
Klippel–Feil Syndrome: The Curious Case of the Skeleton of a Young Slavic Soldier Who Died in 1946



Abstract
:1. Introduction
2. Case Report
3. Materials and Methods
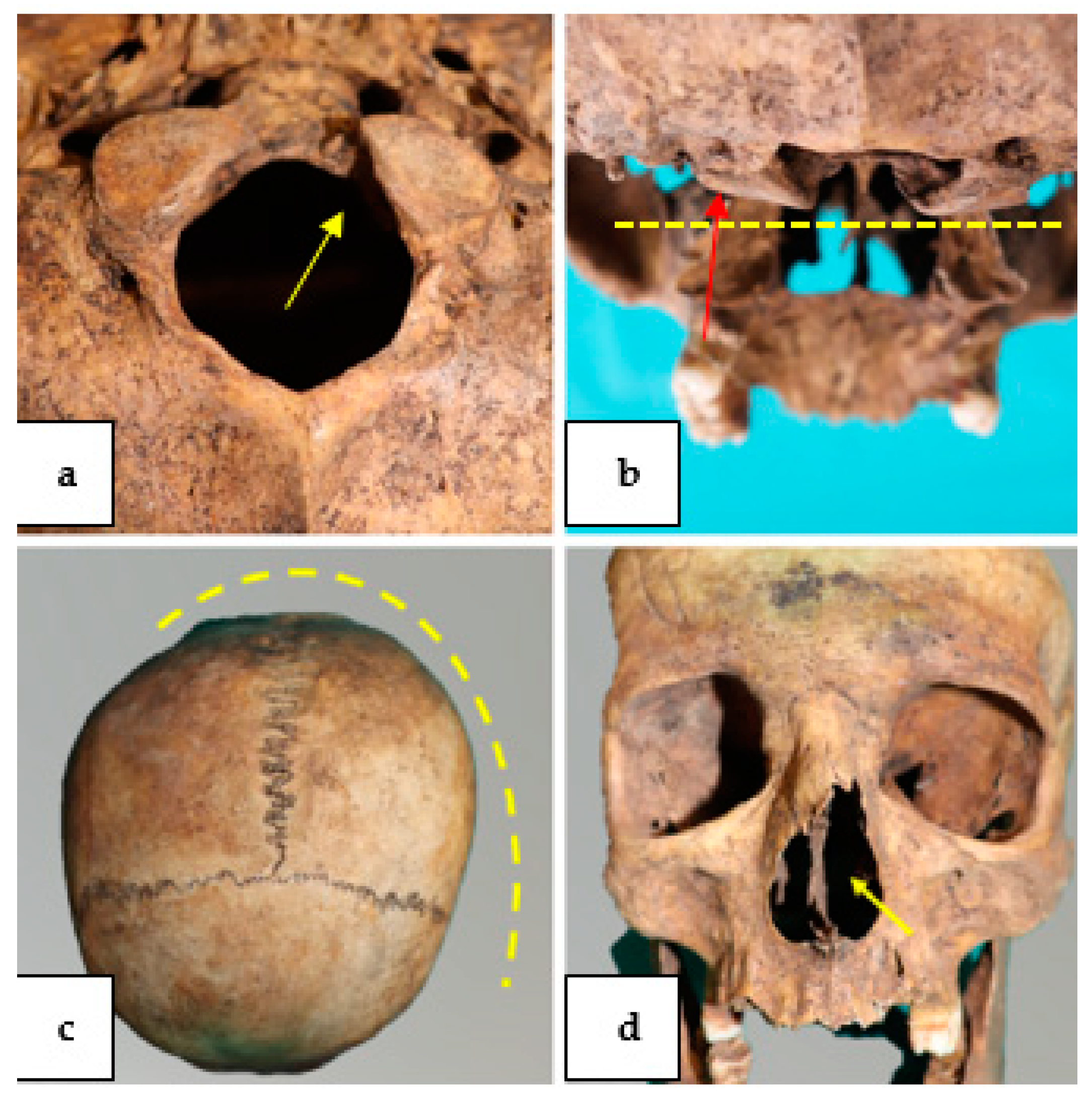
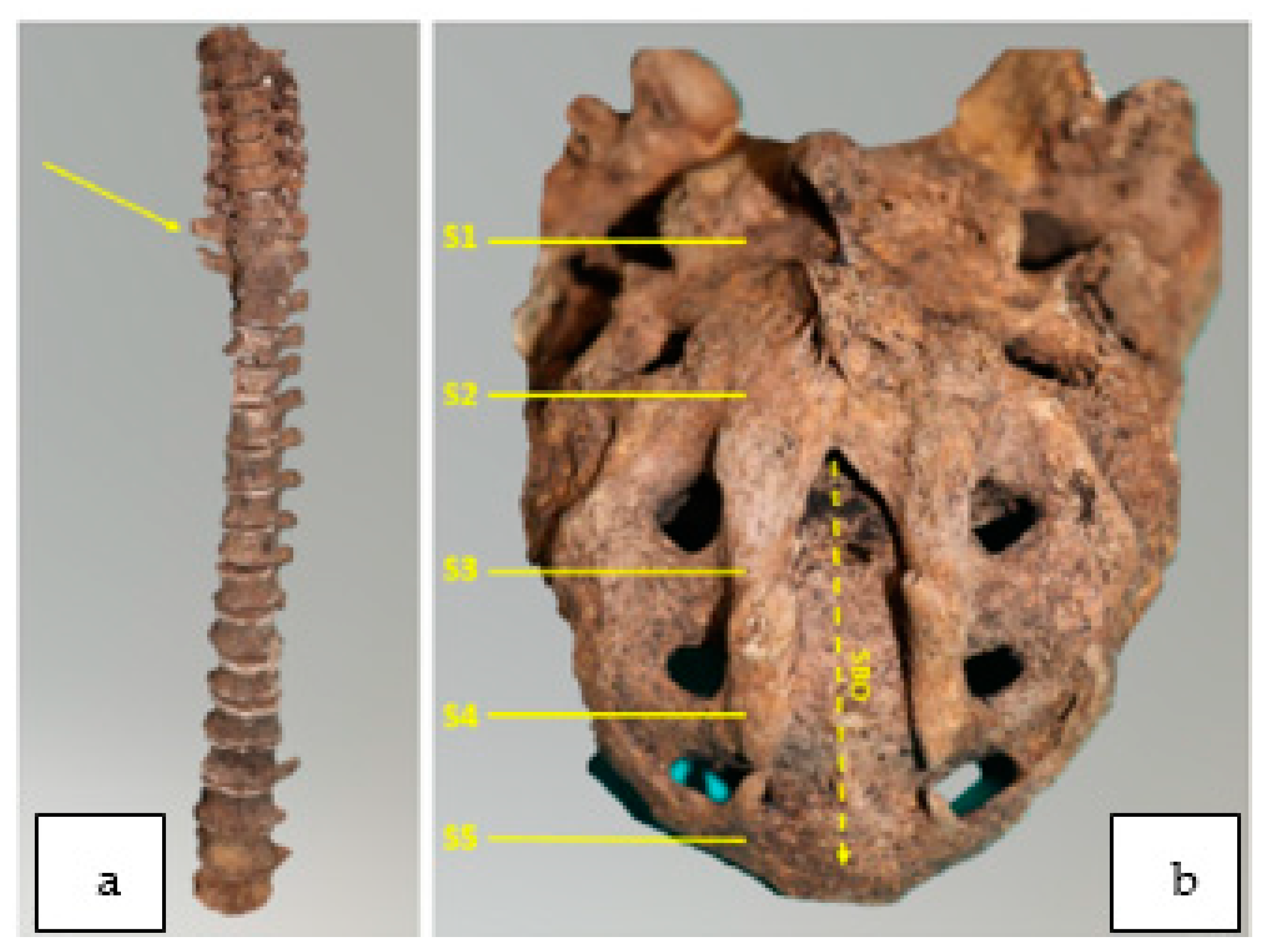
4. Results
5. Discussion
6. Conclusions
7. Impact Statement
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cunha, E. Pathology as a Factor of Personal Identity in Forensic Anthropology. In Forensic Anthropology and Medicine; Humana Press Inc.: Totowa, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Gunderson, C.H.; Greenspan, R.H.; Glaser, G.H.; Lubs, H.A. The Klippel-Feil syndrome. Medicine 1967, 46, 491–512. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E. Developmental Defects of the Axial Skeleton in Paleopathology; University Press Colorado: Boulder, CO, USA, 1994. [Google Scholar]
- Dulić, T. Sentenced “for Ideological and Political Reasons”: The Rehabilitation of Dragoljub “Draža” Mihailović and Social Memory in Serbia. Sociologija 2012, 49, 625–648. [Google Scholar] [CrossRef]
- Resnick, D. Diagnosis of Bone and Joint Disorders; W.B. Saunders: Philadelphia, PA, USA, 2002. [Google Scholar]
- Gladykowska-Rzeczycka, J. A serious defect of two cervical vertebrae from a medieval cemetery in Poland; Klippel-Feil syndrome? Acta Biologica Szegediensis 1997, 42, 49–53. [Google Scholar]
- Samartzis, D.; Herman, J.; Lubicky, J.P.; Shen, F.H. Sprengel’s deformity in Klippel-Feil syndrome. Spine (Phila Pa 1976) 2007, 32, E512–E516. [Google Scholar] [CrossRef]
- Thomsen, M.N.; Schneider, U.; Weber, M.; Johannisson, R.; Niethard, F.U. Scoliosis and congenital anomalies associated with Klippel-Feil syndrome types I-III. Spine (Phila Pa 1976) 1997, 22, 396–401. [Google Scholar] [CrossRef]
- David, K.M.; Copp, A.J.; Stevens, J.M.; Hayward, R.D.; Crockard, H.A. Split cervical spinal cord with Klippel-Feil syndrome: Seven cases. Brain 1996, 119 Pt 6, 1859–1872. [Google Scholar] [CrossRef]
- Roberts, C.A.; Knüsel, C.J.; Race, L. A foot deformity from a Romano-British cemetery at Gloucester, England, and the current evidence for Talipes in paleopathology. Int. J. Osteoarchaeol. 2004, 14, 389–403. [Google Scholar] [CrossRef]
- Scotti, G. Il Bosco Dopo il Mare. Partigiani Italiani in Jugoslavia, 1943–1945; Infinito Edizioni: Castel Gandolfo, Italy, 2009. [Google Scholar]
- Sidell, J.; Thomas, C.; Bayliss, A. Validating and improving archaeological phasing at St. Mary Spital, London. Radiocarbon 2007, 49, 593–610. [Google Scholar] [CrossRef] [Green Version]
- Powers, N. Human Osteology Method Statement. Museum of London. 2012. Available online: https://www.museumoflondon.org.uk/application/files/4814/5633/5269/osteologymethod-statement-revised-2012.pdf (accessed on 14 January 2022).
- Wolff-Heidegger, G. Atlas der systematischen Anatomie des Menschen; Band 1; Karger Publishers: Basel, Switzerland; New York, NY, USA, 1954. [Google Scholar]
- Black, S.; Scheuer, L. Developmental Juvenile Osteology; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Ubelaker, D.H. Human Skeletal Remains: Excavation, Analysis, Interpretation; Smithsonian Institute Press: Washington, DC, USA, 1989. [Google Scholar]
- Acsadi, G.; Nemeskèri, J. History of Human Life Span and Mortality; Akadémiai Kiado: Budapest, Hungary, 1970. [Google Scholar]
- Ferembach, D.; Schwidetzky, I.; Stloukal, M. Empfehlungen für die Alters- und Geschlechtsdiagnose am Skelett. Homo 1979, 30, 1–32. [Google Scholar]
- Krogman, W.M.; Iscan, M.Y. The Human Skeleton in Forensic Medicine; Charles, C., Ed.; Thomas: Springfield, MO, USA, 1986. [Google Scholar]
- Novotny, V. Sex Determination of the Pelvic Bone: A System Approach. Anthropologie 1986, 24, 197–206. [Google Scholar]
- Bruzek, J. A Method for Visual Determination of Sex, Using the Human Hip Bone. Am. J. Phys. Anthropol. 2002, 117, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Tassabehj, M.; Fang, Z.M.; Hilton, E.N.; McGaughran, J.; Zhao, Z.; de Bock, C.E.; Howard, E.; Malass, M.; Donnai, D.; Diwan, A.; et al. Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome. Hum. Mutat. 2008, 29, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, V.; Messeri, P. Antropologia, Il Cranio nel suo Insieme dal Punto di vista Antropologico; Capitolo III: Roma, Italy, 1979; pp. 275–287. [Google Scholar]
- Kucybała, I.; Janik, K.A.; Ciuk, S.; Storman, D.; Urbanik, A. Nasal septal deviation and concha bullosa—Do they have an impact on maxillary sinus volumes and prevalence of maxillary sinusitis? Pol. J. Radiol. 2017, 82, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, W.W.; Winter, R.B.; Morrissy, R.T.; Weinstein, S.L. Lovell and Winter’s Pediatric Orthopaedics; Lippincott Williams & Wilkins: Philadelphia, PA, USA; The British Editorial Society of Bone & Joint Surgery: London, UK, 2006; pp. 693–762. [Google Scholar]
- Sadler, T.W. Langman’s Medical Embryology, 11th ed.; Lippincot Williams and Wilkins: Philadelphia, PA, USA, 2009; pp. 302–303. [Google Scholar]
- Andrews, J.W.; Berger, T.; Elston, D. Andrews’ Diseases of the Skin: Clinical Dermatology, 10th ed.; Saunders: London, UK, 2005; ISBN 0-7216-2921-0. [Google Scholar]
- Aufderheide, A.C.; Rodríguez-Martín, C. The Cambridge Encyclopedia of Human Paleopathology; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Netter, F.H. The CIBA Collection of Medical Illustrations—Muscoloskeletal System: Developmental Disorders, Tumors, Rheumatic Diseases and Joint Replacement; Part II A; Ciba-Geigy: Toms River, NJ, USA, 2002; Volume 8. [Google Scholar]
- Larson, A.R.U.; Josephson, K.D.; Pauli, R.M.; Opitz, J.M.; Williams, M.S. Klippel-Feil anomaly, omovertebral bone, thumb abnormalities, and flexion-crease changes: Novel association or syndrome? Am. J. Med. Genet. 2001, 101, 158–162. [Google Scholar] [CrossRef]
- Hensinger, R.N.; Lang, J.E.; MacEwen, G.D. Klippel-Feil Syndrome: A constellation of associated anomalies. J. Bone Jt. Surg. 1974, 56, 1246–1253. [Google Scholar] [CrossRef]
- Yuksel, M.; Karabiber, H.; Yuksel, K.Z.; Parmaksiz, G. Diagnostic Importance of 3D CT Images in Klippel- Feil Syndrome with Multiple Skeletal Anomalies: A Case Report. Korean J. Radiol. 2005, 6, 278–281. [Google Scholar] [CrossRef] [Green Version]
- Papagrigorakis, M.J.; Synodinos, P.N.; Daliouris, C.P.; Metaxotou, C. De novo inv (2)(p12q34) associated with Klippel-Feil anomaly and hypodontia. Eur. J. Pediatrics 2003, 162, 594–597. [Google Scholar] [CrossRef]
- Tracy, M.R.; Dormans, J.P.; Kusumi, K. Klippel-Feil Syndrome. Clin. Orthop. Relat. Res. 2004, 424, 183–190. [Google Scholar] [CrossRef]
- Brown, M.W.; Templeton, A.W.; Hodges, F.J. The incidence of acquired and congenital fusions in the cervical spine. Am. J. Roentgenol. Radium. Ther. Nucl. Med. 1964, 92, 1255–1259. [Google Scholar]
- Menger, R.P.; Rayi, A.; Notarianni, C. Klippel Feil Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Nagib, M.G.; Maxwell, R.E.; Chou, S.N. Identificazione e gestione dei pazienti ad alto rischio con sindrome di Klippel-Feil. J. Neurochir. 1984, 61, 523–530. [Google Scholar]
- Brokinkel, B.; Wiebe, K.; Hesselmann, V.; Filler, T.J.; Ewelt, C.; Müller-Hofstede, C.; Stummer, W.; Klingenhöfer, M. Trattamento chirurgico in un paziente con sindrome di Klippel-Feil e meningomielocele cervicale anteriore: Un caso clinico e revisione della letteratura. Eur. Spine J. 2013, 22 (Suppl. S3), S517–S520. [Google Scholar] [CrossRef] [PubMed]
- Bayrakli, F.; Guclu, B.; Yakicier, C.; Balaban, H.; Kartal, U.; Erguner, B.; Sagiroglu, M.S.; Yuksel, S.; Ozturk, A.R.; Kazanci, B.; et al. Mutation in MEOX1 gene causes a recessive Klippel-Feil syndrome subtype. BMC Genet. 2013, 14, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, J.Y.; Faqeih, E.; Alsiddiky, A.; Alshammari, M.J.; Ibrahim, N.A.; Alkuraya, F.S. Mutations in MEOX1, encoding mesenchyme homeobox 1, cause Klippel-Feil anomaly. Am. J. Hum. Genet. 2013, 92, 157–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Berry-Wynne, K.M.; Asai-Coakwell, M.; Sundaresan, P.; Footz, T.; French, C.R.; Abitbol, M.; Fleisch, V.C.; Corbett, N.; Allison, W.T.; et al. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum. Mol. Genet. 2010, 19, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Alazami, A.M.; Kentab, A.Y.; Faqeih, E.; Mohamed, J.Y.; Alkhalidi, H.; Hijazi, H.; Alkuraya, F.S.; Abitbol, M.; Fleisch, V.C.; Corbett, N.; et al. A novel syndrome of Klippel-Feil anomaly, myopathy, and characteristic facies is linked to a null mutation in MYO18B. J. Med. Genet. 2015, 52, 400–404. [Google Scholar] [CrossRef]
- Karaca, E.; Yuregir, O.O.; Bozdogan, S.T.; Aslan, H.; Pehlivan, D.; Jhangiani, S.N.; Akdemir, Z.C.; Gambin, T.; Bayram, Y.; Atik, M.M.; et al. rare variants in the notch signaling pathway describe a novel type of autosomal recessive Klippel-Feil syndrome. Am. J. Med. Genet. Part A 2015, 167, 2795–2799. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.; Saller, K. Lehrbuch der Anthropologie; G. Fisher: Stuttgart, Germany, 1957; Volume 62. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Femur Strength Index | Result |
|---|---|
| WEAK (X-12.5) | |
| STRONG (12.6-X) | INDEX 8 |
| PILASTRIC FEMUR INDEX | |
| NULL (X-99) | |
| WEAK (100–109) | |
| MEDIUM (110–119) | |
| STRONG (120-X) | INDEX 102 |
| TOTAL ANALYSIS | WEAK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leggio, A.; Gallieni, M.; Puzo, P.; Introna, F.; Sablone, S. Klippel–Feil Syndrome: The Curious Case of the Skeleton of a Young Slavic Soldier Who Died in 1946. Forensic Sci. 2022, 2, 155-162. https://doi.org/10.3390/forensicsci2010012
Leggio A, Gallieni M, Puzo P, Introna F, Sablone S. Klippel–Feil Syndrome: The Curious Case of the Skeleton of a Young Slavic Soldier Who Died in 1946. Forensic Sciences. 2022; 2(1):155-162. https://doi.org/10.3390/forensicsci2010012
Chicago/Turabian StyleLeggio, Alessia, Massimo Gallieni, Pasquale Puzo, Francesco Introna, and Sara Sablone. 2022. "Klippel–Feil Syndrome: The Curious Case of the Skeleton of a Young Slavic Soldier Who Died in 1946" Forensic Sciences 2, no. 1: 155-162. https://doi.org/10.3390/forensicsci2010012
APA StyleLeggio, A., Gallieni, M., Puzo, P., Introna, F., & Sablone, S. (2022). Klippel–Feil Syndrome: The Curious Case of the Skeleton of a Young Slavic Soldier Who Died in 1946. Forensic Sciences, 2(1), 155-162. https://doi.org/10.3390/forensicsci2010012