
Understanding and Modeling the Pathophysiology of Hydrocephalus: In Search of Better Treatment Options


Abstract
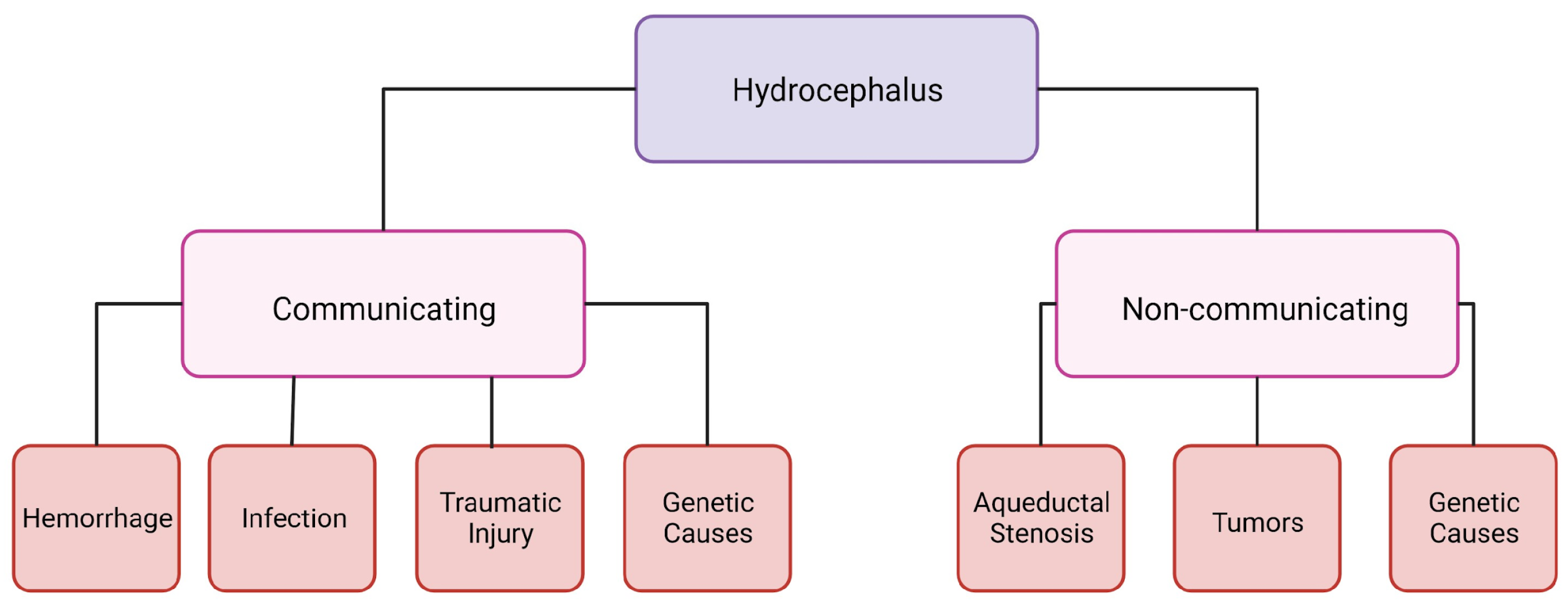
1. Hydrocephalus
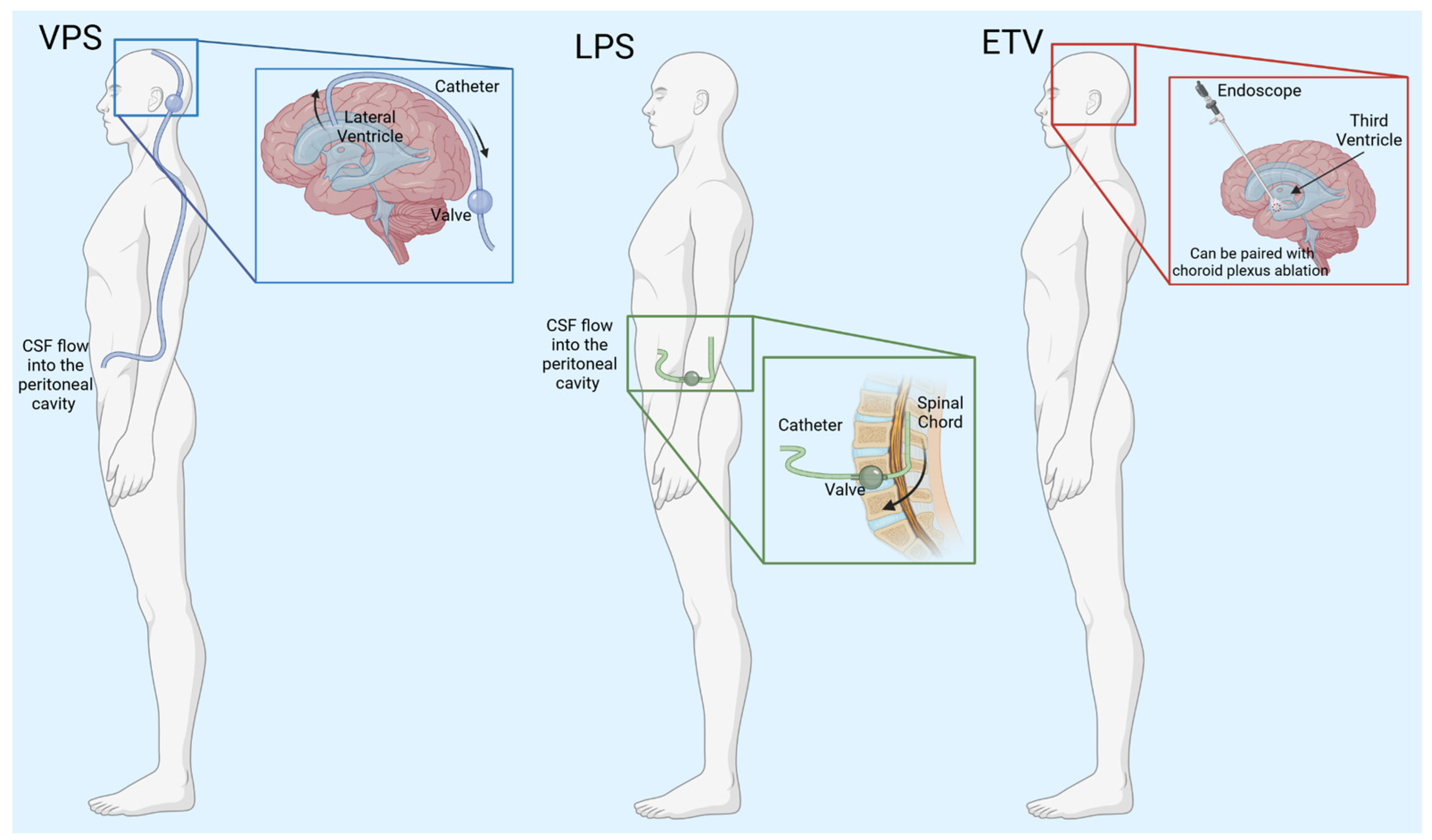
2. Hydrocephalus Treatments
3. Induced Animal Models
4. Genetic Animal Models
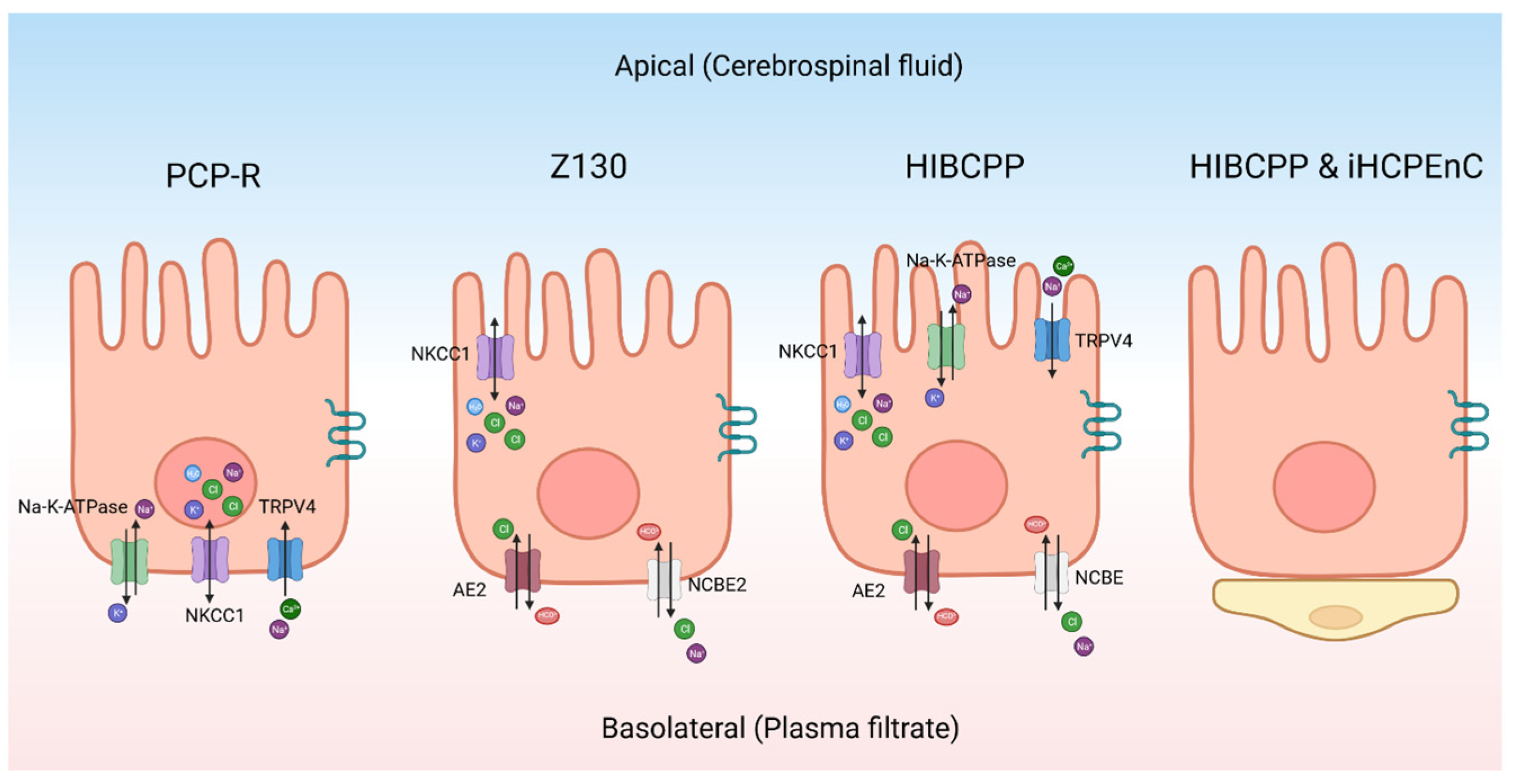
5. The Choroid Plexus Epithelium
6. Cell Culture Models
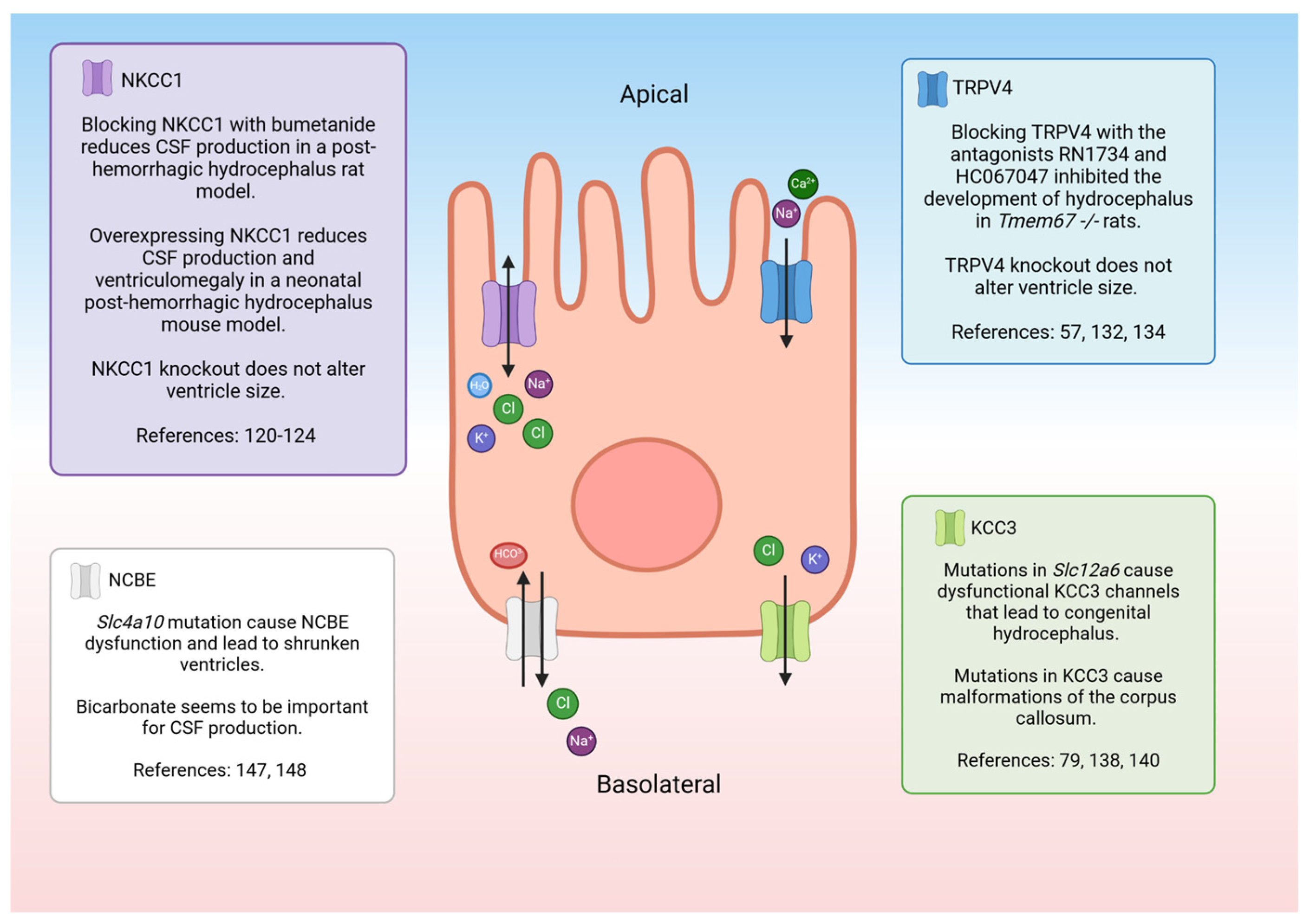
7. Electrolyte Transporters, Channels, and Pumps: Potential Roles in Fluid/Electrolyte Homeostasis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lifshutz, J.I.; Johnson, W.D. History of hydrocephalus and its treatments. Neurosurg. Focus 2001, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Aschoff, A.; Kremer, P.; Hashemi, B.; Kunze, S. The scientific history of hydrocephalus and its treatment. Neurosurg. Rev. 1999, 22, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Mokri, B. The Monro–Kellie hypothesis: Applications in CSF volume depletion. Neurology 2001, 56, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Greitz, D. Radiological assessment of hydrocephalus: New theories and implications for therapy. Neurosurg. Rev. 2004, 27, 145–165. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, ra111–ra147. [Google Scholar] [CrossRef] [PubMed]
- Hochstetler, A.; Raskin, J.; Blazer-Yost, B.L. Hydrocephalus: Historical analysis and considerations for treatment. Eur. J. Med. Res. 2022, 27, 168. [Google Scholar] [CrossRef] [PubMed]
- Koschnitzky, J.E.; Yap, E.; Zhang, Y.; Chau, M.J.; Yerneni, K.; Somera, A.L.; Luciano, M.; Moghekar, A. Inpatient healthcare burden and variables influencing hydrocephalus-related admissions across the lifespan. J. Neurosurg. 2022, 139, 502–511. [Google Scholar] [CrossRef]
- Karimy, J.K.; Reeves, B.C.; Damisah, E.; Duy, P.Q.; Antwi, P.; David, W.; Wang, K.; Schiff, S.J.; Limbrick, D.D., Jr.; Alper, S.L. Inflammation in acquired hydrocephalus: Pathogenic mechanisms and therapeutic targets. Nat. Rev. Neurol. 2020, 16, 285–296. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Mekary, R.; Glancz, L.J.; Yunusa, I.; Baticulon, R.E.; Fieggen, G.; Wellons, J.C.; Park, K.B.; Warf, B.C. Global hydrocephalus epidemiology and incidence: Systematic review and meta-analysis. J. Neurosurg. 2018, 130, 1065–1079. [Google Scholar] [CrossRef]
- Chen, Q.; Feng, Z.; Tan, Q.; Guo, J.; Tang, J.; Tan, L.; Feng, H.; Chen, Z. Post-hemorrhagic hydrocephalus: Recent advances and new therapeutic insights. J. Neurol. Sci. 2017, 375, 220–230. [Google Scholar] [CrossRef]
- Cioca, A.; Gheban, D.; Perju-Dumbrava, D.; Chiroban, O.; Mera, M. Sudden death from ruptured choroid plexus arteriovenous malformation. Am. J. Forensic Med. Pathol. 2014, 35, 100–102. [Google Scholar] [CrossRef]
- Thigpen, M.C.; Whitney, C.G.; Messonnier, N.E.; Zell, E.R.; Lynfield, R.; Hadler, J.L.; Harrison, L.H.; Farley, M.M.; Reingold, A.; Bennett, N.M. Bacterial meningitis in the United States, 1998–2007. N. Engl. J. Med. 2011, 364, 2016–2025. [Google Scholar] [CrossRef]
- Varagur, K.; Sanka, S.A.; Strahle, J.M. Syndromic hydrocephalus. Neurosurg. Clin. 2022, 33, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Stumpel, C.; Vos, Y.J. L1 Syndrome; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Marguet, F.; Vezain, M.; Marcorelles, P.; Audebert-Bellanger, S.; Cassinari, K.; Drouot, N.; Chambon, P.; Gonzalez, B.J.; Horowitz, A.; Laquerriere, A. Neuropathological hallmarks of fetal hydrocephalus linked to CCDC88C pathogenic variants. Acta Neuropathol. Commun. 2021, 9, 104. [Google Scholar] [CrossRef]
- Hale, A.T.; Bastarache, L.; Morales, D.M.; Wellons, J.C.; Limbrick, D.D.; Gamazon, E.R. Multi-omic analysis elucidates the genetic basis of hydrocephalus. Cell Rep. 2021, 35, 109085. [Google Scholar] [CrossRef]
- Isaacs, A.M.; Riva-Cambrin, J.; Yavin, D.; Hockley, A.; Pringsheim, T.M.; Jette, N.; Lethebe, B.C.; Lowerison, M.; Dronyk, J.; Hamilton, M.G. Age-specific global epidemiology of hydrocephalus: Systematic review, metanalysis and global birth surveillance. PLoS ONE 2018, 13, e0204926. [Google Scholar] [CrossRef] [PubMed]
- Tully, H.M.; Dobyns, W.B. Infantile hydrocephalus: A review of epidemiology, classification and causes. Eur. J. Med. Genet. 2014, 57, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Wright, Z.; Larrew, T.W.; Eskandari, R. Pediatric hydrocephalus: Current state of diagnosis and treatment. Pediatr. Rev. 2016, 37, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.; Rosell, M.; Kockum, K.; Lilja-Lund, O.; Söderström, L.; Laurell, K. Prevalence of idiopathic normal pressure hydrocephalus: A prospective, population-based study. PLoS ONE 2019, 14, e0217705. [Google Scholar] [CrossRef]
- Fowler, J.B.; De Jesus, O.; Mesfin, F.B. Ventriculoperitoneal Shunt; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Akyol, M.E.; Cetin, E. Effects of shunt types used in idiopathic normal pressure hydrocephalus on patients’ clinical outcomes. Ann. Med. Res. 2023, 30, 146–281. [Google Scholar] [CrossRef]
- Shannon, C.N.; Carr, K.R.; Tomycz, L.; Wellons, J.C.; Tulipan, N. Time to first shunt failure in pediatric patients over 1 year old: A 10-year retrospective study. Pediatr. Neurosurg. 2015, 49, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.C.; Guo, W. Have we made progress in preventing shunt failure? A critical analysis. J. Neurosurg. Pediatr. 2008, 1, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.J.; Chiang, W.C.; Huang, H.Y.; Lin, S.Z.; Tsai, S.T. Effectiveness and safety of ventriculoperitoneal shunt versus lumboperitoneal shunt for communicating hydrocephalus: A systematic review and meta-analysis with trial sequential analysis. CNS Neurosci. Ther. 2023, 29, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-H.; Chang, C.-S.; Sung, W.-W.; Liu, J.-T. Lumboperitoneal shunt: A new modified surgical technique and a comparison of the complications with ventriculoperitoneal shunt in a single center. Medicina 2019, 55, 643. [Google Scholar] [CrossRef] [PubMed]
- Yadav, Y.R.; Parihar, V.; Pande, S.; Namdev, H.; Agarwal, M. Endoscopic third ventriculostomy. J. Neurosci. Rural Pract. 2012, 3, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Vogel, T.W.; Bahuleyan, B.; Robinson, S.; Cohen, A.R. The role of endoscopic third ventriculostomy in the treatment of hydrocephalus. J. Neurosurg. Pediatr. 2013, 12, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Marano, P.J.; Stone, S.S.; Mugamba, J.; Ssenyonga, P.; Warf, E.B.; Warf, B.C. Reopening of an obstructed third ventriculostomy: Long-term success and factors affecting outcome in 215 infants. J. Neurosurg. Pediatr. 2015, 15, 399–405. [Google Scholar] [CrossRef]
- Kulkarni, A.V.; Drake, J.M.; Mallucci, C.L.; Sgouros, S.; Roth, J.; Constantini, S.; Canadian Pediatric Neurosurgery Study Group. Endoscopic third ventriculostomy in the treatment of childhood hydrocephalus. J. Pediatr. 2009, 155, 254–259.e1. [Google Scholar] [CrossRef]
- Riva-Cambrin, J.; Kestle, J.R.; Rozzelle, C.J.; Naftel, R.P.; Alvey, J.S.; Reeder, R.W.; Holubkov, R.; Browd, S.R.; Cochrane, D.D.; Limbrick, D.D. Predictors of success for combined endoscopic third ventriculostomy and choroid plexus cauterization in a North American setting: A Hydrocephalus Clinical Research Network study. J. Neurosurg. Pediatr. 2019, 24, 128–138. [Google Scholar] [CrossRef]
- Zaben, M.; Manivannan, S.; Sharouf, F.; Hammad, A.; Patel, C.; Bhatti, I.; Leach, P. The efficacy of endoscopic third ventriculostomy in children 1 year of age or younger: A systematic review and meta-analysis. Eur. J. Paediatr. Neurol. 2020, 26, 7–14. [Google Scholar] [CrossRef]
- Warf, B.C.; Weber, D.S.; Day, E.L.; Riordan, C.P.; Staffa, S.J.; Baird, L.C.; Fehnel, K.P.; Stone, S.S. Endoscopic third ventriculostomy with choroid plexus cauterization: Predictors of long-term success and comparison with shunt placement for primary treatment of infant hydrocephalus. J. Neurosurg. Pediatr. 2023, 32, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Schiff, S.J.; Kulkarni, A.V.; Mbabazi-Kabachelor, E.; Mugamba, J.; Ssenyonga, P.; Donnelly, R.; Levenbach, J.; Monga, V.; Peterson, M.; Cherukuri, V. Brain growth after surgical treatment for infant postinfectious hydrocephalus in Sub-Saharan Africa: 2-year results of a randomized trial. J. Neurosurg. Pediatr. 2021, 28, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Bass, N.H.; Fällström, S.; Lundborg, P. Digoxin-induced arrest of the cerebrospinal fluid circulation in the infant rat: Implications for medical treatment of hydrocephalus during early postnatal life. Pediatr. Res. 1979, 13, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.; Winer, J.L.; Kitase, Y.; Brigman, J.L.; Jantzie, L.L. Neonatal administration of erythropoietin attenuates cognitive deficits in adult rats following placental insufficiency. J. Neurosci. Res. 2022, 100, 2112–2126. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.; Conteh, F.S.; Oppong, A.Y.; Yellowhair, T.R.; Newville, J.C.; Demerdash, N.E.; Shrock, C.L.; Maxwell, J.R.; Jett, S.; Northington, F.J. Extended combined neonatal treatment with erythropoietin plus melatonin prevents posthemorrhagic hydrocephalus of prematurity in rats. Front. Cell. Neurosci. 2018, 12, 322. [Google Scholar] [CrossRef] [PubMed]
- Jantzie, L.L.; Muthukumar, S.; Kitase, Y.; Vasan, V.; Fouda, M.A.; Hamimi, S.; Burkhardt, C.; Burton, V.J.; Gerner, G.; Scafidi, J. Infantile Cocktail of Erythropoietin and Melatonin Restores Gait in Adult Rats with Preterm Brain Injury. Dev. Neurosci. 2022, 44, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Safety of Erythropoietin and Melatonin for Very Preterm Infants with Intraventricular Hemorrhage (SCEMPI). Available online: https://clinicaltrials.gov/study/NCT05617833?term=robinson&intr=erythropoietin&rank=1 (accessed on 14 January 2024).
- Zhao, J.; Chen, Z.; Xi, G.; Keep, R.F.; Hua, Y. Deferoxamine attenuates acute hydrocephalus after traumatic brain injury in rats. Transl. Stroke Res. 2014, 5, 586–594. [Google Scholar] [CrossRef]
- Pang, D.; Sclabassi, R.J.; Horton, J.A. Lysis of intraventricular blood clot with urokinase in a canine model: Part 1. Canine intraventricular blood cast model. Neurosurgery 1986, 19, 540–546. [Google Scholar] [CrossRef]
- Chua, C.O.; Chahboune, H.; Braun, A.; Dummula, K.; Chua, C.E.; Yu, J.; Ungvari, Z.; Sherbany, A.A.; Hyder, F.; Ballabh, P. Consequences of intraventricular hemorrhage in a rabbit pup model. Stroke 2009, 40, 3369–3377. [Google Scholar] [CrossRef]
- Kiseleva, Z.N.; Volzhina, N.S. Experimental hydrocephalus in young rats. Arkhiv Patol. 1957, 19, 44–52. [Google Scholar]
- Pudenz, R.H. Experimental and clinical observations on the shunting of cerebrospinal fluid into the circulatory system. Neurosurgery 1958, 5, 98–115. [Google Scholar] [CrossRef] [PubMed]
- McAllister, J.P.; Talcott, M.R.; Isaacs, A.M.; Zwick, S.H.; Garcia-Bonilla, M.; Castaneyra-Ruiz, L.; Hartman, A.L.; Dilger, R.N.; Fleming, S.A.; Golden, R.K. A novel model of acquired hydrocephalus for evaluation of neurosurgical treatments. Fluids Barriers CNS 2021, 18, 49. [Google Scholar] [CrossRef] [PubMed]
- da Silva Lopes, L.; Slobodian, I.; Del Bigio, M.R. Characterization of juvenile and young adult mice following induction of hydrocephalus with kaolin. Exp. Neurol. 2009, 219, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Darder, J.; Barbera, J.; Cerda-Nicolas, M.; Segura, D.; Broseta, J.; Barcia-Salorio, J.L. Sequential morphological and functional changes in kaolin-induced hydrocephalus. J. Neurosurg. 1984, 61, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, D.K.; Harrison, M.R.; Berger, M.S.; Chinn, D.H.; Halks-Miller, M.; Edwards, M.S. Correction of congenital hydrocephalus in utero I. The model: Intracisternal kaolin produces hydrocephalus in fetal lambs and rhesus monkeys. J. Pediatr. Surg. 1983, 18, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Marlin, A.; Wald, A.; Hochwald, G.; Malhan, C. Kaolin-induced hydrocephalus impairs CSF secretion by the choroid plexus. Neurology 1978, 28, 945. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, Y.; Cui, W.; Cao, Y.; Zhao, L.; Wang, H.; Liu, X.; Fan, S.; Huang, K.; Tong, A. Inhibition of neuronal necroptosis mediated by RIP1/RIP3/MLKL provides neuroprotective effects on kaolin-induced hydrocephalus in mice. Cell Prolif. 2021, 54, e13108. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, X.; Guo, J.; Yu, C.; Yang, J. Molecular mechanisms and risk factors for the pathogenesis of hydrocephalus. Front. Genet. 2022, 12, 777926. [Google Scholar] [CrossRef]
- Yamada, H.; Oi, S.; Tamaki, N.; Matsumoto, S.; Taomoto, K. Embryopathoetiology of Congenital Hydrocephalus in Experimental Models: A Comparative Morphological Study in Two Different Models. In Hydrocephalus: Pathogenesis and Treatment; Springer: Tokyo, Japan, 1991; pp. 27–35. [Google Scholar]
- Stambolliu, E.; Ioakeim-Ioannidou, M.; Kontokostas, K.; Dakoutrou, M.; Kousoulis, A.A. The most common comorbidities in Dandy-Walker syndrome patients: A systematic review of case reports. J. Child Neurol. 2017, 32, 886–902. [Google Scholar] [CrossRef]
- Chamberlain, J. Early neurovascular abnormalities underlying 6-aminonicotinamide (6-AN)-induced congenital hydrocephalus in rats. Teratology 1970, 3, 377–387. [Google Scholar] [CrossRef]
- Gattone, V.H.; Tourkow, B.A.; Trambaugh, C.M.; Yu, A.C.; Whelan, S.; Phillips, C.L.; Harris, P.C.; Peterson, R.G. Development of multiorgan pathology in the wpk rat model of polycystic kidney disease. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. Off. Publ. Am. Assoc. Anat. 2004, 277, 384–395. [Google Scholar] [CrossRef]
- Shim, J.W.; Territo, P.R.; Simpson, S.; Watson, J.C.; Jiang, L.; Riley, A.A.; McCarthy, B.; Persohn, S.; Fulkerson, D.; Blazer-Yost, B.L. Hydrocephalus in a rat model of Meckel Gruber syndrome with a TMEM67 mutation. Sci. Rep. 2019, 9, 1069. [Google Scholar] [CrossRef] [PubMed]
- Hochstetler, A.; Smith, H.; Reed, M.; Hulme, L.; Territo, P.; Bedwell, A.; Persohn, S.; Perrotti, N.; D’Antona, L.; Musumeci, F. Inhibition of serum-and glucocorticoid-induced kinase 1 ameliorates hydrocephalus in preclinical models. Fluids Barriers CNS 2023, 20, 61. [Google Scholar] [CrossRef]
- Jones, H.C.; Carter, B.J.; Morel, L. Characteristics of hydrocephalus expression in the LEW/Jms rat strain with inherited disease. Child’s Nerv. Syst. 2003, 19, 11–18. [Google Scholar] [CrossRef]
- Itoh, K.; Fushiki, S. The role of L1 cam in murine corticogenesis, and the pathogenesis of hydrocephalus. Pathol. Int. 2015, 65, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Swetloff, A.; Ferretti, P. Changes in E2F5 intracellular localization in mouse and human choroid plexus epithelium with development. Int. J. Dev. Biol. 2004, 49, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Banizs, B.; Pike, M.M.; Millican, C.L.; Ferguson, W.B.; Komlosi, P.; Sheetz, J.; Bell, P.D.; Schwiebert, E.M.; Yoder, B.K. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 2005, 132, 5329–5339. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, G.J.; Dagnino, L.; Gaubatz, S.; Xu, Y.; Bronson, R.T.; Warren, H.B.; Livingston, D.M. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev. 1998, 12, 1092–1098. [Google Scholar] [CrossRef]
- Lewis, W.R.; Malarkey, E.B.; Tritschler, D.; Bower, R.; Pasek, R.C.; Porath, J.D.; Birket, S.E.; Saunier, S.; Antignac, C.; Knowles, M.R. Mutation of growth arrest specific 8 reveals a role in motile cilia function and human disease. PLoS Genet. 2016, 12, e1006220. [Google Scholar] [CrossRef]
- Hochstetler, A.E.; Whitehouse, L.; Antonellis, P.; Berbari, N.F.; Blazer-Yost, B.L. Characterizing the Expression of TRPV4 in the Choroid Plexus Epithelia as a Prospective Component in the Development of Hydrocephalus in the Gas8GT Juvenile Mouse Model. FASEB J. 2018, 32, C1823–C1842. [Google Scholar] [CrossRef]
- Abdelhamed, Z.; Vuong, S.M.; Hill, L.; Shula, C.; Timms, A.; Beier, D.; Campbell, K.; Mangano, F.T.; Stottmann, R.W.; Goto, J. A mutation in Ccdc39 causes neonatal hydrocephalus with abnormal motile cilia development in mice. Development 2018, 145, dev154500. [Google Scholar] [CrossRef] [PubMed]
- Davy, B.E.; Robinson, M.L. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum. Mol. Genet. 2003, 12, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Bronson, R.T.; Lane, P.W. Hydrocephalus with hop gait (hyh): A new mutation on chromosome 7 in the mouse. Dev. Brain Res. 1990, 54, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Di Curzio, D.L. Animal models of hydrocephalus. Open J. Mod. Neurosurg. 2017, 8, 57–71. [Google Scholar] [CrossRef]
- Jones, H.; Dack, S.; Ellis, C. Morphological aspects of the development of hydrocephalus in a mouse mutant (SUMS/NP). Acta Neuropathol. 1987, 72, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Hochstetler, A.E.; Smith, H.M.; Preston, D.C.; Reed, M.M.; Territo, P.R.; Shim, J.W.; Fulkerson, D.; Blazer-Yost, B.L. TRPV4 antagonists ameliorate ventriculomegaly in a rat model of hydrocephalus. JCI Insight 2020, 5, e137646. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, F.; Draper, C.E.; Bannister, C.M.; Pourghasem, M.; Owen-Lynch, P.J.; Miyan, J.A. Deficient cortical development in the hydrocephalic Texas (H-Tx) rat: A role for CSF. Brain 2002, 125, 1859–1874. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.; Bucknall, R. Inherited prenatal hydrocephalus in the H–Tx rat: A morphological study. Neuropathol. Appl. Neurobiol. 1988, 14, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Kuwamura, M.; Kinoshita, A.; Okumoto, M.; Yamate, J.; Mori, N. Hemorrhagic hydrocephalus (hhy): A novel mutation on mouse chromosome 12. Dev. Brain Res. 2004, 152, 69–72. [Google Scholar] [CrossRef]
- Lin, X.; Liu, B.; Yang, X.; Yue, X.; Diao, L.; Wang, J.; Chang, J. Genetic deletion of Rnd3 results in aqueductal stenosis leading to hydrocephalus through up-regulation of Notch signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 8236–8241. [Google Scholar] [CrossRef]
- Kume, T.; Deng, K.-Y.; Winfrey, V.; Gould, D.B.; Walter, M.A.; Hogan, B.L. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell 1998, 93, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Fernández-Llebrez, P.; Bach, A.; Robert, B.; Soriano, E. Msx1 disruption leads to diencephalon defects and hydrocephalus. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2004, 230, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.; Lehtinen, M.K. Experimental approaches for manipulating choroid plexus epithelial cells. Fluids Barriers CNS 2022, 19, 36. [Google Scholar] [CrossRef] [PubMed]
- Millar, I.D.; Bruce, J.I.; Brown, P.D. Ion channel diversity, channel expression and function in the choroid plexuses. Cerebrospinal Fluid Res. 2007, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Damkier, H.H.; Brown, P.D.; Praetorius, J. Cerebrospinal fluid secretion by the choroid plexus. Physiol. Rev. 2013, 93, 1847–1892. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, Z.; Chen, Y.; Liao, J.; Wang, Y.; Liu, J.; Lin, Z.; Xiao, G. Choroid plexus epithelium and its role in neurological diseases. Front. Mol. Neurosci. 2022, 15, 949231. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; He, J.; Wang, Y.; Wang, C.; Tan, C.; Liao, J.; Tong, L.; Xiao, G. Targeting choroid plexus epithelium as a novel therapeutic strategy for hydrocephalus. J. Neuroinflamm. 2022, 19, 156. [Google Scholar] [CrossRef] [PubMed]
- Sakka, L.; Coll, G.; Chazal, J. Anatomy and physiology of cerebrospinal fluid. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2011, 128, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Oernbo, E.K.; Steffensen, A.B.; Razzaghi Khamesi, P.; Toft-Bertelsen, T.L.; Barbuskaite, D.; Vilhardt, F.; Gerkau, N.J.; Tritsaris, K.; Simonsen, A.H.; Lolansen, S.D. Membrane transporters control cerebrospinal fluid formation independently of conventional osmosis to modulate intracranial pressure. Fluids Barriers CNS 2022, 19, 65. [Google Scholar] [CrossRef]
- Wright, E.M. Mechanisms of ion transport across the choroid plexus. J. Physiol. 1972, 226, 545–571. [Google Scholar] [CrossRef]
- Bouillé, C.; Mesnil, M.; Barriere, H.; Gabrion, J. Gap junctional intercellular communication between cultured ependymal cells, revealed by lucifer yellow CH transfer and freeze-fracture. Glia 1991, 4, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhao, Q.; Graziano, J.H. Primary culture of choroidal epithelial cells: Characterization of an in vitro model of blood-CSF barrier. Vitr. Cell. Dev. Biol.-Anim. 1998, 34, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.E.; Sanders-Bush, E. Sodium-dependent antiporters in choroid plexus epithelial cultures from rabbit. J. Neurochem. 1993, 60, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Baehr, C.; Reichel, V.; Fricker, G. Choroid plexus epithelial monolayers—A cell culture model from porcine brain. Cerebrospinal Fluid Res. 2006, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Harter, D.; Hsu, K.; Rose, H. Immunofluorescence and cytochemical studies of visna virus in cell culture. J. Virol. 1967, 1, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Whittico, M.T.; Hui, A.C.; Giacomini, K.M. Preparation of brush border membrane vesicles from bovine choroid plexus. J. Pharmacol. Methods 1991, 25, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Monnot, A.D.; Zheng, W. Culture of choroid plexus epithelial cells and in vitro model of blood–CSF barrier. Epithel. Cell Cult. Protoc. 2013, 945, 13–29. [Google Scholar]
- Fejes, Z.; Pócsi, M.; Takai, J.; Erdei, J.; Tóth, A.; Balogh, E.; Rusznyák, Á.; Fenyvesi, F.; Nagy, A.; Kappelmayer, J. Preterm intraventricular hemorrhage-induced inflammatory response in human choroid plexus epithelial cells. Int. J. Mol. Sci. 2021, 22, 8648. [Google Scholar] [CrossRef] [PubMed]
- Guy, B.; Zhang, J.S.; Duncan, L.H.; Johnston, R.J., Jr. Human neural organoids: Models for developmental neurobiology and disease. Dev. Biol. 2021, 478, 102–121. [Google Scholar] [CrossRef]
- Schroten, M.; Hanisch, F.-G.; Quednau, N.; Stump, C.; Riebe, R.; Lenk, M.; Wolburg, H.; Tenenbaum, T.; Schwerk, C. A novel porcine in vitro model of the blood-cerebrospinal fluid barrier with strong barrier function. PLoS ONE 2012, 7, e39835. [Google Scholar] [CrossRef]
- Hochstetler, A.; Hulme, L.; Delpire, E.; Schwerk, C.; Schroten, H.; Preston, D.; Simpson, S.; Blazer-Yost, B.L. Porcine choroid plexus-riems cell line demonstrates altered polarization of transport proteins compared with the native epithelium. Am. J. Physiol.-Cell Physiol. 2022, 323, C1–C13. [Google Scholar] [CrossRef] [PubMed]
- Lauer, A.N.; März, M.; Meyer, S.; Meurer, M.; de Buhr, N.; Borkowski, J.; Weiß, C.; Schroten, H.; Schwerk, C. Optimized cultivation of porcine choroid plexus epithelial cells, a blood–cerebrospinal fluid barrier model, for studying granulocyte transmigration. Lab. Investig. 2019, 99, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Hulme, L.; Hochstetler, A.; Schwerk, C.; Schroten, H.; Ishikawa, H.; Tung, C.-Y.; Perrin, B.; Blazer-Yost, B. Characterization of TRPV4-mediated signaling pathways in an optimized human choroid plexus epithelial cell line. Am. J. Physiol.-Cell Physiol. 2022, 323, C1823–C1842. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhao, Q. Establishment and characterization of an immortalized Z310 choroidal epithelial cell line from murine choroid plexus. Brain Res. 2002, 958, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Dziegielewska, K.M.; Ek, C.J.; Habgood, M.D.; Bauer, H.; Bauer, H.-C.; Lindsay, H.; Wakefield, M.J.; Strazielle, N.; Kratzer, I. Mechanisms that determine the internal environment of the developing brain: A transcriptomic, functional and ultrastructural approach. PLoS ONE 2016, 11, e0147680. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Zheng, W. Establishment of an in vitro brain barrier epithelial transport system for pharmacological and toxicological study. Brain Res. 2005, 1057, 37–48. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zheng, W. Toxicology of choroid plexus: Special reference to metal-induced neurotoxicities. Microsc. Res. Tech. 2001, 52, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, I.; Ishiwat, C.; Ishiwata, E.; Sato, Y.; Kiguchi, K.; Tachibana, T.; Hashimoto, H.; Ishikawa, H. Establishment and characterization of a human malignant choroids plexus papilloma cell line (HIBCPP). Hum. Cell 2005, 18, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, W.; Schwerk, C.; Herold, R.; Stump-Guthier, C.; Lampe, M.; Fallier-Becker, P.; Weiß, C.; Sticht, C.; Ishikawa, H.; Schroten, H. Immortalized human choroid plexus endothelial cells enable an advanced endothelial-epithelial two-cell type in vitro model of the choroid plexus. Iscience 2022, 25, 104383. [Google Scholar] [CrossRef]
- Giovannucci, T.A.; Leckey, C.A.; Moncur, E.; Tariq, K.; Thorne, L.; Watkins, L.; Toma, A.; Fox, N.C.; Bateman, R.J.; Mills, K. Choroid plexus protein turnover in human choroid plexus organoids recapitulates turnover in humans measured using stable isotope labeling kinetics (SILK). Alzheimer’s Dement. 2023, 19, e074240. [Google Scholar] [CrossRef]
- Praetorius, J.; Nielsen, S. Distribution of sodium transporters and aquaporin-1 in the human choroid plexus. Am. J. Physiol. -Cell Physiol. 2006, 291, C59–C67. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N.; Toft-Bertelsen, T.L. Dual function of the choroid plexus: Cerebrospinal fluid production and control of brain ion homeostasis. Cell Calcium 2023, 116, 102797. [Google Scholar] [CrossRef] [PubMed]
- Roepke, T.K.; Kanda, V.A.; Purtell, K.; King, E.C.; Lerner, D.J.; Abbott, G.W. KCNE2 forms potassium channels with KCNA3 and KCNQ1 in the choroid plexus epithelium. FASEB J. 2011, 25, 4264. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.; Lu, J.; Mount, D.; Delpire, E. Localization of the K+–Cl− cotransporter, KCC3, in the central and peripheral nervous systems: Expression in the choroid plexus, large neurons and white matter tracts. Neuroscience 2001, 103, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Leefmans, F.J. CrossTalk proposal: Apical NKCC1 of choroid plexus epithelial cells works in the net inward flux mode under basal conditions, maintaining intracellular Cl− and cell volume. J. Physiol. 2020, 598, 4733–4736. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N.; Rose, C. CrossTalk opposing view: NKCC1 in the luminal membrane of choroid plexus is outwardly directed under basal conditions and contributes directly to cerebrospinal fluid secretion. J. Physiol. 2020, 598, 4737–4739. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Leefmans, F.J. Rebuttal from Francisco J. Alvarez-Leefmans. J. Physiol. 2020, 598, 4741–4742. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N.; Rose, C.R. Rebuttal from Nanna MacAulay and Christine R. Rose. J. Physiol. 2020, 598, 4743. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Gagnon, K.B. Elusive role of the Na-K-2Cl cotransporter in the choroid plexus. Am. J. Physiol.-Cell Physiol. 2019, 316, C522–C524. [Google Scholar] [CrossRef]
- Fame, R.M.; Xu, H.; Pragana, A.; Lehtinen, M. Age-appropriate potassium clearance from perinatal cerebrospinal fluid depends on choroid plexus NKCC1. Fluids Barriers CNS 2023, 20, 45. [Google Scholar] [CrossRef]
- Zeuthen, T.; MacAulay, N. Cotransport of water by Na+–K+–2Cl− cotransporters expressed in Xenopus oocytes: NKCC1 versus NKCC2. J. Physiol. 2012, 590, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Zhang, Y.; Liu, T.; Friedel, P.; Zhuo, W.; Somasekharan, S.; Roy, K.; Zhang, L.; Liu, Y. The structural basis of function and regulation of neuronal cotransporters NKCC1 and KCC2. Commun. Biol. 2021, 4, 226. [Google Scholar] [CrossRef] [PubMed]
- Macvicar, B.A.; Feighan, D.; Brown, A.; Ransom, B. Intrinsic optical signals in the rat optic nerve: Role for K+ uptake via NKCC1 and swelling of astrocytes. Glia 2002, 37, 114–123. [Google Scholar] [CrossRef]
- Chou, C.-L.; Yu, M.-J.; Kassai, E.M.; Morris, R.G.; Hoffert, J.D.; Wall, S.M.; Knepper, M.A. Roles of basolateral solute uptake via NKCC1 and of myosin II in vasopressin-induced cell swelling in inner medullary collecting duct. Am. J. Physiol.-Ren. Physiol. 2008, 295, F192–F201. [Google Scholar] [CrossRef]
- Blazer-Yost, B.L. Consideration of Kinase Inhibitors for the Treatment of Hydrocephalus. Int. J. Mol. Sci. 2023, 24, 6673. [Google Scholar] [CrossRef]
- Koumangoye, R.; Bastarache, L.; Delpire, E. NKCC1: Newly found as a human disease-causing ion transporter. Function 2021, 2, zqaa028. [Google Scholar] [CrossRef] [PubMed]
- Gregoriades, J.M.; Madaris, A.; Alvarez, F.J.; Alvarez-Leefmans, F.J. Genetic and pharmacological inactivation of apical Na+-K+-2Cl− cotransporter 1 in choroid plexus epithelial cells reveals the physiological function of the cotransporter. Am. J. Physiol. Cell Physiol. 2019, 316, C525–C544. [Google Scholar] [CrossRef]
- Szymanski, J.; Minichiello, L. NKCC1 deficiency in forming hippocampal circuits triggers neurodevelopmental disorder: Role of BDNF-TrkB signaling. Brain Sci. 2022, 12, 502. [Google Scholar] [CrossRef]
- Karimy, J.K.; Zhang, J.; Kurland, D.B.; Theriault, B.C.; Duran, D.; Stokum, J.A.; Furey, C.G.; Zhou, X.; Mansuri, M.S.; Montejo, J. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat. Med. 2017, 23, 997–1003. [Google Scholar] [CrossRef]
- Sadegh, C.; Xu, H.; Sutin, J.; Fatou, B.; Gupta, S.; Pragana, A.; Taylor, M.; Kalugin, P.N.; Zawadzki, M.E.; Alturkistani, O. Choroid plexus-targeted NKCC1 overexpression to treat post-hemorrhagic hydrocephalus. Neuron 2023, 111, 1591–1608.e4. [Google Scholar] [CrossRef]
- Bothwell, S.W.; Omileke, D.; Patabendige, A.; Spratt, N.J. CSF secretion is not altered by NKCC1 nor TRPV4 antagonism in healthy rats. Brain Sci. 2021, 11, 1117. [Google Scholar] [CrossRef] [PubMed]
- Savardi, A.; Borgogno, M.; De Vivo, M.; Cancedda, L. Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol. Sci. 2021, 42, 1009–1034. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, K. TRPV4 ion channel as important cell sensors. J. Anesth. 2016, 30, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, W.; Choe, Y.; Martí-Renom, M.A.; Bell, A.M.; Denis, C.S.; Hudspeth, A.; Friedman, J.M.; Heller, S. Vanilloid receptor–related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T. TRPV4 calcium entry channel: A paradigm for gating diversity. Am. J. Physiol. Cell Physiol. 2004, 286, C195–C205. [Google Scholar] [CrossRef] [PubMed]
- Strotmann, R.; Harteneck, C.; Nunnenmacher, K.; Schultz, G.; Plant, T.D. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2000, 2, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Darby, W.; Grace, M.; Baratchi, S.; McIntyre, P. Modulation of TRPV4 by diverse mechanisms. Int. J. Biochem. Cell Biol. 2016, 78, 217–228. [Google Scholar] [CrossRef]
- Liedtke, W.; Friedman, J.M. Abnormal osmotic regulation in trpv4-/-mice. Proc. Natl. Acad. Sci. USA 2003, 100, 13698–13703. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, K.; Suzuki, M.; Mizuno, A.; Hara, A. Hearing impairment in TRPV4 knockout mice. Neurosci. Lett. 2005, 382, 304–308. [Google Scholar] [CrossRef]
- Goyal, N.; Skrdla, P.; Schroyer, R.; Kumar, S.; Fernando, D.; Oughton, A.; Norton, N.; Sprecher, D.L.; Cheriyan, J. Clinical pharmacokinetics, safety, and tolerability of a novel, first-in-class TRPV4 ion channel inhibitor, GSK2798745, in healthy and heart failure subjects. Am. J. Cardiovasc. Drugs 2019, 19, 335–342. [Google Scholar] [CrossRef]
- Lawhorn, B.G.; Brnardic, E.J.; Behm, D.J. TRPV4 antagonists: A patent review (2015–2020). Expert Opin. Ther. Pat. 2021, 31, 773–784. [Google Scholar] [CrossRef]
- Park, S.; Ku, S.K.; Ji, H.W.; Choi, J.-H.; Shin, D.M. Ca(2+) is a Regulator of the WNK/OSR1/NKCC Pathway in a Human Salivary Gland Cell Line. Korean J. Physiol. Pharmacol. 2015, 19, 249–255. [Google Scholar] [CrossRef][Green Version]
- Toft-Bertelsen, T.L.; Barbuskaite, D.; Heerfordt, E.K.; Lolansen, S.D.; Andreassen, S.N.; Rostgaard, N.; Olsen, M.H.; Norager, N.H.; Capion, T.; Rath, M.F. Lysophosphatidic acid as a CSF lipid in posthemorrhagic hydrocephalus that drives CSF accumulation via TRPV4-induced hyperactivation of NKCC1. Fluids Barriers CNS 2022, 19, 69. [Google Scholar] [CrossRef]
- Jin, S.C.; Furey, C.G.; Zeng, X.; Allocco, A.; Nelson-Williams, C.; Dong, W.; Karimy, J.K.; Wang, K.; Ma, S.; Delpire, E. SLC12A ion transporter mutations in sporadic and familial human congenital hydrocephalus. Mol. Genet. Genom. Med. 2019, 7, e892. [Google Scholar] [CrossRef]
- Kahle, K.T.; Flores, B.; Bharucha-Goebel, D.; Zhang, J.; Donkervoort, S.; Hegde, M.; Begum, G.; Duran, D.; Liang, B.; Sun, D. Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci. Signal. 2016, 9, ra77. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Alessi, D.R. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signaling pathway. J. Cell Sci. 2008, 121, 3293–3304. [Google Scholar] [CrossRef] [PubMed]
- Lauf, P.K.; Bauer, J. Direct evidence for chloride-dependent volume reduction in macrocytic sheep reticulocytes. Biochem. Biophys. Res. Commun. 1987, 144, 849–855. [Google Scholar] [CrossRef]
- Mercado, A.; Broumand, V.; Zandi-Nejad, K.; Enck, A.H.; Mount, D.B. A C-terminal domain in KCC2 confers constitutive K+-Cl-cotransport. J. Biol. Chem. 2006, 281, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Quinton, P.M. Chloride impermeability in cystic fibrosis. Nature 1983, 301, 421–422. [Google Scholar] [CrossRef]
- Bear, C.E.; Li, C.; Kartner, N.; Bridges, R.J.; Jensen, T.J.; Ramjeesingh, M.; Riordan, J.R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 1992, 68, 809–818. [Google Scholar] [CrossRef]
- Genovese, M.; Borrelli, A.; Venturini, A.; Guidone, D.; Caci, E.; Viscido, G.; Gambardella, G.; di Bernardo, D.; Scudieri, P.; Galietta, L.J. TRPV4 and purinergic receptor signalling pathways are separately linked in airway epithelia to CFTR and TMEM16A chloride channels. J. Physiol. 2019, 597, 5859–5878. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, L.Ø.; Friis, K.A.; Damkier, H.H. In vitro investigation of the effect of proinflammatory cytokines on mouse choroid plexus membrane transporters Ncbe and NKCC1. Fluids Barriers CNS 2023, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, J.; Damkier, H.H. Transport across the choroid plexus epithelium. Am. J. Physiol. Cell Physiol. 2017, 312, C673–C686. [Google Scholar] [CrossRef] [PubMed]
- Damkier, H.H.; Praetorius, J. Genetic ablation of Slc4a10 alters the expression pattern of transporters involved in solute movement in the mouse choroid plexus. Am. J. Physiol. Cell Physiol. 2012, 302, C1452–C1459. [Google Scholar] [CrossRef] [PubMed]
- Christensen, I.B.; Wu, Q.; Bohlbro, A.S.; Skals, M.G.; Damkier, H.H.; Hübner, C.A.; Fenton, R.A.; Praetorius, J. Genetic disruption of slc4a10 alters the capacity for cellular metabolism and vectorial ion transport in the choroid plexus epithelium. Fluids Barriers CNS 2020, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Li, Q.; So, I.; Huang, C.-L.; Ando, H.; Mizutani, A.; Seki, G.; Mikoshiba, K.; Thomas, P.J.; Muallem, S. IRBIT governs epithelial secretion in mice by antagonizing the WNK/SPAK kinase pathway. J. Clin. Investig. 2011, 121, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Ruusuvuori, E.; Sipilä, S.T.; Haapanen, A.; Damkier, H.H.; Kurth, I.; Hentschke, M.; Schweizer, M.; Rudhard, Y.; Laatikainen, L.M. Mice with targeted Slc4a10 gene disruption have small brain ventricles and show reduced neuronal excitability. Proc. Natl. Acad. Sci. USA 2008, 105, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Gurnett, C.A.; Veile, R.; Zempel, J.; Blackburn, L.; Lovett, M.; Bowcock, A. Disruption of sodium bicarbonate transporter SLC4A10 in a patient with complex partial epilepsy and mental retardation. Arch. Neurol. 2008, 65, 550–553. [Google Scholar] [CrossRef]
- Fasham, J.; Huebner, A.K.; Liebmann, L.; Khalaf-Nazzal, R.; Maroofian, R.; Kryeziu, N.; Wortmann, S.B.; Leslie, J.S.; Ubeyratna, N.; Mancini, G.M. SLC4A10 mutation causes a neurological disorder associated with impaired GABAergic transmission. Brain 2023, 146, 4547–4561. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Type | Benefits | Complications | References |
|---|---|---|---|
| Ventriculoperitoneal shunts (VPS) | Treats both communicating and non-communicating hydrocephalus Effective for patients suffering from iNPH | Failure due to mechanical problems, catheter migration, cell overgrowth Fails frequently in pediatric patients; surgery required for shunt revisions Possibility of infection | [21,22,23,24] |
| Lumboperitoneal shunts (LPS) | Does not require brain surgery Decreases chances of brain hemorrhage | Cannot be used for non-communicating hydrocephalus Failure due to mechanical problems, catheter migration, cell overgrowth Possibility of infection | [25,26] |
| Endoscopic third ventriculostomy (ETV) | Does not require a shunt | Pediatric patients do not respond to treatment Requires brain surgery Possibility of infection | [27,28,29,30] |
| Endoscopic third ventriculostomy/choroid plexus cauterization (ETV + CPC) | Lower need for re-operation when compared to ETV alone May be more beneficial in developing countries | Long-term effects of CPC have not been studied Possibility of infection | [31,32,33,34] |
| Disease Model | Animal Models | Type of Hydrocephalus | References |
|---|---|---|---|
| Hydrocephalus caused by traumatic brain injury | Fluid percussion injury model with injection of FeCl3 | Communicating | [40] |
| Post-hemorrhagic hydrocephalus | Induced models using injections of blood, red blood cells, iron, hemoglobin, or glycerol | Communicating | [10,37,41,42] |
| Chemically induced hydrocephalus | Kaolin-injected models, 6-AN rats | Communicating | [43,44,45,46,47,48,49,50,51,52,53,54] |
| Genetic hydrocephalus models | Rat: Wpk, LEW/Jms Mouse: L1CAM, E2F5, Gas8, CCDC39, Hy-3, Hpy, Hyh, Msx1, SUMS/NP | Communicating | [55,56,57,58,59,60,61,62,63,64,65,66,67,68,69] |
| Genetic hydrocephalus models | Rat: H-Tx Mouse: Rnd3, Hhy | Non-communicating | [70,71,72,73,74] |
| Genetic model of post-hemorrhagic hydrocephalus | Mouse: Mf1 Mouse: Hhy | Communicating Non-communicating | [73,74,75] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newland, V.; Jantzie, L.L.; Blazer-Yost, B.L. Understanding and Modeling the Pathophysiology of Hydrocephalus: In Search of Better Treatment Options. Physiologia 2024, 4, 182-201. https://doi.org/10.3390/physiologia4020010
Newland V, Jantzie LL, Blazer-Yost BL. Understanding and Modeling the Pathophysiology of Hydrocephalus: In Search of Better Treatment Options. Physiologia. 2024; 4(2):182-201. https://doi.org/10.3390/physiologia4020010
Chicago/Turabian StyleNewland, Verayna, Lauren L. Jantzie, and Bonnie L. Blazer-Yost. 2024. "Understanding and Modeling the Pathophysiology of Hydrocephalus: In Search of Better Treatment Options" Physiologia 4, no. 2: 182-201. https://doi.org/10.3390/physiologia4020010
APA StyleNewland, V., Jantzie, L. L., & Blazer-Yost, B. L. (2024). Understanding and Modeling the Pathophysiology of Hydrocephalus: In Search of Better Treatment Options. Physiologia, 4(2), 182-201. https://doi.org/10.3390/physiologia4020010