 1977, 35, 1571–1600. [Google Scholar]
- Henderson, C.M.B.; Charnock, J.M.; Bell, A.M.T.; van der Laan, G.C.M.B. X-ray absorption study of 3d transition metals and Mg in glasses and analogue materials in AFe3+Si2O6 and A2X2+Si5O12, where A = K, Rb, or Cs and X = Mg, Mn, Fe, Cu, Ni, Cu, or Zn. J. Non-Cryst. Solids 2016, 451, 23–48. [Google Scholar] [CrossRef]
- Balmer, M.L.; Huang, Q.; Wong-Ng, W.; Roth, R.S.; Santoro, A. Neutron and X-ray diffraction study of the crystal structure of CsTiSi2O6.5. J. Solid State Chem. 1997, 130, 97–102. [Google Scholar] [CrossRef]
- Kobayashi, H.; Sumino, S.; Tamai, S.; Yanase, I. Phase transition and lattice thermal expansion of Cs-deficient pollucite, Cs1-xAl1-xSi2+x (x >= 0.25), compounds. J. Am. Ceram. Soc. 2006, 89, 3157–3161. [Google Scholar] [CrossRef]
- Carmichael, I.S.E. The mineralogy and petrology of the volcanic rocks from the Leucite Hills, Wyoming. Contrib. Miner. Petrol. 1967, 15, 24–66. [Google Scholar] [CrossRef]
- MacKenzie, W.S.; Richardson, D.M.; Wood, B.J. Solid solution of SiO2 in leucite. Bull. Soc. Miner. Cristallogr. 1974, 97, 257–260. [Google Scholar]
- Krzhizhanovskaya, M.G.; Boubnova, R.S.; Filatov, S.K.; Meyer, D.; Pauffler, P. Transformations of the crystal structure in a series of rubidium borosilicate solid solutions from the X-ray powder diffraction data. Glass Phys. Chem. 2003, 29, 599–607. [Google Scholar] [CrossRef]
- Stokes, H.T.; Hatch, D.M. Isotropy Subgroups of the 230 Crystallographic Space Groups; World Scientific: Singapore, 1998. [Google Scholar]
- Bell, A.M.T.; Knight, K.S.; Henderson, C.M.B.; Fitch, A.N. Revision of the structure of CsCuSi5O12 leucite as orthorhombic Pbca. Acta Cryst. 2010, B66, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Henderson, C.M.B. The thermal expansion of the leucite group of minerals. Am. Miner. 1968, 53, 1476–1489. [Google Scholar]
- Palmer, D.C.; Dove, M.T.; Ibberson, R.M.; Powell, B.M. Structural behavior, crystal chemistry, and phase transitions in substituted leucite: High-resolution neutron powder diffraction studies. Am. Miner. 1997, 82, 16–29. [Google Scholar] [CrossRef]
- Bell, A.M.T.; Henderson, C.M.B. Rietveld refinement of the structures of dry-synthesized MFe3+Si2O6 leucites (M = K, Rb, Cs) by synchrotron X-ray powder diffraction. Acta Cryst. 1994, C50, 1531–1536. [Google Scholar] [CrossRef]
- Lange, R.A.; Carmichael, I.S.E.; Stebbins, J.F. Phase transitions in leucite (KAlSi2O6), orthorhombic KAlSiO4, and their iron analogues (KFeSi2O6, KFeSiO4). Am. Miner. 1986, 71, 937–945. [Google Scholar]
- Palmer, D.C.; Salje, E.K.H.; Schmahl, W.W. Phase transitions in leucite: X-ray diffraction studies. Phys. Chem. Miner. 1989, 16, 714–719. [Google Scholar] [CrossRef]
- Xu, H.; Navrotsky, A.; Balmer, M.L.; Su, Y. Crystal chemistry and phase transitions in substituted pollucites along the CsAlSi2O6–CsTiSi2O6.5 join: A powder synchrotron X-ray diffractometry study. J. Am. Ceram. Soc. 2002, 85, 1235–1242. [Google Scholar] [CrossRef]
- Bell, A.M.T.; Redfern, S.A.T.; Henderson, C.M.B.; Kohn, S.C. Structures of synthetic K2MgSi5O12 leucites by integrated X-ray powder diffraction, electron diffraction and 29Si MAS NMR methods. Acta Cryst. 1994, B50, 31–41. [Google Scholar] [CrossRef]
- Bell, A.M.T.; Redfern, S.A.T.; Henderson, C.M.B.; Kohn, S.C. Structural relations and tetrahedral ordering pattern of synthetic orthorhombic Cs2CdSi5O12 leucite: A combined synchrotron X-ray powder diffraction and multinuclear MAS NMR study. Acta Cryst. 1994, 50, 560–566. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).