
Comparison of Univariate and Multivariate Applications of GBLUP and Artificial Neural Network for Genomic Prediction of Growth and Carcass Traits in the Brangus Heifer Population


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotypes, SNP Markers and Genomic Relationship Matrix
2.2. Genomic Best Linear Unbiased Prediction
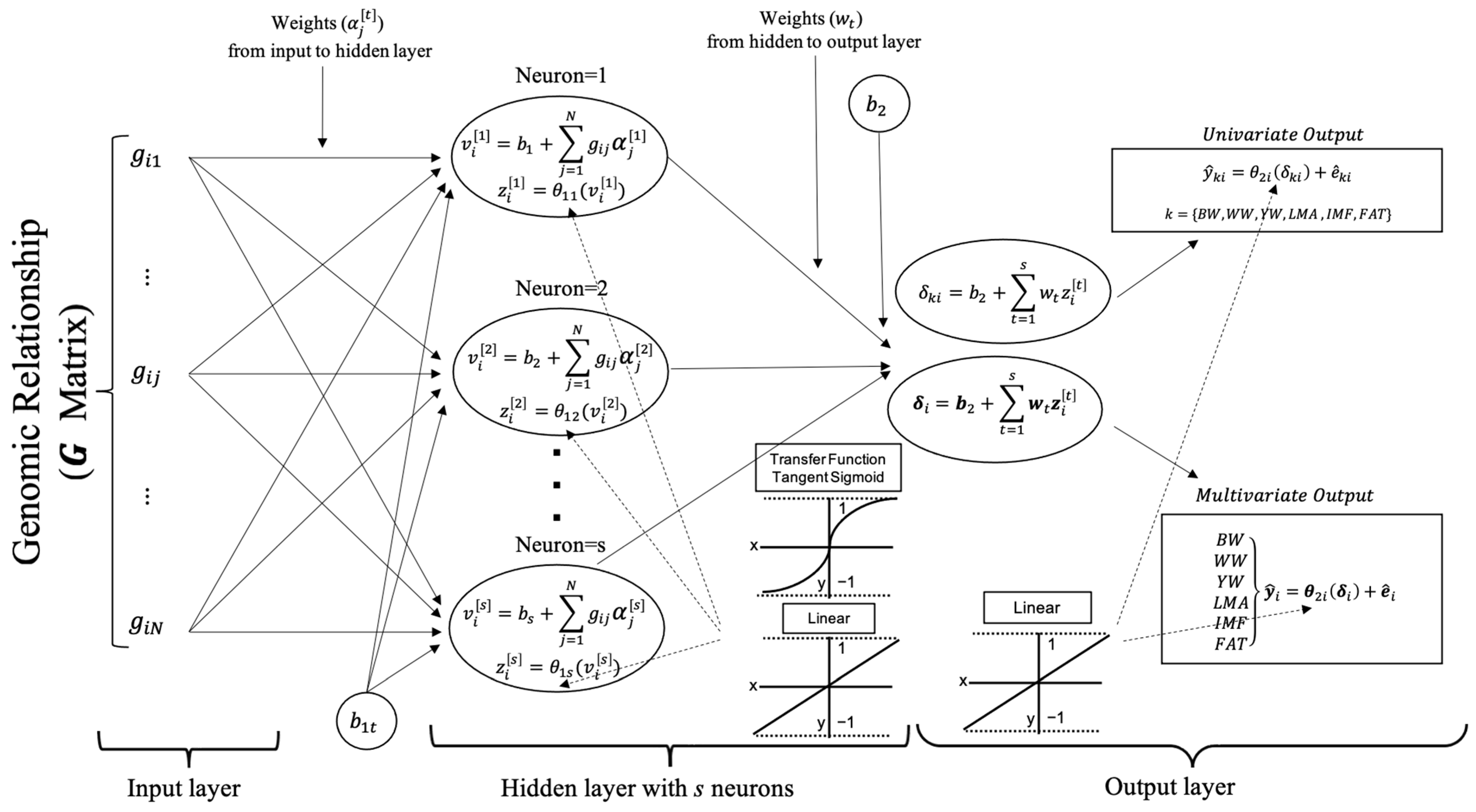
2.3. Artificial Neural Networks
2.3.1. Number of Neurons in Hidden Layer
2.3.2. Learning Algorithm in Hidden Layer
2.3.3. Transfer Functions in Hidden Layer
- The tangent sigmoid transfer function produces the scaled output over the −1 to +1 closed range, which is obtained for and , respectively [22,23]. Because there is a non-linear association between inputs and outputs, the TanSig function is widely used to determine these characteristics of the MLPANN model.
- Linear transfer function produces an output in the range of to [24,25]. The association between inputs and outputs in the MLPANN models could not be non-linear and is determined by the purelin transfer function, which can be an acceptable representation of the input/output behavior in the MLPANN models.
2.3.4. Univariate or Multivariate Outputs in Output Layer
- The univariate output of neurons in the output layer for the growth (BW, WW and YW) and carcass (FAT, IMF and LMA) traits is as follows:where and ,
2.4. Cross-Validation and Predictive Performance of Artificial Neural Networks
2.5. Analyses of Univariate and Multivariate GBLUP and MLPANN Models
3. Results and Discussion
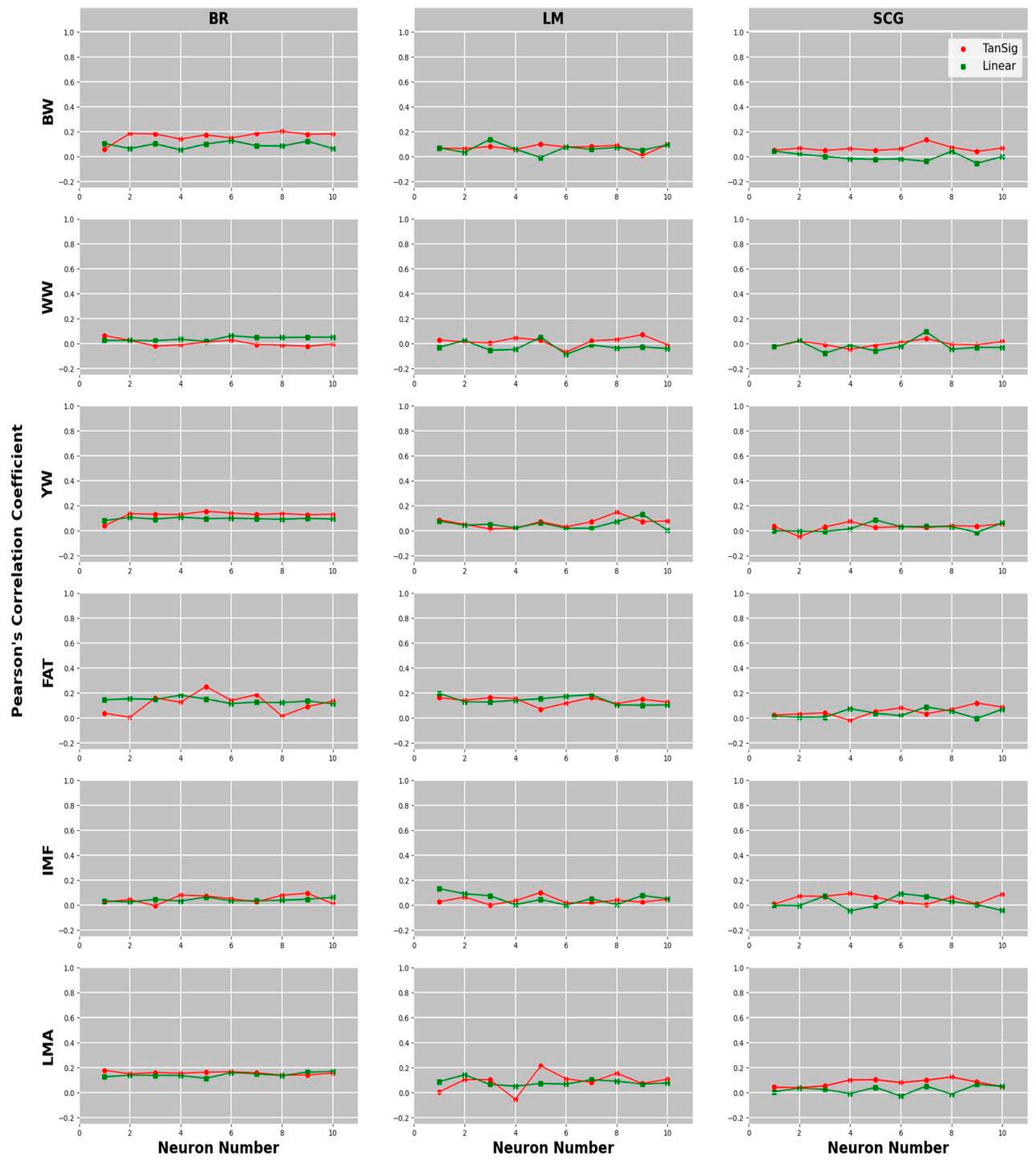
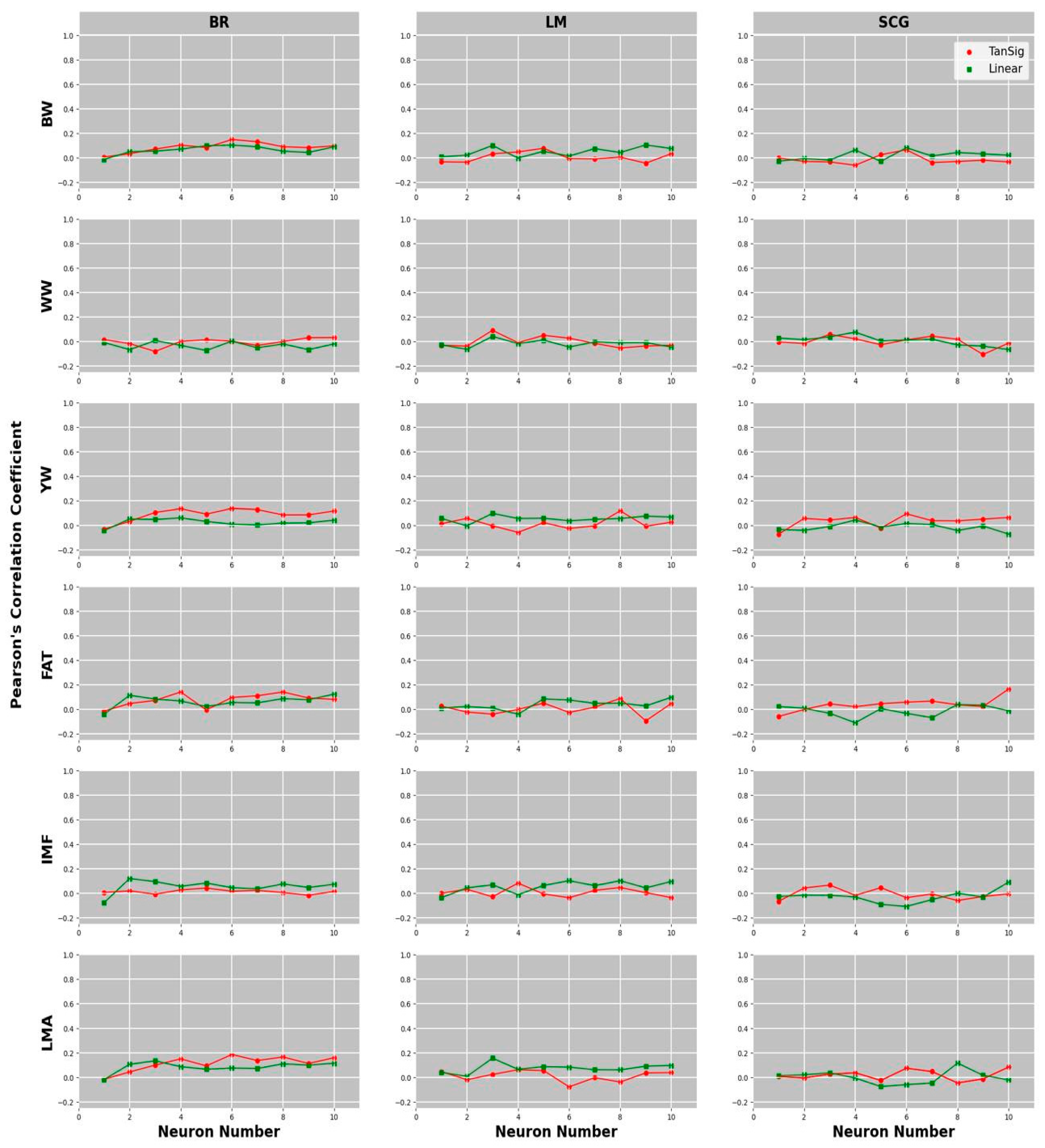
3.1. Comparison of Predictive Abilities of Univariate and Multivariate GBLUP and MLPANN Models
3.2. Comparison of Learning Algorithms and Transfer Functions for the Predictive Ability of MLPANN Models
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.L.; Sonstegard, T.S.; et al. Development and characterization of high density SNP genotyping assay for cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [PubMed]
- Applied Biosystems. Axiom Bovine Genotyping v3 Array (384HT Format). 2019. Available online: https://www.thermofisher.com/order/catalog/product/55108%209#/551089 (accessed on 15 January 2022).
- Illumina. Infinium iSelect Custom Genotyping Assays. 2016. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/technote_iselect_design.pdf (accessed on 15 January 2022).
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef]
- Pérez, P.; de los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.d.B.; Rao, C.R.; Rao, M. Data Mining Using Neural Networks: A Guide for Statisticians, 1st ed.; Chapman and Hall/CRC: London, UK, 2009. [Google Scholar]
- Kononenko, I.; Kukar, M. Machine Learning and Data Mining: Introduction to Principles and Algorithms; Horwood Publishing: London, UK, 2007. [Google Scholar]
- Howard, R.; Carriquiry, A.L.; and Beavis, W.D. Parametric and Nonparametric Statistical Methods for Genomic Selection of Traits with Additive and Epistatic Genetic Architectures. G3 Genes Genomes Genet. 2014, 4, 1027–1046. [Google Scholar] [CrossRef]
- Luna-Nevarez, P.; Bailey, D.W.; Bailey, C.C.; VanLeeuwen, D.M.; Enns, R.M.; Silver, G.A.; DeAtley, K.L.; Thomas, M.G. Growth characteristics, reproductive performance, and evaluation of their associative relationships in Brangus cattle managed in a Chihuahuan Desert production system. J. Anim. Sci. 2010, 88, 1891–1904. [Google Scholar] [CrossRef]
- Fortes, M.R.S.; Snelling, W.M.; Reverter, A.; Nagaraji, S.H.; Lehnert, S.A.; Hawken, R.J.; DeAtley, K.L.; Peters, S.O.; Silver, G.A.; Rincon, G.; et al. Gene network analyses of first service conception in Brangus heifers: Use of genome and trait associations, hypothalamic-transcriptome information, and transcription factors. J. Anim. Sci. 2012, 90, 2894–2906. [Google Scholar] [CrossRef]
- Peters, S.O.; Kizilkaya, K.; Garrick, D.J.; Fernando, R.L.; Reecy, J.M.; Weaber, R.L.; Silver, G.A.; Thomas, M.G. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in Brangus heifers. J. Anim. Sci. 2012, 90, 3398–3409. [Google Scholar] [CrossRef]
- Granato, I.S.C.; Galli, G.; de Oliveira Couto, E.G.; Souza, M.B.E.; Mendonça, L.F.; Fritsche-Neto, R. snpReady: A tool to assist breeders in genomic analysis. Mol. Breed. 2018, 38, 102. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. The impact of genetic relationship information on genome-assisted breeding values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef]
- vanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Cybenko, G. Approximation by superpositions of a sigmoidal function. Math. Control Signals Syst. 1989, 2, 303–314. [Google Scholar] [CrossRef]
- Hornik, K.; Stinchcombe, M.; White, H. Multilayer feedforward networks are universal approximators. Neural Netw. 1989, 2, 359–366. [Google Scholar] [CrossRef]
- Ehret, A.; Hochstuhl, D.; Gianola, D.; Thaller, G. Application of neural networks with back-propagation to genome-enabled prediction of complex traits in Holstein-Friesian and German Fleckvieh cattle. Genet. Sel. Evol. 2015, 47, 22. [Google Scholar] [CrossRef] [PubMed]
- Haykin, S.S. Neural Networks: A Comprehensive Foundation; Prentice Hall: Hoboken, NJ, USA, 1999; 842p. [Google Scholar]
- Kröse, B.; Smagt Patrick, V.D. An Introduction to Neural Networks, 8th ed.; The University of Amsterdam: Amsterdam, The Netherlands, 1996; 135p. [Google Scholar]
- Christodoulou, C.G.; Georgiopoulos, M. Applications of Neural Networks in Electromagnetics, 1st ed.; Artech House: Norwood, MA, USA, 2001; 512p. [Google Scholar]
- Civco, D.L. Artificial neural networks for land cover classification and mapping. Int. J. Geogr. Inf. Syst. 1993, 7, 173–186. [Google Scholar] [CrossRef]
- Kaminsky, E.J.; Barad, H.; Brown, W. Textural neural network and version space classifiers for remote sensing. Int. J. Remote Sens. 1997, 18, 741–762. [Google Scholar] [CrossRef]
- Bouabaz, M.; Hamami, M. A Cost Estimation Model for Repair Bridges Based on Artificial Neural Network. Am. J. Appl. Sci. 2008, 5, 334–339. [Google Scholar] [CrossRef]
- Dorofki, M.; Elshafie, A.; Jaafar, O.; Karim, O.A.; Mastura, S. Comparison of Artificial Neural Network Transfer Functions Abilities to Simulate Extreme Runoff Data. Int. Proc. Chem. Biol. Environ. Eng. 2012, 33, 39–44. [Google Scholar]
- Saatchi, M.; McClure, M.C.; McKay, S.D.; Rolf, M.M.; Kim, J.; Decker, J.E.; Taxis, T.M.; Chapple, R.H.; Ramey, H.R.; Northcutt, S.L.; et al. Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet. Sel. Evol. 2011, 43, 40. [Google Scholar] [CrossRef]
- Hartigan, J.A.; Wong, M.A.; Algorithm, A.S. 136: A k-means clustering algorithm. Appl. Stat. 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Gorjanc, G.; Henderson, D.A.; Kinghorn, B.; Percy, A. GeneticsPed: Pedigree and Genetic Relationship Functions. R Package Version 1.52.0. 2020. Available online: https://rgenetics.org (accessed on 15 March 2023).
- Kassambara, A.; Mundt, F. factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version 1.0.5. 2017. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 15 March 2023).
- Demuth, H.; Beale, M. Neural Network Toolbox User’s Guide Version 4.0; The Mathworks: Natick, MA, USA, 2000. [Google Scholar]
- Su, G.; Christensen, O.F.; Janss, L.; Lund, M.S. Comparison of genomic predictions using genomic relationship matrices built with different weighting factors to account for locus-specific variances. J. Dairy Sci. 2014, 97, 6547–6559. [Google Scholar] [CrossRef]
- Peters, S.; Sinecen, M.; Kizilkaya, K.; Thomas, M. Genomic prediction with different heritability, QTL and SNP panel scenarios using artificial neural network. IEEE Access 2020, 8, 147995–148006. [Google Scholar] [CrossRef]
- Peters, S.O.; Kizilkaya, K.; Sinecen, M.; Metav, B.; Thiruvenkadan, A.K.; Thomas, M.G. Genomic Prediction Accuracies for Growth and Carcass Traits in a Brangus Heifer Population. Animals 2023, 13, 1272. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Pong-Wong, R.; Villanueva, B.; Woolliams, J.A. The impact of genetic architecture on genome-wide evaluation methods. Genetics 2010, 185, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, J.; Ding, X.; Bijma, P.; de Koning, D.-J.; Zhang, Q. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS ONE 2010, 5, e12648. [Google Scholar] [CrossRef]
- Peters, S.O.; Kizilkaya, K.; Sinecen, M.; Thomas, M.G. Multivariate Application of Artificial Neural Networks for Genomic Prediction. In Proceedings of the World Congress on Genetics Applied to Livestock Production, Rotterdam, The Netherlands, 3–8 July 2022. [Google Scholar]
- Okut, H.; Gianola, D.; Rosa, G.J.M.; Weigel, K.A. Prediction of body mass index in mice using dense molecular markers and a regularized neural network. Genet. Res. 2011, 93, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Herring, W.; What Have We Learned About Trait Relationships. 2003. 1–8. Available online: https://animal.ifas.ufl.edu/beef_extension/bcsc/2002/pdf/herring.pdf (accessed on 15 March 2023).
- Peters, S.O.; Kizilkaya, K.; Garrick, D.; Reecy, J. PSX-9 Comparison of single- and multiple-trait genomic predictions of cattle carcass quality traits in multibreed and purebred populations. J. Anim. Sci. 2024, 102 (Suppl. S3), 453–454. [Google Scholar] [CrossRef]
- Rostamzadeh Mahdabi, E.; Tian, R.; Li, Y.; Wang, X.; Zhao, M.; Li, H.; Yang, D.; Zhang, H.; Li, S.; Esmailizadeh, A. Genomic heritability and correlation between carcass traits in Japanese Black cattle evaluated under different ceilings of relatedness among individuals. Front. Genet. 2023, 14, 1053291. [Google Scholar] [CrossRef]
- Weik, F.; Hickson, R.E.; Morris, S.T.; Garrick, D.J.; Archer, J.A. Genetic Parameters for Growth, Ultrasound and Carcass Traits in New Zealand Beef Cattle and Their Correlations with Maternal Performance. Animals 2022, 12, 25. [Google Scholar] [CrossRef]
- Caetano, S.L.; Savegnago, R.P.; Boligon, A.A.; Ramos, S.B.; Chud, T.C.S.; Lôbo, R.B.; Munari, D.P. Estimates of genetic parameters for carcass, growth and reproductive traits in Nellore cattle. Livest. Sci. 2013, 155, 1–7. [Google Scholar] [CrossRef]
- Gianola, D.; Okut, H.; Weigel, K.; Rosa, G. Predicting complex quantitative traits with Bayesian neural networks: A case study with Jersey cows and wheat. BMC Genet. 2011, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Sinecen, M. Comparison of genomic best linear unbiased prediction and Bayesian regularization neural networks for genomic selection. IEEE Access 2019, 7, 79199–79210. [Google Scholar] [CrossRef]
- Peters, S.O.; Sinecen, M.; Gallagher, G.R.; Lauren, A.P.; Jacob, S.; Hatfield, J.S.; Kizilkaya, K. Comparison of Linear Model and Artificial Neural Network Using Antler Beam Diameter and Length of White-Tailed Deer (Odocoileus virginianus) Dataset. PLoS ONE 2019, 14, e0212545. [Google Scholar] [CrossRef]
- Kayri, M. Predictive Abilities of Bayesian Regularization and LevenbergMarquardt Algorithms in Artificial Neural Networks: A Comparative Empirical Study on Social Data. Math. Comput. Appl. 2016, 21, 20. [Google Scholar] [CrossRef]
- Okut, H.; Wu, X.L.; Rosa, G.J.M.; Bauck, S.; Woodward, B.W.; Schnabel, R.D.; Taylor, J.F.; Gianola, D. Predicting expected progeny difference for marbling score in Angus cattle using artificial neural networks and Bayesian regression models. Genet. Sel. Evolut. 2013, 45, 34. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, P.; McElroy, N.R. LogD7.4 modeling using Bayesian regularized neural networks assessment and correction of the errors of prediction. J. Chem. Inf. Model. 2006, 46, 1379–1387. [Google Scholar] [CrossRef]
- Saini, L.M. Peak load forecasting using Bayesian regularization, Resilient and adaptive backpropagation learning based artificial neural networks. Electr. Power Syst. Res. 2008, 78, 1302–1310. [Google Scholar] [CrossRef]
- Lauret, P.; Fock, F.; Randrianarivony, R.N.; Manicom-Ramsamy, J.F. Bayesian Neural Network approach to short time load forecasting. Energy Convers. Manag. 2008, 5, 1156–1166. [Google Scholar] [CrossRef]
- Ticknor, J.L. A Bayesian regularized artificial neural network for stock market forecasting. Expert Syst. Appl. 2013, 14, 5501–5506. [Google Scholar] [CrossRef]
- Gurney, K. An Introduction to Neural Networks, 1st ed.; CRC Press: London, UK, 1997. [Google Scholar]
- Sharma, S.; Sharma, S.; Athaiya, A. Activation functions in neural networks. Int. J. Eng. Appl. Sci. Technol. 2020, 4, 310–316. [Google Scholar] [CrossRef]
- Maier, H.R.; Dandy, C.G. Neural networks for the prediction and forecasting of water resources variables: A review of modelling issues and applications. Environ. Model. Softw. 2000, 15, 101–124. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Univariate Analysis | |||||||
|---|---|---|---|---|---|---|---|
| Trait | MLPANN-BR | MLPANN-LM | MLPANN-SCG | GBLUP | |||
| TanSig—N | Linear—N | TanSig—N | Linear—N | TanSig—N | Linear—N | ||
| BW | 0.201–8 | 0.128–6 | 0.099–5 | 0.137–3 | 0.133–7 | 0.043–1 | 0.169 |
| WW | 0.064–1 | 0.062–6 | 0.070–9 | 0.049–5 | 0.039–7 | 0.094–7 | 0.032 |
| YW | 0.155–5 | 0.108–4 | 0.148–8 | 0.130–9 | 0.074–4 | 0.085–5 | 0.130 |
| FAT | 0.250–5 | 0.180–4 | 0.162–7 | 0.194–1 | 0.118–9 | 0.086–7 | 0.164 |
| IMF | 0.094–9 | 0.063–5 | 0.100–5 | 0.131–1 | 0.092–4 | 0.092–4 | 0.121 |
| LMA | 0.178–1 | 0.168–10 | 0.212–5 | 0.141–2 | 0.124–8 | 0.065–9 | 0.183 |
| Multivariate Analysis | |||||||
| Trait | MLPANN-BR | MLPANN-LM | MLPANN-SCG | GBLUP | |||
| TanSig—N | Linear—N | TanSig—N | Linear—N | TanSig—N | Linear—N | ||
| BW | 0.148–6 | 0.103–6 | 0.076–5 | 0.103–9 | 0.064–6 | 0.081–6 | 0.154 |
| WW | 0.031–10 | 0.006–3 | 0.090–3 | 0.040–3 | 0.057–3 | 0.075–4 | 0.032 |
| YW | 0.137–6 | 0.060–4 | 0.118–8 | 0.097–3 | 0.094–6 | 0.043–4 | 0.126 |
| FAT | 0.140–4 | 0.122–10 | 0.087–8 | 0.096–10 | 0.164–10 | 0.037–8 | 0.161 |
| IMF | 0.041–5 | 0.119–2 | 0.081–4 | 0.101–6 | 0.065–3 | 0.089–10 | 0.110 |
| LMA | 0.186–6 | 0.134–3 | 0.062–4 | 0.156–3 | 0.083–10 | 0.115–8 | 0.178 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peters, S.O.; Kızılkaya, K.; Sinecen, M.; Thomas, M.G. Comparison of Univariate and Multivariate Applications of GBLUP and Artificial Neural Network for Genomic Prediction of Growth and Carcass Traits in the Brangus Heifer Population. Ruminants 2025, 5, 16. https://doi.org/10.3390/ruminants5020016
Peters SO, Kızılkaya K, Sinecen M, Thomas MG. Comparison of Univariate and Multivariate Applications of GBLUP and Artificial Neural Network for Genomic Prediction of Growth and Carcass Traits in the Brangus Heifer Population. Ruminants. 2025; 5(2):16. https://doi.org/10.3390/ruminants5020016
Chicago/Turabian StylePeters, Sunday O., Kadir Kızılkaya, Mahmut Sinecen, and Milt G. Thomas. 2025. "Comparison of Univariate and Multivariate Applications of GBLUP and Artificial Neural Network for Genomic Prediction of Growth and Carcass Traits in the Brangus Heifer Population" Ruminants 5, no. 2: 16. https://doi.org/10.3390/ruminants5020016
APA StylePeters, S. O., Kızılkaya, K., Sinecen, M., & Thomas, M. G. (2025). Comparison of Univariate and Multivariate Applications of GBLUP and Artificial Neural Network for Genomic Prediction of Growth and Carcass Traits in the Brangus Heifer Population. Ruminants, 5(2), 16. https://doi.org/10.3390/ruminants5020016