Abstract
Background: Colorectal cancer (CRC) is the third most diagnosed cancer globally and the second leading cause of cancer-related deaths. Despite advancements, metastatic CRC (mCRC) has a five-year survival rate below 20%. Next-generation sequencing (NGS) is recommended nowadays to guide mCRC treatment; however, its clinical utility when compared with traditional molecular testing in mCRC is debated due to limited survival improvement and cost-effectiveness concerns. Methods: This retrospective study included mCRC patients (≥18 years) treated at a single oncology centre who underwent NGS during treatment planning. Tumour samples were analysed using either a 52-gene Oncomine™ Focus Assay or a 500+-gene Oncomine™ Comprehensive Assay Plus. Variants were classified by clinical significance (ESMO ESCAT) and potential benefit (ESMO-MCBS and OncoKBTM). The Mann–Whitney and Chi square tests were used to compare characteristics of different groups, with significance at p < 0.05. Results: Eighty-six metastatic colorectal cancer (mCRC) patients were analysed, all being MMR proficient. Most cases (73.3%) underwent sequencing at diagnosis of metastatic disease, using primary tumour samples (74.4%) and a focused NGS assay (75.6%). A total of 206 somatic variants were detected in 86.0% of patients, 31.1% of which were classified as clinically significant, predominantly KRAS mutations (76.6%), with G12D and G12V variants as the most frequent. Among 33.7% RAS/BRAF wild-type patients, 65.5% received anti-EGFR therapies. Eleven patients (12.8%) had other actionable variants which were ESCAT level I-II, including four identified as TMB-high, four KRAS G12C, two BRAF V600E, and one HER2 amplification. Four received therapies classified as OncoKbTM level 1–2 and ESMO-MCBS score 4, leading to disease control in three cases. Conclusions: NGS enables the detection of rare variants, supports personalised treatments, and expands therapeutic options. As new drugs emerge and genomic data integration improves, NGS is poised to enhance real-world mCRC management.
1. Introduction
Colorectal cancer is the third most commonly diagnosed cancer globally and the second leading cause of cancer-related mortality [1]. Despite significant advancements in treatment over the past decade, 20% of patients are diagnosed with upfront metastatic colorectal cancer (mCRC), and up to 40% of those initially treated for localised disease will eventually develop metastatic disease. Despite recent improvements in systemic therapies for mCRC, the five-year survival rate remains below 20% [2,3]. Prognosis and treatment approaches for mCRC are heavily influenced by clinical and pathological factors, including tumour location (right-sided or left-sided colon) and the presence of RAS or BRAF variants [3].
Currently, in terms of molecular profiling, the first-line treatment for mCRC patients requires only the determination of RAS/BRAF mutational status and microsatellite instability (MSI) status [4]. Testing for KRAS and NRAS variants at codons 12, 13, 61, 117, and 146, as well as BRAF variants at codon 600, is considered the standard of care [5]. However, for these studies, many experts and professional societies recommend multigene next-generation sequencing (NGS) testing at the time of mCRC diagnosis, especially if it incurs no additional cost compared with traditional gene-directed polymerase chain reaction (PCR) techniques [6,7,8]. Larger NGS panels allow not only the calculation of tumour mutational burden (TMB) and determination of MSI status but also the detection of HER2 amplifications and potentially rare actionable gene variants [5,9].
The Princess Margaret IMPACT/COMPACT multitumour trial demonstrated that patients undergoing NGS and enrolled in genotype-matched clinical trials had a 10% higher treatment response rate compared with those included in genotype-unmatched trials [10]. Subsequently, the OCTANE study, which enrolled over 4500 patients, found that NGS results altered drug treatment in 15.7% of cases; however, there was no observed difference in overall survival for patients receiving genotype-matched therapies [11]. Specifically for colorectal cancer, patients who underwent NGS in the OCTANE study experienced higher general health-associated costs and were more likely to receive supportive care rather than inclusion in clinical trials [12].
Although broad genomic analyses are uncovering the genetic landscape of colorectal cancer and highlighting the prognostic and clinical implications of various gene signatures, the methodologies employed in these studies often diverge from what is applicable in routine clinical practice [7,13]. Some cohorts have used more targeted sequencing approaches to analyse real-world patients, but these efforts are typically retrospective or reliant on surgical samples, failing to demonstrate the real-world impact of NGS on treatment selection [14,15].
Up to 53% of colorectal cancer patients harbour KRAS/NRAS variants. BRAF V600E variants are present in approximately 8.5% of patients, and these hotspot mutations can be reliably detected using more traditional techniques, like PCR, faster and with high sensitivity, when compared with NGS [6,16,17]. Similarly, high levels of MSI (MSI-high) are found in 8.5% of patients and can be identified through an immunohistochemical surrogate biomarker, namely mismatch repair (MMR) protein expression, and even PCR platforms [4,6,18]. Thus, beyond the RAS/BRAF status and MSI-high findings, other actionable variants are rare but detectable by NGS [6]. Given the uncertain cost-effectiveness and real-world implications of NGS in mCRC, our aim is to explore the clinical impact of sequencing in a real-world cohort of mCRC patients, focusing on the actionability of identified gene variants and their influence on disease management and prognosis.
2. Materials and Methods
2.1. Patient Selection and Characterisation
Metastatic CRC patients aged 18 years or older, treated at the same oncological centre, were selected for NGS analysis between 2022 and 2023. Patients with newly diagnosed and untreated mCRC were included, as well as previously treated mCRC patients requiring further therapeutic strategies as per physician evaluation. Only patients eligible for systemic or locoregional treatment were selected. Patients with multiple active cancers or missing clinical information regarding previous treatment lines or precise diagnostic dates were excluded from the analysis.
All clinical, histological, and radiological data were collected retrospectively from electronic medical records. Clinical data included the patient’s age and gender, all systemic and locoregional cancer treatments received, and the time of death. Histological data were retrieved from pathology reports, including tumour stage, anatomical location, and the origin of the sample used for sequencing (primary tumour specimen/biopsy or metastasis specimen/biopsy). Radiological data were used to determine the pattern of metastatic spread at mCRC diagnosis, response to treatments, and the time of disease progression.
2.2. Molecular Studies
Each tumour was sequenced once, using the most recent biological tumour sample available at the time of NGS prescription. Surgical samples and biopsies of either the primary tumour or metastatic tissue were utilised, as re-biopsy in real-world patients is often impractical or unfeasible.
Two sequencing platforms were used, chosen by the physician based on patient evaluation: (1) a focused assay, the Oncomine™ Focus Assay (Thermo Fisher Scientific, Waltham, MA, USA), enabling DNA and RNA analysis across 52 genes; and (2) a comprehensive assay, the Oncomine™ Comprehensive Assay Plus (Thermo Fisher Scientific, Waltham, MA, USA), for DNA analysis of over 500 genes, coupled with the Oncomine™ Focus Assay for RNA analysis. Both assays detect single-nucleotide variants (SNVs), insertions and deletions (indels), copy number variations (CNVs), and gene fusions. The comprehensive assay also enables tumour mutational burden (TMB) determination. The only non-overlapping genes between the two assays were ERG, ETV1, ETV4, ETV5, and PPARG, which were exclusive to the focused assay. The characteristics of both assays are detailed in Table S1.
MSI status was assessed using a surrogate marker, namely mismatch repair (MMR) protein expression. This was performed using an immunohistochemical panel of four antibodies: MLH1, MLH2, PMS2, and MSH6. TMB was classified as high if greater than 10 mutations per megabase (mut/Mb) [19].
2.3. Variant and Targeted Treatment Classification
Variants were classified using three approaches. Firstly, variants were categorised according to the Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists as variants of clinical significance (CSVs) or variants of uncertain clinical significance (VUSs) [20]. Benign variants were not reported.
The clinical actionability of variants was further classified using the European Society for Medical Oncology (ESMO) Scale for Clinical Actionability of Molecular Targets (ESCAT) into tiers. The ESCAT scale defines six levels of clinical evidence for molecular targets: tiers I to V and X (lack of evidence). Tier I represents the highest level of clinical evidence [21]. In this study, only ESCAT tier I/II molecular alterations were considered, as the use of tier III/IV alterations is not recommended by ESMO in clinical practice [6]. Definitions of ESCAT tiers and the variants within each category are detailed in Table S2.
The potential clinical benefit of targeted therapies was evaluated using the ESMO Magnitude of Clinical Benefit Scale (ESMO-MCBS) for the non-curative setting, ranging from level 1 to 5, and the OncoKB™ system, which includes levels 1 to 4 and level R (resistance). Level 5 in ESMO-MCBS denotes the highest clinical benefit, while level 1 in OncoKB™ represents the strongest evidence for therapeutic efficacy [22,23,24]. Variant classifications according to OncoKB™ can be accessed at https://www.oncokb.org/actionable-genes, acessed on 30 September 2024.
2.4. Statistical Considerations
Sample characteristics were summarised using descriptive statistics. The Mann–Whitney test was employed to compare median TMB values between groups with different clinical characteristics. The chi-square test was used to evaluate the influence of clinical factors on the capability of NGS to detect genetic variants. Stratification factors included tumour stage, primary tumour location, prior treatments, the biological sample used for sequencing, the sequencing panel employed, and genetic variants detected. Results were considered statistically significant at p < 0.05.
Overall survival (OS) was defined as the time from mCRC diagnosis to death. Progression-free survival (PFS) was defined as the interval between the first treatment cycle and clinical or radiological disease progression.
3. Results
3.1. Sample Characteristics
A total of 86 patients were included, 66.3% of whom were male. Of these, 57% had colon cancer and 43% had rectal cancer, with the majority presenting with left-sided disease (83.7%). Sequencing was performed predominantly at the time of metastatic disease diagnosis (73.3%), utilising biological material from the primary tumour (74.4%) and a focused NGS assay (75.6%). Among the 23 patients who underwent NGS for previously treated mCRC, 6 were not re-biopsied, with sequencing performed on primary tumour samples. Table 1 provides baseline characteristics according to the sequencing assay performed.

Table 1.
Patients and disease characterisation by sequencing assay.
3.2. Molecular Profile and Detected Variants
Figure 1 summarises the included patients and overall results.
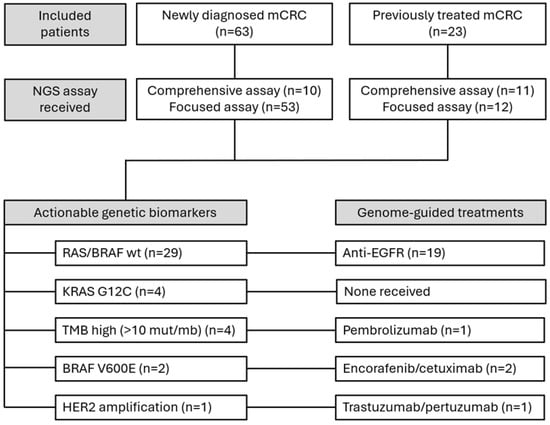
Figure 1.
Diagram illustrating the patient selection process and the main results influencing treatment strategies.
A total of 206 somatic variants were identified. Sixty-four variants (31.1%) were classified as clinically significant (CSVs). Of these, 49 occurred in KRAS (76.6%), 4 in NRAS (6.3%), and 2 in BRAF (3.1%). Additional clinically significant variants were found in PIK3CA, PTEN, HER2, and TSC2. All CSVs were single-nucleotide variants except for one HER2 amplification and two TSC2 deletions. The frequency, prevalence, and characterisation of detected variants are shown in Figure 2.
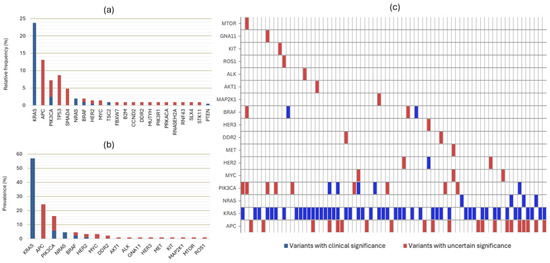
Figure 2.
(a) Relative frequency of all detected variants (n = 206). Forty-nine variants of uncertain significance (VUSs), which occurred only once in the sample and in different genes, are excluded from this graph. (b) Prevalence of variants in the 86 included patients. (c) Heatmap showing the distribution of variants based on their clinical significance for each of the 74 patients with detectable variants. Graphs (b,c) include only genes that overlap between the two NGS assays.
When considering only overlapping variants between the two assays, 116 variants were identified, of which, 61 (52.6%) were CSVs and 55 were VUSs. Three patients had two simultaneous APC VUSs, while one patient had two simultaneous PIK3CA VUSs.
Among the 49 KRAS variants, all were classified as CSVs and occurred in exons 2, 3, and 4. The most frequent variants were G12D and G12V, each accounting for 24.5%. Detailed information regarding KRAS variants is presented in Figure 3. The median allele frequency (MAF) for KRAS variants was 31% (IQR 17). The detection of RAS variants was independent of primary tumour location (p = 0.592), stage at diagnosis (p = 0.852), origin of biological material (p = 0.516), or technique used for sample collection (p = 0.209). Regarding actionable variants, KRAS G12C represented 8.2% of KRAS CSVs and was found in 4.7% of patients.
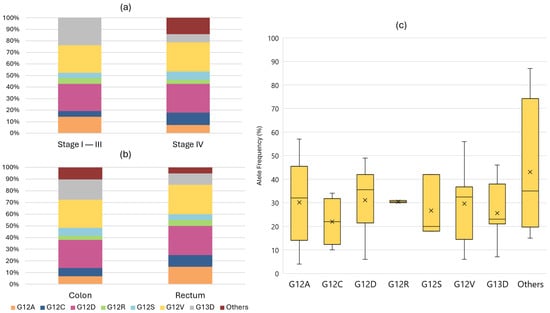
Figure 3.
Relative frequencies of each KRAS variant according to stage at diagnosis (a) or location of the primary tumour (b). Graphic (c) displays a box plot showing the distribution of the median allele frequency for each KRAS variant.
Besides KRAS, APC and PIK3CA exhibited the highest frequency of detected variants. Among the 15 PIK3CA variants, the majority were in exon 10, with 5 (33.3%) classified as CSVs. All APC variants were classified as VUSs, most of which occurred in exon 16. Two BRAF CSVs were identified as BRAF V600E, present in 2.3% of patients. Figure 4 provides details regarding the most frequently detected variants and their impact on the amino acid sequence.
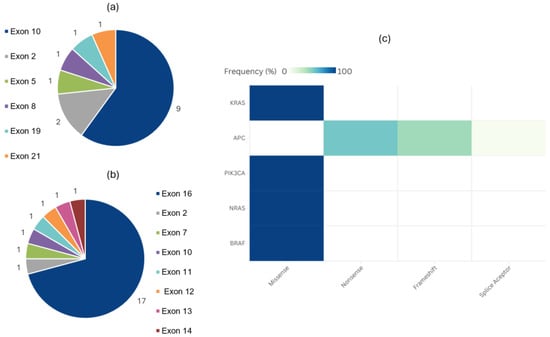
Figure 4.
Absolute frequency of PIK3CA (a) and APC (b) variants distributed by affected exon. Heatmap (c) showing relative frequency of variants, classified according to amino acid change, for each of the most commonly altered genes overlapping between the two panels.
TMB was assessable only for patients undergoing the comprehensive assay. The median TMB was 5.7 mutations/megabase (mut/Mb) (IQR 4.73). Four patients were classified as TMB-high (>10 mut/Mb) [19]. The median TMB did not differ by tumour sidedness (p = 0.153), stage at diagnosis (p = 0.382), or tissue origin (primary or metastasis) (p = 0.554).
3.3. Actionability and Therapeutic Implications
The scoring of actionable variants based on ESCAT is shown in Table 2. Twenty-nine patients (33.7%) were RAS/BRAF wild-type. Of these, 19 patients received anti-EGFR therapies (65.5%), with 17 (89.5%) receiving them as part of first-line treatment. Eight patients experienced disease progression, with a median duration of response to anti-EGFR therapies combined with fluoropyrimidine-based chemotherapy of 20.9 months (95% CI: 9.6–32.1). This combination represents therapeutic level 1 in OncoKB™ and ESMO-MCBS score 4 in the non-curative setting.
Table 2.
Clinical actionability of different tier I and II variants and molecular profiles according to the Scale for Clinical Actionability of Molecular Targets (ESCAT).
In later treatment lines, two RAS/BRAF wild-type patients received irinotecan/cetuximab in the third line and achieved partial responses, despite the absence of ESCAT or ESMO-MCBS scoring for this strategy. One patient received panitumumab monotherapy beyond the third line (ESMO-MCBS score 3) but showed progression at the first radiological evaluation. Subsequent lines of anti-EGFR therapy were guided by liquid biopsy to confirm RAS/BRAF status.
In addition to RAS/BRAF wild-type patients, 11 patients (12.8%) harboured other actionable variants, including 4 patients classified as TMB-high. During a median follow-up of 22.8 months (IQR 18.4), 4 of these 11 patients (36.4%) received targeted therapies in subsequent lines of treatment. Figure 5 provides detailed information on therapies received, treatment outcomes, and clinical benefit scores for these four patients.
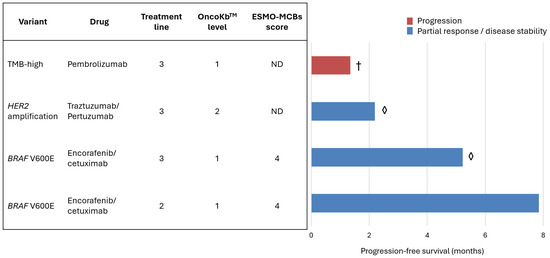
Figure 5.
The table (left) presents four patients treated with targeted therapies in subsequent lines of systemic treatment for mCRC. The variant-matched treatment is scored according to its potential clinical benefit using the ESMO Magnitude of Clinical Benefit Scale (ESMO-MCBS) in the non-curative setting and OncoKBTM therapeutic level. The bars (right) display the median progression-free survival (in months) and the best response to treatment: progression (red) or partial response/disease stability (blue). One patient died (†) and two are still on treatment (◊). ND—not defined.
Three out of four patients achieved either partial responses or stable disease with genotype-matched therapies. One patient with a BRAF V600E mutation achieved 7.8 months of progression-free survival (PFS) in the second line, while another with the same mutation remained on third-line treatment after 5.2 months of PFS. For comparison, the median PFS for the approved third-line mCRC treatment, irrespective of genotype (trifluridine–tipiracil plus bevacizumab), is 5.6 months.
As of the data cut-off, 29 patients (33.7%) had died. The median overall survival (OS) was 39.5 months (95% CI: 25.5–53.6). The median OS for patients receiving NGS-guided treatment beyond the first line (excluding RAS/BRAF wild-type patients) had not yet been reached.
3.4. Factors Impacting NGS Results
Given the retrospective nature of the study and the well-documented heterogeneity of colorectal cancer both within the tumour and across different metastatic sites, we analysed how various clinical decisions and factors may have influenced the detection of variants [26]. The origin of biological material (p = 0.556) and collection technique (p = 0.699) did not influence the ability of NGS to detect clinically significant variants. The detection of any variant was influenced by the sequencing platform used (p = 0.034), though the detection of pathogenic variants was not (p = 0.100).
Other clinical factors, including primary tumour location (p = 0.300), sidedness (p = 0.537), stage at diagnosis (p = 0.475), and the use of chemotherapy in the curative setting (p = 0.921), had no significant impact on the detection of pathogenic variants.
4. Discussion
Beyond the lack of real-world data on the clinical utility of NGS in metastatic colorectal cancer (mCRC), there is a need to evaluate its clinical impact across different populations. Variations in national regulations significantly influence the accessibility of NGS-directed cancer treatments [27]. In this Portuguese cohort, aside from RAS/BRAF wild-type profiles, NGS identified actionable variants or molecular characteristics in 11 out of 86 patients (12.8%), classified as ESCAT tiers 1 and 2 [21]. To date, four of these patients have accessed targeted therapies with high potential clinical benefit, classified as OncoKB™ levels 1 or 2. Among them, three achieved disease control [24].
Our study detected a slightly lower proportion of targetable alterations (classified by ESCAT) compared to recent larger real-world cohorts. Notably, our sample lacked representation of MMR-deficient cancers, and we limited the analysis to ESCAT tier 1 and 2 genomic alterations. Hypothetical targets (tier III and beyond), such as PIK3CA- and HER2-activating mutations, were excluded due to the practical difficulty of obtaining matched treatments outside clinical trials [28].
NGS enhances tumour genome characterisation, particularly with the use of comprehensive assays. In this study, 206 somatic variants were identified, including 64 classified as clinically significant variants (CSVs). The RAS mutation rate was 57%, with variants occurring in exons 2, 3, and 4, consistent with previously published data [6]. Emerging anticancer drugs targeting KRAS variants other than G12C announce an expansion in genome-targeted treatments for mCRC, further highlighting the clinical value of sequencing assays [29,30].
When considering the most important molecular alterations for treatment selection in mCRC, namely RAS, BRAF, and MSI status, conventional techniques such as immunohistochemistry and PCR demonstrate high sensitivity [16,17,31]. However, when PCR is employed for each biomarker individually, a substantial amount of specimen may be required. Newly automated and combined analyses, however, reduce both the amount of tissue needed and the turnaround time [17,18]. NGS, on the other hand, is associated with higher costs and longer turnaround times compared with the one-time use of conventional techniques [8]. While these traditional techniques were not redundantly applied in this study for comparison, NGS has already demonstrated the ability to detect other rare targetable variants, rare RAS mutations, and other anti-EGFR resistance-related variants in mCRC, providing a more complete platform when choosing the first-line treatment in mCRC [27]. For example, one patient (1.2%) exhibited an HER2 amplification, which may have influenced resistance to anti-EGFR therapy and also facilitated the selection of an NGS-directed treatment in a subsequent line of therapy [32,33].
Beyond rare variant detection, NGS provides new biomarkers with potential prognostic and therapeutic significance, such as median allele frequency (MAF) and tumour mutational burden (TMB). Although MAF’s prognostic value appears restricted to circulating tumour DNA (ctDNA) rather than tissue samples, the detection of low-MAF variants via NGS may enhance treatment selection, especially for RAS-mutant mCRC [34,35,36]. TMB, a predictive biomarker for immunotherapy, requires comprehensive NGS panels to be calculated [6,19]. In our cohort, only a quarter of patients underwent comprehensive assays, identifying four TMB-high cases (19%), one of whom has been treated with pembrolizumab to date [37].
The decision to perform comprehensive assays in our study was based on clinical judgement, factoring in cost and limited availability. Table 1 illustrates this bias, since comprehensive panels were more frequently offered to previously treated and younger mCRC patients. While this impacts the results of our study, it suggests that more patients with targetable biomarkers, such as TMB-high status, could have been identified if broader sequencing was offered to more patients.
This study has several limitations, including its short follow-up period and small sample size. The use of two different NGS platforms resulted in variability in genomic information between samples. While this discrepancy influenced the detection of variants overall (p = 0.034), it did not affect the identification of pathogenic variants, as the most common pathogenic mutations in mCRC are included in both panels.
Moreover, the choice of primary tumour or metastatic tissue did not impact variant detection. However, the high genetic intratumoral and inter-metastatic heterogeneity in colorectal cancer, as well as the impact of treatment on mutational profiles, are well documented [26]. These factors are particularly relevant for RAS mutations and anti-EGFR resistance variants, which are often monitored through liquid biopsies during mCRC treatment [38,39,40,41]. Thus, potential bias related to sequencing timing and tissue source cannot be excluded in our sample.
Additionally, as a retrospective real-world cohort, selection bias due to clinical decision-making likely influenced our findings. For instance, the absence of MMR-deficient cancers in our sample is likely attributable to the current standard of care, which recommends first-line immunotherapy for these patients, which does not require comprehensive molecular testing [4].
In advanced cancers, earlier studies demonstrated limited clinical benefits from comprehensive sequencing, with only a small proportion of patients deriving advantages from genome-matched therapies [5,42,43]. However, recent reviews suggest that NGS-informed treatments may provide benefits across all tumour types, including mCRC [44]. While much of this evidence is still largely based on small cohorts and expert opinions [6,45,46], NGS—particularly smaller targeted platforms—is emerging as a cost-effective method for therapeutic selection when compared with single-gene testing. It also expands treatment options and increases inclusion in clinical trials [8,47]. Therefore, the clinical relevance of NGS in mCRC is anticipated to grow over the coming years, driven by decreasing costs of NGS and the increasing availability of targeted treatments [8].
Future directions for this study should include a longer follow-up and an expanded cohort size to better elucidate the real-world benefits of routine NGS and NGS-matched therapies in advanced mCRC. These efforts could provide valuable insights into optimising the clinical application of NGS in this setting.
5. Conclusions
Our study demonstrates that standard NGS testing is both feasible and effective in real-world settings, enabling a more personalised therapeutic approach and expanding access to additional lines of treatment. The significance of sequencing as a biomarker, along with its clinical benefit, is expected to grow in the future as new drugs are developed and greater genomic data are gathered and analysed in molecular tumour boards. This will help generate real-world evidence to support the utility of NGS-directed treatments.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/dna5010004/s1. Table S1: Characteristics of the sequencing assays used with overlapping genes marked in bold; Table S2: Classification of relevant variants in colorectal cancer according to the European Society for Medical Oncology (ESMO) Scale for Clinical Actionability of Molecular Targets (ESCAT).
Author Contributions
Conceptualization, R.R. and N.B.; methodology, R.R., R.S. and L.G.S.; formal analysis, R.R., R.C., I.F., G.C. and M.G.; writing—original draft preparation, R.R., R.S. and L.G.S.; writing—review and editing, R.R., T.F. and J.P.; visualisation, R.R., T.F., J.P. and N.B.; supervision, N.B.; project administration, R.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Ethics Committee of Coimbra Portuguese Institute of Oncology. The code is TI 26/2024, and the date of approval is 7 November 2024.
Informed Consent Statement
Patient consent was waived due to the anonymized, retrospective collection of data, the high number of included subjects, and the high mortality rate.
Data Availability Statement
Anonymized data will be available upon request to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Doleschal, B.; Petzer, A.; Rumpold, H. Current Concepts of Anti-EGFR Targeting in Metastatic Colorectal Cancer. Front. Oncol. 2022, 12, 1048166. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, A.; Adam, R.; Roselló, S.; Arnold, D.; Normanno, N.; Taïeb, J.; Seligmann, J.; De Baere, T.; Osterlund, P.; Yoshino, T.; et al. Metastatic Colorectal Cancer: ESMO Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2023, 34, 10–32. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Chee, C.E.; Wong, W.; Lam, R.C.T.; Tan, I.B.H.; Ma, B.B.Y. Current Advances in Targeted Therapy for Metastatic Colorectal Cancer—Clinical Translation and Future Directions. Cancer Treat. Rev. 2024, 125, 102700. [Google Scholar] [CrossRef] [PubMed]
- Mosele, M.F.; Westphalen, C.B.; Stenzinger, A.; Barlesi, F.; Bayle, A.; Bièche, I.; Bonastre, J.; Castro, E.; Dienstmann, R.; Krämer, A.; et al. Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Advanced Cancer in 2024: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2024, 35, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Cornish, A.J.; Gruber, A.J.; Kinnersley, B.; Chubb, D.; Frangou, A.; Caravagna, G.; Noyvert, B.; Lakatos, E.; Wood, H.M.; Thorn, S.; et al. The Genomic Landscape of 2023 Colorectal Cancers. Nature 2024, 633, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.; Goerke, L.; Anderson, A.; Wilsdon, T. Assessing the Cost-Effectiveness of Next-Generation Sequencing as a Biomarker Testing Approach in Oncology and Policy Implications: A Literature Review. Value Health 2024, 27, 1300–1309. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network Colon Cancer (Version 5.2024). Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1428 (accessed on 30 September 2024).
- Stockley, T.L.; Oza, A.M.; Berman, H.K.; Leighl, N.B.; Knox, J.J.; Shepherd, F.A.; Chen, E.X.; Krzyzanowska, M.K.; Dhani, N.; Joshua, A.M.; et al. Molecular Profiling of Advanced Solid Tumors and Patient Outcomes with Genotype-Matched Clinical Trials: The Princess Margaret IMPACT/COMPACT Trial. Genome Med. 2016, 8, 109. [Google Scholar] [CrossRef]
- Bogdan, L.; Saleh, R.R.; Avery, L.; Del Rossi, S.; Yu, C.; Bedard, P.L. Clinical Utility of Tumor Next-Generation Sequencing Panel Testing to Inform Treatment Decisions for Patients with Advanced Solid Tumors in a Tertiary Care Center. JCO Precis. Oncol. 2024, 8, e2400092. [Google Scholar] [CrossRef]
- Hernando-Calvo, A.; Nguyen, P.; Bedard, P.L.; Chan, K.K.W.; Saleh, R.R.; Weymann, D.; Yu, C.; Amir, E.; Regier, D.A.; Gyawali, B.; et al. Impact on Costs and Outcomes of Multi-Gene Panel Testing for Advanced Solid Malignancies: A Cost-Consequence Analysis Using Linked Administrative Data. EClinicalMedicine 2024, 69, 102443. [Google Scholar] [CrossRef] [PubMed]
- Nunes, L.; Li, F.; Wu, M.; Luo, T.; Hammarström, K.; Torell, E.; Ljuslinder, I.; Mezheyeuski, A.; Edqvist, P.-H.; Löfgren-Burström, A.; et al. Prognostic Genome and Transcriptome Signatures in Colorectal Cancers. Nature 2024, 633, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.H.; Yu, G.Y.; Hong, Y.G.; Lian, W.; Chouhan, H.; Xu, Y.; Liu, L.J.; Bai, C.G.; Zhang, W. Clinical Significance of Multiple Gene Detection with a 22-Gene Panel in Formalin-Fixed Paraffin-Embedded Specimens of 207 Colorectal Cancer Patients. Int. J. Clin. Oncol. 2019, 24, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Song, I.H.; Lee, A.; Kang, J.; Lee, Y.S.; Lee, I.K.; Song, Y.S.; Lee, S.H. Enhancing the Landscape of Colorectal Cancer Using Targeted Deep Sequencing. Sci. Rep. 2021, 11, 8154. [Google Scholar] [CrossRef]
- Su, W.-C.; Tsai, Y.-C.; Tsai, H.-L.; Chang, T.-K.; Yin, T.-C.; Huang, C.-W.; Chen, Y.-C.; Li, C.-C.; Chen, P.-J.; Liu, Y.-R.; et al. Comparison of Next-Generation Sequencing and Polymerase Chain Reaction for Personalized Treatment-Related Genomic Status in Patients with Metastatic Colorectal Cancer. Curr. Issues Mol. Biol. 2022, 44, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Zekri, J.; Baghdadi, M.A.; Alardati, H.; Khallaf, H.; Kabanja, J.H. Evaluation of the Idylla KRAS and NRAS Mutation Test in Colorectal Cancer Tissue. Exp. Mol. Pathol. 2019, 110, 104270. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, J.; Li, L.; Mu, X.; Wang, Y.; Li, X. Evaluation of a Fully Automated Idylla Test System for Microsatellite Instability in Colorectal Cancer. Clin. Color. Cancer 2019, 18, e316–e323. [Google Scholar] [CrossRef]
- Mo, S.-F.; Cai, Z.-Z.; Kuai, W.-H.; Li, X.; Chen, Y.-T. Universal Cutoff for Tumor Mutational Burden in Predicting the Efficacy of Anti-PD-(L)1 Therapy for Advanced Cancers. Front. Cell Dev. Biol. 2023, 11, 1209243. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.K.Y.; Bedard, P.L.; Tortora, G.; Douillard, J.-Y.; et al. A Framework to Rank Genomic Alterations as Targets for Cancer Precision Medicine: The ESMO Scale for Clinical Actionability of Molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the Expanding Landscape of Clinical Actionability for Patients with Cancer. Cancer Discov. 2024, 14, 49–65. [Google Scholar] [CrossRef]
- Cherny, N.I.; Dafni, U.; Bogaerts, J.; Latino, N.J.; Pentheroudakis, G.; Douillard, J.-Y.; Tabernero, J.; Zielinski, C.; Piccart, M.J.; de Vries, E.G.E. ESMO-Magnitude of Clinical Benefit Scale Version 1.1. Ann. Oncol. 2017, 28, 2340–2366. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef] [PubMed]
- Weiser, M.R. AJCC 8th Edition: Colorectal Cancer. Ann. Surg. Oncol. 2018, 25, 1454–1455. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Genetic Alterations of Metastatic Colorectal Cancer. Biomedicines 2020, 8, 414. [Google Scholar] [CrossRef]
- Bayle, A.; Basile, D.; Garinet, S.; Rance, B.; Laurent-Puig, P.; Blons, H.; Taieb, J.; Perkins, G. Next-Generation Sequencing Targeted Panel in Routine Care for Metastatic Colon Cancers. Cancers 2021, 13, 5750. [Google Scholar] [CrossRef] [PubMed]
- Mulet Margalef, N.; Castillo, C.; Mosteiro, M.; Pérez, X.; Aguilar, S.; Ruíz-Pace, F.; Gil, M.; Cuadra, C.; Ruffinelli, J.C.; Martínez, M.; et al. Genomically Matched Therapy in Refractory Colorectal Cancer According to ESMO Scale for Clinical Actionability of Molecular Targets: Experience of a Comprehensive Cancer Centre Network. Mol. Oncol. 2023, 17, 1908–1916. [Google Scholar] [CrossRef]
- Mustachio, L.M.; Chelariu-Raicu, A.; Szekvolgyi, L.; Roszik, J. Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers 2021, 13, 1204. [Google Scholar] [CrossRef]
- Boilève, A.; Smolenschi, C.; Lambert, A.; Boige, V.; Delaye, M.; Camilleri, G.M.; Tarabay, A.; Valéry, M.; Fuerea, A.; Pudlarz, T.; et al. KRAS, a New Target for Precision Medicine in Colorectal Cancer? Cancers 2024, 16, 3455. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Frankel, W.L. A Practical Guide to Biomarkers for the Evaluation of Colorectal Cancer. Mod. Pathol. 2019, 32, 1–15. [Google Scholar] [CrossRef]
- Spiekman, I.A.C.; Zeverijn, L.J.; Geurts, B.S.; Verkerk, K.; Haj Mohammad, S.F.; van der Noort, V.; Roepman, P.; de Leng, W.W.J.; Jansen, A.M.L.; Gootjes, E.C.; et al. Trastuzumab plus Pertuzumab for HER2-Amplified Advanced Colorectal Cancer: Results from the Drug Rediscovery Protocol (DRUP). Eur. J. Cancer 2024, 202, 113988. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus Trastuzumab for HER2-Amplified Metastatic Colorectal Cancer (MyPathway): An Updated Report from a Multicentre, Open-Label, Phase 2a, Multiple Basket Study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Manca, P.; Corallo, S.; Lonardi, S.; Fucà, G.; Busico, A.; Leone, A.G.; Corti, F.; Antoniotti, C.; Procaccio, L.; Smiroldo, V.; et al. Variant Allele Frequency in Baseline Circulating Tumour DNA to Measure Tumour Burden and to Stratify Outcomes in Patients with RAS Wild-Type Metastatic Colorectal Cancer: A Translational Objective of the Valentino Study. Br. J. Cancer 2022, 126, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Shen, M.-Y.; Chen, W.T.-L.; Yang, C.-A. Evaluation of the Prognostic Value of Low-Frequency KRAS Mutation Detection in Circulating Tumor DNA of Patients with Metastatic Colorectal Cancer. J. Pers. Med. 2023, 13, 1051. [Google Scholar] [CrossRef]
- Jin, K.; Bao, Q.; Zhao, T.; Wang, H.; Huang, L.; Wang, K.; Xing, B. Comparing Baseline VAF in Circulating Tumor DNA and Tumor Tissues Predicting Prognosis of Patients with Colorectal Cancer Liver Metastases after Curative Resection. Clin. Res. Hepatol. Gastroenterol. 2024, 48, 102464. [Google Scholar] [CrossRef] [PubMed]
- Duvivier, H.L.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, E.; Ahn, E.R.; Al Baghdadi, T.; Alva, A.S.; Dublis, S.A.; Cannon, T.L.; Calfa, C.J.; et al. Pembrolizumab in Patients with Tumors with High Tumor Mutational Burden: Results from the Targeted Agent and Profiling Utilization Registry Study. J. Clin. Oncol. 2023, 41, 5140–5150. [Google Scholar] [CrossRef]
- Ciardiello, D.; Martinelli, E.; Troiani, T.; Mauri, G.; Rossini, D.; Martini, G.; Napolitano, S.; Famiglietti, V.; Del Tufo, S.; Masi, G.; et al. Anti-EGFR Rechallenge in Patients with Refractory CtDNA RAS/BRAF Wt Metastatic Colorectal Cancer. JAMA Netw. Open 2024, 7, e245635. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, D.; Boscolo Bielo, L.; Napolitano, S.; Martinelli, E.; Troiani, T.; Nicastro, A.; Latiano, T.P.; Parente, P.; Maiello, E.; Avallone, A.; et al. Comprehensive Genomic Profiling by Liquid Biopsy Captures Tumor Heterogeneity and Identifies Cancer Vulnerabilities in Patients with RAS/BRAF Wild-Type Metastatic Colorectal Cancer in the CAPRI 2-GOIM Trial. Ann. Oncol. 2024, 35, 1105–1115. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Nakamura, Y.; Esaki, T.; Yasui, H.; Taniguchi, H.; Satake, H.; Sunakawa, Y.; Komatsu, Y.; Kagawa, Y.; et al. Clinical Features Associated with NeoRAS Wild-Type Metastatic Colorectal Cancer A SCRUM-Japan GOZILA Substudy. Nat. Commun. 2024, 15, 5885. [Google Scholar] [CrossRef]
- Albuquerque, J.; Neto da Silva, D.; Padrão, T.; Leal-Costa, L.; Bizarro, R.; Correia, J.; Baptista, C.; Machete, M.; Prazeres, G.; Margarido, I.; et al. Loss of RAS Mutations in Liquid Biopsies of Patients with Multi-Treated Metastatic Colorectal Cancer. Oncologist 2024, 29, e337–e344. [Google Scholar] [CrossRef]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.-C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly Targeted Therapy Based on Tumour Molecular Profiling versus Conventional Therapy for Advanced Cancer (SHIVA): A Multicentre, Open-Label, Proof-of-Concept, Randomised, Controlled Phase 2 Trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, S.N.; Peneva, D.; Cuyun Carter, G.; Palomares, M.R.; Thakkar, S.; Hall, D.W.; Dalglish, H.; Campos, C.; Yermilov, I. Comprehensive Review on the Clinical Impact of Next-Generation Sequencing Tests for the Management of Advanced Cancer. JCO Precis. Oncol. 2023, 7, e2200715. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Schwaederlé, M.C.; Fanta, P.T.; Okamura, R.; Leichman, L.; Lippman, S.M.; Lanman, R.B.; Raymond, V.M.; Talasaz, A.; Kurzrock, R. Genomic Assessment of Blood-Derived Circulating Tumor DNA in Patients with Colorectal Cancers: Correlation with Tissue Sequencing, Therapeutic Response, and Survival. JCO Precis. Oncol. 2019, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Alifrangis, C.; Chandrasinghe, P.; Cereser, B.; Del Bel Belluz, L.; Leo, C.A.; Moderau, N.; Tabassum, N.; Warusavitarne, J.; Krell, J.; et al. The Benefit of Tumor Molecular Profiling on Predicting Treatments for Colorectal Adenocarcinomas. Oncotarget 2018, 9, 11371–11376. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ortendahl, J.D.; Cuyun Carter, G.; Thakkar, S.G.; Bognar, K.; Hall, D.W.; Abdou, Y. Value of next Generation Sequencing (NGS) Testing in Advanced Cancer Patients. J. Med. Econ. 2024, 27, 519–530. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).