Human Pulmonary Artery Endothelial Cells Increased Glycolysis and Decreased Nitric Oxide Synthase O-GlcNAcylation in Pulmonary Arterial Hypertension


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Metabolic Analysis
2.3. Protein Analysis
2.4. Nitric Oxide
2.5. Statistical Analysis
3. Results
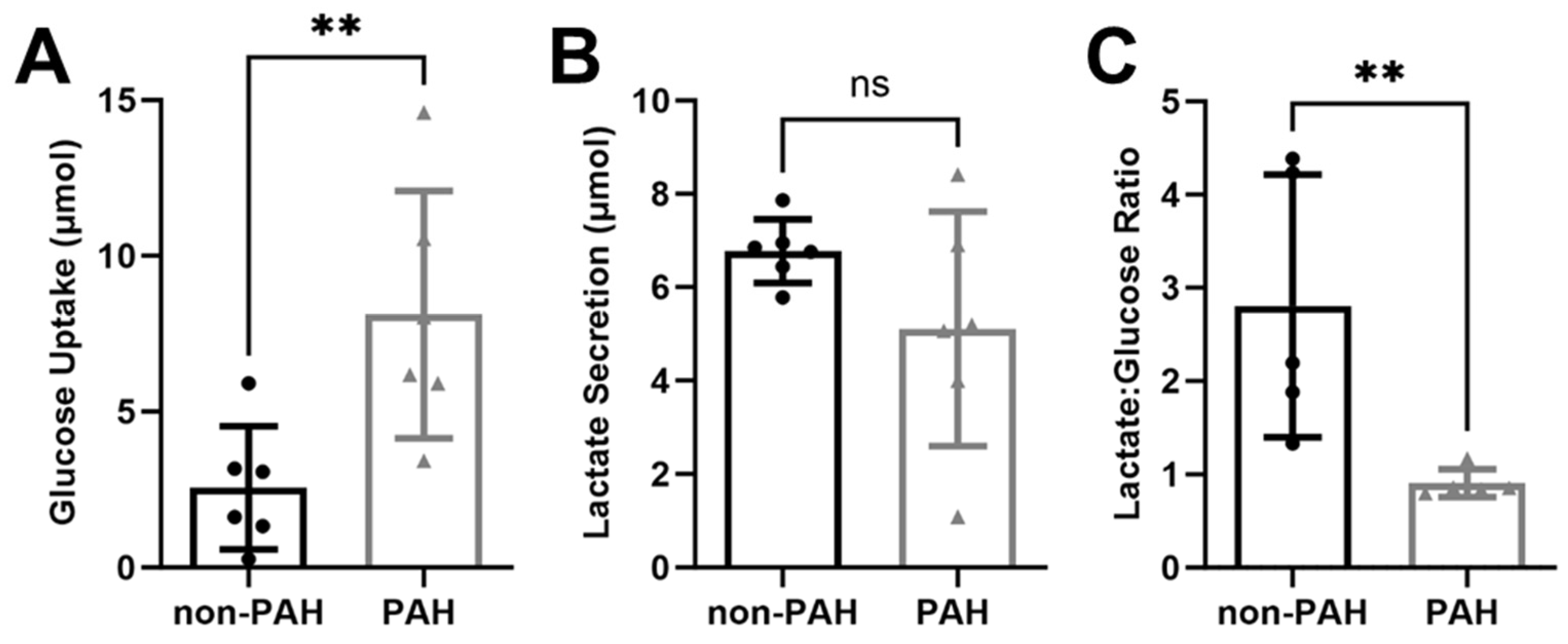
3.1. PAH HPAECs Had Higher Glucose Uptake with Reduced Lactate Secretion
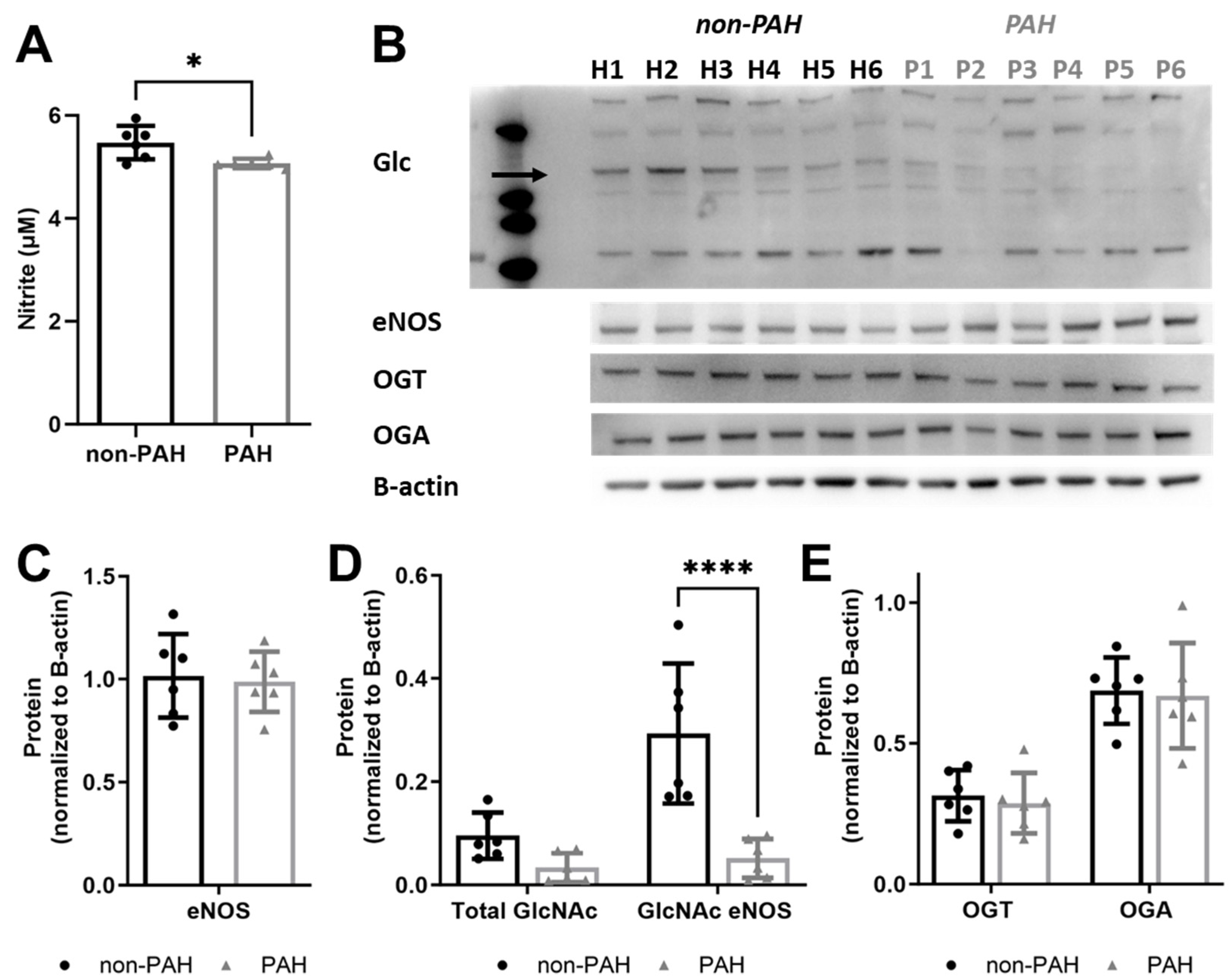
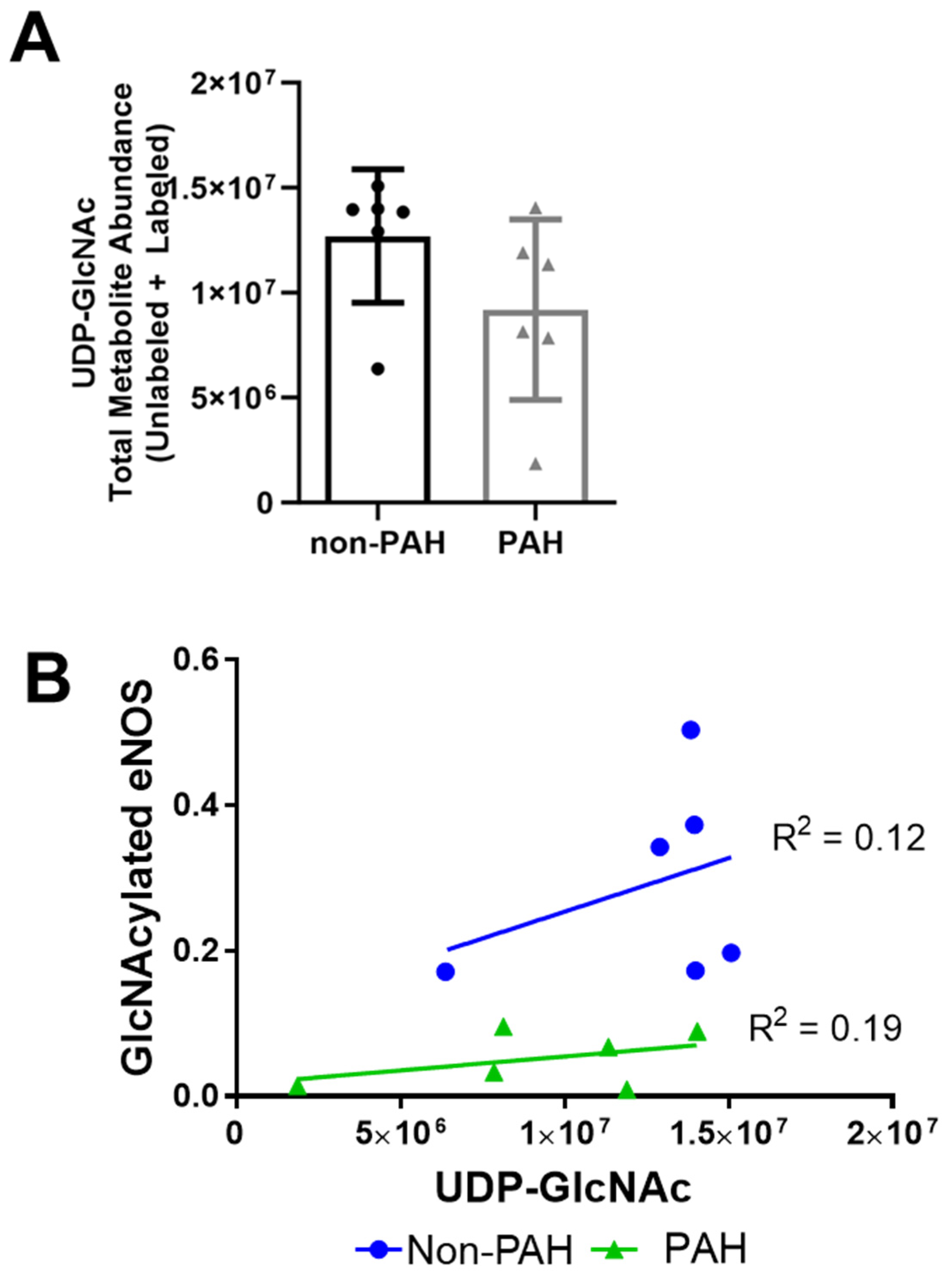
3.2. PAH HPAECs Had Lower Nitric Oxide Production Yet Lower eNOS O-GlcNAcylation
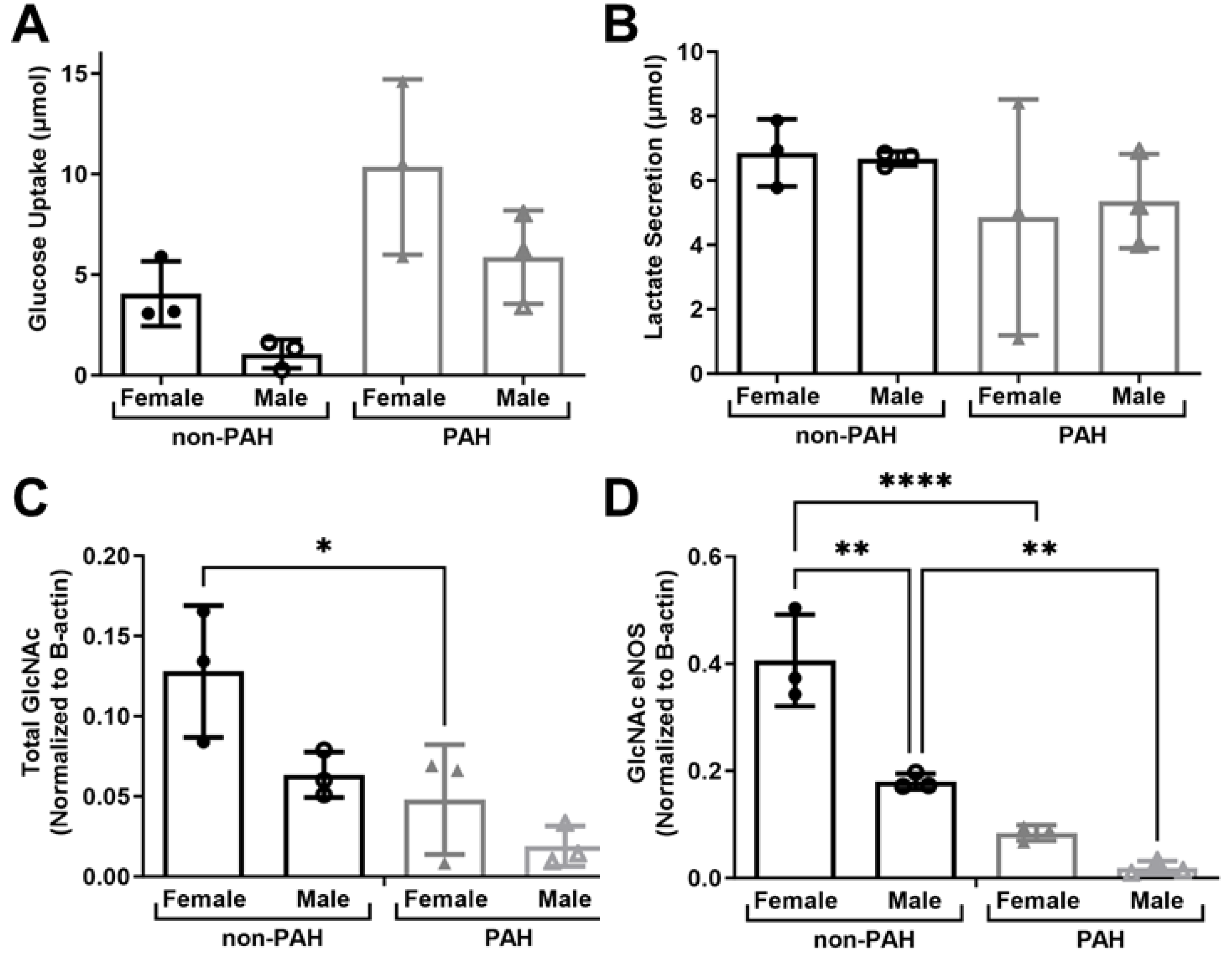
3.3. HPAEC High Donor Variability Could Be Linked to Sex Differences
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Montani, D.; Günther, S.; Dorfmüller, P.; Perros, F.; Girerd, B.; Garcia, G.; Jaïs, X.; Savale, L.; Artaud-Macari, E.; Price, L.C.; et al. Pulmonary arterial hypertension. Orphanet J. Rare Dis. 2013, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.P.; Humbert, M.P.; Souza, R.P.; Idrees, M.P.; Kawut, S.M.P.; Sliwa-Hahnle, K.P.; Jing, Z.-C.P.; Gibbs, J.S.R.P. A global view of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Pugh, M.E.; Hemnes, A.R. Pulmonary hypertension in women. Expert Rev. Cardiovasc. Ther. 2010, 8, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Sakao, S.; Tatsumi, K.; Voelkel, N.F. Endothelial cells and pulmonary arterial hypertension: Apoptosis, proliferation, interaction and transdifferentiation. Respir. Res. 2009, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Cool, C.D.; Stewart, J.S.; Werahera, P.; Miller, G.J.; Williams, R.L.; Voelkel, N.F.; Tuder, R.M. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am. J. Pathol. 1999, 155, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Abman, S.H.; Gladwin, M.T. Nitric Oxide Deficiency and Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Giaid, A.; Saleh, D. Reduced Expression of Endothelial Nitric Oxide Synthase in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef]
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of Endothelin-1 in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef]
- Botney, M.D. Role of Hemodynamics in Pulmonary Vascular Remodeling. Am. J. Respir. Crit. Care Med. 1999, 159, 361–364. [Google Scholar] [CrossRef]
- Tuder, R.M.; Davis, L.A.; Graham, B.B. Targeting Energetic Metabolism A New Frontier in the Pathogenesis and Treatment of Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 260–266. [Google Scholar] [CrossRef]
- Dromparis, P.; Sutendra, G.; Michelakis, E.D. The role of mitochondria in pulmonary vascular remodeling. J. Mol. Med. 2010, 88, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Jonigk, D.; Golpon, H.; Bockmeyer, C.L.; Maegel, L.; Hoeper, M.M.; Gottlieb, J.; Nickel, N.; Hussein, K.; Maus, U.; Lehmann, U.; et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 2011, 179, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Cool, C.D.; Kennedy, D.; Voelkel, N.F.; Tuder, R.M. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum. Pathol. 1997, 28, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.D.; Shroyer, K.R.; Markham, N.E.; Cool, C.D.; Voelkel, N.F.; Tuder, R.M. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J. Clin. Investig. 1998, 101, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Girgis, R.E.; Champion, H.C.; Diette, G.B.; Johns, R.A.; Permutt, S.; Sylvester, J.T. Decreased exhaled nitric oxide in pulmonary arterial hypertension: Response to bosentan therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Khoo Jeffrey, P.; Zhao, L.; Alp Nicholas, J.; Bendall Jennifer, K.; Nicoli, T.; Rockett, K.; Wilkins Martin, R.; Channon Keith, M. Pivotal Role for Endothelial Tetrahydrobiopterin in Pulmonary Hypertension. Circulation 2005, 111, 2126–2133. [Google Scholar] [CrossRef]
- Bauer, P.M.; Bauer, E.M.; Rogers, N.M.; Yao, M.; Feijoo-Cuaresma, M.; Pilewski, J.M.; Champion, H.C.; Zuckerbraun, B.S.; Calzada, M.J.; Isenberg, J.S. Activated CD47 promotes pulmonary arterial hypertension through targeting caveolin-1. Cardiovasc. Res. 2012, 93, 682–693. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Archer, S.L.; Badesch, D.B.; Barst, R.J.; Farber, H.W.; Lindner, J.R.; Mathier, M.A.; McGoon, M.D.; Park, M.H.; Rosenson, R.S.; et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J. Am. Coll. Cardiol. 2009, 53, 1573–1619. [Google Scholar] [CrossRef]
- Barnes, J.W.; Tian, L.; Heresi, G.A.; Farver, C.F.; Asosingh, K.; Comhair, S.A.; Aulak, K.S.; Dweik, R.A. O-linked beta-N-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation 2015, 131, 1260–1268. [Google Scholar] [CrossRef]
- Fijalkowska, I.; Xu, W.; Comhair, S.A.A.; Janocha, A.J.; Mavrakis, L.A.; Krishnamachary, B.; Zhen, L.; Mao, T.; Richter, A.; Erzurum, S.C.; et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am. J. Pathol. 2010, 176, 1130. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.L.; Kaneko, F.T.; Zheng, S.; Comhair, S.A.A.; Janocha, A.J.; Goggans, T.; Thunnissen, F.; Farver, C.; Hazen, S.L.; Jennings, C.; et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004, 18, 1746. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, M.; Li, L.; Chen, L. Involvement of the Warburg effect in non-tumor diseases processes. J. Cell. Physiol. 2018, 233, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Gurtu, V.; Michelakis, E.D. Emerging therapies and future directions in pulmonary arterial hypertension. Can. J. Cardiol. 2015, 31, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.R.; Cool, C.D.; King, J.A.; Stevens, T.; Burns, N.; Winn, R.A.; Kasper, M.; Voelkel, N.F. The cancer paradigm of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Archer, S.L. Pulmonary hypertension begets pulmonary hypertension: Mutually reinforcing roles for haemodynamics, inflammation, and cancer-like phenotypes. Cardiovasc. Res. 2016, 111, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Marsboom, G.; Wietholt, C.; Haney, C.R.; Toth, P.T.; Ryan, J.J.; Morrow, E.; Thenappan, T.; Bache-Wiig, P.; Piao, L.; Paul, J.; et al. Lung (1)(8)F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 670–679. [Google Scholar] [CrossRef]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef]
- Karunakaran, U.; Jeoung, N.H. O-GlcNAc Modification: Friend or Foe in Diabetic Cardiovascular Disease. Korean Diabetes J. 2010, 34, 211–219. [Google Scholar] [CrossRef]
- Mäkimattila, S.; Virkamäki, A.; Groop, P.H.; Cockcroft, J.; Utriainen, T.; Fagerudd, J.; Yki-Järvinen, H. Chronic hyperglycemia impairs endothelial function and insulin sensitivity via different mechanisms in insulin-dependent diabetes mellitus. Circulation 1996, 94, 1276–1282. [Google Scholar] [CrossRef]
- Medford, H.M.; Chatham, J.C.; Marsh, S.A. Chronic ingestion of a Western diet increases O-linked-beta-N-acetylglucosamine (O-GlcNAc) protein modification in the rat heart. Life Sci. 2012, 90, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Musicki, B.; Kramer, M.F.; Becker, R.E.; Burnett, A.L. Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GIcNAc in diabetes-associated erectile dysfunction. Proc. Natl. Acad. Sci. USA 2005, 102, 11870–11875. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.-R.; Hart, G.-W. opography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H.; Vogt, C.; Iozzo, P.; Pipek, R.; Daniels, M.C.; Virkamäki, A.; Mäkimattila, S.; Mandarino, L.; DeFronzo, R.A.; McClain, D.; et al. UDP-N-acetylglucosamine transferase and glutamine: Fructose 6-phosphate amidotransferase activities in insulin-sensitive tissues. Diabetologia 1997, 40, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.A.; Dell’Italia, L.J.; Chatham, J.C. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids 2011, 40, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Federici, M.; Menghini, R.; Mauriello, A.; Hribal, M.L.; Ferrelli, F.; Lauro, D.; Sbraccia, P.; Spagnoli, L.G.; Sesti, G.; Lauro, R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation 2002, 106, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; McDonough, P.M.; Swanson, E.; Trost, S.U.; Suzuki, M.; Fukuda, M.; Dillmann, W.H. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J. Biol. Chem. 2003, 278, 44230–44237. [Google Scholar] [CrossRef]
- Hu, Y.; Belke, D.; Suarez, J.; Swanson, E.; Clark, R.; Hoshijima, M.; Dillmann Wolfgang, H. Adenovirus-Mediated Overexpression of O-GlcNAcase Improves Contractile Function in the Diabetic Heart. Circ. Res. 2005, 96, 1006–1013. [Google Scholar] [CrossRef]
- Luo, B.; Soesanto, Y.; McClain, D.A. Protein modification by O-linked GlcNAc reduces angiogenesis by inhibiting Akt activity in endothelial cells. Arter. Thromb. Vasc. Biol. 2008, 28, 651–657. [Google Scholar] [CrossRef]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef]
- Yao, D.; Taguchi, T.; Matsumura, T.; Pestell, R.; Edelstein, D.; Giardino, I.; Suske, G.; Rabbani, N.; Thornalley, P.J.; Sarthy, V.P.; et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J. Biol. Chem. 2007, 282, 31038–31045. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.D.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Basehore, S.E.; Bohlman, S.; Weber, C.; Swaminathan, S.; Zhang, Y.; Jang, C.; Arany, Z.; Clyne, A.M. Laminar Flow on Endothelial Cells Suppresses eNOS O-GlcNAcylation to Promote eNOS Activity. Circ. Res. 2021, 129, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sud, N.; Wiseman, D.A.; Carter, A.L.; Kumar, S.; Hou, Y.; Rau, T.; Wilham, J.; Harmon, C.; Oishi, P.; et al. Altered carnitine homeostasis is associated with decreased mitochondrial function and altered nitric oxide signaling in lambs with pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L46–L56. [Google Scholar] [CrossRef] [PubMed]
- Clyne, A.M. Endothelial response to glucose: Dysfunction, metabolism, and transport. Biochem. Soc. Trans. 2021, 49, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Moiz, B.; Garcia, J.; Basehore, S.; Sun, A.; Li, A.; Padmanabhan, S.; Albus, K.; Jang, C.; Sriram, G.; Clyne, A.M. (13)C Metabolic Flux Analysis Indicates Endothelial Cells Attenuate Metabolic Perturbations by Modulating TCA Activity. Metabolites 2021, 11, 226. [Google Scholar] [CrossRef]
- Sedlak, J.M.; Clyne, A.M. A Modified Parallel Plate Flow Chamber to Study Local Endothelial Response to Recirculating Disturbed Flow. J. Biomech. Eng. 2020, 142, 0410031. [Google Scholar] [CrossRef]
- Murata, T.; Sato, K.; Hori, M.; Ozaki, H.; Karaki, H. Decreased endothelial nitric-oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia-induced pulmonary hypertension. J. Biol. Chem. 2002, 277, 44085–44092. [Google Scholar] [CrossRef]
- Ghosh, S.; Gupta, M.; Xu, W.; Mavrakis, D.A.; Janocha, A.J.; Comhair, S.A.A.; Haque, M.M.; Stuehr, D.J.; Yu, J.; Polgar, P.; et al. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L1199–L1205. [Google Scholar] [CrossRef]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, e68–e75. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Busse, R. Signal transduction of eNOS activation. Cardiovasc. Res. 1999, 43, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, M.; Hayashi, N.; Jing, T.; Titani, K. Regulation of endothelial nitric oxide synthase by protein kinase C. J. Biochem. 2003, 133, 773–781. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.J.; Facundo, H.T.; Ngoh, G.A.; Ameen, M.; Brainard, R.E.; Lemma, K.M.; Long, B.W.; Prabhu, S.D.; Xuan, Y.-T.; Jones, S.P. O-linked β-N-acetylglucosamine transferase is indispensable in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Belke, D.D. Swim-exercised mice show a decreased level of protein O-GlcNAcylation and expression of O-GlcNAc transferase in heart. J. Appl. Physiol. 2011, 111, 157–162. [Google Scholar] [CrossRef]
- da Costa, R.M.; da Silva, J.F.; Alves, J.V.; Dias, T.B.; Rassi, D.M.; Garcia, L.V.; Lobato, N.d.S.; Tostes, R.C. Increased O-GlcNAcylation of Endothelial Nitric Oxide Synthase Compromises the Anti-contractile Properties of Perivascular Adipose Tissue in Metabolic Syndrome. Front. Physiol. 2018, 9, 341. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Yan, L.-J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Models Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef]
- Williamson, J.R.; Chang, K.; Frangos, M.; Hasan, K.S.; Ido, Y.; Kawamura, T.; Nyengaard, J.R.; van den Enden, M.; Kilo, C.; Tilton, R.G. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993, 42, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.M.; Ho, E.C.M.; Lam, K.S.L.; Chung, S.K. Contribution of Polyol Pathway to Diabetes-Induced Oxidative Stress. J. Am. Soc. Nephrol. 2003, 14, S233. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Tilton, R.G.; Williamson, J.R. Glucose-induced metabolic imbalances in the pathogenesis of diabetic vascular disease. Diabetes/Metab. Rev. 1991, 7, 35–59. [Google Scholar] [CrossRef] [PubMed]
- Fresquet, F.; Pourageaud, F.; Leblais, V.; Brandes, R.P.; Savineau, J.P.; Marthan, R.; Muller, B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br. J. Pharmacol. 2006, 148, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Hoshikawa, Y.; Ono, S.; Suzuki, S.; Tanita, T.; Chida, M.; Song, C.; Noda, M.; Tabata, T.; Voelkel, N.F.; Fujimura, S. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J. Appl. Physiol. 2001, 90, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L2–L10. [Google Scholar] [CrossRef] [PubMed]
- Olivier-Van Stichelen, S.; Abramowitz, L.K.; Hanover, J.A. X marks the spot: Does it matter that O-GlcNAc transferase is an X-linked gene? Biochem. Biophys. Res. Commun. 2014, 453, 201–207. [Google Scholar] [CrossRef]
- Weber, C.M.; Clyne, A.M. Sex differences in the blood-brain barrier and neurodegenerative diseases. APL Bioeng. 2021, 5, 011509. [Google Scholar] [CrossRef]
- Weber, C.M.; Harris, M.N.; Zic, S.M.; Sangha, G.S.; Arnold, N.S.; Dluzen, D.F.; Clyne, A.M. Angiotensin II Increases Oxidative Stress and Inflammation in Female, But Not Male, Endothelial Cells. Cell. Mol. Bioeng. 2023, 16, 127–141. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Gender | Age | Race | |
|---|---|---|---|---|
| H1 | Donor | Female | 36 | White |
| H2 | Donor | Female | 50 | White |
| H3 | Donor | Female | 34 | White |
| H4 | Donor | Male | 47 | White |
| H5 | Donor | Male | 51 | White |
| H6 | Donor | Male | 49 | White |
| P1 | IPAH | Male | 40 | White |
| P2 | IPAH | Male | 51 | White |
| P3 | IPAH | Male | 16 | White |
| P4 | IPAH | Female | 32 | White |
| P5 | IPAH | Female | 16 | White |
| P6 | IPAH | Female | 45 | Unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basehore, S.E.; Clyne, A.M. Human Pulmonary Artery Endothelial Cells Increased Glycolysis and Decreased Nitric Oxide Synthase O-GlcNAcylation in Pulmonary Arterial Hypertension. Int. J. Transl. Med. 2024, 4, 140-151. https://doi.org/10.3390/ijtm4010007
Basehore SE, Clyne AM. Human Pulmonary Artery Endothelial Cells Increased Glycolysis and Decreased Nitric Oxide Synthase O-GlcNAcylation in Pulmonary Arterial Hypertension. International Journal of Translational Medicine. 2024; 4(1):140-151. https://doi.org/10.3390/ijtm4010007
Chicago/Turabian StyleBasehore, Sarah E., and Alisa Morss Clyne. 2024. "Human Pulmonary Artery Endothelial Cells Increased Glycolysis and Decreased Nitric Oxide Synthase O-GlcNAcylation in Pulmonary Arterial Hypertension" International Journal of Translational Medicine 4, no. 1: 140-151. https://doi.org/10.3390/ijtm4010007
APA StyleBasehore, S. E., & Clyne, A. M. (2024). Human Pulmonary Artery Endothelial Cells Increased Glycolysis and Decreased Nitric Oxide Synthase O-GlcNAcylation in Pulmonary Arterial Hypertension. International Journal of Translational Medicine, 4(1), 140-151. https://doi.org/10.3390/ijtm4010007