
Research Themes in KAT6A Syndrome: A Scoping Review


Abstract
1. Introduction
2. Materials and Methods
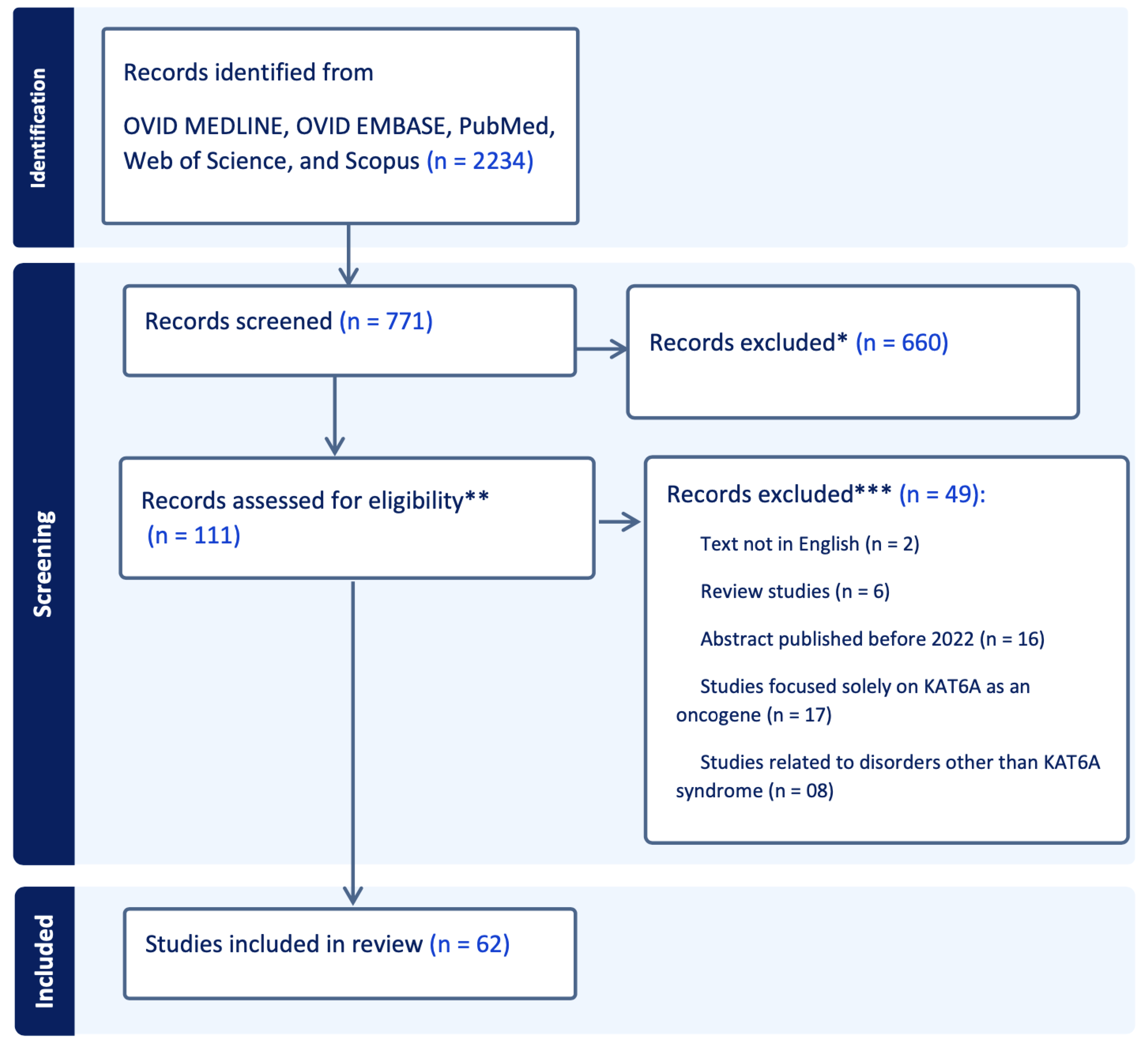
2.1. Database Search
2.2. Inclusion and Exclusion Criteria
2.3. Article Selection
2.4. Data Charting
2.5. Data Synthesis
2.6. Ethics
3. Results
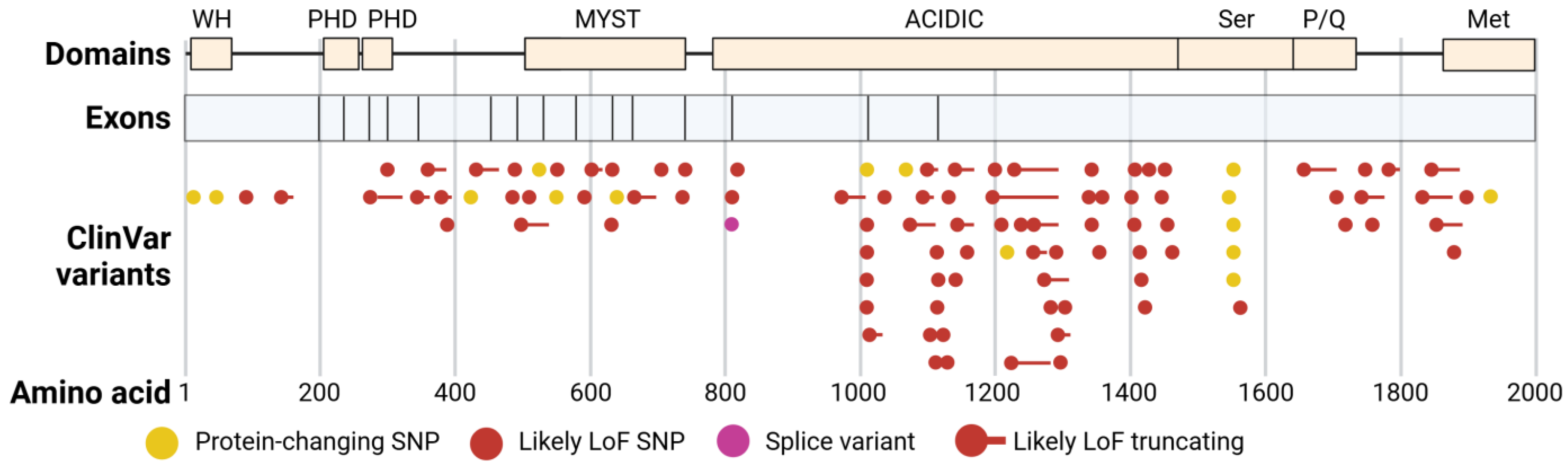
3.1. Theme 1—Genotype and Phenotype Map in KAT6A Syndrome
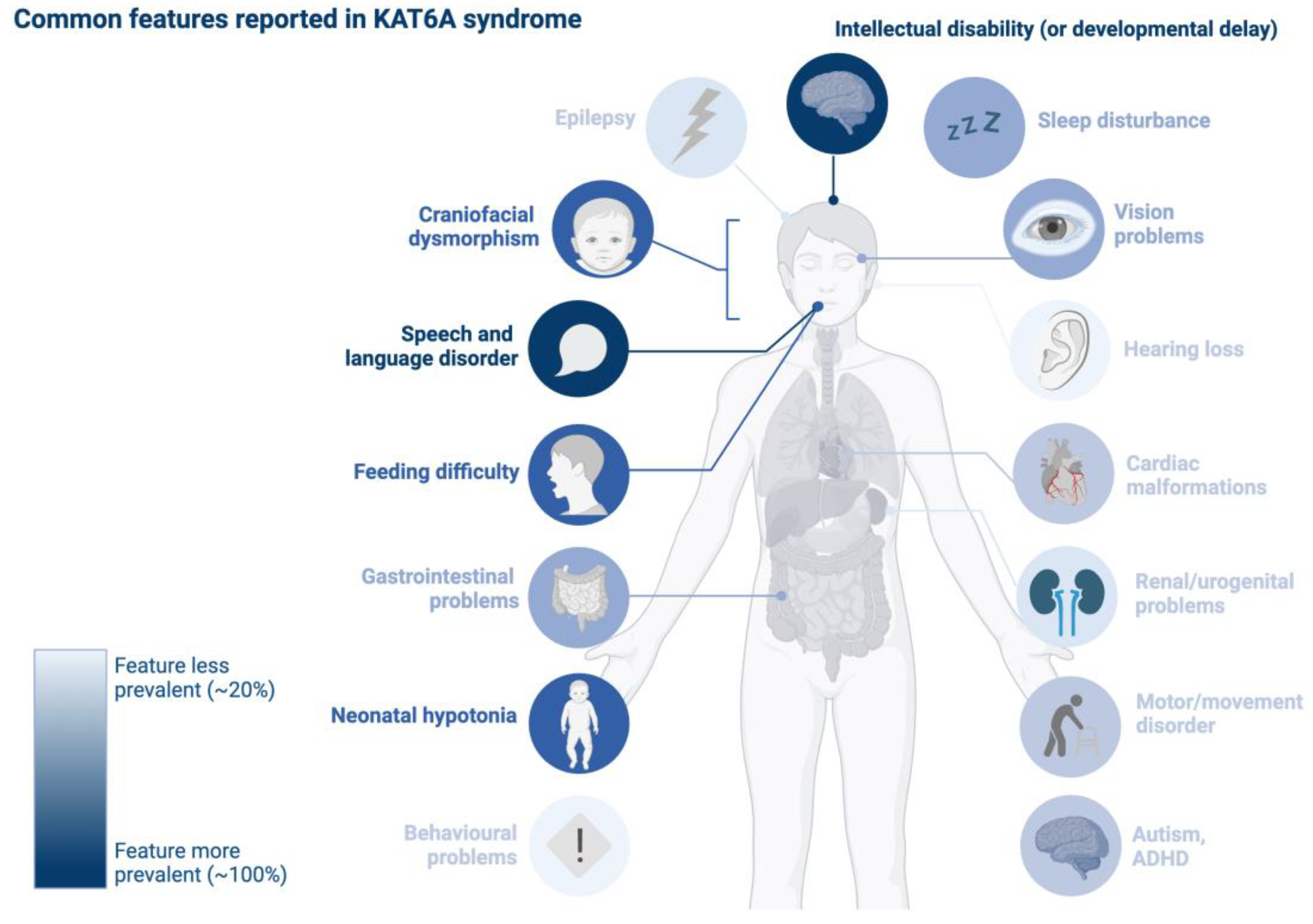
3.2. Theme 2—Neurodevelopmental Profile in KAT6A Syndrome
3.3. Theme 3—Epigenetic and Developmental Roles of KAT6A
3.4. Theme 4—Molecular Biomarkers Derived from Individuals with KAT6A Syndrome
3.5. Theme 5—Drug Discovery and Development
3.6. Theme 6—Phenotypic Overlaps Between KAT6A Syndrome and Related Disorders
4. Discussion
Limitations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAC | augmentative and alternative communication |
| CPAP | continuous positive airway pressure |
| DNA | deoxyribonucleic acid |
| Dlx | distal-less homeobox |
| HDACs | histone deacetylases |
| iPSCs | induced pluripotent stem cells |
| MDEMs | Mendelian disorders of the epigenetic machinery |
| PCDHs | protocadherins |
| RNA | ribonucleic acid |
Appendix A
- Search strategy
- Pubmed(“KAT6A”[Title/Abstract] OR ((“lysine acetyltransferase*”[Title/Abstract] OR “lysine acetyl transferase*”[Title/Abstract] OR “histone acetyltransferase*”[Title/Abstract] OR “histone acetyl transferase*”[Title/Abstract] OR “MOZ”[Title/Abstract] OR “MYST”[Title/Abstract] OR “MYST3”[Title/Abstract]) AND (“KAT”[Title/Abstract] OR “KATs”[Title/Abstract]))) AND (“NOTNLM”[All Fields] OR “publisher”[Filter] OR “inprocess”[Filter] OR “pubmednotmedline”[Filter] OR “indatareview”[Filter] OR “pubstatusaheadofprint”[All Fields]) = 341
- Scopus#1 Title/AbstractKAT6A OR OR KAT-6A#2 Title/Abstractlysine-acetyltransferase* OR lysine-acetyl-transferase* OR histone-acetyltransferase* OR histone-acetyl-transferase* OR MOZ OR MYST OR MYST3#3 Title/AbstractKAT or KATs = 2665#4 #2 AND #3 = 245#5 #1 OR #4 = 470
- Web of Science#1 Title/AbstractKAT6A OR KAT-6A = 203#2 Title/Abstractlysine-acetyltransferase* OR lysine-acetyl-transferase* OR histone-acetyltransferase* OR histone- acetyl-transferase* OR MOZ OR MYST OR MYST3 = 8927#3 Title/AbstractKAT or KATs#4 #2 AND #3#5 #1 OR #4 = 407
- Medline-Ovid(KAT6A or KAT-6A).tw,kf. = 188lysine acetyltransferases/or exp histone acetyltransferases/ = 13,692(lysine-acetyltransferase* or lysine-acetyl-transferase* or histone-acetyltransferase* or histone-acetyl-transferase* or MOZ or MYST or MYST3).tw,kf,hw. = 9411(KAT or KATs).tw,kf. = 1276(2 or 3) and 4 = 2201 or 5 = 400
- EMBASE-Ovid(KAT6A or KAT-6A).tw,kw,dq. = 347lysine acetyltransferase/ = 539histone acetyltransferase/ = 7894(lysine-acetyltransferase* or lysine-acetyl-transferase* or histone-acetyltransferase* or histone-acetyl-transferase* or MOZ or MYST or MYST3).tw,kw,dq,hw. = 11056(KAT or KATs).tw,kw,dq. = 1567(2 or 3 or 4) and 5 = 2841 or 6 = 616
References
- National Organization for Rare Disorders. Available online: https://rarediseases.org/rare-diseases/kat6a-syndrome/ (accessed on 21 January 2025).
- Wiesel-Motiuk, N.; Assaraf, Y.G. The key roles of the lysine acetyltransferases KAT6A and KAT6B in physiology and pathology. Drug Resist. Updates 2020, 53, 100729. [Google Scholar] [CrossRef]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian disorders of the epigenetic machinery: Postnatal malleability and therapeutic prospects. Hum. Mol. Genet. 2019, 28, R254–R264, Erratum in Hum. Mol. Genet. 2020, 29, 876. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, S.; Wright, J.; Voss, A.K.; Lockhart, P.J.; Amor, D.J. The Mendelian disorders of chromatin machinery: Harnessing metabolic pathways and therapies for treatment. Mol. Genet. Metab. 2024, 142, 108360. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, J.M.; Malleret, G.; Touzani, K.; Vronskaya, S.; Ishii, S.; Kandel, E.R.; Barco, A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: A model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 2004, 42, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Korzus, E.; Rosenfeld, M.G.; Mayford, M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 2004, 42, 961–972. [Google Scholar] [CrossRef]
- Benjamin, J.S.; Pilarowski, G.O.; Carosso, G.A.; Zhang, L.; Huso, D.L.; Goff, L.A.; Vernon, H.J.; Hansen, K.D.; Bjornsson, H.T. Aketogenic diet rescues hippocampal memory defects in a mouse model of Kabuki syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 125–130. [Google Scholar] [CrossRef]
- KAT6 Foundation. Available online: https://kat6a.org/about-kat6a/ (accessed on 21 January 2025).
- Zeng, F.; Yang, Y.; Xu, Z.; Wang, Z.; Ke, H.; Zhang, J.; Dong, T.; Yang, W.; Wang, J. Clinical manifestations and genetic analysis of a newborn with Arboleda-Tham syndrome. Front. Genet. 2022, 13, 990098. [Google Scholar] [CrossRef]
- Trinh, J.; Hüning, I.; Yüksel, Z.; Baalmann, N.; Imhoff, S.; Klein, C.; Rolfs, A.; Gillessen-Kaesbach, G.; Lohmann, K. A KAT6A variant in a family with autosomal dominantly inherited microcephaly and developmental delay. J. Hum. Genet. 2018, 63, 997–1001. [Google Scholar] [CrossRef]
- Covidence Systematic Review Software, Veritas Health Innovation, Melbourne, Australia. Available online: www.covidence.org (accessed on 21 January 2025).
- Tham, E.; Lindstrand, A.; Santani, A.; Malmgren, H.; Nesbitt, A.; Dubbs, H.A.; Zackai, E.H.; Parker, M.J.; Millan, F.; Rosenbaum, K.; et al. Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am. J. Hum. Genet. 2015, 96, 507–513. [Google Scholar] [CrossRef]
- Arboleda, V.A.; Lee, H.; Dorrani, N.; Zadeh, N.; Willis, M.; Macmurdo, C.F.; Manning, M.A.; Kwan, A.; Hudgins, L.; Barthelemy, F.; et al. De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. Am. J. Hum. Genet. 2015, 96, 498–506. [Google Scholar] [CrossRef]
- Millan, F.; Cho, M.T.; Retterer, K.; Monaghan, K.G.; Bai, R.; Vitazka, P.; Everman, D.B.; Smith, B.; Angle, B.; Roberts, V.; et al. Whole exome sequencing reveals de novo pathogenic variants in KAT6A as a cause of a neurodevelopmental disorder. Am. J. Med. Genet. Part A 2016, 170, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.R.; Abel, S.N.; McClure, M.B.; Foster, J., 2nd; Walke, M.I.; Jayakar, P.; Bademci, G.; Tekin, M. Novel causative variants in DYRK1A, KARS, and KAT6A associated with intellectual disability and additional phenotypic features. J. Pediatr. Genet. 2017, 6, 77–83. [Google Scholar] [CrossRef]
- Elenius, V.; Lähdesmäki, T.; Hietala, M.; Jartti, T. Food allergy in a child with de novo KAT6A mutation. Clin. Transl. Allergy 2017, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Zwaveling-Soonawala, N.; Maas, S.M.; Alders, M.; Majoie, C.B.; Fliers, E.; van Trotsenburg, A.S.P.; Hennekam, R.C.M. Variants in KAT6A and pituitary anomalies. Am. J. Med. Genet. Part A 2017, 173, 2562–2565. [Google Scholar] [CrossRef]
- Zwaveling-Soonawala, N.; Alders, M.; Jongejan, A.; Kovacic, L.; Duijkers, F.A.; Maas, S.M.; Fliers, E.; van Trotsenburg, A.S.P.; Hennekam, R.C. Clues for polygenic inheritance of pituitary stalk interruption syndrome from exome sequencing in 20 patients. J. Clin. Endocrinol. Metab. 2018, 103, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Satoh, C.; Maekawa, R.; Kinoshita, A.; Mishima, H.; Doi, M.; Miyazaki, M.; Fukuda, M.; Takahashi, H.; Kondoh, T.; Yoshiura, K.I. Three brothers with a nonsense mutation in KAT6A caused by parental germline mosaicism. Hum. Genome Var. 2017, 4, 17045. [Google Scholar] [CrossRef]
- Efthymiou, S.; Salpietro, V.; Bettencourt, C.; Houlden, H. Paroxysmal movement disorder and epilepsy caused by a de novo truncating mutation in KAT6A. J. Pediatr. Genet. 2018, 7, 114–116. [Google Scholar] [CrossRef]
- Alkhateeb, A.; Alazaizeh, W. A novel de novo frameshift mutation in KAT6A identified by whole exome sequencing. J. Pediatr. Genet. 2019, 8, 10–14. [Google Scholar]
- Urreizti, R.; Lopez-Martin, E.; Martinez-Monseny, A.; Pujadas, M.; Castilla-Vallmanya, L.; Pérez-Jurado, L.A.; Serrano, M.; Natera-de Benito, D.; Martínez-Delgado, B.; Posada-de-la-Paz, M.; et al. Five new cases of syndromic intellectual disability due to KAT6A mutations: Widening the molecular and clinical spectrum. Orphanet J. Rare Dis. 2020, 15, 44. [Google Scholar] [CrossRef]
- Lin, Y.F.; Lin, T.C.; Kirby, R.; Weng, H.Y.; Liu, Y.M.; Niu, D.M.; Tsai, S.F.; Yang, C.F. Diagnosis of Arboleda-Tham syndrome by whole genome sequencing in an Asian boy with severe developmental delay. Mol. Genet. Metab. Rep. 2020, 25, 100686. [Google Scholar] [CrossRef]
- Wang, D.; Lai, P. Global retardation and hereditary spherocytosis associated with a novel deletion of chromosome 8p11.21 encompassing KAT6A and ANK1. Eur. J. Med. Genet. 2020, 63, 104082. [Google Scholar] [CrossRef]
- Bae, S.; Yang, A.; Kim, J.; Lee, H.J.; Park, H.K. Identification of a novel KAT6A variant in an infant presenting with facial dysmorphism and developmental delay: A case report and literature review. BMC Med. Genom. 2021, 14, 297. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Yang, L.; Wu, J.; Xiong, F.; Jinrong, L. A de novo heterozygous variant in KAT6A is associated with a newly named neurodevelopmental disorder Arboleda-Tham syndrome—A case report. Transl. Pediatr. 2021, 10, 2224–4344. [Google Scholar] [CrossRef] [PubMed]
- Marji, F.P.; Hall, J.A.; Anstadt, E.; Madan-Khetarpal, S.; Goldstein, J.A.; Losee, J.E. A novel frameshift mutation in KAT6A is associated with pancraniosynostosis. J. Pediatr. Genet. 2021, 10, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Young, L.; Brooks, B.; Traboulsi, E.I. Ocular findings in a patient with KAT6A mutation. J. Pediatr. Ophthalmol. Strabismus 2021, 58, e9–e11. [Google Scholar] [CrossRef]
- Korakavi, N.; Bupp, C.; Grysko, B.; Juusola, J.; Borta, C.; Madura, C. First case of pan-suture craniosynostosis due to de novo mosaic KAT6A mutation. Child’s Nerv. Syst. 2022, 38, 173–177. [Google Scholar] [CrossRef]
- Troisi, S.; Maitz, S.; Severino, M.; Spano, A.; Cappuccio, G.; Brunetti-Pierri, N.; Torella, A.; Nigro, V.; Bilo, L.; Coppola, A. Epilepsy in KAT6A syndrome: Description of two individuals and revision of the literature. Eur. J. Med. Genet. 2022, 65, 104380. [Google Scholar] [CrossRef]
- Velinder, M.; Viskochil, D.; Palumbos, J.; Bentley, A.; Botto, L. Reanalysis of commercial exome trio data reveals a de novo loss of function variant in KAT6A. Genet. Med. 2022, 24, S170. [Google Scholar] [CrossRef]
- Wang, D.; He, J.; Li, X.; Yan, S.; Pan, L.; Wang, T.; Zhou, L.; Liu, J.; Peng, X. The clinical spectrum of a nonsense mutation in KAT6A: A case report. J. Int. Med. Res. 2022, 50, 3000605221140304. [Google Scholar] [CrossRef]
- Agarwal, U.; Lim, J.; Pottinger, C.; Suk, E.K.; Chaoui, R. Prenatal diagnosis of KAT6A syndrome in two fetuses with congenital heart disease. Ultrasound Obstet. Gynecol. 2023, 61, 114–116. [Google Scholar] [CrossRef]
- Ai, Q.; Jiang, L.; Chen, Y.; Yao, X.; Yin, J.; Chen, S. A case of KAT6A syndrome with a newly discovered mutation in the KAT6A gene, mainly manifested as bone marrow failure syndrome. Hematology 2023, 28, 2182159. [Google Scholar] [CrossRef] [PubMed]
- Bukvic, N.; Chetta, M.; Bagnulo, R.; Leotta, V.; Pantaleo, A.; Palumbo, O.; Palumbo, P.; Oro, M.; Rivieccio, M.; Laforgia, N.; et al. What have we learned from patients who have Arboleda-Tham syndrome due to a de novo KAT6A pathogenic variant with impaired histone acetyltransferase function? Genes 2023, 14, 165. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.H.; Chang, J.G. Novel de novo mutation in KAT6A gene in a child with severe aplastic anemia. Pediatr. Blood Cancer 2023, 70, e30417. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.G.; Ionescu, R.O.; Perez, C.C.; Forte, H.; Ron, A.G.; Cortes, M.F.; Gonzalez, M.N. Arboleda-Tham Syndrome: A case report. Clin. Chem. Lab. Med. 2023, 61, S1851. [Google Scholar]
- Di Caprio, A.; Rossi, C.; Bertucci, E.; Bedetti, L.; Bertoncelli, N.; Miselli, F.; Corso, L.; Bondi, C.; Iughetti, L.; Berardi, A.; et al. Fetal hepatic calcification in severe KAT6A (Arboleda-Tham) syndrome. Eur. J. Med. Genet. 2024, 67, 104906. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Li, L.; Yang, N. Diagnosis of Arboleda-Tham syndrome by whole-exome sequencing in an Asian girl with severe developmental delay. Mol. Genet. Genom. Med. 2024, 12, e2420. [Google Scholar] [CrossRef]
- Gauthier-Vasserot, A.; Thauvin-Robinet, C.; Bruel, A.L.; Duffourd, Y.; St-Onge, J.; Jouan, T.; Rivière, J.B.; Heron, D.; Donadieu, J.; Bellanné-Chantelot, C.; et al. Application of whole-exome sequencing to unravel the molecular basis of undiagnosed syndromic congenital neutropenia with intellectual disability. Am. J. Med. Genet. Part A 2017, 173, 62–71. [Google Scholar] [CrossRef]
- Duran, D.; Zeng, X.; Jin, S.C.; Choi, J.; Nelson-Williams, C.; Yatsula, B.; Gaillard, J.; Furey, C.G.; Lu, Q.; Timberlake, A.T.; et al. Mutations in chromatin modifier and ephrin signaling genes in vein of Galen malformation. Neuron 2019, 101, 429–443. [Google Scholar] [CrossRef]
- Eising, E.; Carrion-Castillo, A.; Vino, A.; Strand, E.A.; Jakielski, K.J.; Scerri, T.S.; Hildebrand, M.S.; Webster, R.; Ma, A.; Mazoyer, B.; et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol. Psychiatry 2019, 24, 1065–1078. [Google Scholar] [CrossRef]
- Kennedy, J.; Goudie, D.; Blair, E.; Chandler, K.; Joss, S.; McKay, V.; Green, A.; Armstrong, R.; Lees, M.; Kamien, B.; et al. KAT6A syndrome: Genotype-phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. 2019, 21, 850–860. [Google Scholar] [CrossRef]
- Timberlake, A.T.; Jin, S.C.; Nelson-Williams, C.; Wu, R.; Furey, C.G.; Islam, B.; Haider, S.; Loring, E.; Galm, A.; Steinbacher, D.M.; et al. Mutations in TFAP2B and previously unimplicated genes of the BMP, Wnt, and Hedgehog pathways in syndromic craniosynostosis. Proc. Natl. Acad. Sci. USA 2019, 116, 15116–15121. [Google Scholar] [CrossRef]
- Bardi, F.; Bosschieter, P.; Verheij, J.; Go, A.; Haak, M.; Bekker, M.; Sikkel, E.; Coumans, A.; Pajkrt, E.; Bilardo, C. Is there still a role for nuchal translucency measurement in the changing paradigm of first trimester screening? Prenat. Diagn. 2020, 40, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Rochtus, A.; Olson, H.E.; Smith, L.; Keith, L.G.; El Achkar, C.; Taylor, A.; Mahida, S.; Park, M.; Kelly, M.; Shain, C.; et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia 2020, 61, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Kritioti, E.; Theodosiou, A.; Parpaite, T.; Alexandrou, A.; Nicolaou, N.; Papaevripidou, I.; Séjourné, N.; Coste, B.; Christophidou-Anastasiadou, V.; Tanteles, G.A.; et al. Unravelling the genetic causes of multiple malformation syndromes: A whole exome sequencing study of the Cypriot population. PLoS ONE 2021, 16, e0253562. [Google Scholar] [CrossRef] [PubMed]
- St John, M.; Amor, D.J.; Morgan, A.T. Speech and language development and genotype–phenotype correlation in 49 individuals with KAT6A syndrome. Am. J. Med. Genet. Part A 2022, 188, 3389–3400. [Google Scholar] [CrossRef]
- Anabusi, S.; Van Mieghem, T.; Ryan, G.; Shinar, S. Elevated middle cerebral artery peak systolic velocity (MCA PSV) in fetuses with unexplained anemia. Am. J. Obstet. Gynecol. 2024, 230, S210. [Google Scholar] [CrossRef]
- Kunisetty, B.; Martin-Giacalone, B.A.; Zhao, X.; Luna, P.N.; Brooks, B.P.; Hufnagel, R.B.; Shaw, C.A.; Rosenfeld, J.A.; Agopian, A.J.; Lupo, P.J.; et al. High clinical exome sequencing diagnostic rates and novel phenotypic expansions for nonisolated microphthalmia, anophthalmia, and coloboma. Investig. Ophthalmol. Vis. Sci. 2024, 65, 25. [Google Scholar] [CrossRef]
- Ng, R.; Kalinousky, A.J.; Harris, J. Neuropsychological profile associated with KAT6A syndrome: Emergent genotype-phenotype trends. Orphanet J. Rare Dis. 2024, 19, 196. [Google Scholar] [CrossRef]
- Topa, A.; Rohlin, A.; Fehr, A.; Lovmar, L.; Stenman, G.; Tarnow, P.; Maltese, G.; Bhatti-Søfteland, M.; Kölby, L. The value of genome-wide analysis in craniosynostosis. Front. Genet. 2024, 14, 1322462. [Google Scholar] [CrossRef]
- Smith, C.; Harris, J. Sleep, behaviour, and adaptive function in KAT6A syndrome. Brain Sci. 2021, 11, 966. [Google Scholar] [CrossRef]
- Baker, E.K.; St John, M.; Hearps, S.J.C.; Amor, D.J.; Morgan, A.T. Abstracts for the 45th Human Genetics Society of Australasia Annual Scientific Meeting, Perth, Western Australia, 24–27 November 2022. Twin Res. Hum. Genet. 2023, 26, 49–126. [Google Scholar]
- Voss, A.K.; Collin, C.; Dixon, M.P.; Thomas, T. Moz and retinoic acid coordinately regulate H3K9 acetylation, Hox gene expression, and segment identity. Dev. Cell 2009, 17, 674–686. [Google Scholar] [CrossRef]
- Kong, Y.; Grimaldi, M.; Curtin, E.; Dougherty, M.; Kaufman, C.; White, R.M.; Zon, L.I.; Liao, E.C. Neural crest development and craniofacial morphogenesis is coordinated by nitric oxide and histone acetylation. Chem. Biol. 2014, 21, 488–501. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Downer, N.L.; Kueh, A.J.; Thomas, T.; Voss, A.K. Excessive versus physiologically relevant levels of retinoic acid in embryonic stem cell differentiation. Stem Cells 2014, 32, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Dreveny, I.; Deeves, S.E.; Fulton, J.; Yue, B.; Messmer, M.; Bhattacharya, A.; Collins, H.M.; Heery, D.M. The double PHD finger domain of MOZ/MYST3 induces α-helical structure of the histone H3 tail to facilitate acetylation and methylation sampling and modification. Nucleic Acids Res. 2014, 42, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Heair, H.M.; Kemper, A.G.; Roy, B.; Lopes, H.B.; Rashid, H.; Clarke, J.C.; Afreen, L.K.; Ferraz, E.P.; Kim, E.; Javed, A.; et al. MicroRNA 665 regulates dentinogenesis through microRNA-mediated silencing and epigenetic mechanisms. Mol. Cell. Biol. 2015, 35, 3116–3130. [Google Scholar] [CrossRef]
- Vanyai, H.K.; Thomas, T.; Voss, A.K. Mesodermal expression of Moz is necessary for cardiac septum development. Dev. Biol. 2015, 403, 22–29. [Google Scholar] [CrossRef]
- Han, Y.; Tanios, F.; Reeps, C.; Zhang, J.; Schwamborn, K.; Eckstein, H.H.; Zernecke, A.; Pelisek, J. Histone acetylation and histone acetyltransferases show significant alterations in human abdominal aortic aneurysm. Clin. Epigenet. 2016, 8, 3. [Google Scholar] [CrossRef]
- Vanyai, H.K.; Garnham, A.; May, R.E.; McRae, H.M.; Collin, C.; Wilcox, S.; Smyth, G.K.; Thomas, T.; Voss, A.K. MOZ directs the distal-less homeobox gene expression program during craniofacial development. Development 2019, 146, dev175042. [Google Scholar] [CrossRef]
- Nava, A.A.; Jops, C.T.; Vuong, C.K.; Niles-Jensen, S.L.; Bondhus, L.; Ong, C.J.; de la Torre-Ubieta, L.; Gandal, M.J.; Arboleda, V.A. KAT6A mutations drive transcriptional dysregulation of cell cycle and Autism risk genes in an Arboleda-Tham syndrome cerebral organoid model. bioRxiv 2023. [Google Scholar] [CrossRef]
- Singh, M.; Spendlove, S.J.; Wei, A.; Bondhus, L.M.; Nava, A.A.; de L. Vitorino, F.N.; Amano, S.; Lee, J.; Echeverria, G.; Gomez, D.; et al. KAT6A mutations in Arboleda-Tham syndrome drive epigenetic regulation of posterior HOXC cluster. Hum. Genet. 2023, 142, 1705–1720. [Google Scholar] [PubMed]
- Weber, L.M.; Jia, Y.; Stielow, B.; Gisselbrecht, S.S.; Cao, Y.; Ren, Y.; Rohner, I.; King, J.; Rothman, E.; Fischer, S.; et al. The histone acetyltransferase KAT6A is recruited to unmethylated CpG islands via a DNA binding winged helix domain. Nucleic Acids Res. 2023, 51, 574–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fan, M.; Yang, J.; Mihaljević, L.; Chen, K.H.; Ye, Y.; Sun, S.; Qiu, Z. KAT6A deficiency impairs cognitive functions through suppressing RSPO2/Wnt signaling in hippocampal CA3. Sci. Adv. 2024, 10, eadm9326. [Google Scholar] [CrossRef] [PubMed]
- Munuera-Cabeza, M.; Álvarez-Córdoba, M.; Suárez-Rivero, J.M.; Povea-Cabello, S.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Reche-López, D.; Cilleros-Holgado, P.; Piñero-Pérez, R.; et al. Pantothenate and L-Carnitine supplementation improves pathological alterations in cellular models of KAT6A syndrome. Genes 2022, 13, 2300. [Google Scholar] [CrossRef]
- Vos, N.; Reilly, J.; Elting, M.W.; Campeau, P.M.; Coman, D.; Stark, Z.; Tan, T.Y.; Amor, D.J.; Kaur, S.; StJohn, M.; et al. DNA methylation episignatures are sensitive and specific biomarkers for detection of patients with KAT6A/KAT6B variants. Epigenomics 2023, 15, 351–367. [Google Scholar] [CrossRef]
- Kaur, S.; Van Bergen, N.J.; Ben-Zeev, B.; Leonardi, E.; Tan, T.Y.; Coman, D.; Kamien, B.; White, S.M.; St John, M.; Phelan, D.; et al. Expanding the genetic landscape of Rett syndrome to include lysine acetyltransferase 6A (KAT6A). J. Genet. Genom. 2020, 47, 650–654. [Google Scholar] [CrossRef]
- Ng, R.; Kalinousky, A.; Harris, J. Expanding the neuropsychological phenotype of KAT6B disorders: Overlapping features with KAT6A syndrome. J. Autism Dev. Disord. 2024, 2, 101196. [Google Scholar]
- KAT6A/KAT6B Patient Registry. Available online: https://kat6a.iamrare.org/ (accessed on 26 January 2025).

{kind=link}
{kind=link}
{kind=link}
| Theme | Description |
|---|---|
| Genotype and phenotype map in KAT6A syndrome | This theme includes case reports, case series, and cohort studies that expand the genotype and phenotypic spectrum of KAT6A syndrome. These studies either report novel KAT6A variants and their associated clinical phenotypes, validate previously reported variants, or identify correlations between the type, location, and nature of KAT6A variants and the clinical manifestations in affected individuals. |
| Neurodevelopmental profile in KAT6A syndrome | This theme focuses on studies that investigate specific phenotypic aspects of individuals already diagnosed with KAT6A syndrome. Unlike Theme 1, which centres on identifying new variants and expanding genotype–phenotype correlations, Theme 2 delves into the detailed characterisation of particular phenotypic features, such as intellectual disability, speech and language disorders, motor development, and behavioural traits. Studies may report both strengths and deficits and explore links between specific KAT6A variants and the severity of the phenotype. The purpose is to better understand the phenotypic spectrum of KAT6A variants, to support personalised interventions. |
| Epigenetic and developmental roles of KAT6A | This theme explores how KAT6A influences gene expression through epigenetic mechanisms, such as histone acetylation. Studies focus on understanding KAT6A’s role in regulating key developmental processes, including cell differentiation, growth, and neurodevelopment. Researchers aim to uncover how variants in KAT6A disrupt these processes and contribute to developmental disorders, with a particular focus on the impact on brain development and function. |
| Molecular biomarkers derived from individuals with KAT6A syndrome | This theme includes studies aimed at identifying reliable molecular biomarkers linked to variations in KAT6A. By analysing biological samples (such as blood, tissue, or saliva) from affected individuals, the researchers aim to better understand how KAT6A variants influence molecular pathways and contribute to the clinical phenotype. |
| Drug discovery and development | This theme includes studies focused on validating potential therapeutic targets, addressing dysregulated pathways, and testing the effectiveness of compounds in correcting the effects of KAT6A variants. These include preclinical studies using animal models, cellular systems, and patient-derived samples to explore pharmacological and metabolic interventions that may mitigate the effects of KAT6A dysfunction. |
| Phenotypic overlap between KAT6A syndrome and related disorders | This theme includes studies identifying shared clinical features between KAT6A syndrome and other genetic disorders. By comparing the phenotypic and molecular characteristics of these conditions, the researchers aim to uncover shared pathways and mechanisms, which can inform differential diagnosis, therapeutic strategies, and a deeper understanding of the biology underlying these disorders. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripathi, T.; St John, M.; Wright, J.; Esber, N.; Amor, D.J. Research Themes in KAT6A Syndrome: A Scoping Review. DNA 2025, 5, 21. https://doi.org/10.3390/dna5020021
Tripathi T, St John M, Wright J, Esber N, Amor DJ. Research Themes in KAT6A Syndrome: A Scoping Review. DNA. 2025; 5(2):21. https://doi.org/10.3390/dna5020021
Chicago/Turabian StyleTripathi, Tanya, Miya St John, Jordan Wright, Natacha Esber, and David J. Amor. 2025. "Research Themes in KAT6A Syndrome: A Scoping Review" DNA 5, no. 2: 21. https://doi.org/10.3390/dna5020021
APA StyleTripathi, T., St John, M., Wright, J., Esber, N., & Amor, D. J. (2025). Research Themes in KAT6A Syndrome: A Scoping Review. DNA, 5(2), 21. https://doi.org/10.3390/dna5020021