1. Introduction
Zeolites are crystalline, microporous materials renowned for their molecular sieving and catalytic applications [
1,
2]. The aluminosilicate framework is assembled from silica and alumina tetrahedra linked through apical oxygen atoms. Charge-compensating cations reside in the zeolite pores to render an overall neutral framework. Existing zeolite materials are often modified to optimise their performance in many applications. For example, cation exchange can be used to tailor the pore architecture of a zeolite to significantly improve its selectivity, adsorption and diffusion properties. We have previously shown that changing the nature of the extra-framework cations in zeolites A (
LTA) and Y (
FAU) can cause structural modifications, such as altering the size of the unit-cell and pore windows and changing the local environment of aluminium atoms in the framework [
3,
4]. In this work, we explore the effect of cation exchange on the pore dimensions, cation locations and water distribution within the 12-ring (12R) channel systems of
LTL-type zeolites. Zeolites with the
LTL topology have attracted particular attention because of their unique, quasi-1D, 12R channel systems, with free access diameters of 7 Å [
5,
6,
7]. The geometric constraints imparted by the
LTL framework, and the electrostatic fields generated by non-framework cations, create an ideal host environment for a variety of polar guest materials. For example, donor and acceptor chromophores are readily ordered into supramolecular arrays within the 12R channels, allowing efficient resonance energy transfer to occur. These host–guest materials are of particular interest as artificial antenna systems for light harvesting, with applications in photochemical and optoelectronic devices [
8,
9,
10,
11,
12]. The cylindrical shape of the 1D channels not only suppresses the aggregation of adsorbed dye molecules, but can further enhance their photochemical properties. An example can be given for encapsulated proflavine dye molecules which align within the 1D channels and are forced to adopt a bent configuration due to the confinement effects of the framework. This alters the fluorescent properties of the material in comparison to the isolated dye [
13]. The importance of non-framework cations in generating composite materials that are stable and water-resistant has also been shown. The stability of fluoroenone-KL materials has been attributed to strong interactions between non-framework potassium cations in the 12R channels and the carbonyl oxygen atoms of the organic dye molecules [
14]. The applications currently being investigated for
LTL composite materials, ranging from catalysis and optoelectronics, to drug delivery, all make use of the available void space and host–guest interactions within the channel systems [
9,
15,
16]. Tuning the zeolite pore dimensions and electrostatic fields via cation-exchange is one way to alter the physical and chemical properties of the material. It has previously been shown that the catalytic activity of platinum-L zeolites can be improved by increasing the size of non-framework cations. Selectivity for the dehydrocyclisation of n-hexane and hydrogenation of benzene increases from Li
to Cs
exchanged Pt/L zeolites [
17]. On the other hand, exchange with smaller Mg
cations leads to a greater ethanol adsorption capacity. The small ionic radius and high charge density of non-framework Mg
cations in the pores allows them to bind more strongly to ethanol molecules which restricts their motion [
18]. Here, we explore the effect of cation exchange on the pore geometry of zeolite L. Non-framework K
cations are exchanged with larger Cs
and smaller Li
ions to physically alter pore apertures, cation locations and water distribution within the 12R channels. Advanced structural characterisation provides insight into the locations of these extra-framework cations and water molecules. This is fundamental in understanding and exploiting the characteristics of these materials. This work is a step towards tuning
LTL-zeolites for better use as functional materials.
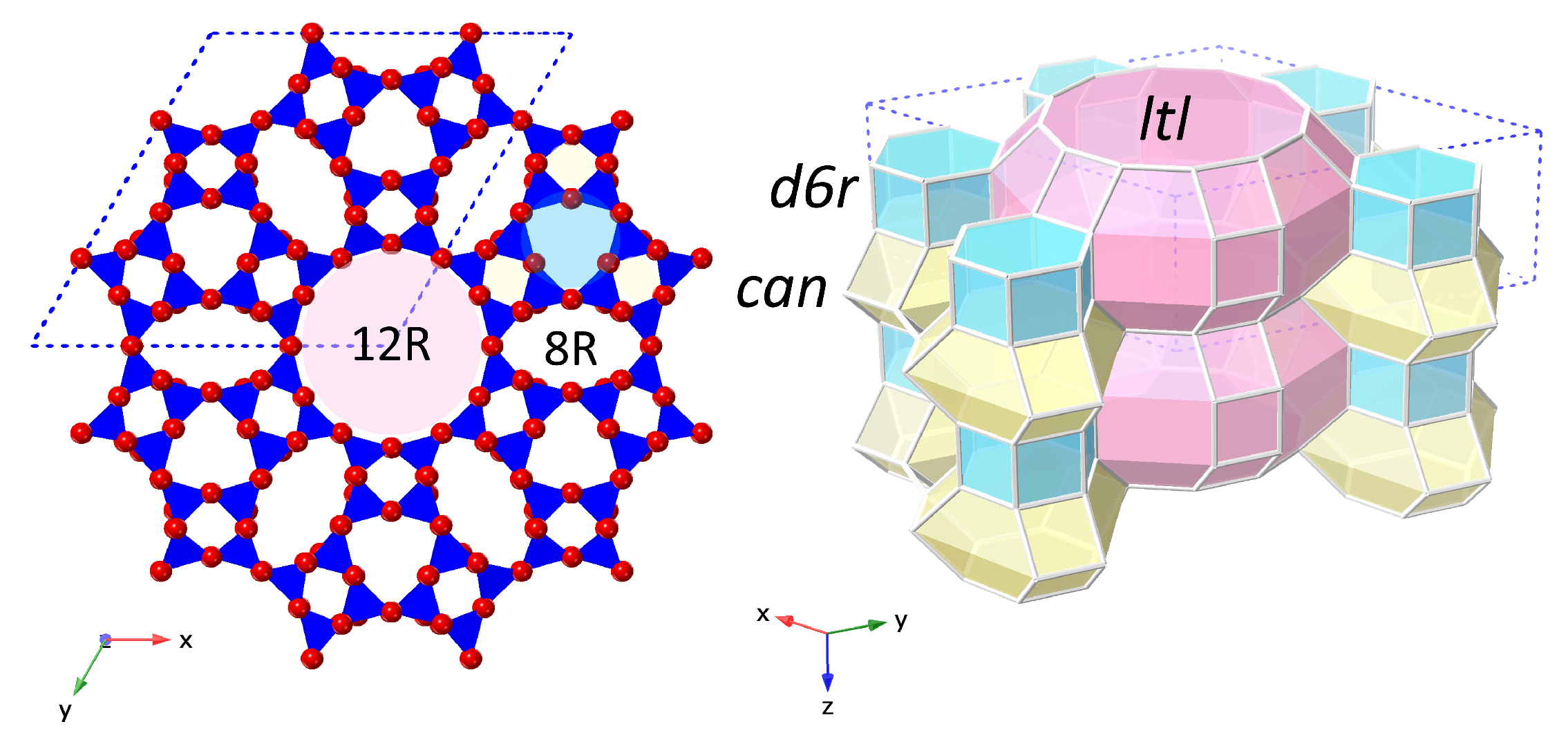
The 12R, 1D channel of the
LTL framework is orientated along the crystallographic
c-axis, enclosed by vertical stacks of alternating cancrinite cages (
can) and double-6 ring units (
d6rs), with smaller, elliptical 8R channels alternating between these columns,
Figure 1. K
cations have previously been shown to reside at four distinct cation sites, labelled A–D in
Figure 2 [
6,
7,
19]. Site A is located at the centre of the
d6r units (z = 0), site B at the centre of the
can cages (z = 1/2), site C in the window between the elliptical 8R channels and walls of the 12R channel (z = 1/2) and site D at the periphery of the 12R channel (z = 0).
3. Results and Discussion
Rietveld refinement of high-resolution, powder X-ray diffraction data for our
LTL zeolites reveal that extra-framework cations are located at sites B-D. Site B is at the centre of the
can cages, site C is at the windows of the elliptical 8R channels and site D is at the periphery of the 12R channels, as shown in
Figure 4.
Site A, reported by Barrer at the centre of the
d6rs is vacant in our as-synthesised K-L and cation exchanged Li and Cs-L zeolites [
19]. The occupancy at site A depends on the synthesis conditions, particularly the Si/Al ratio, where additional cations may be required to render an overall neutral structure. Analysis by energy dispersive X-ray spectroscopy (EDX) gave an average Si/Al ratio of 3.4(2) for the parent K-L sample. Refinement of cation occupancies for the parent K-L zeolite reveal that sites B and C are fully occupied by K cations, whereas site D is statistically occupied by 0.63 K
ions per unit-cell. This is consistent with a Si/Al of 3.1, which is in close agreement to the average value obtained from EDX data. Cation exchange at these sites occur in variable degrees to produce partially exchanged Cs and Li-L zeolites. Site B remains fully occupied by K
ions in both cases, as the small windows of the
can cages restrict the exchange pathway. Site B can, therefore, be regarded as a locked site and is inert to cation exchange with both Li
and Cs
ions. Cations are preferentially exchanged at sites D and C, decreasing in the order D > C > B.
Figure 5 shows the refined, partially exchanged Cs-L structure. Site D undergoes a complete exchange of 0.63 K
ions per unit-cell and is repopulated by 0.72 Cs
ions per unit-cell. In comparison to the parent K-L structure (
Figure 4), Cs
ions located at site D are inset further into the 12R channels so that stability is achieved by accommodating their higher steric demands. Cs
ions can, therefore, be considered stable at site D and a 100% exchange rate proves that this is an open site. Refined occupancies reveal that site C is partially occupied by 0.45 and 0.38 K
or Cs
ions, respectively. Refinement models with K
solely populating site C result in occupancies > 1 and this excess electron density indicates the presence of Cs
. Site C is, in fact, not a closed site, as previously suggested by Newell and Rees, and can exchange K
ions with Cs
ions [
24]. Our refinement models show that site C is susceptible to exchange with Cs
ions and is similarly populated to its gallosilicate analogue, reported by Seoung et al., to have a 50:50 fractional occupancy at this same site [
25]. It is also important to note that the overall occupancy at site C by K
and Cs
ions combined is 0.83 per unit cell compared to the full occupancy by K
ions in the parent K-L zeolite. To render an overall neutral structure, this is compensated for by the increased overall occupancy at site D which is 0.72 Cs
per unit-cell compared to the 0.63 K
in the parent K-L zeolite. This emphasises that Cs
ions preferentially occupy site D over site C as the free volume available is greater here to accommodate larger Cs
cations. Exchange of the parent K-L zeolite with Li
ions leads to a partially exchanged Li-L zeolite. Again, site B is inert to exchange and remains fully occupied by K
ions. Interestingly, unlike with Cs
ion exchange, site C is also locked and remains fully populated by K
ions. Only site D is shown to be susceptible to Li
exchange and our refinement models show it to be partially occupied by 0.11 and 0.57 Li
and K
ions, respectively. Refinement models with only K
ions at site D again show excess electron density, separate to that associated with the K
ions, and this identifies as Li
ions that sit further towards the periphery of the 12R channel.
Figure 6 illustrates the positions of Li
and K
ions at site D which vary in their y coordinates, sitting at y = 0.36 and y = 0.29, respectively. Partial exchange of K
ions for Li
ions at site D implies that Li
ions are less stable here than larger K
or Cs
ions. Because of their small ionic radii, Li
ions must sit much closer to the zeolite framework in order to achieve stability through electrostatic interactions with AlO
tetrahedra. However, even when sitting at the very periphery of the channel walls, Li
ions still struggle to fully exploit the free volume available and cannot maximise their interactions with the framework at this site. K
and Cs
ions have larger ionic radii and are much better matched to the volume of this site. They benefit from stabilising interactions from the surrounding framework. Similarly, at site C, the windows of the elliptical 8R channels are large in comparison to the small Li
ions. Larger K
ions are more stable at this site and it would be unfavourable for K
ions to exchange with Li
ions here. The partial occupancy of Cs
and K
ions at site C indicates that Cs
ions are somewhat stable here, but K
ions are still preferable. Cs
ions are too large to achieve 100% occupancy at site C. The topology of the site also plays a role. For example, site D is more versatile, and cations can be displaced along the
a/
b-axis, either into the channel centre or towards the wall periphery. This is not possible at site C because it is a symmetric site. AlO
tetrahedra may be randomly distributed surrounding the cation and electrostatic interactions can occur in multiple directions. Such detailed information of site occupancies is not attainable from semi-qualitative analysis, such as EDX. Refining site occupancies is, therefore, important in order to identify low exchange rates which could compromise the efficacy of the 1D chemical reactor for applications in catalysis or gas adsorption.
Table 1 shows how cation exchange at site D affects the 12R channel apertures (measured from O1-O1 and O2-O2). This can be used to control diffusion into and out of the channel.
Steric and electronic effects at site D cause smaller Li cations to retreat along the a/b direction towards the periphery of the channel and larger Cs cations are displaced further into the centre of the channel. The free-access diameter is, therefore, smaller for the Cs-exchanged sample. This could be envisaged as a tool to control the entrance and exit of reactant and product molecules to the ‘1D reactor’ during catalytic reactions. Water molecules are exclusively confined within the 1D channels of LTL zeolites.
Figure 7 illustrates the refined water sites in K-L that are fractionally occupied. The O-O distances suggest that water molecules form discrete, hydrogen-bonded layers and cation-stabilised water clusters, in agreement with those nanostructures in the analogous gallosilicate reported by Lee et al. [
23]. Cation exchange can be utilised as a tool to manipulate the geometries of the water nanostructures within the 1-D channel system of
LTL-type zeolites. Water content fluctuates with the nature of the cation at site D. This could be particularly useful for host–guest systems which depend on variable degrees of hydrogen bonding for stabilisation.
Table 1 shows that the overall water content of the channel decreases upon exchange of K
ions with both Li
and Cs
ions. For Cs-L, this can be rationalised by the decrease in free volume available to accommodate water molecules. Large Cs
ions are further displaced into the centre of channel and there is a drastic reduction in the number of water molecules per unit-cell that can be supported in a cation-stabilised cluster. Water content also decreases when K
is partially exchanged for Li
. Again, the decrease in water content can be accounted for by the position of the cations at site D. Smaller Li
ions are displaced towards the channel walls and cannot interact as efficiently with water molecules to form cation clusters. Furthermore, the reduced K
occupancy at site D means that there is also a reduction in stabilising interactions from K
ions with water in the cation-stabilised water cluster.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}