
Hypoxia Increases Nitric Oxide-Dependent Inhibition of Angiogenic Growth


 ,
,  and
and 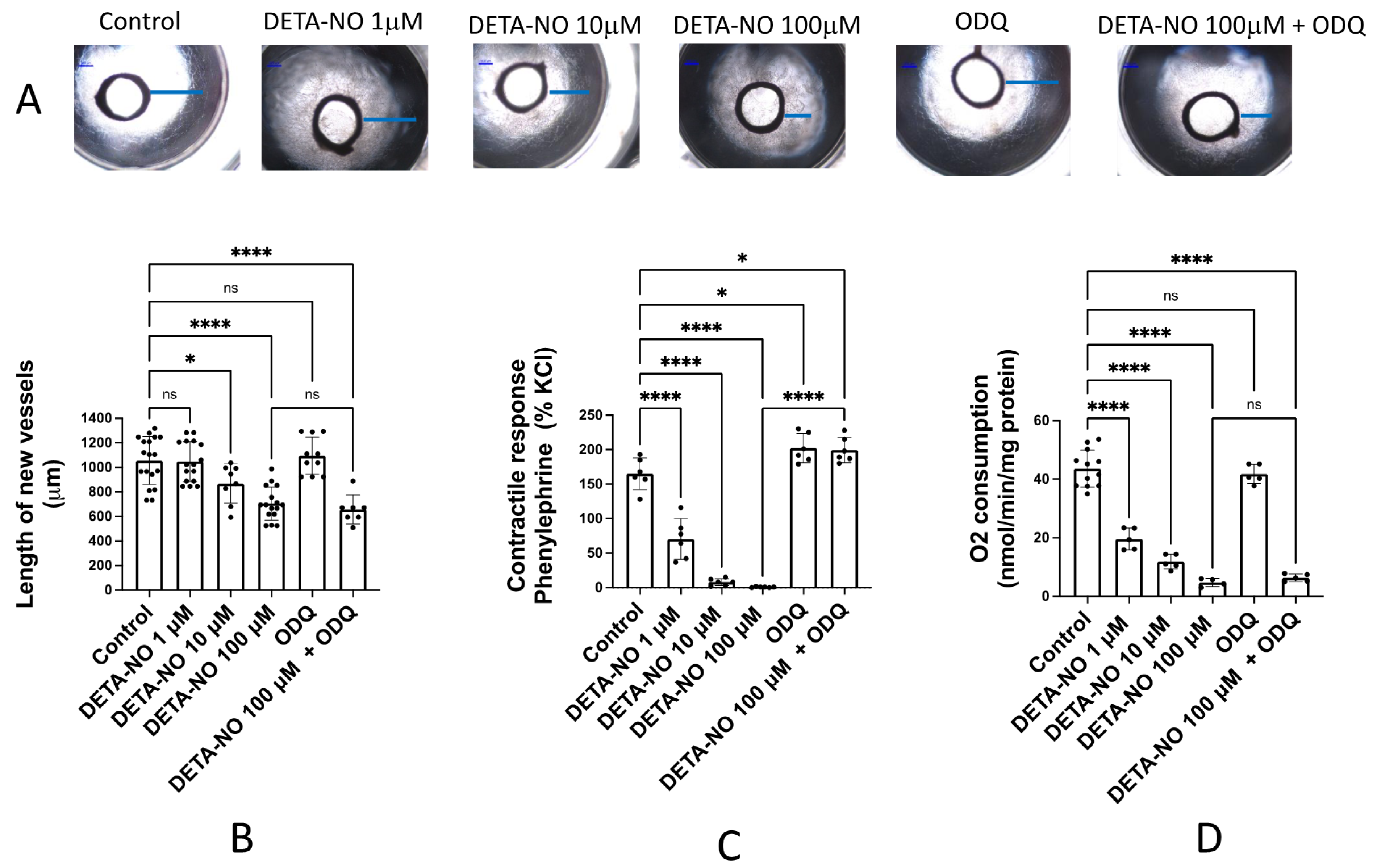{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Tissues
2.2. Arterial Ring Model of Angiogenesis
2.3. Tube Formation Assay
2.4. Functional Experiments
2.5. Oxygen Consumption Measurements
2.6. qRT-PCR
2.7. Statistical Analysis
3. Results
3.1. The NO Donor DETA-NO Inhibits Angiogenic Growth by a Mechanism Independent of sGC Activation
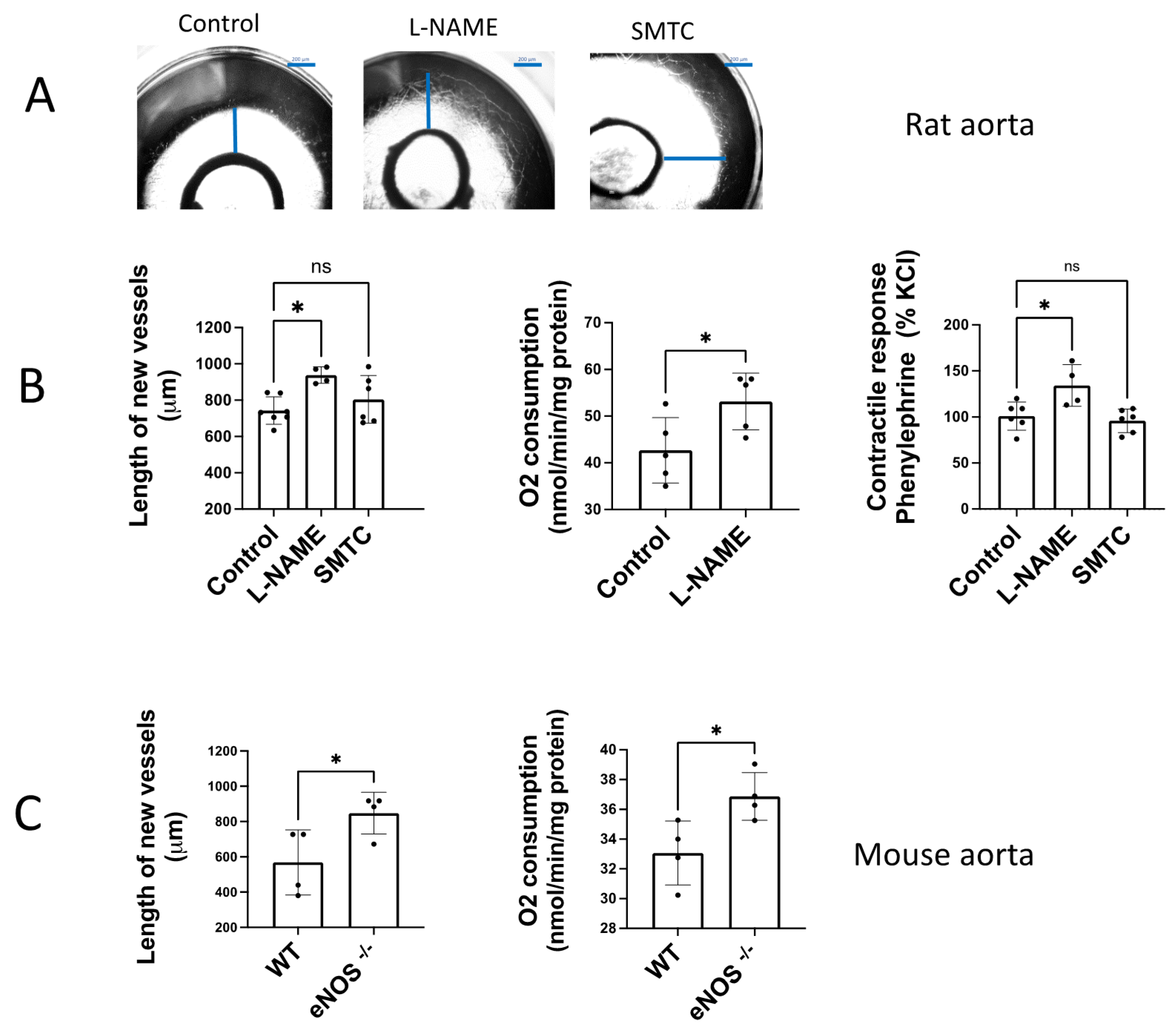
3.2. NO Endogenously Released by eNOS Inhibits Angiogenic Growth and O2 Consumption
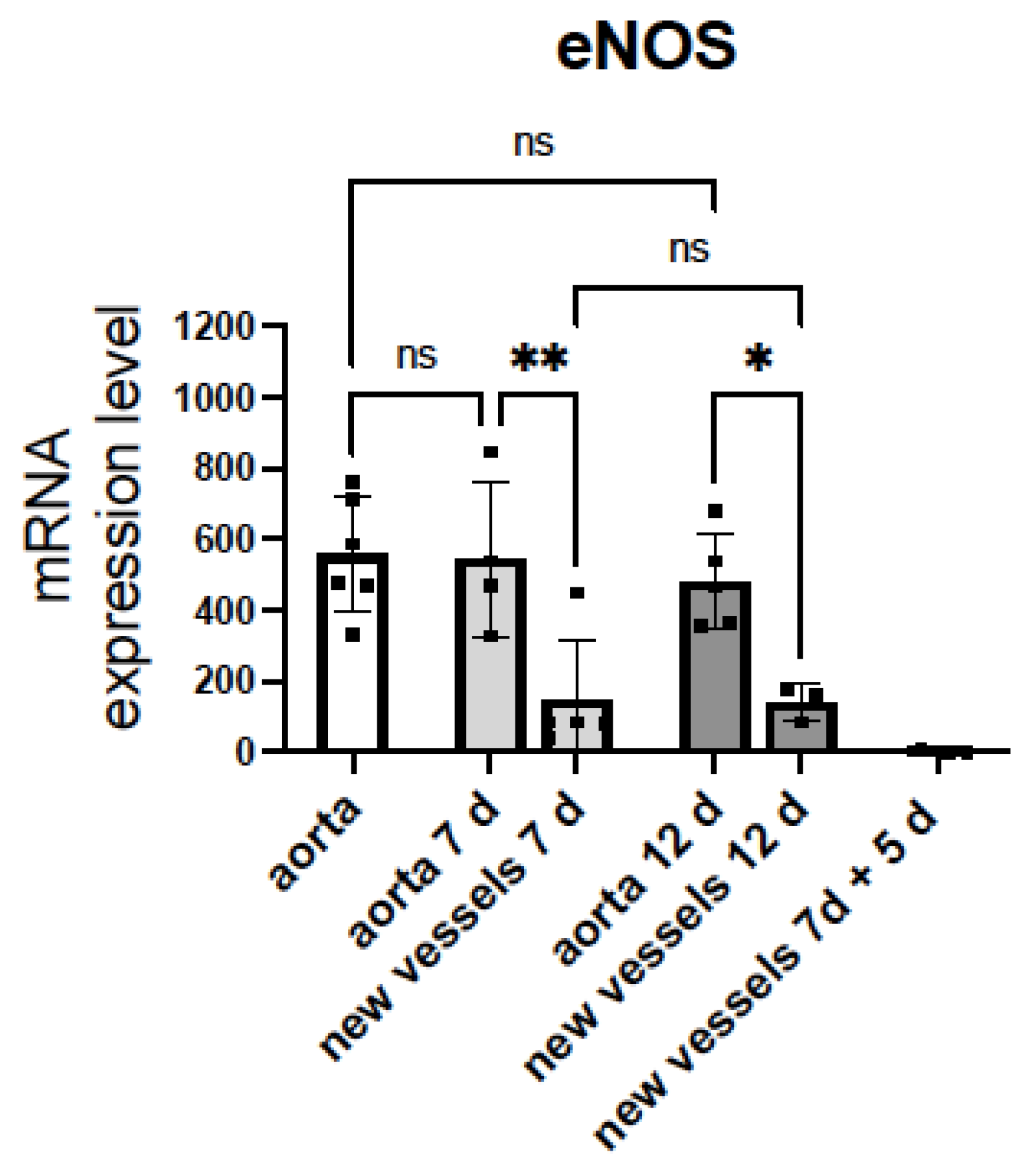
3.3. eNOS Expression Decreases in Sprouting Microvessels vs. the Aortic Ring
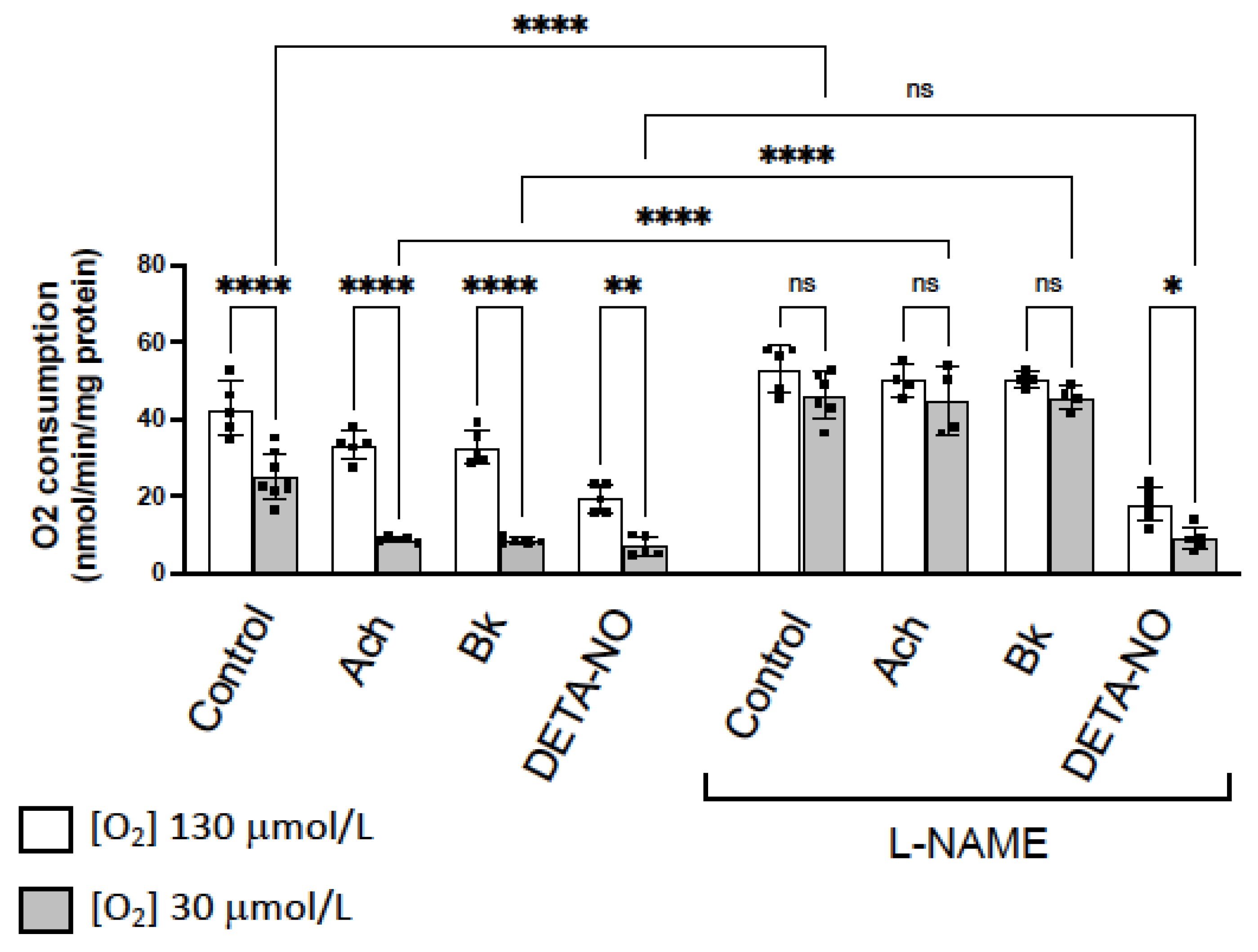
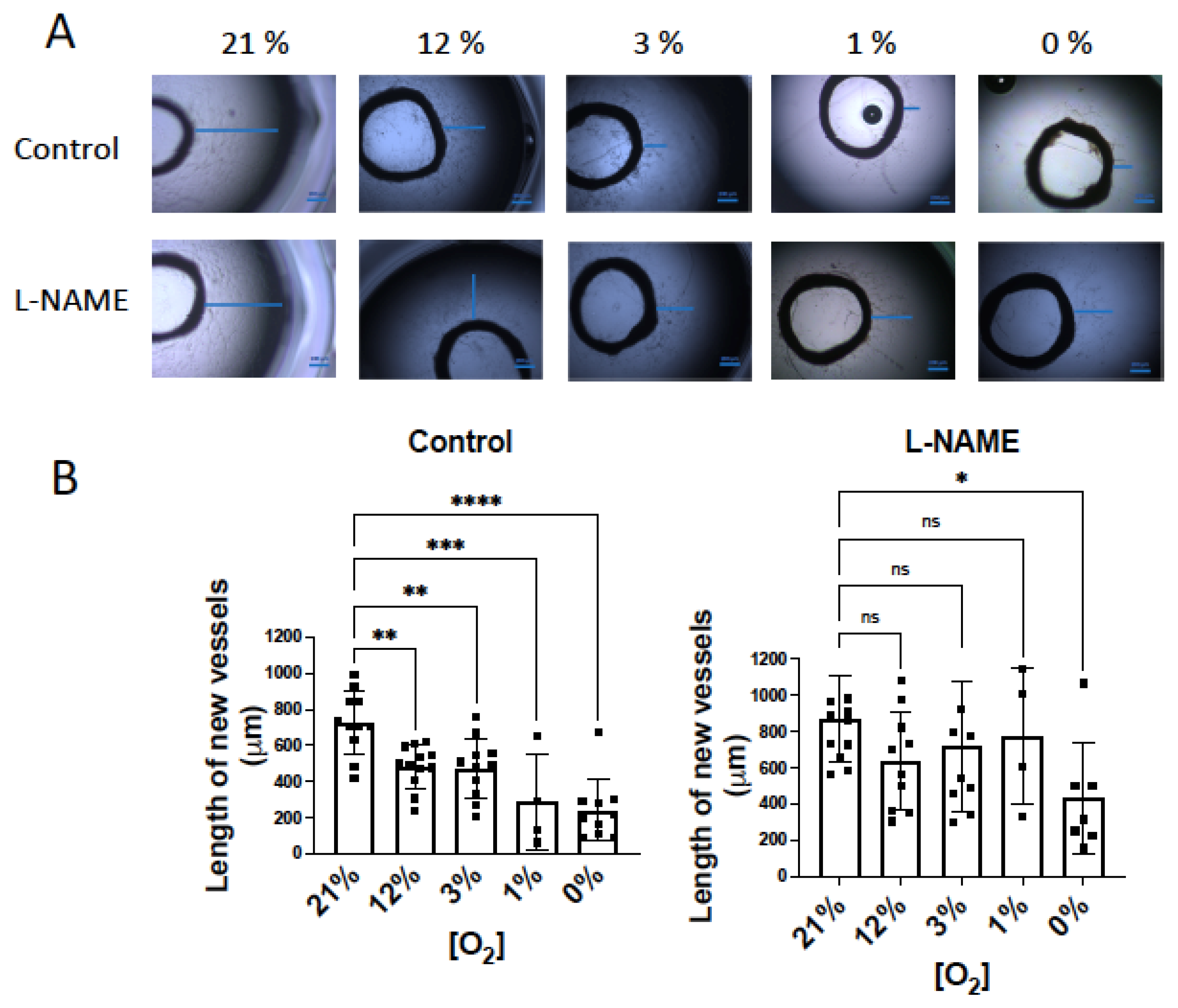
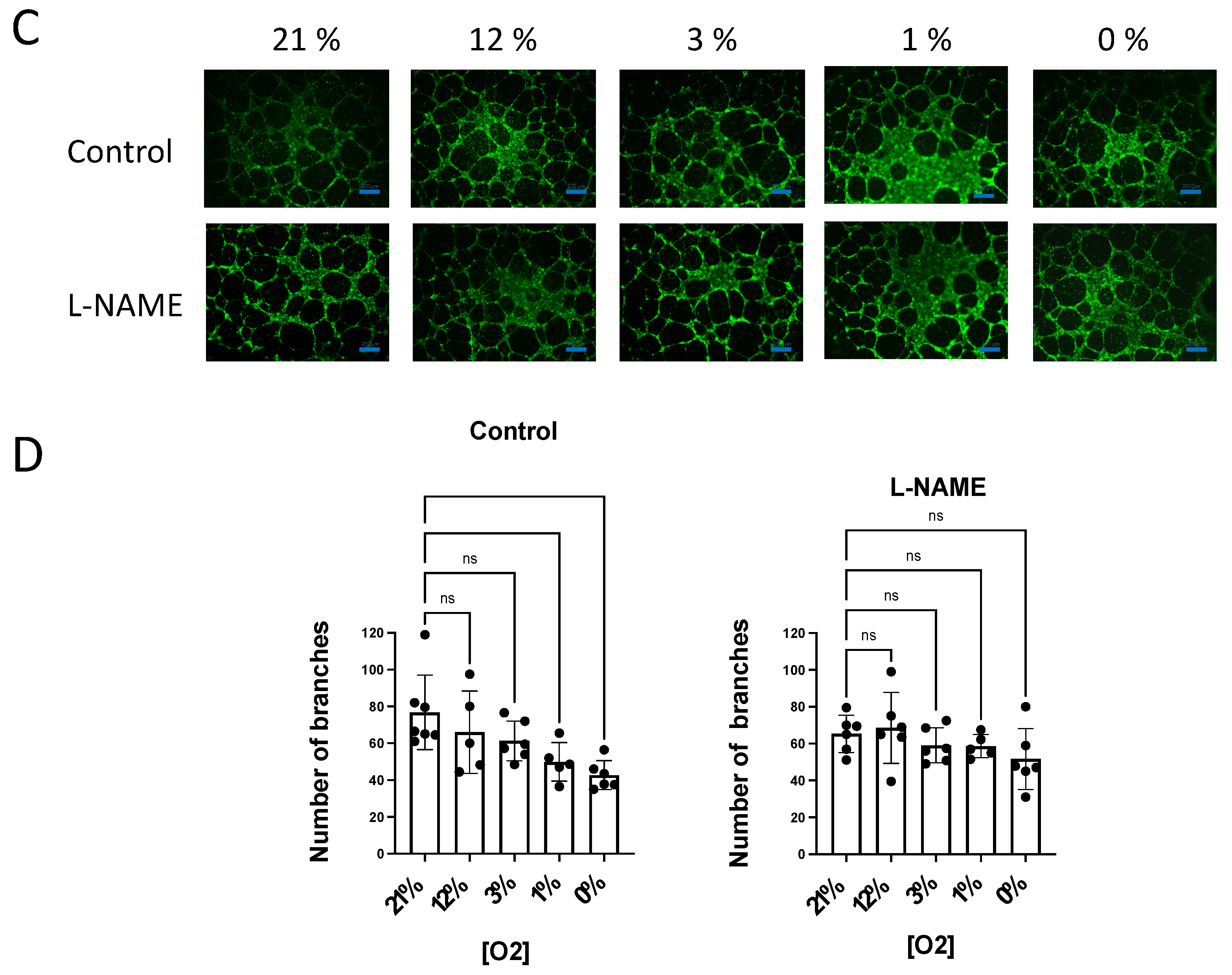
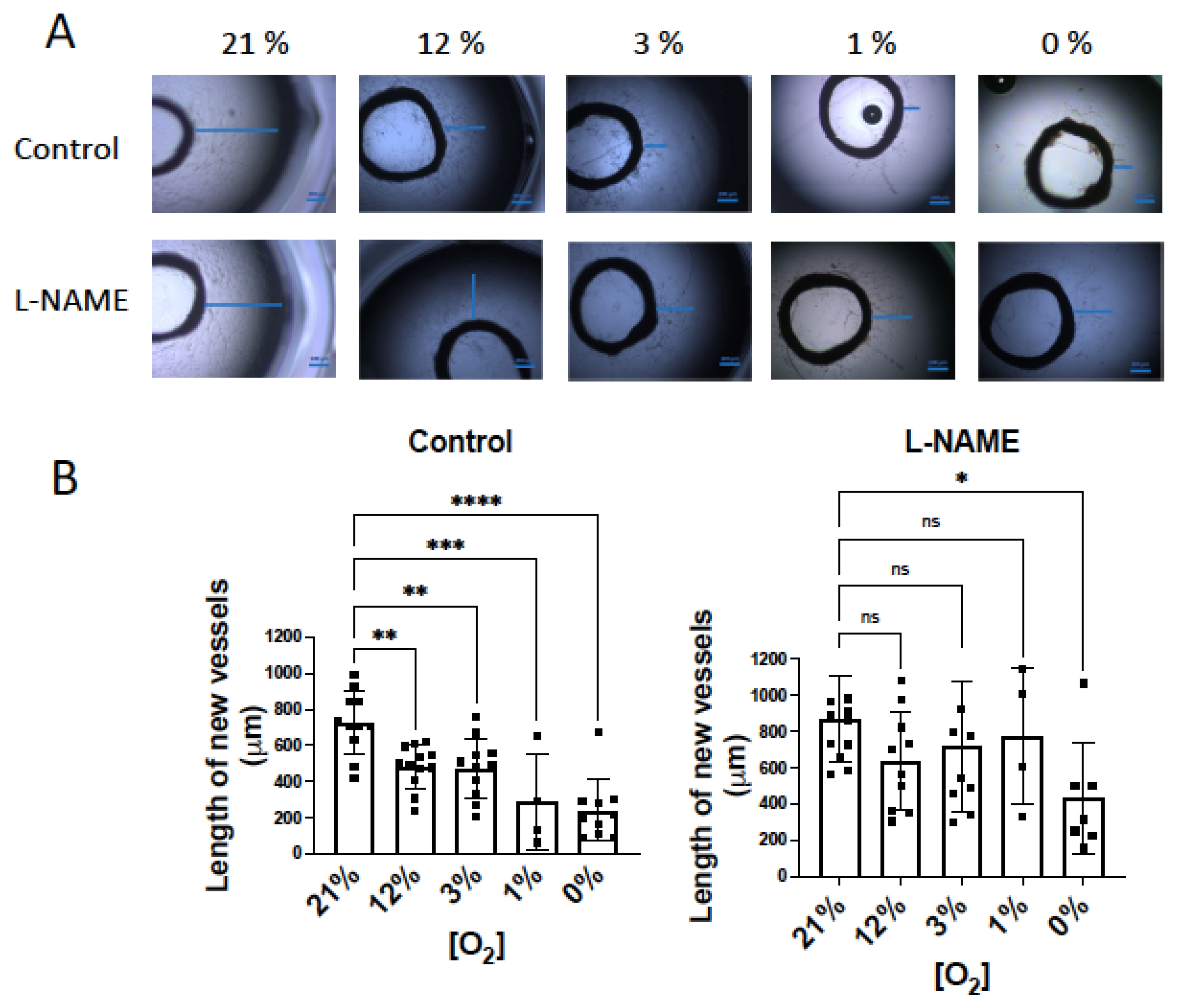
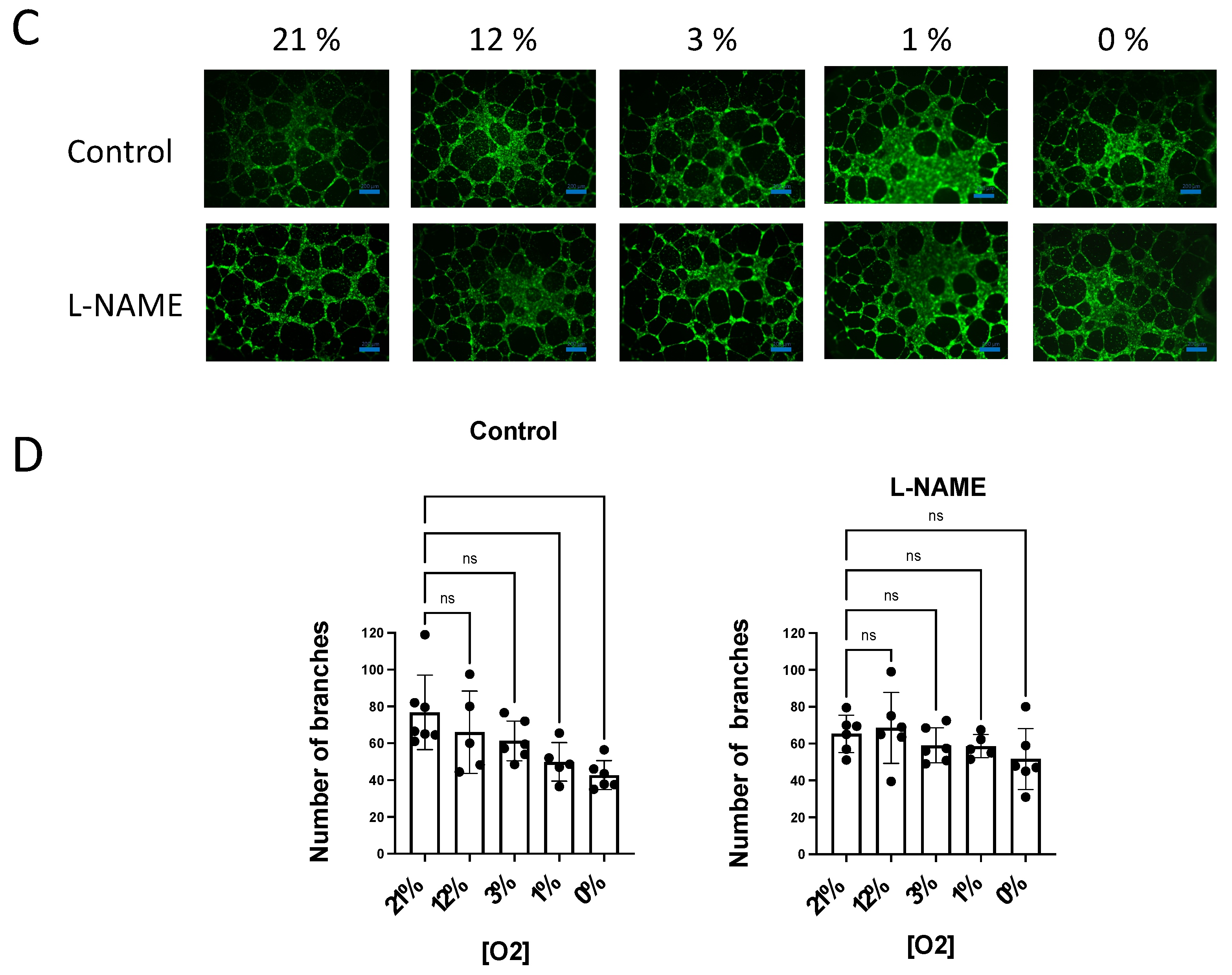
3.4. NO Inhibition of O2 Consumption and Angiogenic Growth Is More Pronounced in Hypoxic Vessels
4. Discussion
- (1)
- eNOS-derived NO inhibits angiogenic growth by a mechanism related to inhibition of CcO and independent of the sGC pathway
- (2)
- NO inhibition of the angiogenic growth and O2 consumption is more evident in hypoxic vessels
4.1. NO Inhibits Angiogenic Growth by a Mechanism Related to Inhibition of CcO and Independent of the sGC Pathway
4.2. NO-Induced Inhibition of Angiogenic Growth and O2 Consumption Depends on the O2 Concentration and Is More Pronounced in Hypoxic Vessels
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Schiffmann, L.M.; Werthenbach, J.P.; Heintges-Kleinhofer, F.; Seeger, J.M.; Fritsch, M.; Günther, S.D.; Willenborg, S.; Brodesser, S.; Lucas, C.; Jüngst, C.; et al. Mitochondrial respiration controls neoangiogenesis during wound healing and tumour growth. Nat. Commun. 2020, 11, 3653. [Google Scholar] [CrossRef]
- Eelen, G.; De Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The yin and yang of nitric oxide in cancer progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Mistry, R.K.; Brewer, A.C. Redox regulation of gasotransmission in the vascular system: A focus on angiogenesis. Free. Radic. Biol. Med. 2017, 108, 500–516. [Google Scholar] [CrossRef] [Green Version]
- Alsharabasy, A.M.; Glynn, S.A.; Pandit, A. The role of extracellular matrix in tumour angiogenesis: The throne has NOx servants. Biochem. Soc. Trans. 2020, 48, 2539–2555. [Google Scholar] [CrossRef]
- Rajendran, S.; Shen, X.; Glawe, J.; Kolluru, G.K.; Kevil, C.G. Nitric Oxide and Hydrogen Sulfide Regulation of Ischemic Vascular Growth and Remodeling. Compr. Physiol. 2019, 9, 1213–1247. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.H.; Dervan, E.; Bhattacharyya, D.D.; McAuliffe, J.D.; Miranda, K.M.; Glynn, S.A. The Role of Nitric Oxide in Cancer: Master Regulator or Not? Int. J. Mol. Sci. 2020, 21, 9393. [Google Scholar] [CrossRef]
- Lehners, M.; Dobrowinski, H.; Feil, S.; Feil, R. cGMP Signaling and Vascular Smooth Muscle Cell Plasticity. J. Cardiovasc. Dev. Dis. 2018, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Ziche, M.; Morbidelli, L. Nitric Oxide and Angiogenesis. J. Neuro-Oncol. 2000, 50, 139–148. [Google Scholar] [CrossRef]
- Poderoso, J.J.; Helfenberger, K.; Poderoso, C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 2019, 88, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.; Moncada, S. Nitric Oxide, Cytochrome C Oxidase, and the Cellular Response to Hypoxia. Arter. Thromb. Vasc. Biol. 2010, 30, 643–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Núnez, C.; Víctor, V.M.; Tur, R.; Alvarez-Barrientos, A.; Moncada, S.; Esplugues, J.V.; D’Ocón, P. Discrepancies Between Nitroglycerin and NO-Releasing Drugs on Mitochondrial Oxygen Consumption, Vasoactivity, and the Release of NO. Circ. Res. 2005, 97, 1063–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor, V.M.; Nuñez, C.; D’Ocón, P.; Taylor, C.T.; Esplugues, J.V.; Moncada, S. Regulation of oxygen distribution in tissues by endothelial nitric oxide. Circ. Res. 2009, 104, 1178–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuñez, C.; Victor, V.M.; Martí, M.; D’Ocon, P. Role of endothelial nitric oxide in pulmonary and systemic arteries during hypoxia. Nitric Oxide 2014, 37, 17–27. [Google Scholar] [CrossRef]
- Vicente, D.; Hernández, B.; Segura, V.; Pascual, D.; Fornaciari, G.; Monto, F.; Mirabet, V.; Montesinos, M.C.; D’Ocon, P. Methodological Approach to Use Fresh and Cryopreserved Vessels as Tools to Analyze Pharmacological Modulation of the Angiogenic Growth. J. Cardiovasc. Pharmacol. 2016, 68, 230–240. [Google Scholar] [CrossRef]
- Muedra, V.; Moreno, L.; Rodilla, V.; Arce, C.; Montó, F.; Blázquez, Á.; Pérez, P.; D’Ocon, P. Dexamethasone Preconditioning in Cardiac Procedures Reduces Decreased Antithrombin Activity and Is Associated to Beneficial Outcomes: Role of Endothelium. Front. Pharmacol. 2018, 9, 1014. [Google Scholar] [CrossRef]
- Arce, C.; Vicente, D.; Segura, V.; Flacco, N.; Montó, F.; Almenar, L.; Agüero, J.; Rueda, J.; Jiménez-Altayó, F.; Vila, E.; et al. Activation of α1A-adrenoceptors desensitizes the rat aorta response to phenylephrine through a neuronal NOS pathway, A mechanism lost with ageing. Br. J. Pharmacol. 2017, 174, 2015–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncada, S.; Erusalimsky, J. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 2002, 3, 214–220. [Google Scholar] [CrossRef]
- Hagen, T.; Taylor, C.T.; Lam, F.; Moncada, S. Redistribution of Intracellular Oxygen in Hypoxia by Nitric Oxide: Effect on HIF1alpha. Science 2003, 302, 1975–1978. [Google Scholar] [CrossRef]
- Cooper, C.E.; Giulivi, C. Nitric oxide regulation of mitochondrial oxygen consumption II: Molecular mechanism and tissue physiology. Am. J. Physiol. Cell Physiol. 2007, 292, C1993–C2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; García-Lecea, M.; Cadenas, S.; Hernández, C.; Moncada, S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003, 376, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine kinase 1 is critically involved in nitric oxide-mediated human endothelial cell migration and tube formation. Br. J. Pharmacol. 2010, 160, 1641–1651. [Google Scholar] [CrossRef] [Green Version]
- Pipili-Synetos, E.; Sakkoula, E.; Haralabopoulos, G.; Andriopoulou, P.; Peristeris, P.; Maragoudakis, M. Evidence that nitric oxide is an endogenous antiangiogenic mediator. Br. J. Pharmacol. 1994, 111, 894–902. [Google Scholar] [CrossRef] [Green Version]
- Pipili-Synetos, E.; Papageorgiou, A.; Sakkoula, E.; Sotiropoulou, G.; Fotsis, T.; Karakiulakis, G.; Maragoudakis, M. Inhibition of angiogenesis, tumour growth and metastasis by the NO-releasing vasodilators, isosorbide mononitrate and dinitrate. Br. J. Pharmacol. 1995, 116, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccone, V.; Monti, M.; Monzani, E.; Casella, L.; Morbidelli, L. The metal-nonoate Ni(SalPipNONO) inhibits in vitro tumor growth, invasiveness and angiogenesis. Oncotarget 2018, 9, 13353–13365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Hwang, H.H.; Jeong, S.; Seo, D.; Jeong, Y.; Lee, D.Y.; Lee, K. Inducing angiogenesis with the controlled release of nitric oxide from biodegradable and biocompatible copolymeric nanoparticles. Int. J. Nanomed. 2018, 13, 6517–6530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potente, M.; Carmeliet, P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef]
- Diebold, L.P.; Gil, H.J.; Gao, P.; Martinez, C.A.; Weinberg, S.; Chandel, N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019, 1, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, E.A. Nitric Oxide and the Vascular Endothelium. In The Vascular Endothelium I. Handbook of Experimental Pharmacology; Moncada, S., Higgs, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 176, pp. 213–254. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Ratcliffe, P. Oxygen sensors and angiogenesis. Semin. Cell Dev. Biol. 2002, 13, 29–37. [Google Scholar] [CrossRef]
- Aplin, A.; Nicosia, R.F. Hypoxia paradoxically inhibits the angiogenic response of isolated vessel explants while inducing overexpression of vascular endothelial growth factor. Angiogenesis 2016, 19, 133–146. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arce, C.; Vicente, D.; Monto, F.; González, L.; Nuñez, C.; Victor, V.M.; Jiménez-Altayó, F.; D’Ocon, P. Hypoxia Increases Nitric Oxide-Dependent Inhibition of Angiogenic Growth. Int. J. Transl. Med. 2021, 1, 366-380. https://doi.org/10.3390/ijtm1030022
Arce C, Vicente D, Monto F, González L, Nuñez C, Victor VM, Jiménez-Altayó F, D’Ocon P. Hypoxia Increases Nitric Oxide-Dependent Inhibition of Angiogenic Growth. International Journal of Translational Medicine. 2021; 1(3):366-380. https://doi.org/10.3390/ijtm1030022
Chicago/Turabian StyleArce, Cristina, Diana Vicente, Fermí Monto, Laura González, Cristina Nuñez, Víctor M. Victor, Francesc Jiménez-Altayó, and Pilar D’Ocon. 2021. "Hypoxia Increases Nitric Oxide-Dependent Inhibition of Angiogenic Growth" International Journal of Translational Medicine 1, no. 3: 366-380. https://doi.org/10.3390/ijtm1030022
APA StyleArce, C., Vicente, D., Monto, F., González, L., Nuñez, C., Victor, V. M., Jiménez-Altayó, F., & D’Ocon, P. (2021). Hypoxia Increases Nitric Oxide-Dependent Inhibition of Angiogenic Growth. International Journal of Translational Medicine, 1(3), 366-380. https://doi.org/10.3390/ijtm1030022