
Thermodynamic Overview of Bioconjugation Reactions Pertinent to Lysine and Cysteine Peptide and Protein Residues



Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
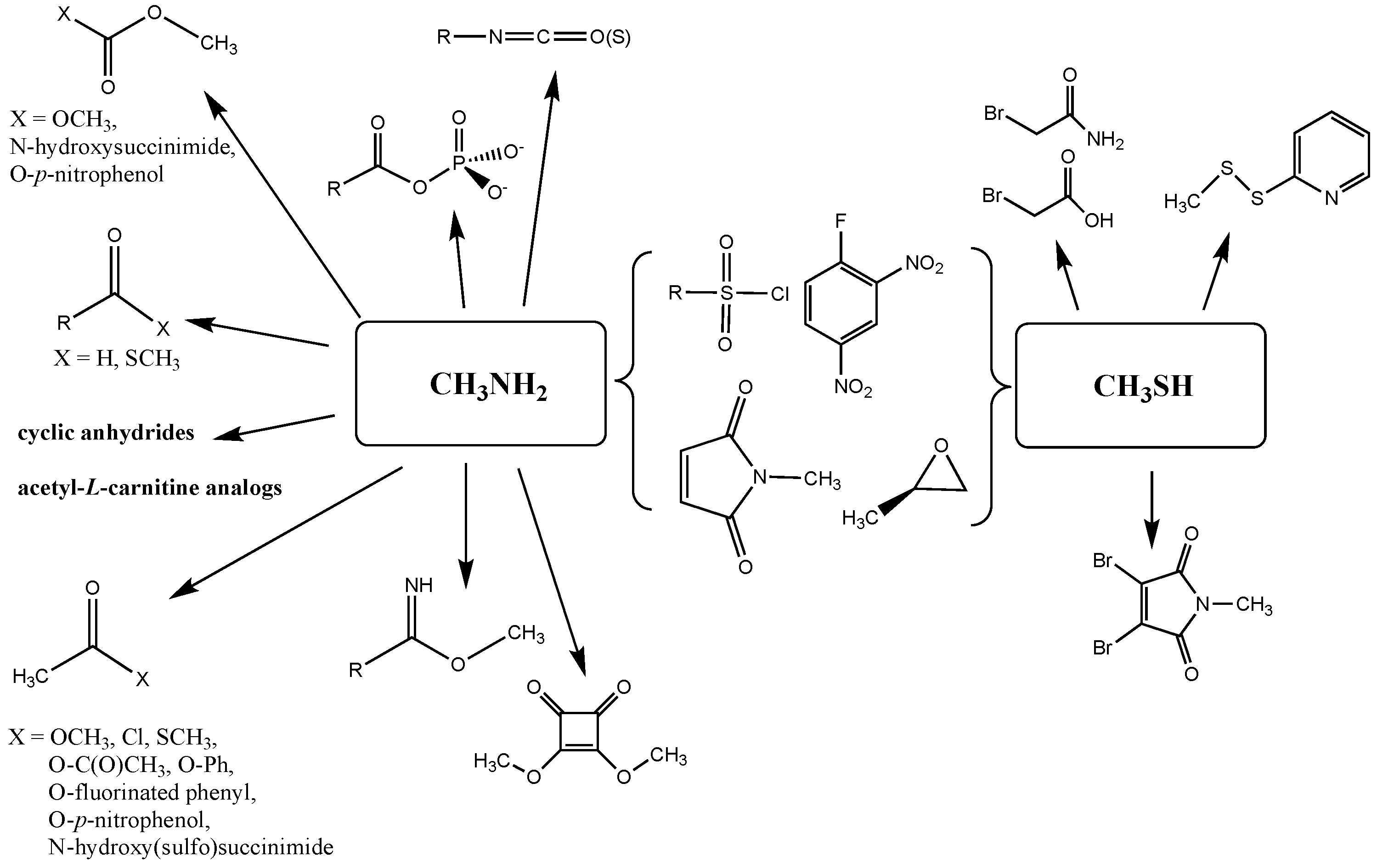
3.1. Lysine Bioconjugation Reactions
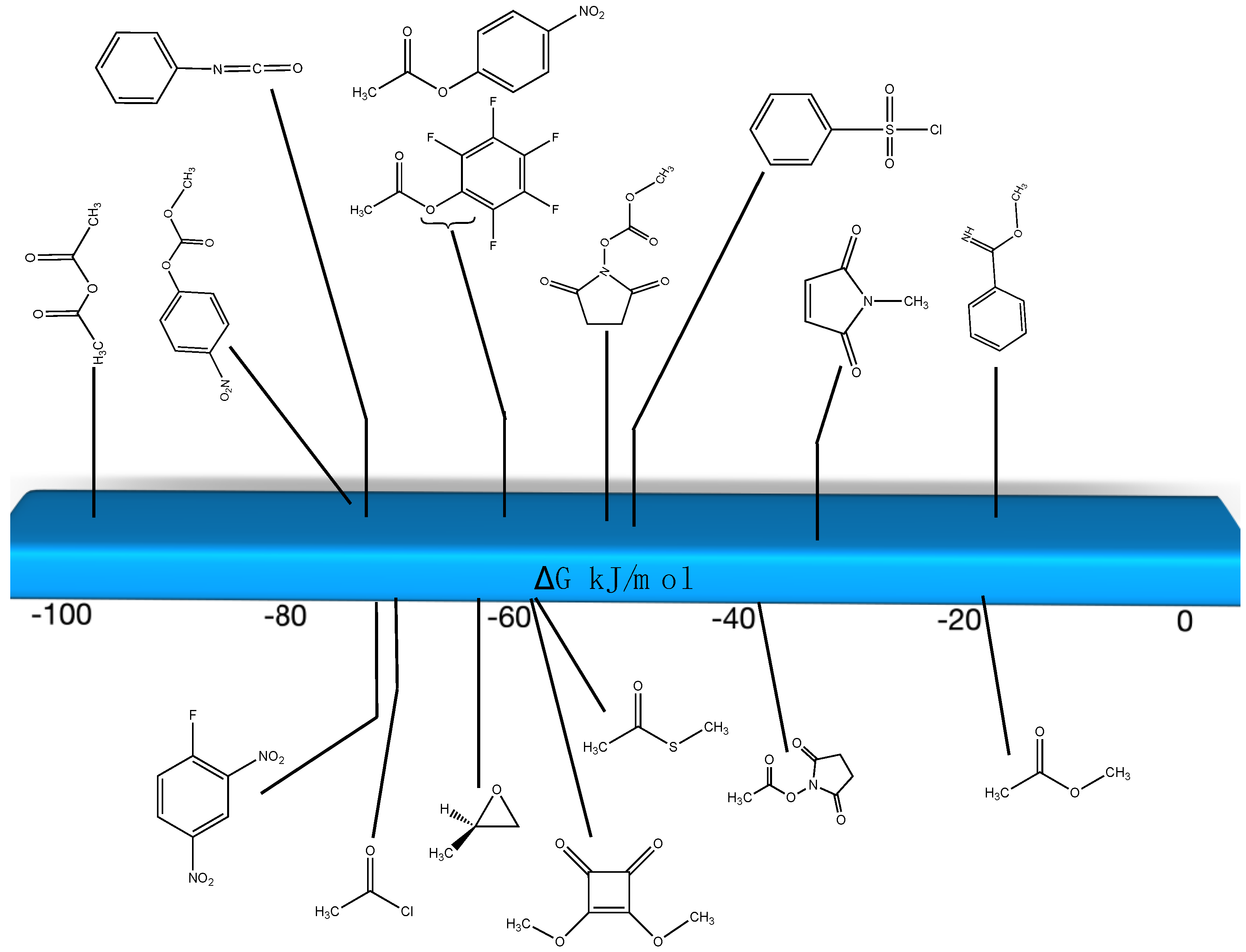
3.1.1. Alkyl Esters and Thioesters, Anhydrides, and Acyl Chlorides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | T1 ΔE (kJ/mol) | G3(MP2) ΔE (kJ/mol) |
|---|---|---|
 | 19.45 | 22.65 |
 | −2.28 | −2.87 |
 | −48.45 | −45.34 |
 | −70.18 | −70.86 |
 | −43.00 | −38.25 |
 | −64.73 | −63.77 |
 | −10.29 | −5.82 |
 | −32.02 | −31.34 |
3.1.2. Phenyl and N-hydroxysuccinimide Esters
3.1.3. Carbonates, Isocyanates, and Isothiocyanates
3.1.4. Additional Modification Reactions
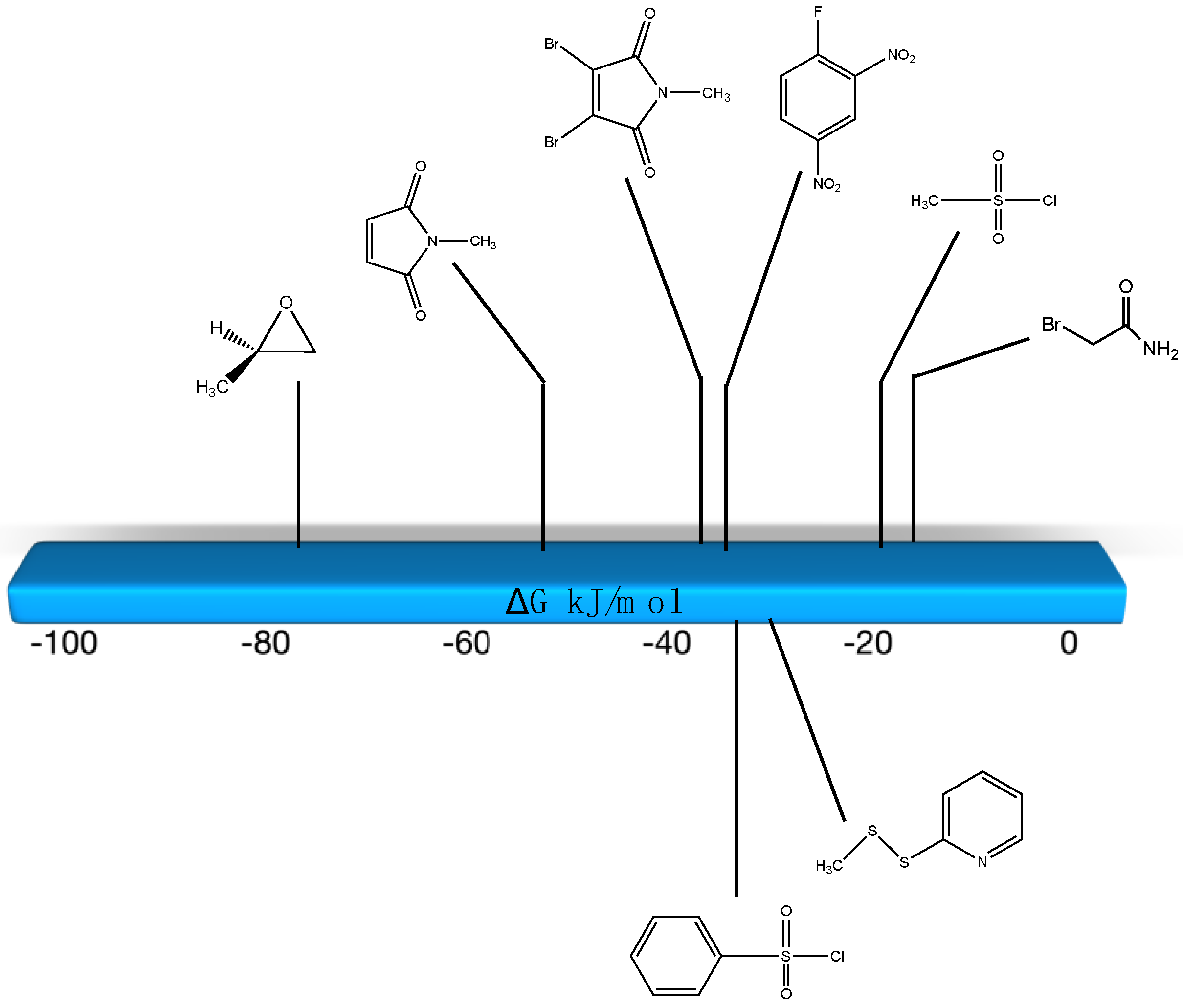
3.2. Cysteine Bioconjugation Reactions
Sulfonyl Chlorides, Epoxides, Maleimides, 2,4-Dinitrofluorobenzene, Disulfides, α-Bromoacids and α-Bromoamides
3.3. Click Reactions
3.4. Reactive Cellular Intermediates
3.4.1. Thioesters, Cyclic Anhydrides, and Acylphosphates
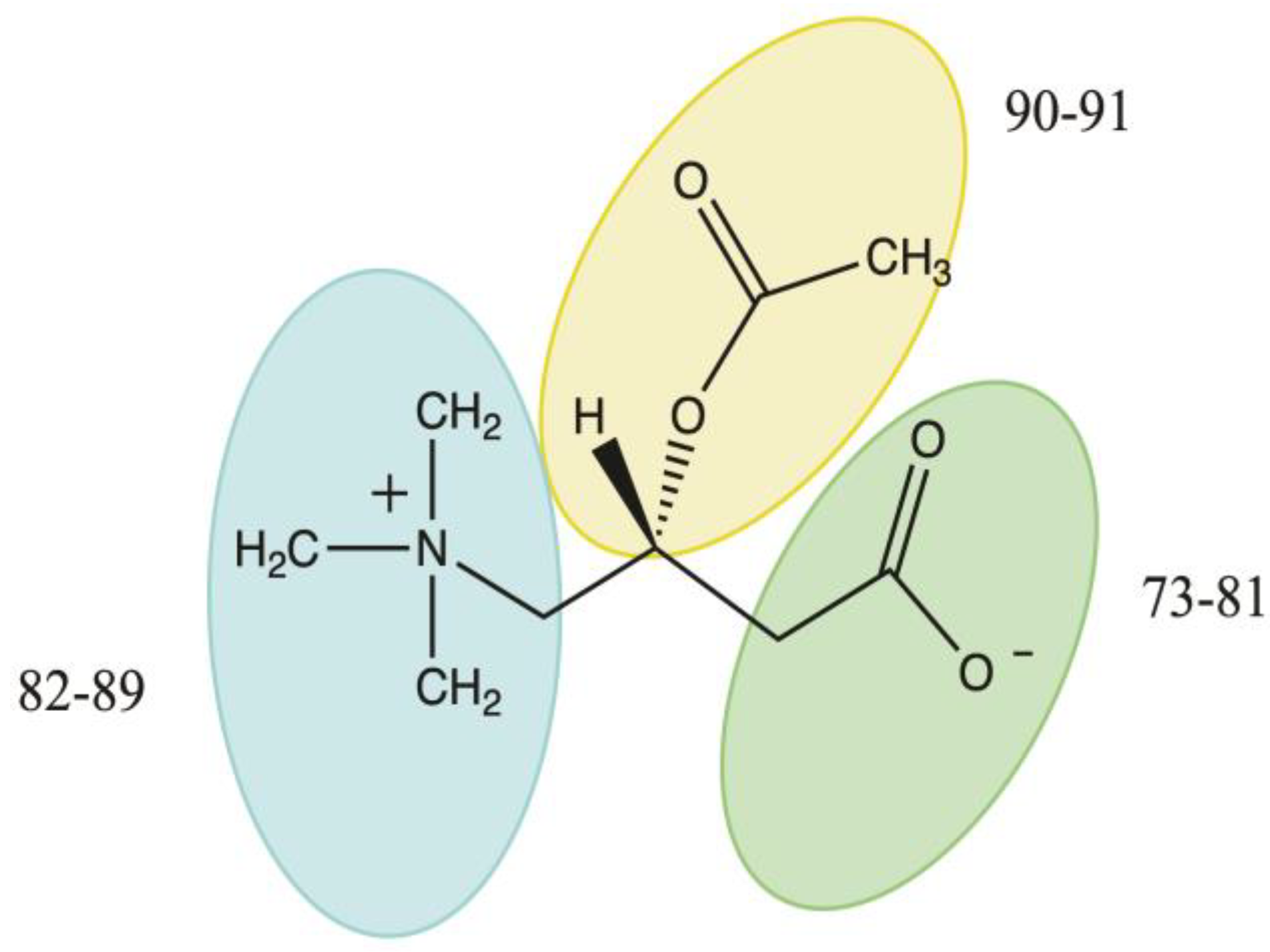
3.4.2. Acetyl-L-Carnitine
3.5. Predictive Value and Cautionary Notes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cserep, G.B.; Herner, A.; Kele, P. Bioorthogonal fluorescent labels: A review on combined forces. Methods Appl. Fluoresc. 2015, 3, 042001. [Google Scholar] [CrossRef]
- Ossadnik, D.; Kuzin, S.; Qi, M.; Yulikov, M.; Godt, A. A Gd(III)-Based Spin Label at the Limits for Linewidth Reduction through Zero-Field Splitting Optimization. Inorg. Chem. 2023, 62, 408–432. [Google Scholar] [CrossRef]
- Lundblad, R.L. Chemical Reagents for Protein Modification, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Hermanson, G.T. Bioconjugate Techniques, 3rd ed.; Academic Press: London, UK, 2013. [Google Scholar]
- Chen, X.; Wu, Y.W. Selective chemical labeling of proteins. Org. Biomol. Chem. 2016, 14, 5417–5439. [Google Scholar] [CrossRef]
- Fischer, N.H.; Oliveira, M.T.; Diness, F. Chemical modification of proteins—Challenges and trends at the start of the 2020s. Biomater. Sci. 2023, 11, 719–748. [Google Scholar] [CrossRef]
- Sornay, C.; Vaur, V.; Wagner, A.; Chaubet, G. An overview of chemo- and site-selectivity aspects in the chemical conjugation of proteins. R. Soc. Open Sci. 2022, 9, 211563. [Google Scholar] [CrossRef]
- DeGruyter, J.N.; Malins, L.R.; Baran, P.S. Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry 2017, 56, 3863–3873. [Google Scholar] [CrossRef]
- Reddy, N.C.; Kumar, M.; Molla, R.; Rai, V. Chemical methods for modification of proteins. Org. Biomol. Chem. 2020, 18, 4669–4691. [Google Scholar] [CrossRef]
- Kohn, M. Immobilization strategies for small molecule, peptide and protein microarrays. J. Pept. Sci. 2009, 15, 393–397. [Google Scholar] [CrossRef]
- Lim, C.Y.; Owens, N.A.; Wampler, R.D.; Ying, Y.; Granger, J.H.; Porter, M.D.; Takahashi, M.; Shimazu, K. Succinimidyl ester surface chemistry: Implications of the competition between aminolysis and hydrolysis on covalent protein immobilization. Langmuir 2014, 30, 12868–12878. [Google Scholar] [CrossRef]
- O’Connell, L.; Marcoux, P.R.; Roupioz, Y. Strategies for Surface Immobilization of Whole Bacteriophages: A Review. ACS Biomater. Sci. Eng. 2021, 7, 1987–2014. [Google Scholar] [CrossRef]
- Adumeau, P.; Sharma, S.K.; Brent, C.; Zeglis, B.M. Site-Specifically Labeled Immunoconjugates for Molecular Imaging—Part 1: Cysteine Residues and Glycans. Mol. Imaging Biol. 2016, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Liu, W.; Jin, S.; Zhao, W.; Xu, Y.; Zhou, Z.; Chen, S.; Pan, L. Facile Generation of Potent Bispecific Fab via Sortase A and Click Chemistry for Cancer Immunotherapy. Cancers 2021, 13, 4540. [Google Scholar] [CrossRef] [PubMed]
- Matikonda, S.S.; McLaughlin, R.; Shrestha, P.; Lipshultz, C.; Schnermann, M.J. Structure-Activity Relationships of Antibody-Drug Conjugates: A Systematic Review of Chemistry on the Trastuzumab Scaffold. Bioconjug. Chem. 2022, 33, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Dozier, J.K.; Distefano, M.D. Site-Specific PEGylation of Therapeutic Proteins. Int. J. Mol. Sci. 2015, 16, 25831–25864. [Google Scholar] [CrossRef] [PubMed]
- Vaillard, V.A.; Menegon, M.; Neuman, N.I.; Vaillard, S.E. mPEG-NHS carbonates: Effect of alkyl spacers on the reactivity: Kinetic and mechanistic insights. J. Appl. Polym. Sci. 2019, 136, 47028. [Google Scholar] [CrossRef]
- Jayachandran, B.; Parvin, T.N.; Alam, M.M.; Chanda, K.; Mm, B. Insights on Chemical Crosslinking Strategies for Proteins. Molecules 2022, 27, 8124. [Google Scholar] [CrossRef] [PubMed]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef]
- Reinkemeier, C.D.; Koehler, C.; Sauter, P.F.; Shymanska, N.V.; Echalier, C.; Rutkowska, A.; Will, D.W.; Schultz, C.; Lemke, E.A. Synthesis and Evaluation of Novel Ring-Strained Noncanonical Amino Acids for Residue-Specific Bioorthogonal Reactions in Living Cells. Chemistry 2021, 27, 6094–6099. [Google Scholar] [CrossRef]
- Kim, S.; Ko, W.; Sung, B.H.; Kim, S.C.; Lee, H.S. Direct protein-protein conjugation by genetically introducing bioorthogonal functional groups into proteins. Bioorg. Med. Chem. 2016, 24, 5816–5822. [Google Scholar] [CrossRef]
- Kuhlemann, A.; Beliu, G.; Janzen, D.; Petrini, E.M.; Taban, D.; Helmerich, D.A.; Doose, S.; Bruno, M.; Barberis, A.; Villmann, C.; et al. Genetic Code Expansion and Click-Chemistry Labeling to Visualize GABA-A Receptors by Super-Resolution Microscopy. Front. Synaptic Neurosci. 2021, 13, 727406. [Google Scholar] [CrossRef]
- Elia, N. Using unnatural amino acids to selectively label proteins for cellular imaging: A cell biologist viewpoint. FEBS J. 2021, 288, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Kang, D.; Park, H.S. Site-Specific Labeling of Proteins Using Unnatural Amino Acids. Mol. Cells 2019, 42, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, T.; Daub, E.; Honek, J.F. Bioorthogonal Modification of the Major Sheath Protein of Bacteriophage M13: Extending the Versatility of Bionanomaterial Scaffolds. Bioconjug. Chem. 2016, 27, 2276–2280. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, T.; Howie, B.; Zhang, L.; Leung, K.T.; Honek, J.F. Bioconjugation of Bacteriophage Pf1 and Extension to Pf1-Based Bionanomaterials. Curr. Nanosci. 2021, 17, 139–150. [Google Scholar] [CrossRef]
- Battigelli, A.; Almeida, B.; Shukla, A. Recent Advances in Bioorthogonal Click Chemistry for Biomedical Applications. Bioconjug. Chem. 2022, 33, 263–271. [Google Scholar] [CrossRef]
- Taiariol, L.; Chaix, C.; Farre, C.; Moreau, E. Click and Bioorthogonal Chemistry: The Future of Active Targeting of Nanoparticles for Nanomedicines? Chem. Rev. 2022, 122, 340–384. [Google Scholar] [CrossRef]
- Stump, B. Click Bioconjugation: Modifying Proteins Using Click-Like Chemistry. Chembiochem 2022, 23, e202200016. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Baek, M.; Monda, F.; Olsen, C.A. Chiral Posttranslational Modification to Lysine epsilon-Amino Groups. Acc. Chem. Res. 2022, 55, 1456–1466. [Google Scholar] [CrossRef]
- Christensen, D.G.; Xie, X.; Basisty, N.; Byrnes, J.; McSweeney, S.; Schilling, B.; Wolfe, A.J. Post-translational Protein Acetylation: An Elegant Mechanism for Bacteria to Dynamically Regulate Metabolic Functions. Front. Microbiol. 2019, 10, 1604. [Google Scholar] [CrossRef]
- Trub, A.G.; Hirschey, M.D. Reactive Acyl-CoA Species Modify Proteins and Induce Carbon Stress. Trends Biochem. Sci. 2018, 43, 369–379. [Google Scholar] [CrossRef]
- Kuhn, M.L.; Zemaitaitis, B.; Hu, L.I.; Sahu, A.; Sorensen, D.; Minasov, G.; Lima, B.P.; Scholle, M.; Mrksich, M.; Anderson, W.F.; et al. Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS ONE 2014, 9, e94816. [Google Scholar] [CrossRef]
- Pettegrew, J.W.; Levine, J.; McClure, R.J. Acetyl-L-carnitine physical-chemical, metabolic, and therapeutic properties: Relevance for its mode of action in Alzheimer’s disease and geriatric depression. Mol. Psychiatry 2000, 5, 616–632. [Google Scholar] [CrossRef]
- Swamy-Mruthinti, S.; Carter, A.L. Acetyl- L -carnitine decreases glycation of lens proteins: In vitro studies. Exp. Eye Res. 1999, 69, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Lu, L.L. Can B3LYP be Improved by Optimization of the Proportions of Exchange and Correlation Functionals? Int. J. Quantum Chem. 2015, 115, 502–509. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef]
- Ohlinger, W.S.; Klunzinger, P.E.; Deppmeier, B.J.; Hehre, W.J. Efficient Calculation of Heats of Formation. J. Phys. Chem. A 2009, 113, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K.; Rassolov, V.; Pople, J.A. Gaussian-3 theory using reduced Moller-Plesset order. J. Chem. Phys. 1999, 110, 4703–4709. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. 5. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. 1. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- NIST. NIST Computational Chemistry Comparison and Benchmark Database; Johnson, R.D., III, Ed.; NIST: Gaithersburg, MD, USA, 2022. [Google Scholar]
- Hehre, W.J. A Guide to Molecular Mechanics and Quantum Chemical Calculations; Wavefunction, Inc.: Irvine, CA, USA, 2003. [Google Scholar]
- Costa, A.M.; Bosch, L.; Petit, E.; Vilarrasa, J. Computational Study of the Addition of Methanethiol to 40+ Michael Acceptors as a Model for the Bioconjugation of Cysteines. J. Org. Chem. 2021, 86, 7107–7118. [Google Scholar] [CrossRef] [PubMed]
- Wavefunction, I. Spartan’20: Tutorial and User’s Guide; Wavefunction, Inc.: Irvine, CA, USA, 2022. [Google Scholar]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Skyner, R.E.; McDonagh, J.L.; Groom, C.R.; van Mourik, T.; Mitchell, J.B.O. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Cao, Y.; Halls, M.D.; Vadicherla, T.R.; Friesner, R.A. Pseudospectral implementations of long-range corrected density functional theory. J. Comput. Chem. 2021, 42, 2089–2102. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Parish, C.A.; Still, W.C. MacroModel. The computational chemists molecular modeling tool. Abstr. Pap. Am. Chem. S 1996, 211, 90-Comp. [Google Scholar]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods. 1. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods. 2. Applications. J. Comput. Chem. 1989, 10, 221–264. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc. Perkin Trans. 2 1993, 5, 799–805. [Google Scholar] [CrossRef]
- Bondi, A. Van Der Waals Volumes + Radii. J. Phys. Chem 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Moellering, R.E.; Cravatt, B.F. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 2013, 341, 549–553. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Smith, C.L.; Smith, A.C.; Robinson, A.J.; Hoogewijs, K.; Murphy, M.P. The Causes and Consequences of Nonenzymatic Protein Acylation. Trends Biochem. Sci. 2018, 43, 921–932. [Google Scholar] [CrossRef]
- Fraenkel-Conrat, H. Methods for investigating the essential groups for enzyme activity. In Meth Enzymol; Colowick, S.P., Kaplan, N.O., Eds.; Academic Press: New York, NY, USA, 1959; Volume 4, pp. 247–269. [Google Scholar]
- Smyth, D.G. Acetylation of amino and tyrosine hydroxyl groups. Preparation of inhibitors of oxytocin with no intrinsic activity on the isolated uterus. J. Biol. Chem. 1967, 242, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Spanedda, M.V.; Bourel-Bonnet, L. Cyclic Anhydrides as Powerful Tools for Bioconjugation and Smart Delivery. Bioconjug. Chem. 2021, 32, 482–496. [Google Scholar] [CrossRef]
- Hori, K. Theoretical study of a reaction path via a hydrogen-bonded intermediate for the alkaline hydrolysis of esters in the gas phase. J. Chem. Soc. Perkin Trans. 2 1992, 1992, 1629–1633. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Landry, D.W.; Ornstein, R.L. Theoretical Studies of Fundamental Pathways for Alkaline Hydrolysis of Carboxylic Acid Esters in Gas Phase. J. Am. Chem. Soc. 2000, 122, 1522–1530. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Landry, D.W.; Ornstein, R.L. Reaction Pathways and Energy Barriers for Alkaline Hydrolysis of Carboxylic Acid Esters in Water Studied by a Hybrid Supermolecule-Polarizable Continuum Approach. J. Am. Chem. Soc. 2000, 122, 2621–2627. [Google Scholar] [CrossRef]
- Hori, K.; Ikenaga, Y.; Arata, K.; Takahashi, T.; Kasai, K.; Noguchi, Y.; Sumimoto, M.; Yamamoto, H. Theoretical study on the reaction mechanism for the hydrolysis of esters and amides under acidic conditions. Tetrahedron 2007, 63, 1264–1269. [Google Scholar] [CrossRef]
- Martin, R.B. Mechanisms of Acid Hydrolysis of Carboxylic Acid Esters and Amides. J. Am. Chem. Soc. 1962, 84, 4130–4136. [Google Scholar] [CrossRef]
- Limpanuparb, T.; Punyain, K.; Tantirungrotechai, Y. A DFT investigation of methanolysis and hydrolysis of triacetin. J. Mol. Struct. THEOCHEM 2010, 955, 23–32. [Google Scholar] [CrossRef]
- Kallies, B.; Mitzner, R. Models of water-assisted hydrolyses of methyl formate, formamide, and urea from combined DFT-SCRF calculations. J. Mol. Model. 1998, 4, 183–196. [Google Scholar] [CrossRef]
- Allen, S.E.; Hsieh, S.Y.; Gutierrez, O.; Bode, J.W.; Kozlowski, M.C. Concerted Amidation of Activated Esters: Reaction Path and Origins of Selectivity in the Kinetic Resolution of Cyclic Amines via N-Heterocyclic Carbenes and Hydroxamic Acid Cocatalyzed Acyl Transfer. J. Am. Chem. Soc. 2014, 136, 11783–11791. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, Y.; Hua, R. Acid-catalyzed carboxylic acid esterification and ester hydrolysis mechanism: Acylium ion as a sharing active intermediate via a spontaneous trimolecular reaction based on density functional theory calculation and supported by electrospray ionization-mass spectrometry. Phys. Chem. Chem. Phys. 2015, 17, 30279–30291. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Dmitrenko, O.; Liao, L.A.; Bach, R.D. Computational studies of nucleophilic substitution at carbonyl carbon: The S(N)2 mechanism versus the tetrahedral intermediate in organic synthesis. J. Org. Chem. 2004, 69, 7317–7328. [Google Scholar] [CrossRef]
- Yang, W.; Drueckhammer, D.G. Computational studies of the aminolysis of oxoesters and thioesters in aqueous solution. Org. Lett. 2000, 2, 4133–4136. [Google Scholar] [CrossRef]
- Yang, W.; Drueckhammer, D.G. Understanding the relative acyl-transfer reactivity of oxoesters and thioesters: Computational analysis of transition state delocalization effects. J. Am. Chem. Soc. 2001, 123, 11004–11009. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Zipse, H. Bifunctional catalysis of ester aminolysis—A computational and experimental study. Liebigs. Ann. Recl. 1996, 1996, 1501–1509. [Google Scholar] [CrossRef]
- Galabov, B.; Atanasov, Y.; Ilieva, S.; Schaefer, H.F. Mechanism of the Aminolysis of Methyl Benzoate: A Computational Study. J. Phys. Chem. A 2005, 109, 11470–11474. [Google Scholar] [CrossRef] [PubMed]
- Ikhazuangbe, P.M.O.; Adama, K.K. Experimental Investigation of the Kinetics and Thermodynamics of the Production of Methanol from the Hydrolysis of Methyl Acetate Catalyzed with Hydrochloric Acid. Int. J. Chem. Chem. Process. 2021, 7, 13–25. [Google Scholar]
- Shi, Z.; Hsieh, Y.H.; Weinberg, N.; Wolfe, S. The neutral hydrolysis of methyl acetate—Part 2. Is there a tetrahedral intermediate? Can. J. Chem. 2009, 87, 544–555. [Google Scholar] [CrossRef]
- Kruger, H.G. Ab initio mechanistic study of the protection of alcohols and amines with anhydrides. J. Mol. Struc. THEOCHEM 2002, 577, 281–285. [Google Scholar] [CrossRef]
- Petrova, T.; Okovytyy, S.; Gorb, L.; Leszczynski, J. Computational study of the aminolysis of anhydrides: Effect of the catalysis to the reaction of succinic anhydride with methylamine in gas phase and nonpolar solution. J. Phys. Chem. A 2008, 112, 5224–5235. [Google Scholar] [CrossRef]
- Ruff, F.; Farkas, O. Concerted S(N)2 mechanism for the hydrolysis of acid chlorides: Comparisons of reactivities calculated by the density functional theory with experimental data. J. Phys. Org. Chem. 2011, 24, 480–491. [Google Scholar] [CrossRef]
- Vlasov, V.M. Substituent effects in substrates on activation parameters in the bimolecular nucleophilic reactions in solution. New J. Chem. 2010, 34, 2962–2970. [Google Scholar] [CrossRef]
- Chandru, K.; Gilbert, A.; Butch, C.; Aono, M.; Cleaves, H.J. The Abiotic Chemistry of Thiolated Acetate Derivatives and the Origin of Life. Sci. Rep. 2016, 6, 29883. [Google Scholar] [CrossRef] [PubMed]
- Agouridas, V.; El Mahdi, O.; Diemer, V.; Cargoet, M.; Monbaliu, J.M.; Melnyk, O. Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev. 2019, 119, 7328–7443. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G.; Pratt, C.W. Fundamentals of Biochemistry, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Kulkarni, R.A.; Worth, A.J.; Zengeya, T.T.; Shrimp, J.H.; Garlick, J.M.; Roberts, A.M.; Montgomery, D.C.; Sourbier, C.; Gibbs, B.K.; Mesaros, C.; et al. Discovering Targets of Non-enzymatic Acylation by Thioester Reactivity Profiling. Cell Chem. Biol. 2017, 24, 231–242. [Google Scholar] [CrossRef]
- Wagner, G.R.; Bhatt, D.P.; O’Connell, T.M.; Thompson, J.W.; Dubois, L.G.; Backos, D.S.; Yang, H.; Mitchell, G.A.; Ilkayeva, O.R.; Stevens, R.D.; et al. A Class of Reactive Acyl-CoA Species Reveals the Non-enzymatic Origins of Protein Acylation. Cell Metab. 2017, 25, 823–837.e8. [Google Scholar] [CrossRef]
- Guthrie, J.P. Hydration of Thioesters—Evaluation of Free-Energy Changes for Addition of Water to Some Thioesters, Rate-Equilibrium Correlations over Very Wide Ranges in Equilibrium-Constants, and a New Mechanistic Criterion. J. Am. Chem. Soc. 1978, 100, 5892–5904. [Google Scholar] [CrossRef]
- Jencks, W.P.; Gilchrist, M. Free Energies of Hydrolysis of Some Esters + Thiol Esters of Acetic Acid. J. Am. Chem. Soc. 1964, 86, 4651–4654. [Google Scholar] [CrossRef]
- Slosarczyk, A.T.; Ramapanicker, R.; Norberg, T.; Baltzer, L. Mixed pentafluorophenyl and o-fluorophenyl esters of aliphatic dicarboxylic acids: Efficient tools for peptide and protein conjugation. RSC Adv. 2012, 2, 908–914. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, R.Y.; Wang, Y.C.; Chen, X.Z.; Yin, X.G.; Du, J.J.; Lei, Z.; Xin, L.M.; Gao, X.F.; Liu, Z.; et al. Polyfluorophenyl Ester-Terminated Homobifunctional Cross-Linkers for Protein Conjugation. Synlett 2017, 28, 1934–1938. [Google Scholar] [CrossRef]
- Lomant, A.J.; Fairbanks, G. Chemical probes of extended biological structures: Synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate). J. Mol. Biol. 1976, 104, 243–261. [Google Scholar] [CrossRef] [PubMed]
- Cuatrecasas, P.; Parikh, I. Adsorbents for affinity chromatography. Use of N-hydroxysuccinimide esters of agarose. Biochemistry 1972, 11, 2291–2299. [Google Scholar] [CrossRef]
- Rao, H.B.; Wang, Y.Y.; Zeng, X.Y.; Xue, Y.; Li, Z.R. Theoretical study on the aminolysis of p-substituted phenyl acetates with dimeric ammonia in vacuo and acetonitrile. Comput. Theor. Chem. 2013, 1008, 8–14. [Google Scholar] [CrossRef]
- Sung, D.D.; Koo, I.S.; Yang, K.; Lee, I. DFT studies on the structure and stability of zwitterionic tetrahedral intermediate in the aminolysis of esters. Chem. Phys. Lett. 2006, 426, 280–284. [Google Scholar] [CrossRef]
- Yi, G.Q.; Zeng, Y.; Xia, X.F.; Xue, Y.; Kim, C.K.; Yan, G.S. The substituent effects of the leaving groups on the aminolysis of phenyl acetates: DFT studies. Chem. Phys. 2008, 345, 73–81. [Google Scholar] [CrossRef]
- Andres, G.O.; Pierini, A.B.; de Rossi, R.H. Kinetic and theoretical studies on the mechanism of intramolecular catalysis in phenyl ester hydrolysis. J. Org. Chem. 2006, 71, 7650–7656. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhang, H. Density functional theory study of selective deacylation of aromatic acetate in the presence of aliphatic acetate under ammonium acetate mediated conditions. J. Org. Chem. 2014, 79, 6135–6142. [Google Scholar] [CrossRef]
- Xie, D.; Zhou, Y.; Xu, D.; Guo, H. Solvent effect on concertedness of the transition state in the hydrolysis of p-nitrophenyl acetate. Org. Lett. 2005, 7, 2093–2095. [Google Scholar] [CrossRef] [PubMed]
- Elzahhar, P.; Belal, A.S.F.; Elamrawy, F.; Helal, N.A.; Nounou, M.I. Bioconjugation in Drug Delivery: Practical Perspectives and Future Perceptions. Methods Mol. Biol. 2019, 2000, 125–182. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, D.R.; Jiang, H.; Kalin, J.H.; Chen, Z.; Cole, P.A. Site-Specific Protein Labeling with N-Hydroxysuccinimide-Esters and the Analysis of Ubiquitin Ligase Mechanisms. J. Am. Chem. Soc. 2018, 140, 9374–9378. [Google Scholar] [CrossRef]
- Staros, J.V. N-hydroxysulfosuccinimide active esters: Bis(N-hydroxysulfosuccinimide) esters of two dicarboxylic acids are hydrophilic, membrane-impermeant, protein cross-linkers. Biochemistry 1982, 21, 3950–3955. [Google Scholar] [CrossRef]
- Bu, J.; Fisher, C.M.; Gilbert, J.D.; Prentice, B.M.; McLuckey, S.A. Selective Covalent Chemistry via Gas-Phase Ion/ion Reactions: An Exploration of the Energy Surfaces Associated with N-Hydroxysuccinimide Ester Reagents and Primary Amines and Guanidine Groups. J. Am. Soc. Mass. Spectrom. 2016, 27, 1089–1098. [Google Scholar] [CrossRef]
- Morpurgo, M.; Bayer, E.A.; Wilchek, M. N-hydroxysuccinimide carbonates and carbamates are useful reactive reagents for coupling ligands to lysines on proteins. J. Biochem. Biophys. Methods 1999, 38, 17–28. [Google Scholar] [CrossRef]
- Zalipsky, S.; Seltzer, R.; Menon-Rudolph, S. Evaluation of a new reagent for covalent attachment of polyethylene glycol to proteins. Biotechnol. Appl. Biochem. 1992, 15, 100–114. [Google Scholar] [CrossRef]
- Hu, H.; Luo, C.; Wang, B.; Lai, T.; Zhang, G.; Gao, G. NaCl catalyzed transesterification and hydrolysis of ethylene carbonate. Mol. Catal. 2023, 538, 113010. [Google Scholar] [CrossRef]
- Chen, R.; Luo, X.L.; Liang, G.M. Theoretical studies on the aminolysis mechanism of propylene carbonate with ammonia. Theor. Chem. Acc. 2015, 134, 32. [Google Scholar] [CrossRef]
- Zabalov, M.V.; Tiger, R.P.; Berlin, A.A. Mechanism of urethane formation from cyclocarbonates and amines: A quantum chemical study. Russ. Chem. B 2012, 61, 518–527. [Google Scholar] [CrossRef]
- Zabalov, M.V.; Levina, M.A.; Tiger, R.P. Molecular Organization of Reagents in the Kinetics and Catalysis of Liquid-Phase Reactions: XIII. Cyclic Transition States Involving Solvent Molecules in the Mechanism of Aminolysis of Cyclocarbonates in an Alcohol Medium. Kinet. Catal. 2020, 61, 721–729. [Google Scholar] [CrossRef]
- Karlsson, I.; Samuelsson, K.; Ponting, D.J.; Tornqvist, M.; Ilag, L.L.; Nilsson, U. Peptide Reactivity of Isothiocyanates—Implications for Skin Allergy. Sci. Rep. 2016, 6, 21203. [Google Scholar] [CrossRef]
- Topuz, F.; Bartneck, M.; Pan, Y.; Tacke, F. One-Step Fabrication of Biocompatible Multifaceted Nanocomposite Gels and Nanolayers. Biomacromolecules 2017, 18, 386–397. [Google Scholar] [CrossRef]
- Wang, K.; Wang, D.; Ji, K.; Chen, W.; Zheng, Y.; Dai, C.; Wang, B. Post-synthesis DNA modifications using a trans-cyclooctene click handle. Org. Biomol. Chem. 2015, 13, 909–915. [Google Scholar] [CrossRef]
- Longo, B.; Zanato, C.; Piras, M.; Dall’Angelo, S.; Windhorst, A.D.; Vugts, D.J.; Baldassarre, M.; Zanda, M. Design, Synthesis, Conjugation, and Reactivity of Novel trans,trans-1,5-Cyclooctadiene-Derived Bioorthogonal Linkers. Bioconjug. Chem. 2020, 31, 2201–2210. [Google Scholar] [CrossRef]
- Yuan, Y.; Cao, J.P.; Liu, Y.L.; Shi, A.J.; Zhang, Q.; Lin, X.X.; Wang, M.L. Catalytic Effects of Water Clusters on the Hydrolysis of Toluene-2,4-diisocyanate: A DFT Study. B Chem. Soc. Jpn. 2016, 89, 74–91. [Google Scholar] [CrossRef]
- Davies, G.E.; Stark, G.R. Use of dimethyl suberimidate, a cross-linking reagent, in studying the subunit structure of oligomeric proteins. Proc. Natl. Acad. Sci. USA 1970, 66, 651–656. [Google Scholar] [CrossRef]
- Uchiumi, T.; Terao, K.; Ogata, K. Identification of neighboring protein pairs in rat liver 60S ribosomal subunits cross-linked with dimethyl suberimidate or dimethyl 3,3′-dithiobispropionimidate. J. Biochem. 1980, 88, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Seong, H.; Park, J.; Bae, M.; Shin, S. Rapid and Efficient Extraction of Cell-Free DNA Using Homobifunctional Crosslinkers. Biomedicines 2022, 10, 1883. [Google Scholar] [CrossRef]
- Begum, M.F.; Varghese, H.T.; Mary, Y.S.; Panicker, C.Y.; Salim, M.A. Structural defects in imidates: An ab initio Study. Int. J. Chem. Sci. 2011, 9, 1763–1767. [Google Scholar]
- Mossberg, K.; Ericsson, M. Detection of doubly stained fluorescent specimens using confocal microscopy. J. Microsc. 1990, 158, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Yamabe, S.; Zeng, G.X.; Guan, W.; Sakaki, S. SN1-SN2 and SN2-SN3 Mechanistic Changes Revealed by Transition States of the Hydrolyses of Benzyl Chlorides and Benzenesulfonyl Chlorides. J. Comput. Chem. 2014, 35, 1140–1148. [Google Scholar] [CrossRef]
- Ivanov, S.N.; Kislov, V.V.; Gnedin, B.G. Solvation effects in hydrolysis of 2-toluenesulfonyl halides in aqueous dioxane. Catalysis with water molecules in cyclic transition states. Russ. J. Gen. Chem. 2004, 74, 95–100. [Google Scholar] [CrossRef]
- Ivanov, S.N.; Kislov, V.V.; Gnedin, B.G. Simulation of benzenesulfonyl chloride hydrolysis. Influence of the size and structural ordering of aqueous clusters on thermodynamic and activation parameters of the process. Russ. J. Gen. Chem. 2004, 74, 86–94. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Berenguer-Murcia, A.; Carballares, D.; Morellon-Sterling, R.; Fernandez-Lafuente, R. Stabilization of enzymes via immobilization: Multipoint covalent attachment and other stabilization strategies. Biotechnol. Adv. 2021, 52, 107821. [Google Scholar] [CrossRef]
- Lundin, A.; Panas, I.; Ahlberg, E. A mechanistic investigation of ethylene oxide hydrolysis to ethanediol. J. Phys. Chem. A 2007, 111, 9087–9092. [Google Scholar] [CrossRef]
- Muniz Filho, R.C.D.; Sousa, S.A.A.d.; Pereira, F.d.S.; Ferreira, M.M.C. Theoretical Study of Acid-Catalyzed Hydrolysis of Epoxides. J. Phys. Chem. A 2010, 114, 5187–5194. [Google Scholar] [CrossRef]
- Piletic, I.R.; Edney, E.O.; Bartolotti, L.J. A computational study of acid catalyzed aerosol reactions of atmospherically relevant epoxides. Phys. Chem. Chem. Phys. 2013, 15, 18065–18076. [Google Scholar] [CrossRef]
- Renault, K.; Fredy, J.W.; Renard, P.Y.; Sabot, C. Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjug. Chem. 2018, 29, 2497–2513. [Google Scholar] [CrossRef] [PubMed]
- Ravasco, J.; Faustino, H.; Trindade, A.; Gois, P.M.P. Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chemistry 2019, 25, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.S.; Jameson, D.M. Chemistry of Protein and Nucleic Acid Cross-Linking and Conjugation, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Nakane, P.K. Recent progress in the peroxidase-labeled antibody method. Ann. N. Y. Acad. Sci. 1975, 254, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Jose, K.B.; Cyriac, J.; Moolayil, J.T.; Sebastian, V.S.; George, M. The mechanism of aromatic nucleophilic substitution reaction between ethanolamine and fluoro-nitrobenzenes: An investigation by kinetic measurements and DFT calculations. J. Phys. Org. Chem. 2011, 24, 714–719. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. A Universal Approach to Solvation Modeling. Acc. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Ratto, A.; Honek, J.F. Oxocarbon Acids and their Derivatives in Biological and Medicinal Chemistry. Curr. Med. Chem. 2023. [Google Scholar] [CrossRef]
- Storer, R.I.; Aciro, C.; Jones, L.H. Squaramides: Physical properties, synthesis and applications. Chem. Soc. Rev. 2011, 40, 2330–2346. [Google Scholar] [CrossRef]
- Chasak, J.; Slachtova, V.; Urban, M.; Brulikova, L. Squaric acid analogues in medicinal chemistry. Eur. J. Med. Chem. 2021, 209, 112872. [Google Scholar] [CrossRef]
- Agnew-Francis, K.A.; Williams, C.M. Squaramides as Bioisosteres in Contemporary Drug Design. Chem. Rev. 2020, 120, 11616–11650. [Google Scholar] [CrossRef]
- Dingels, C.; Wurm, F.; Wagner, M.; Klok, H.A.; Frey, H. Squaric acid mediated chemoselective PEGylation of proteins: Reactivity of single-step-activated alpha-amino poly(ethylene glycol)s. Chemistry 2012, 18, 16828–16835. [Google Scholar] [CrossRef] [PubMed]
- Wurm, F.R.; Klok, H.A. Be squared: Expanding the horizon of squaric acid-mediated conjugations. Chem. Soc. Rev. 2013, 42, 8220–8236. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Kelly, M.; Vann, W.F.; Qadri, F.; Ryan, E.T.; Kovac, P. Conjugate Vaccines from Bacterial Antigens by Squaric Acid Chemistry: A Closer Look. Chembiochem 2017, 18, 799–815. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Trinh, M.N.; Kovac, P. Conjugation of carbohydrates to proteins using di(triethylene glycol monomethyl ether) squaric acid ester revisited. Carbohydr. Res. 2018, 456, 24–29. [Google Scholar] [CrossRef]
- Suleymanoglu, N.; Ustabas, R.; Alpaslan, Y.B.; Eyduran, F.; Ozyurek, C.; Iskeleli, N.O. Experimental (C-13 NMR, H-1 NMR, FT-IR, single-crystal X-ray diffraction) and DFT studies on 3,4-bis(isoproylamino)cyclobut-3-ene-1,2-dione. Spectrochim. Acta A 2011, 83, 472–477. [Google Scholar] [CrossRef]
- Silva, C.E.; Dos Santos, H.F.; Speziali, N.L.; Diniz, R.; de Oliveira, L.F.C. Role of the Substituent Effect over the Squarate Oxocarbonic Ring: Spectroscopy, Crystal Structure, and Density Functional Theory Calculations of 1,2-Dianilinosquairane. J. Phys. Chem. A 2010, 114, 10097–10109. [Google Scholar] [CrossRef]
- Ximenis, M.; Bustelo, E.; Algarra, A.G.; Vega, M.; Rotger, C.; Basallote, M.G.; Costa, A. Kinetic Analysis and Mechanism of the Hydrolytic Degradation of Squaramides and Squaramic Acids. J. Org. Chem. 2017, 82, 2160–2170. [Google Scholar] [CrossRef]
- Jiao, T.Y.; Wu, G.C.; Zhang, Y.; Shen, L.B.; Lei, Y.; Wang, C.Y.; Fahrenbach, A.C.; Li, H. Self-Assembly in Water with N-Substituted Imines. Angew. Chem. Int. Ed. 2020, 59, 18350–18367. [Google Scholar] [CrossRef]
- Ortega-Castro, J.; Adrover, M.; Frau, J.; Salva, A.; Donoso, J.; Munoz, F. DFT studies on Schiff base formation of vitamin B6 analogues. Reaction between a pyridoxamine-analogue and carbonyl compounds. J. Phys. Chem. A 2010, 114, 4634–4640. [Google Scholar] [CrossRef]
- Salva, A.; Donoso, J.; Frau, J.; Munoz, F. DFT studies on Schiff base formation of vitamin B-6 analogues. J. Phys. Chem. A 2003, 107, 9409–9414. [Google Scholar] [CrossRef]
- Afonso, C.F.; Marques, M.C.; Antonio, J.P.M.; Cordeiro, C.; Gois, P.M.P.; Cal, P.; Bernardes, G.J.L. Cysteine-Assisted Click-Chemistry for Proximity-Driven, Site-Specific Acetylation of Histones. Angew. Chem. Int. Ed. Engl. 2022, 61, e202208543. [Google Scholar] [CrossRef] [PubMed]
- Ahangarpour, M.; Kavianinia, I.; Brimble, M.A. Thia-Michael addition: The route to promising opportunities for fast and cysteine-specific modification. Org. Biomol. Chem. 2023, 21, 3057–3072. [Google Scholar] [CrossRef] [PubMed]
- Harel, O.; Jbara, M. Posttranslational Chemical Mutagenesis Methods to Insert Posttranslational Modifications into Recombinant Proteins. Molecules 2022, 27, 4389. [Google Scholar] [CrossRef] [PubMed]
- Abdelsalam, A.M.; Somaida, A.; Ayoub, A.M.; Alsharif, F.M.; Preis, E.; Wojcik, M.; Bakowsky, U. Surface-Tailored Zein Nanoparticles: Strategies and Applications. Pharmaceutics 2021, 13, 1354. [Google Scholar] [CrossRef] [PubMed]
- Stuparu, M.C.; Khan, A. Thiol-epoxy “click” chemistry: Application in preparation and postpolymerization modification of polymers. J. Polym. Sci. Pol. Chem. 2016, 54, 3057–3070. [Google Scholar] [CrossRef]
- Liao, R.Z.; Thiel, W. Determinants of Regioselectivity and Chemoselectivity in Fosfomycin Resistance Protein FosA from QM/MM Calculations. J. Phys. Chem. B 2013, 117, 1326–1336. [Google Scholar] [CrossRef]
- Senger, N.A.; Bo, B.; Cheng, Q.; Keeffe, J.R.; Gronert, S.; Wu, W.M. The Element Effect Revisited: Factors Determining Leaving Group Ability in Activated Nucleophilic Aromatic Substitution Reactions. J. Org. Chem. 2012, 77, 9535–9540. [Google Scholar] [CrossRef]
- Northrop, B.H.; Frayne, S.H.; Choudhary, U. Thiol-maleimide “click” chemistry: Evaluating the influence of solvent, initiator, and thiol on the reaction mechanism, kinetics, and selectivity. Polym. Chem. 2015, 6, 3415–3430. [Google Scholar] [CrossRef]
- Raycroft, M.A.R.; Racine, K.E.; Rowley, C.N.; Keillor, J.W. Mechanisms of Alkyl and Aryl Thiol Addition to N-Methylmaleimide. J. Org. Chem. 2018, 83, 11674–11685. [Google Scholar] [CrossRef]
- Moran, D.; Sukcharoenphon, K.; Puchta, R.; Schaefer, H.F.; Schleyer, P.V.; Hoff, C.D. 2-pyridinethiol/2-pyridinethione tautomeric equilibrium. A comparative experimental and computational study. J. Org. Chem. 2002, 67, 9061–9069. [Google Scholar] [CrossRef]
- Dahl, K.H.; McKinley-McKee, J.S. The reactivity of affinity labels: A kinetic study of the reaction of alkyl halides with thiolate anions—A model reaction for protein alkylation. Bioorg. Chem. 1981, 10, 329–341. [Google Scholar] [CrossRef]
- Wang, H.; Vath, G.M.; Gleason, K.J.; Hanna, P.E.; Wagner, C.R. Probing the mechanism of hamster arylamine N-acetyltransferase 2 acetylation by active site modification, site-directed mutagenesis, and pre-steady state and steady state kinetic studies. Biochemistry 2004, 43, 8234–8246. [Google Scholar] [CrossRef]
- Pals, J.A.; Wagner, E.D.; Plewa, M.J. Energy of the Lowest Unoccupied Molecular Orbital, Thiol Reactivity, and Toxicity of Three Monobrominated Water Disinfection Byproducts. Environ. Sci. Technol. 2016, 50, 3215–3221. [Google Scholar] [CrossRef]
- Desai, K.K.; Miller, B.G. Recruitment of genes and enzymes conferring resistance to the nonnatural toxin bromoacetate. Proc. Natl. Acad. Sci. USA 2010, 107, 17968–17973. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.F.C.; Oliveira, B.L.; D’Onofrio, A.; Farinha, C.M.; Gano, L.; Paulo, A.; Bernardes, G.J.L.; Mendes, F. In Vivo Pretargeting Based on Cysteine-Selective Antibody Modification with IEDDA Bioorthogonal Handles for Click Chemistry. Bioconjug. Chem. 2021, 32, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Ochtrop, P.; Hackenberger, C.P.R. Recent advances of thiol-selective bioconjugation reactions. Curr. Opin. Chem. Biol. 2020, 58, 28–36. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Smith, M.E.; Schumacher, F.F.; Ryan, C.P.; Tedaldi, L.M.; Papaioannou, D.; Waksman, G.; Caddick, S.; Baker, J.R. Protein modification, bioconjugation, and disulfide bridging using bromomaleimides. J. Am. Chem. Soc. 2010, 132, 1960–1965. [Google Scholar] [CrossRef]
- Feuillatre, O.; Gely, C.; Huvelle, S.; Baltus, C.B.; Juen, L.; Joubert, N.; Desgranges, A.; Viaud-Massuard, M.C.; Martin, C. Impact of Maleimide Disubstitution on Chemical and Biological Characteristics of HER2 Antibody-Drug Conjugates. ACS Omega 2020, 5, 1557–1565. [Google Scholar] [CrossRef]
- Tedaldi, L.M.; Smith, M.E.; Nathani, R.I.; Baker, J.R. Bromomaleimides: New reagents for the selective and reversible modification of cysteine. Chem. Commun. 2009, 45, 6583–6585. [Google Scholar] [CrossRef]
- Karabacak, M.; Coruh, A.; Kurt, M. FT-IR, FT-Raman, NMR spectra, and molecular structure investigation of 2,3-dibromo-N-methylmaleimide: A combined experimental and theoretical study. J. Mol. Struct. 2008, 892, 125–131. [Google Scholar] [CrossRef]
- Scinto, S.L.; Bilodeau, D.A.; Hincapie, R.; Lee, W.; Nguyen, S.S.; Xu, M.; Am Ende, C.W.; Finn, M.G.; Lang, K.; Lin, Q.; et al. Bioorthogonal chemistry. Nat. Rev. Methods Primers 2021, 1, 30. [Google Scholar] [CrossRef] [PubMed]
- Aarjane, M.; Slassi, S.; Amine, A. Novel series of N-acylhydrazone based on acridone: Synthesis, conformational and theoretical studies. J. Mol. Struct. 2021, 1225, 129079. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, N.K.; Finn, M.G. Introduction: Click Chemistry. Chem. Rev. 2021, 121, 6697–6698. [Google Scholar] [CrossRef]
- Adumeau, P.; Sharma, S.K.; Brent, C.; Zeglis, B.M. Site-Specifically Labeled Immunoconjugates for Molecular Imaging—Part 2: Peptide Tags and Unnatural Amino Acids. Mol. Imaging Biol. 2016, 18, 153–165. [Google Scholar] [CrossRef]
- Kim, C.H.; Axup, J.Y.; Schultz, P.G. Protein conjugation with genetically encoded unnatural amino acids. Curr. Opin. Chem. Biol. 2013, 17, 412–419. [Google Scholar] [CrossRef]
- Sarrett, S.M.; Keinanen, O.; Dayts, E.J.; Dewaele-Le Roi, G.; Rodriguez, C.; Carnazza, K.E.; Zeglis, B.M. Inverse electron demand Diels-Alder click chemistry for pretargeted PET imaging and radioimmunotherapy. Nat. Protoc. 2021, 16, 3348–3381. [Google Scholar] [CrossRef]
- Beck, S.; Schultze, J.; Rader, H.J.; Holm, R.; Schinnerer, M.; Barz, M.; Koynov, K.; Zentel, R. Site-Specific DBCO Modification of DEC205 Antibody for Polymer Conjugation. Polymers 2018, 10, 141. [Google Scholar] [CrossRef]
- Spampinato, A.; Kuzmova, E.; Pohl, R.; Sykorova, V.; Vrabel, M.; Kraus, T.; Hocek, M. trans-Cyclooctene- and Bicyclononyne-Linked Nucleotides for Click Modification of DNA with Fluorogenic Tetrazines and Live Cell Metabolic Labeling and Imaging. Bioconjug. Chem. 2023, 34, 772–780. [Google Scholar] [CrossRef]
- Wagner, J.A.; Mercadante, D.; Nikic, I.; Lemke, E.A.; Grater, F. Origin of Orthogonality of Strain-Promoted Click Reactions. Chemistry 2015, 21, 12431–12435. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aznar, P.; Escorihuela, J. Computational insights into the inverse electron-demand Diels-Alder reaction of norbornenes with 1,2,4,5-tetrazines: Norbornene substituents’ effects on the reaction rate. Org. Biomol. Chem. 2022, 20, 6400–6412. [Google Scholar] [CrossRef]
- Battisti, U.M.; Garcia-Vazquez, R.; Svatunek, D.; Herrmann, B.; Loffler, A.; Mikula, H.; Herth, M.M. Synergistic Experimental and Computational Investigation of the Bioorthogonal Reactivity of Substituted Aryltetrazines. Bioconjug. Chem. 2022, 33, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Svatunek, D.; Denk, C.; Mikula, H. A computational model to predict the Diels-Alder reactivity of aryl/alkyl-substituted tetrazines. Monatsh. Chem. 2018, 149, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Sundhoro, M.; Houk, K.N.; Yan, M. Electrophilic Azides for Materials Synthesis and Chemical Biology. Acc. Chem. Res. 2020, 53, 937–948. [Google Scholar] [CrossRef]
- Baldensperger, T.; Glomb, M.A. Pathways of Non-enzymatic Lysine Acylation. Front. Cell Dev. Biol. 2021, 9, 664553. [Google Scholar] [CrossRef]
- Lammers, M. Post-translational Lysine Ac(et)ylation in Bacteria: A Biochemical, Structural, and Synthetic Biological Perspective. Front. Microbiol. 2021, 12, 757179. [Google Scholar] [CrossRef]
- Wang, M.M.; You, D.; Ye, B.C. Site-specific and kinetic characterization of enzymatic and nonenzymatic protein acetylation in bacteria. Sci. Rep. 2017, 7, 14790. [Google Scholar] [CrossRef]
- Simic, Z.; Weiwad, M.; Schierhorn, A.; Steegborn, C.; Schutkowski, M. The epsilon-Amino Group of Protein Lysine Residues Is Highly Susceptible to Nonenzymatic Acylation by Several Physiological Acyl-CoA Thioesters. Chembiochem 2015, 16, 2337–2347. [Google Scholar] [CrossRef]
- Carrico, C.; Cruz, A.; Walter, M.; Meyer, J.; Wehrfritz, C.; Shah, S.; Wei, L.; Schilling, B.; Verdin, E. Coenzyme A binding sites induce proximal acylation across protein families. Sci. Rep. 2023, 13, 5029. [Google Scholar] [CrossRef]
- Faulkner, S.; Maksimovic, I.; David, Y. A chemical field guide to histone nonenzymatic modifications. Curr. Opin. Chem. Biol. 2021, 63, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Graf, L.G.; Vogt, R.; Blasl, A.T.; Qin, C.; Schulze, S.; Zuhlke, D.; Sievers, S.; Lammers, M. Assays to Study Enzymatic and Non-Enzymatic Protein Lysine Acetylation In Vitro. Curr. Protoc. 2021, 1, e277. [Google Scholar] [CrossRef]
- Maksimovic, I.; David, Y. Non-enzymatic Covalent Modifications as a New Chapter in the Histone Code. Trends Biochem. Sci. 2021, 46, 718–730. [Google Scholar] [CrossRef]
- Baeza, J.; Smallegan, M.J.; Denu, J.M. Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem. Biol. 2015, 10, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Indiveri, C. Post-translational modification by acetylation regulates the mitochondrial carnitine/acylcarnitine transport protein. Mol. Cell. Biochem. 2017, 426, 65–73. [Google Scholar] [CrossRef]
- Hong, S.Y.; Ng, L.T.; Ng, L.F.; Inoue, T.; Tolwinski, N.S.; Hagen, T.; Gruber, J. The Role of Mitochondrial Non-Enzymatic Protein Acylation in Ageing. PLoS ONE 2016, 11, e0168752. [Google Scholar] [CrossRef]
- Kerner, J.; Yohannes, E.; Lee, K.; Virmani, A.; Koverech, A.; Cavazza, C.; Chance, M.R.; Hoppel, C. Acetyl-L-carnitine increases mitochondrial protein acetylation in the aged rat heart. Mech. Ageing Dev. 2015, 145, 39–50. [Google Scholar] [CrossRef]
- Martin, W.F.; Thauer, R.K. Energy in Ancient Metabolism. Cell 2017, 168, 953–955. [Google Scholar] [CrossRef] [PubMed]
- Semenov, S.N.; Kraft, L.J.; Ainla, A.; Zhao, M.; Baghbanzadeh, M.; Campbell, V.E.; Kang, K.; Fox, J.M.; Whitesides, G.M. Autocatalytic, bistable, oscillatory networks of biologically relevant organic reactions. Nature 2016, 537, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Shalayel, I.; Leqraa, N.; Blandin, V.; Vallée, Y. Catalysis before Enzymes: Thiol-Rich Peptides as Molecular Diversity Providers on the Early Earth. Diversity 2023, 15, 256. [Google Scholar] [CrossRef]
- Chevallot-Beroux, E.; Gorges, J.; Moran, J. Energy Conservation via Thioesters in a Non-enzymatic Metabolism-like Reaction Network. ChemRxiv 2019, 1–10. [Google Scholar] [CrossRef]
- De Duve, C. A research proposal on the origin of life. Orig. Life Evol. Biosph. 2003, 33, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Harmel, R.; Fiedler, D. Features and regulation of non-enzymatic post-translational modifications. Nat. Chem. Biol. 2018, 14, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years... and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef]
- Honek, J.F. Glyoxalase biochemistry. Biomol. Concepts 2015, 6, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Varner, E.L.; Trefely, S.; Bartee, D.; von Krusenstiern, E.; Izzo, L.; Bekeova, C.; O’Connor, R.S.; Seifert, E.L.; Wellen, K.E.; Meier, J.L.; et al. Quantification of lactoyl-CoA (lactyl-CoA) by liquid chromatography mass spectrometry in mammalian cells and tissues. Open Biol. 2020, 10, 200187. [Google Scholar] [CrossRef]
- Wolfenden, R.; Liang, Y.L. Contributions of Solvent Water to Biological Group-Transfer Potentials: Mixed Anhydrides of Phosphoric and Carboxylic Acids. Bioorg. Chem. 1989, 17, 486–489. [Google Scholar] [CrossRef]
- Weinert, B.T.; Iesmantavicius, V.; Wagner, S.A.; Scholz, C.; Gummesson, B.; Beli, P.; Nystrom, T.; Choudhary, C. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell 2013, 51, 265–272. [Google Scholar] [CrossRef]
- Kluger, R.; Tsui, W.C. Methyl Acetyl Phosphate—A Small Anionic Acetylating Agent. J. Org. Chem. 1980, 45, 2723–2724. [Google Scholar] [CrossRef]
- Ueno, H.; Pospischil, M.A.; Kluger, R.; Manning, J.M. Methyl Acetyl Phosphate—A Novel Acetylating Agent Its Site-Specific Modification of Human Hemoglobin-A. J. Chromatogr. 1986, 359, 193–201. [Google Scholar] [CrossRef]
- Ueno, H.; Pospischil, M.A.; Manning, J.M.; Kluger, R. Site-Specific Modification of Hemoglobin by Methyl Acetyl Phosphate. Arch. Biochem. Biophys. 1986, 244, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Kluger, R. 1994 Syntex Award Lecture—Anionic Electrophiles, Protein Modification, and Artificial Blood. Can. J. Chem. 1994, 72, 2193–2197. [Google Scholar] [CrossRef]
- Kataoka, K.; Tanizawa, K.; Fukui, T.; Ueno, H.; Yoshimura, T.; Esaki, N.; Soda, K. Identification of active site lysyl residues of phenylalanine dehydrogenase by chemical modification with methyl acetyl phosphate combined with site-directed mutagenesis. J. Biochem. 1994, 116, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Fritz, I.B.; Schultz, S.K.; Srere, P.A. Properties of partially purified carnitine acetyltransferase. J. Biol. Chem. 1963, 238, 2509–2517. [Google Scholar] [CrossRef]
- Muller, D.M.; Strack, E. The binding energy of the ester group in O-acylcarnitines and some carboxyl derivatives, III. Hydrolysis enthalpy of O-acylcarnitines and betaine esters (author’s transl). Hoppe Seylers Z. Physiol. Chem. 1973, 354, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Pieklik, J.R.; Guynn, R.W. Equilibrium constants of the reactions of choline acetyltransferase, carnitine acetyltransferase, and acetylcholinesterase under physiological conditions. J. Biol. Chem. 1975, 250, 4445–4450. [Google Scholar] [CrossRef]
- Colucci, W.J.; Gandour, R.D. Carnitine Acetyltransferase—A Review of Its Biology, Enzymology, and Bioorganic Chemistry. Bioorg. Chem. 1988, 16, 307–334. [Google Scholar] [CrossRef]
- Gandour, R.D. Rationalizing the solution properties of zwitterions by means of computational chemistry. Chem. Biodivers. 2005, 2, 1580–1594. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Klamt, A.; Moya, C.; Palomar, J. A comprehensive comparison of the IEFPCM and SS(V)PE contimuum solvation methods with the COSMO approach. J. Chem. Theory. Comput. 2015, 11, 4220–4225. [Google Scholar] [CrossRef]
- Rosas-Garcia, V.M.; Gandour, R.D. Conformationally-Dependent Free Energies of Solvation. An Explanation for the Large Group-Transfer Potential of Acetylcarnitine. J. Am. Chem. Soc. 1997, 119, 7587–7588. [Google Scholar] [CrossRef]
- Ferri, L.; Jocelyn, P.C.; Siliprandi, N. The mitochondrial handling of D,L-thiocarnitine and its S-acetyl derivative. FEBS Lett. 1980, 121, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.R.; Drummond, G.I. Enzymes of ketone body metabolism. III. Enzymic thiolysis of acetoacetyl coenzyme A and acetoacetyl-pantetheine by mono- and dithiol compounds. J. Biol. Chem. 1961, 236, 2892–2897. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Botting, R.M. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- Thomas, G. Medicinal Chemistry, 2nd ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2007. [Google Scholar]
- Palmer, M.; Chan, A.; Dieckmann, T.; Honek, J. Biochemical Pharmacology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Alfonso, L.F.; Srivenugopal, K.S.; Bhat, G.J. Does aspirin acetylate multiple cellular proteins? (Review). Mol. Med. Rep. 2009, 2, 533–537. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Zhang, C.J.; Zhang, J.; He, Y.; Lee, Y.M.; Chen, S.; Lim, T.K.; Ng, S.; Shen, H.M.; Lin, Q. Mapping sites of aspirin-induced acetylations in live cells by quantitative acid-cleavable activity-based protein profiling (QA-ABPP). Sci. Rep. 2015, 5, 7896. [Google Scholar] [CrossRef] [PubMed]
- Bateman, L.A.; Zaro, B.W.; Miller, S.M.; Pratt, M.R. An alkyne-aspirin chemical reporter for the detection of aspirin-dependent protein modification in living cells. J. Am. Chem. Soc. 2013, 135, 14568–14573. [Google Scholar] [CrossRef]
- Mikkelsen, S.R.; Cortón, E. Bioanalytical Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Bisswanger, H. Practical Enzymology, 2nd ed.; Wiley-Blackwell: Weinheim, Germany, 2011. [Google Scholar]
- Breidenbach, J.; Bartz, U.; Gutschow, M. Coumarin as a structural component of substrates and probes for serine and cysteine proteases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140445. [Google Scholar] [CrossRef]
- Means, G.E.; Bender, M.L. Acetylation of human serum albumin by p-nitrophenyl acetate. Biochemistry 1975, 14, 4989–4994. [Google Scholar] [CrossRef]
- Bhatt, A.N.; Rai, Y.; Verma, A.; Pandey, S.; Kaushik, K.; Parmar, V.S.; Arya, A.; Prasad, A.K.; Dwarakanath, B.S. Non-Enzymatic Protein Acetylation by 7-Acetoxy-4-Methylcoumarin: Implications in Protein Biochemistry. Protein Pept. Lett. 2020, 27, 736–743. [Google Scholar] [CrossRef]
- Raj, H.G.; Parmar, V.S.; Jain, S.C.; Goel, S.; Singh, A.; Tyagi, Y.K.; Jha, H.N.; Olsen, C.E.; Wengel, J. Mechanism of biochemical action of substituted 4-methylbenzopyran-2-ones. Part 4: Hyperbolic activation of rat liver microsomal NADPH cytochrome C reductase by the novel acetylator 7,8-diacetoxy-4-methylcoumarin. Bioorg. Med. Chem. 1999, 7, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Raj, H.G.; Parmar, V.S.; Jain, S.C.; Kohli, E.; Ahmad, N.; Goel, S.; Tyagi, Y.K.; Sharma, S.K.; Wengel, J.; Olsen, C.E. Mechanism of biochemical action of substituted 4-methylbenzopyran-2-ones. Part 7: Assay and characterization of 7,8-diacetoxy-4-methylcoumarin: Protein transacetylase from rat liver microsomes based on the irreversible inhibition of cytosolic glutathione S-transferase. Bioorg. Med. Chem. 2000, 8, 1707–1712. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Flury, P.; Kruger, N.; Su, H.; Schakel, L.; Barbosa Da Silva, E.; Eppler, O.; Kronenberger, T.; Nie, T.; Luedtke, S.; et al. Small-Molecule Thioesters as SARS-CoV-2 Main Protease Inhibitors: Enzyme Inhibition, Structure-Activity Relationships, Antiviral Activity, and X-ray Structure Determination. J. Med. Chem. 2022, 65, 9376–9395. [Google Scholar] [CrossRef] [PubMed]




| Reaction | Gas Phase (kJ/mol) | Water (kJ/mol) |
|---|---|---|
 | 15.04 (ΔE) 19.26 (ΔG) 18.74 (ΔH) | 16.42 (ΔE) 21.70 (ΔG) 20.11 (ΔH) |
 | −3.16 (ΔE) −4.25 (ΔG) −3.81 (ΔH) | −19.63 (ΔE) −20.73 (ΔG) −20.28 (ΔH) |
 | −54.65 (ΔE) −55.85 (ΔG) −46.34 (ΔH) | −47.55 (ΔE) −49.83 (ΔG) −40.20 (ΔH) |
 | −77.34 (ΔE) −85.40 (ΔG) −74.34 (ΔH) | −92.15 (ΔE) −98.13 (ΔG) −88.83 (ΔH) |
 | −43.74 (ΔE) −41.72 (ΔG) −45.17 (ΔH) | −37.94 (ΔE) −35.92 (ΔG) −39.37 (ΔH) |
 | −61.99 (ΔE) −58.07 (ΔG) −68.19 (ΔH) | −74.08 (ΔE) −71.01 (ΔG) −80.35 (ΔH) |
 | −17.17 (ΔE) −26.96 (ΔG) −15.46 (ΔH) | −10.03 (ΔE) −19.81 (ΔG) −8.32 (ΔH) |
 | −35.41 (ΔE) −43.31 (ΔG) −38.48 (ΔH) | −46.19 (ΔE) −59.97 (ΔG) −48.79 (ΔH) |
| Reaction | Gas Phase (kJ/mol) | Water (kJ/mol) |
|---|---|---|
 | −17.88 (ΔE) −14.68 (ΔG) −12.18 (ΔH) | −18.79 (ΔE) −15.49 ΔG) −13.08 (ΔH) |
 | −36.13 (ΔE) −31.03 (ΔG) −35.20 (ΔH) | −54.90 (ΔE) −49.72 (ΔG) −53.96 (ΔH) |
 | −25.68 (ΔE) −25.75 (ΔG) −18.69 (ΔH) | −27.60 (ΔE) −27.67 (ΔG) −20.62 (ΔH) |
 | −43.93 (ΔE) −42.10 (ΔG) −41.71 (ΔH) | −63.71 (ΔE) −61.89 (ΔG) −61.50 (ΔH) |
 | −15.19 (ΔE) −11.16 (ΔG) −9.42 (ΔH) | −17.30 (ΔE) −12.94 (ΔG) −11.46 (ΔH) |
 | −33.44 (ΔE) −27.51 (ΔG) −32.43 (ΔH) | −53.41 (ΔE) −47.16 (ΔG) −52.35 (ΔH) |
 | −21.21 (ΔE) −20.56 (ΔG) −14.48 (ΔH) | −17.25 (ΔE) −16.59 (ΔG) −10.52 (ΔH) |
 | −39.46 (ΔE) −36.91 (ΔG) −37.50 (ΔH) | −53.36 (ΔE) −50.82 (ΔG) −51.4 (ΔH) |
 | −23.23 (ΔE) −20.91 (ΔG) −16.26 (ΔH) | −22.50 (ΔE) −20.50 (ΔG) −15.57(ΔH) |
 | −41.48 (ΔE) −37.26 (ΔG) −39.27 (ΔH) | −58.62 (ΔE) −54.73 (ΔG) −56.45 (ΔH) |
 | −18.28 (ΔE) −19.27 (ΔG) −11.64 (ΔH) | −15.74 (ΔE) −16.72 (ΔG) −9.09 (ΔH) |
 | −36.48 (ΔE) −43.13 (ΔG) −34.48 (ΔH) | −51.82 (ΔE) −54.68 (ΔG) −49.33 (ΔH) |
 | −20.56 (ΔE) −20.62 (ΔG) −13.94 (ΔH) | −21.01 (ΔE) −20.67 (ΔG) −14.41 (ΔH) |
 | −38.76 (ΔE) −44.47 (ΔG) −36.78 (ΔH) | −57.16 (ΔE) −59.27 (ΔG) −54.78 (ΔH) |
 | −23.72 (ΔE) −24.67 (ΔG) −16.87 (ΔH) | −22.05 (ΔE) −22.53 (ΔG) −15.19 (ΔH) |
 | −41.95 (ΔE) −47.41 (ΔG) −39.51 (ΔH) | −58.20 (ΔE) −61.14 (ΔG) −55.56 (ΔH) |
 | −20.57 (ΔE) −23.08 (ΔG) −13.89 (ΔH) | −20.57 (ΔE) −22.56 (ΔG) −13.84 (ΔH) |
 | −38.80 (ΔE) −45.82 (ΔG) −36.53 (ΔH) | −56.72 (ΔE) −61.16 (ΔG) −54.20 (ΔH) |
| Reaction | Gas Phase (kJ/mol) | Water (kJ/mol) |
|---|---|---|
 | −6.53 (ΔE) −7.48 (ΔG) 0.28 (ΔH) | −4.21 (ΔE) −4.94 (ΔG) 2.68 (ΔH) |
 | −24.78 (ΔE) −23.84 (ΔG) −22.74 (ΔH) | −40.37 (ΔE) −39.15 (ΔG) −38.19 (ΔH) |
 | −7.58 (ΔE) −6.84 (ΔG) −1.77 (ΔH) | −6.06 (ΔE) −5.32 (ΔG) −0.25 (ΔH) |
 | −25.83 (ΔE) −23.19 (ΔG) −24.78 (ΔH) | −41.82 (ΔE) −39.19 (ΔG) −40.78 (ΔH) |
| Reaction | Gas Phase (kJ/mol) | Water (kJ/mol) |
|---|---|---|
 | 14.36 (ΔE) 12.93 (ΔG) 17.9 (ΔH) | 15.21 (ΔE) 13.78 (ΔG) 18.75 (ΔH) |
 | −16.30 (ΔE) −21.36 (ΔG) −17.51 (ΔH) | −23.31 (ΔE) −28.55 (ΔG) −24.69 (ΔH) |
 | −15.43 (ΔE) −12.89 (ΔG) −9.04 (ΔH) | −14.31 (ΔE) −12.58 (ΔG) −8.29 (ΔH) |
 | −45.89 (ΔE) −47.63 (ΔG) −44.56 (ΔH) | −52.83 (ΔE) −53.28 (ΔG) −51.48 (ΔH) |
 | −27.58 (ΔE) −30.84 (ΔG) −20.93 (ΔH) | −31.78 (ΔE) −35.22 (ΔG) −25.18 (ΔH) |
 | −58.05 (ΔE) −63.91 (ΔG) −56.11 (ΔH) | −70.42 (ΔE) −76.11 (ΔG) −68.40 (ΔH) |
| Reaction | Gas Phase (kJ/mol) | Water (kJ/mol) |
|---|---|---|
 | −107.59 (ΔE) −43.88 (ΔG) −94.78 (ΔH) | −100.33 (ΔE) −36.62 (ΔG) −87.52 (ΔH) |
 | −110.34 (ΔE) −48.79 (ΔG) −104.15 (ΔH) | −121.49 (ΔE) −59.39 (ΔG) −113.27(ΔH) |
 | −108.46 (ΔE) −51.20 (ΔG) −95.44 (ΔH) | −103.03 (ΔE) −45.76 (ΔG) −90.01 (ΔH) |
 | −114.32 (ΔE) −61.84 (ΔG) −106.66 (ΔH) | −127.76 (ΔE) −75.48 (ΔG) −120.13(ΔH) |
 | −84.23 (ΔE) −27.27 (ΔG) −71.00 (ΔH) | −71.93 (ΔE) −19.17 (ΔG) −58.22 (ΔH) |
 | −99.68 (ΔE) −40.26 (ΔG) −90.44 (ΔH) | −114.08 (ΔE) −60.29 (ΔG) −105.58(ΔH) |
 | −65.37(ΔE) −9.07 (ΔG) −52.74 (ΔH) | −60.63 (ΔE) −6.42 (ΔG) −47.63 (ΔH) |
 | −82.39 (ΔE) −28.24 (ΔG) −75.12 (ΔH) | −108.74 (ΔE) −53.16 (ΔG) −100.6 (ΔH) |
| Reaction | Gas Phase (kJ/mol) | Water (kJ/mol) |
|---|---|---|
 | −2.39 (ΔE) −4.89 (ΔG) −4.11 (ΔH) | −15.16 (ΔE) −17.65 (ΔG) −16.88 (ΔH) |
 | −6.63 (ΔE) −6.95 (ΔG) −7.26 (ΔH) | −17.78 (ΔE) −18.98 (ΔG) −18.45 (ΔH) |
 | −35.49 (ΔE) −36.36 (ΔG) −43.22 (ΔH) | −42.86 (ΔE) −43.73 (ΔG) −50.58 (ΔH) |
 | −34.50 (ΔE) −34.85 (ΔG) −42.20 (ΔH) | −53.06 (ΔE) −51.03 (ΔG) −59.92 (ΔH) |
 | −120.41 (ΔE) −66.04 (ΔG) −113.51 (ΔH) | −116.93 (ΔE) −62.56 (ΔG) −110.02(ΔH) |
 | −105.01 (ΔE) −46.27 (ΔG) −97.70 (ΔH) | −91.93 (ΔE) −34.75 (ΔG) −84.41 (ΔH) |
 | −62.60 (ΔE) −52.69 (ΔG) −64.26 (ΔH) | −83.95 (ΔE) −73.96 (ΔG) −85.60 (ΔH) |
| Reaction | Gas Phase (kJ/mol) | Water (kJ/mol) |
|---|---|---|
 | −44.02 (ΔE) −46.26(ΔG) −44.44 (ΔH) | −57.84 (ΔE) −60.08 (ΔG) −58.26 (ΔH) |
 | −23.70 (ΔE) −25.78 (ΔG) −25.03 (ΔH) | −44.02 (ΔE) −46.10 (ΔG) −45.35 (ΔH) |
 | −12.05 (ΔE) −9.55 (ΔG) −15.37(ΔH) | −16.64 (ΔE) −14.41 (ΔG) −19.91 (ΔH) |
 | −3.47 (ΔE) −2.3 (ΔG) −9.8 (ΔH) | −9.67 (ΔE) −8.51 (ΔG) −15.98 (ΔH) |
| Reaction | Gas Phase (kJ/mol) | Water (kJ/mol) |
|---|---|---|
 | −19.34 (ΔE) −11.28 (ΔG) −22.26 (ΔH) | −27.45 (ΔE) −19.39 (ΔG) −30.37 (ΔH) |
 | −28.32 (ΔE) −24.75 (ΔG) −30.8 (ΔH) | −38.26 (ΔE) −34.48 (ΔG) −40.50 (ΔH) |
 | −134.89(ΔE) −77.60(ΔG) −123.47(ΔH) | −135.13 (ΔE) −77.84 (ΔG) −123.71(ΔH) |
 | −109.96(ΔE) −43.36 (ΔG) −98.95 (ΔH) | −111.84 (ΔE) −55.92 (ΔG) −101.03(ΔH) |
 | −36.56 (ΔE) −26.20 (ΔG) −35.74 (ΔH) | −46.04 (ΔE) −35.59 (ΔG) −44.95 (ΔH) |
| Reaction | Gas Phase (kJ/mol) | Water (kJ/mol) |
|---|---|---|
 | −5.41 (ΔE) −8.27 (ΔG) −4.53 (ΔH) | −6.04 (ΔE) −8.90 (ΔG) −5.16 (ΔH) |
 | −11.42 (ΔE) −3.03 (ΔG) −1.46 (ΔH) | −40.19 (ΔE) −31.80 (ΔG) −30.24 (ΔH) |
 | −17.63 (ΔE) −16.47 (ΔG) −22.03 (ΔH) | −22.70 (ΔE) −18.64 (ΔG) −25.60 (ΔH) |
 | −20.21 (ΔE) −14.59 (ΔG) −24.44 (ΔH) | −22.15 (ΔE) −16.19 (ΔG) −25.16 (ΔH) |
 | −43.49 (ΔE) −36.36 (ΔG) −46.60 (ΔH) | −43.97 (ΔE) −36.84 (ΔG) −47.08 (ΔH) |
 | −32.88 (ΔE) −26.76 (ΔG) −37.22 (ΔH) | −32.06 (ΔE) −25.79 (ΔG) −36.24 (ΔH) |
 | −23.65 (ΔE) −18.70 (ΔG) −28.63 (ΔH) | −25.97 (ΔE) −21.02 (ΔG) −30.95 (ΔH) |
| Reaction | M06-2X/cc-PVTZ(-f)++ in Implicit Water (kJ/mol) |
|---|---|
 | −19.54 (ΔE) −22.22 (ΔG) −19.96 (ΔH) |
 | −8.49 (ΔE) −13.81 (ΔG) −9.41 (ΔH) |
 | −44.22 (ΔE) −49.20 (ΔG) −47.91 (ΔH) |
 | −113.68 (ΔE) −44.56 (ΔG) −99.70 (ΔH) |
 | −311.29 (ΔE) −238.03 (ΔG) −295.56 (ΔH) |
 | −338.61 (ΔE) −259.83 (ΔG) −324.59 (ΔH) |
 | −348.32 (ΔE) −269.03 (ΔG) −333.13 (ΔH) |
 | −310.62 (ΔE) −286.65 (ΔG) −301.33 (ΔH) |
| Reaction | M06-2X/cc-PVTZ(-f)++ in Implicit Water (kJ/mol) |
|---|---|
 | −44.22 (ΔE) −49.20 (ΔG) −47.91 (ΔH) |
 | −58.32 (ΔE) −61.17 (ΔG) −62.63 (ΔH) |
 | −54.60 (ΔE) −58.58 (ΔG) −58.99 (ΔH) |
 | −54.06 (ΔE) −58.62 (ΔG) −57.95 (ΔH) |
 | −61.09 (ΔE) −61.13 (ΔG) −63.76 (ΔH) |
 | −98.49 (ΔE) −41.25 (ΔG) −91.09 (ΔH) |
 | −105.69 (ΔE) −47.24 (ΔG) −97.15 (ΔH) |
 | −105.35 (ΔE) −47.66 (ΔG) −97.15 (ΔH) |
 | −104.56 (ΔE) −43.26 (ΔG) −98.28 (ΔH) |
 | −40.67 (ΔE) −46.90 (ΔG) −43.76 (ΔH) |
 | −46.82 (ΔE) −50.50 (ΔG) −52.00 (ΔH) |
 | −28.70 (ΔE) −33.47 (ΔG) −28.83 (ΔH) |
 | −53.76 (ΔE) −63.93 (ΔG) −50.42 (ΔH) |
 | −60.50 (ΔE) −65.19 (ΔG) −63.89 (ΔH) |
 |  |  |
| −28.83 (ΔE); −31.30 (ΔG); −31.09 (ΔH) | −33.39 (ΔE); −36.86 (ΔG); −35.73 (ΔH) | −29.41 (ΔE); −32.84 (ΔG); −31.71 (ΔH) |
 |  |  |
| −40.79 (ΔE); −43.39 (ΔG); −42.84 (ΔH) | −37.95 (ΔE); −41.17 (ΔG); −40.00 (ΔH) | −43.05 (ΔE); −46.07 (ΔG); −45.56 (ΔH) |
 |  |  |
| −45.65 (ΔE); −49.58 (ΔG); −45.69 (ΔH) | −72.43 (ΔE); −78.95 (ΔG); −75.19 (ΔH) | −38.07 (ΔE); −41.67 (ΔG); −39.71 (ΔH) |
 |  |  |
| −21.76 (ΔE); −25.06 (ΔG); −22.59 (ΔH) | −37.91 (ΔE); −41.51 (ΔG); −41.67 (ΔH) | −40.12 (ΔE); −43.30 (ΔG); −42.97 (ΔH) |
 |  |  |
| −61.59 (ΔE); −67.28 (ΔG); −62.93 (ΔH) | −36.78 (ΔE); −38.20 (ΔG); −39.66 (ΔH) | −32.51 (ΔE); −36.15 (ΔG); −33.43 (ΔH) |
 |  |  |
| −52.93 (ΔE); −55.52 (ΔG); −53.26 (ΔH) | −35.27 (ΔE); −35.31 (ΔG); −37.07 (ΔH) | −71.55 (ΔE); −74.73 (ΔG); −74.73 (ΔH) |
 |  |  |
| −59.75 (ΔE); −59.75 (ΔG); −60.71 (ΔH) | −38.49 (ΔE); −42.01 (ΔG); −37.95 (ΔH) | −30.67 (ΔE); −35.77 (ΔG); −30.46 (ΔH) |
 |  |  |
| −34.39 (ΔE); −37.66 (ΔG); −34.77 (ΔH) | −50.96 (ΔE); −60.00 (ΔG); −54.89 (ΔH) | −46.32 (ΔE); −55.94 (ΔG); −49.79 (ΔH) |
 |  |  |
| −23.43 (ΔE); −26.15 (ΔG); −23.81 (ΔH) | −21.30 (ΔE); −23.35 (ΔG); −21.59 (ΔH) | −36.19 (ΔE); −39.41 (ΔG); −36.57 (ΔH) |
 |  |  |
| −27.15 (ΔE); −30.84 (ΔG); −29.54 (ΔH) | −52.30 (ΔE); −55.10 (ΔG); −55.27 (ΔH) | −34.27 (ΔE); −37.45 (ΔG); −36.48 (ΔH) |
 | ||
| −53.26 (ΔE); −55.27 (ΔG); −56.23 (ΔH) |
| Reaction | M06-2X/cc-PVTZ(-f)++ in Implicit Water (kJ/mol) |
|---|---|
 | −73.76 (ΔE) −75.77 (ΔG) −72.80 (ΔH) |
 | −60.17 (ΔE) −63.76 (ΔG) −59.33 (ΔH) |
 | −74.98 (ΔE) −86.23 (ΔG) −78.74 (ΔH) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopandic, M.; Merza, F.; Honek, J.F. Thermodynamic Overview of Bioconjugation Reactions Pertinent to Lysine and Cysteine Peptide and Protein Residues. Compounds 2023, 3, 464-503. https://doi.org/10.3390/compounds3030035
Lopandic M, Merza F, Honek JF. Thermodynamic Overview of Bioconjugation Reactions Pertinent to Lysine and Cysteine Peptide and Protein Residues. Compounds. 2023; 3(3):464-503. https://doi.org/10.3390/compounds3030035
Chicago/Turabian StyleLopandic, Maja, Fatima Merza, and John F. Honek. 2023. "Thermodynamic Overview of Bioconjugation Reactions Pertinent to Lysine and Cysteine Peptide and Protein Residues" Compounds 3, no. 3: 464-503. https://doi.org/10.3390/compounds3030035
APA StyleLopandic, M., Merza, F., & Honek, J. F. (2023). Thermodynamic Overview of Bioconjugation Reactions Pertinent to Lysine and Cysteine Peptide and Protein Residues. Compounds, 3(3), 464-503. https://doi.org/10.3390/compounds3030035