
Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges



Abstract
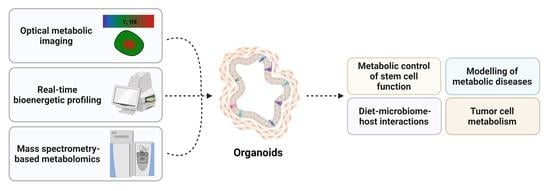
1. Introduction
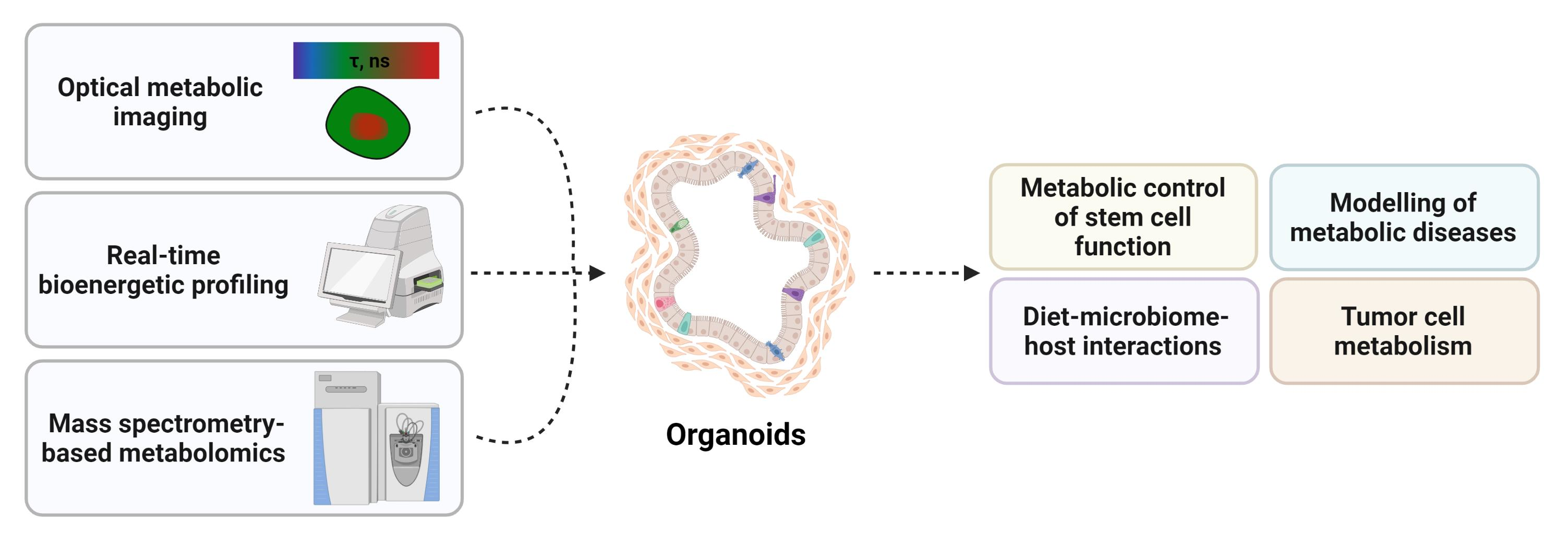
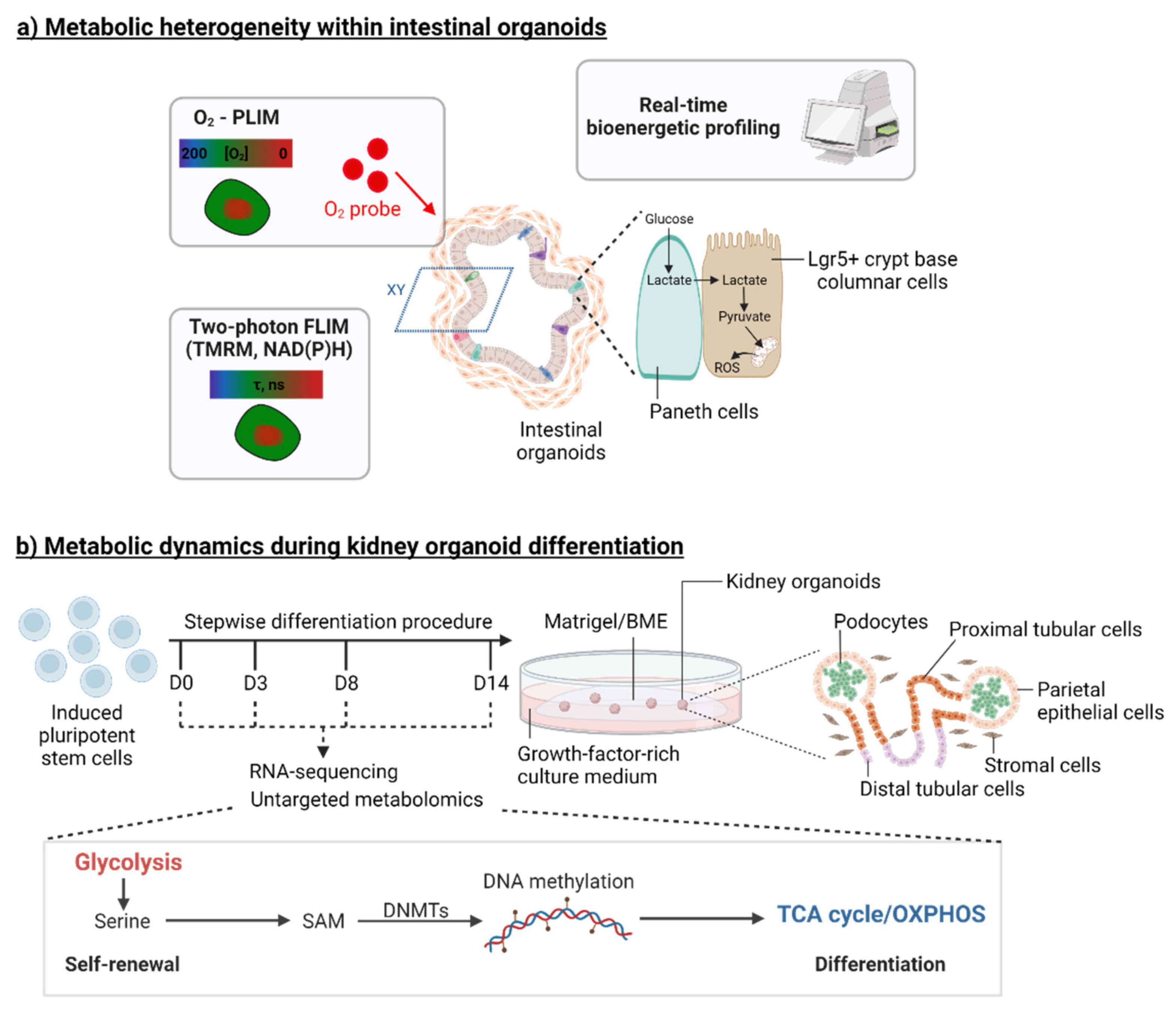
2. Organoid Models to Study the Metabolic Control of Stem Cell Function
3. Organoids and Metabolic Diseases
3.1. Organoids to Model Liver Metabolic Diseases
3.1.1. Alpha-1 Antitrypsin Deficiency and Citrullinemia Type-1
3.1.2. Wilson’s and Wolman’s Diseases
3.1.3. Non-Alcoholic Fatty Liver Disease
3.2. Organoids to Model Pancreatic and Renal Metabolic Diseases
3.2.1. Diabetes
3.2.2. Kidney Diseases
4. Organoids for Modelling Diet–Microbiome–Host Interactions
5. Organoids and Tumor Metabolism
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kretzschmar, K.; Clevers, H. Organoids: Modeling Development and the Stem Cell Niche in a Dish. Dev. Cell 2016, 38, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.H.; Karlsson, K.; Kuo, C.J. Applications of Organoids for Cancer Biology and Precision Medicine. Nat. Cancer 2020, 1, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef]
- Ootani, A.; Li, X.; Sangiorgi, E.; Ho, Q.T.; Ueno, H.; Toda, S.; Sugihara, H.; Fujimoto, K.; Weissman, I.L.; Capecchi, M.R.; et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 2009, 15, 701–706. [Google Scholar] [CrossRef]
- Katano, T.; Ootani, A.; Mizoshita, T.; Tanida, S.; Tsukamoto, H.; Ozeki, K.; Ebi, M.; Mori, Y.; Kataoka, H.; Kamiya, T.; et al. Establishment of a long-term three-dimensional primary culture of mouse glandular stomach epithelial cells within the stem cell niche. Biochem. Biophys. Res. Commun. 2013, 432, 558–563. [Google Scholar] [CrossRef]
- Flores, A.; Schell, J.; Krall, A.S.; Jelinek, D.; Miranda, M.; Grigorian, M.; Braas, D.; White, A.C.; Zhou, J.L.; Graham, N.A.; et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat. Cell Biol. 2017, 19, 1017–1026. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Glasauer, A.; Hoover, P.; Yang, S.; Blatt, H.; Mullen, A.R.; Getsios, S.; Gottardi, C.J.; DeBerardinis, R.J.; Lavker, R.M.; et al. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci. Signal. 2013, 6, ra8. [Google Scholar] [CrossRef]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef]
- Corbet, C. Stem Cell Metabolism in Cancer and Healthy Tissues: Pyruvate in the Limelight. Front. Pharmacol. 2017, 8, 958. [Google Scholar] [CrossRef]
- Okkelman, I.A.; Foley, T.; Papkovsky, D.B.; Dmitriev, R.I. Multi-Parametric Imaging of Hypoxia and Cell Cycle in Intestinal Organoid Culture. Adv. Exp. Med. Biol. 2017, 1035, 85–103. [Google Scholar] [CrossRef]
- Okkelman, I.A.; Foley, T.; Papkovsky, D.B.; Dmitriev, R.I. Live cell imaging of mouse intestinal organoids reveals heterogeneity in their oxygenation. Biomaterials 2017, 146, 86–96. [Google Scholar] [CrossRef]
- Okkelman, I.A.; Neto, N.; Papkovsky, D.B.; Monaghan, M.G.; Dmitriev, R.I. A deeper understanding of intestinal organoid metabolism revealed by combining fluorescence lifetime imaging microscopy (FLIM) and extracellular flux analyses. Redox. Biol. 2020, 30, 101420. [Google Scholar] [CrossRef]
- Okkelman, I.A.; Papkovsky, D.B.; Dmitriev, R.I. Estimation of the Mitochondrial Membrane Potential Using Fluorescence Lifetime Imaging Microscopy. Cytom. A 2020, 97, 471–482. [Google Scholar] [CrossRef]
- Bas, T.; Augenlicht, L.H. Real time analysis of metabolic profile in ex vivo mouse intestinal crypt organoid cultures. J. Vis. Exp. 2014, 93, e52026. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Davidson, L.A.; Callaway, E.S.; Wright, G.A.; Safe, S.; Chapkin, R.S. A bioassay to measure energy metabolism in mouse colonic crypts, organoids, and sorted stem cells. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 309, G1–G9. [Google Scholar] [CrossRef]
- Ludikhuize, M.C.; Meerlo, M.; Burgering, B.M.T.; Rodriguez Colman, M.J. Protocol to profile the bioenergetics of organoids using Seahorse. STAR Protoc. 2021, 2, 100386. [Google Scholar] [CrossRef]
- Rodriguez-Colman, M.J.; Schewe, M.; Meerlo, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Hornsveld, M.; Oost, K.C.; Snippert, H.J.; et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef]
- Schell, J.C.; Wisidagama, D.R.; Bensard, C.; Zhao, H.; Wei, P.; Tanner, J.; Flores, A.; Mohlman, J.; Sorensen, L.K.; Earl, C.S.; et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 2017, 19, 1027–1036. [Google Scholar] [CrossRef]
- Moore, S.R.; Guedes, M.M.; Costa, T.B.; Vallance, J.; Maier, E.A.; Betz, K.J.; Aihara, E.; Mahe, M.M.; Lima, A.A.; Oria, R.B.; et al. Glutamine and alanyl-glutamine promote crypt expansion and mTOR signaling in murine enteroids. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 308, G831–G839. [Google Scholar] [CrossRef]
- Saito, Y.; Iwatsuki, K.; Hanyu, H.; Maruyama, N.; Aihara, E.; Tadaishi, M.; Shimizu, M.; Kobayashi-Hattori, K. Effect of essential amino acids on enteroids: Methionine deprivation suppresses proliferation and affects differentiation in enteroid stem cells. Biochem. Biophys. Res. Commun. 2017, 488, 171–176. [Google Scholar] [CrossRef]
- Wang, B.; Rong, X.; Palladino, E.N.D.; Wang, J.; Fogelman, A.M.; Martin, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206–220.e4. [Google Scholar] [CrossRef]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Cheng, C.W.; Cao, A.Q.; Tripathi, S.; Mana, M.D.; Bauer-Rowe, K.E.; Abu-Remaileh, M.; Clavain, L.; Erdemir, A.; Lewis, C.A.; et al. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 2018, 22, 769–778.e4. [Google Scholar] [CrossRef]
- Mana, M.D.; Hussey, A.M.; Tzouanas, C.N.; Imada, S.; Barrera Millan, Y.; Bahceci, D.; Saiz, D.R.; Webb, A.T.; Lewis, C.A.; Carmeliet, P.; et al. High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Rep. 2021, 35, 109212. [Google Scholar] [CrossRef]
- Chen, L.; Vasoya, R.P.; Toke, N.H.; Parthasarathy, A.; Luo, S.; Chiles, E.; Flores, J.; Gao, N.; Bonder, E.M.; Su, X.; et al. HNF4 Regulates Fatty Acid Oxidation and Is Required for Renewal of Intestinal Stem Cells in Mice. Gastroenterology 2020, 158, 985–999.e9. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Goktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef]
- Merlos-Suarez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Cespedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Munoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Beyaz, S.; Chung, C.; Mou, H.; Bauer-Rowe, K.E.; Xifaras, M.E.; Ergin, I.; Dohnalova, L.; Biton, M.; Shekhar, K.; Eskiocak, O.; et al. Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis. Cell Stem Cell 2021, 28, 1922–1935.e25. [Google Scholar] [CrossRef]
- Sebastian, C.; Ferrer, C.; Serra, M.; Choi, J.E.; Ducano, N.; Mira, A.; Shah, M.S.; Stopka, S.A.; Perciaccante, A.J.; Isella, C.; et al. A non-dividing cell population with high pyruvate dehydrogenase kinase activity regulates metabolic heterogeneity and tumorigenesis in the intestine. Nat. Commun. 2022, 13, 1503. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.J.; Titmarsh, D.M.; Koenig, X.; Parker, B.L.; Ryall, J.G.; Quaife-Ryan, G.A.; Voges, H.K.; Hodson, M.P.; Ferguson, C.; Drowley, L.; et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl. Acad. Sci. USA 2017, 114, E8372–E8381. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xiong, Y.; Zhang, S.; Sui, Y.; Yu, C.; Liu, P.; Li, H.; Guo, W.; Gao, Y.; Przepiorski, A.; et al. The Dynamics of Metabolic Characterization in iPSC-Derived Kidney Organoid Differentiation via a Comparative Omics Approach. Front. Genet. 2021, 12, 632810. [Google Scholar] [CrossRef] [PubMed]
- Browne, A.W.; Arnesano, C.; Harutyunyan, N.; Khuu, T.; Martinez, J.C.; Pollack, H.A.; Koos, D.S.; Lee, T.C.; Fraser, S.E.; Moats, R.A.; et al. Structural and Functional Characterization of Human Stem-Cell-Derived Retinal Organoids by Live Imaging. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3311–3318. [Google Scholar] [CrossRef]
- Mun, S.J.; Ryu, J.S.; Lee, M.O.; Son, Y.S.; Oh, S.J.; Cho, H.S.; Son, M.Y.; Kim, D.S.; Kim, S.J.; Yoo, H.J.; et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 2019, 71, 970–985. [Google Scholar] [CrossRef] [PubMed]
- Bustos, J.F.; Alvarado Gonzalez, J.C.; de Abreu, D.A.R.; Liebisch-Rey, H.; Silva, A.; Ortiz, D.; Ramirez, L.B.; Ortega, J.; Celis Regalado, L.G. Modeling Metabolic Diseases with Organoids: A Review. J. Biomed. Res. Environ. Sci. 2021, 2, 272–279. [Google Scholar] [CrossRef]
- Rauth, S.; Karmakar, S.; Batra, S.K.; Ponnusamy, M.P. Recent advances in organoid development and applications in disease modeling. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188527. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, G.; Shen, C.; Uygun, K.; Yarmush, M.L.; Meng, Q. A novel 3D liver organoid system for elucidation of hepatic glucose metabolism. Biotechnol. Bioeng. 2012, 109, 595–604. [Google Scholar] [CrossRef]
- Park, E.; Kim, H.K.; Jee, J.; Hahn, S.; Jeong, S.; Yoo, J. Development of organoid-based drug metabolism model. Toxicol. Appl. Pharmacol. 2019, 385, 114790. [Google Scholar] [CrossRef]
- Nuciforo, S.; Heim, M.H. Organoids to model liver disease. JHEP Rep. 2021, 3, 100198. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Huch, M. Disease modelling in human organoids. Dis. Model. Mech. 2019, 12, dmm039347. [Google Scholar] [CrossRef]
- Nantasanti, S.; de Bruin, A.; Rothuizen, J.; Penning, L.C.; Schotanus, B.A. Concise Review: Organoids Are a Powerful Tool for the Study of Liver Disease and Personalized Treatment Design in Humans and Animals. Stem Cells Transl. Med. 2016, 5, 325–330. [Google Scholar] [CrossRef]
- Kim, J.W.; Kim, H.W.; Nam, S.A.; Lee, J.Y.; Cho, H.J.; Kim, T.M.; Kim, Y.K. Human kidney organoids reveal the role of glutathione in Fabry disease. Exp. Mol. Med. 2021, 53, 1580–1591. [Google Scholar] [CrossRef]
- Gomez-Mariano, G.; Matamala, N.; Martinez, S.; Justo, I.; Marcacuzco, A.; Jimenez, C.; Monzon, S.; Cuesta, I.; Garfia, C.; Martinez, M.T.; et al. Liver organoids reproduce alpha-1 antitrypsin deficiency-related liver disease. Hepatol. Int. 2020, 14, 127–137. [Google Scholar] [CrossRef]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef]
- Akbari, S.; Sevinc, G.G.; Ersoy, N.; Basak, O.; Kaplan, K.; Sevinc, K.; Ozel, E.; Sengun, B.; Enustun, E.; Ozcimen, B.; et al. Robust, Long-Term Culture of Endoderm-Derived Hepatic Organoids for Disease Modeling. Stem Cell Rep. 2019, 13, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef]
- Haaker, M.W.; Kruitwagen, H.S.; Vaandrager, A.B.; Houweling, M.; Penning, L.C.; Molenaar, M.R.; van Wolferen, M.E.; Oosterhoff, L.A.; Spee, B.; Helms, J.B. Identification of potential drugs for treatment of hepatic lipidosis in cats using an in vitro feline liver organoid system. J. Vet. Intern. Med. 2020, 34, 132–138. [Google Scholar] [CrossRef]
- Kruitwagen, H.S.; Oosterhoff, L.A.; Vernooij, I.; Schrall, I.M.; van Wolferen, M.E.; Bannink, F.; Roesch, C.; van Uden, L.; Molenaar, M.R.; Helms, J.B.; et al. Long-Term Adult Feline Liver Organoid Cultures for Disease Modeling of Hepatic Steatosis. Stem Cell Rep. 2017, 8, 822–830. [Google Scholar] [CrossRef]
- Kruitwagen, H.S.; Oosterhoff, L.A.; van Wolferen, M.E.; Chen, C.; Nantasanti Assawarachan, S.; Schneeberger, K.; Kummeling, A.; van Straten, G.; Akkerdaas, I.C.; Vinke, C.R.; et al. Long-Term Survival of Transplanted Autologous Canine Liver Organoids in a COMMD1-Deficient Dog Model of Metabolic Liver Disease. Cells 2020, 9, 410. [Google Scholar] [CrossRef] [PubMed]
- Nantasanti, S.; Spee, B.; Kruitwagen, H.S.; Chen, C.; Geijsen, N.; Oosterhoff, L.A.; van Wolferen, M.E.; Pelaez, N.; Fieten, H.; Wubbolts, R.W.; et al. Disease Modeling and Gene Therapy of Copper Storage Disease in Canine Hepatic Organoids. Stem Cell Rep. 2015, 5, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.K.; Aboussouan, L.S. Alpha1-antitrypsin deficiency. Lancet 2005, 365, 2225–2236. [Google Scholar] [CrossRef]
- Favier, R.P.; Spee, B.; Schotanus, B.A.; van den Ingh, T.S.; Fieten, H.; Brinkhof, B.; Viebahn, C.S.; Penning, L.C.; Rothuizen, J. COMMD1-deficient dogs accumulate copper in hepatocytes and provide a good model for chronic hepatitis and fibrosis. PLoS ONE 2012, 7, e42158. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Z.; Guardamagna, O.; Nair, D.; Soran, H.; Hovingh, K.; Bertolini, S.; Jones, S.; Coric, M.; Calandra, S.; Hamilton, J.; et al. Lysosomal acid lipase deficiency—An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014, 235, 21–30. [Google Scholar] [CrossRef]
- Larsen, H.L.; Grapin-Botton, A. The molecular and morphogenetic basis of pancreas organogenesis. Semin. Cell Dev. Biol. 2017, 66, 51–68. [Google Scholar] [CrossRef]
- Tsakmaki, A.; Fonseca Pedro, P.; Bewick, G.A. Diabetes through a 3D lens: Organoid models. Diabetologia 2020, 63, 1093–1102. [Google Scholar] [CrossRef]
- Bittenglova, K.; Habart, D.; Saudek, F.; Koblas, T. The Potential of Pancreatic Organoids for Diabetes Research and Therapy. Islets 2021, 13, 85–105. [Google Scholar] [CrossRef]
- Jiang, L.; Shen, Y.; Liu, Y.; Zhang, L.; Jiang, W. Making human pancreatic islet organoids: Progresses on the cell origins, biomaterials and three-dimensional technologies. Theranostics 2022, 12, 1537–1556. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Z.; Song, E.; Xu, T. Islet organoid as a promising model for diabetes. Protein Cell 2022, 13, 239–257. [Google Scholar] [CrossRef]
- Wassmer, C.H.; Lebreton, F.; Bellofatto, K.; Perez, L.; Cottet-Dumoulin, D.; Andres, A.; Bosco, D.; Berney, T.; Othenin-Girard, V.; De Tejada, B.M.; et al. Bio-Engineering of Pre-Vascularized Islet Organoids for the Treatment of Type 1 Diabetes. Transpl. Int. 2021, 35, 10214. [Google Scholar] [CrossRef]
- Lebreton, F.; Lavallard, V.; Bellofatto, K.; Bonnet, R.; Wassmer, C.H.; Perez, L.; Kalandadze, V.; Follenzi, A.; Boulvain, M.; Kerr-Conte, J.; et al. Insulin-producing organoids engineered from islet and amniotic epithelial cells to treat diabetes. Nat. Commun. 2019, 10, 4491. [Google Scholar] [CrossRef]
- Yoshihara, E.; O’Connor, C.; Gasser, E.; Wei, Z.; Oh, T.G.; Tseng, T.W.; Wang, D.; Cayabyab, F.; Dai, Y.; Yu, R.T.; et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature 2020, 586, 606–611. [Google Scholar] [CrossRef]
- Morizane, R.; Lam, A.Q.; Freedman, B.S.; Kishi, S.; Valerius, M.T.; Bonventre, J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015, 33, 1193–1200. [Google Scholar] [CrossRef]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; de Sousa Lopes, S.M.C.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef]
- Taguchi, A.; Kaku, Y.; Ohmori, T.; Sharmin, S.; Ogawa, M.; Sasaki, H.; Nishinakamura, R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 2014, 14, 53–67. [Google Scholar] [CrossRef]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef]
- Taguchi, A.; Nishinakamura, R. Higher-Order Kidney Organogenesis from Pluripotent Stem Cells. Cell Stem Cell 2017, 21, 730–746.e6. [Google Scholar] [CrossRef]
- Liu, M.; Cardilla, A.; Ngeow, J.; Gong, X.; Xia, Y. Studying Kidney Diseases Using Organoid Models. Front. Cell Dev. Biol. 2022, 10, 845401. [Google Scholar] [CrossRef]
- Romero-Guevara, R.; Ioannides, A.; Xinaris, C. Kidney Organoids as Disease Models: Strengths, Weaknesses and Perspectives. Front. Physiol. 2020, 11, 563981. [Google Scholar] [CrossRef]
- Islam, M.; Nishinakamura, R. How to rebuild the kidney: Recent advances in kidney organoids. J. Biochem. 2019, 166, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Nishinakamura, R. Human kidney organoids: Progress and remaining challenges. Nat. Rev. Nephrol. 2019, 15, 613–624. [Google Scholar] [CrossRef]
- Cruz, N.M.; Song, X.; Czerniecki, S.M.; Gulieva, R.E.; Churchill, A.J.; Kim, Y.K.; Winston, K.; Tran, L.M.; Diaz, M.A.; Fu, H.; et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat. Mater. 2017, 16, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Czerniecki, S.M.; Cruz, N.M.; Harder, J.L.; Menon, R.; Annis, J.; Otto, E.A.; Gulieva, R.E.; Islas, L.V.; Kim, Y.K.; Tran, L.M.; et al. High-Throughput Screening Enhances Kidney Organoid Differentiation from Human Pluripotent Stem Cells and Enables Automated Multidimensional Phenotyping. Cell Stem Cell 2018, 22, 929–940.e4. [Google Scholar] [CrossRef] [PubMed]
- Forbes, T.A.; Howden, S.E.; Lawlor, K.; Phipson, B.; Maksimovic, J.; Hale, L.; Wilson, S.; Quinlan, C.; Ho, G.; Holman, K.; et al. Patient-iPSC-Derived Kidney Organoids Show Functional Validation of a Ciliopathic Renal Phenotype and Reveal Underlying Pathogenetic Mechanisms. Am. J. Hum. Genet. 2018, 102, 816–831. [Google Scholar] [CrossRef] [PubMed]
- Dvela-Levitt, M.; Kost-Alimova, M.; Emani, M.; Kohnert, E.; Thompson, R.; Sidhom, E.H.; Rivadeneira, A.; Sahakian, N.; Roignot, J.; Papagregoriou, G.; et al. Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell 2019, 178, 521–535.e3. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, S.; Islam, M.; Sharmin, S.; Naganuma, H.; Yoshimura, Y.; Haque, F.; Era, T.; Nakazato, H.; Nakanishi, K.; Sakuma, T.; et al. Organoids from Nephrotic Disease-Derived iPSCs Identify Impaired NEPHRIN Localization and Slit Diaphragm Formation in Kidney Podocytes. Stem Cell Rep. 2018, 11, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Hale, L.J.; Howden, S.E.; Phipson, B.; Lonsdale, A.; Er, P.X.; Ghobrial, I.; Hosawi, S.; Wilson, S.; Lawlor, K.T.; Khan, S.; et al. 3D organoid-derived human glomeruli for personalised podocyte disease modelling and drug screening. Nat. Commun. 2018, 9, 5167. [Google Scholar] [CrossRef]
- Garreta, E.; Prado, P.; Stanifer, M.L.; Monteil, V.; Marco, A.; Ullate-Agote, A.; Moya-Rull, D.; Vilas-Zornoza, A.; Tarantino, C.; Romero, J.P.; et al. A diabetic milieu increases ACE2 expression and cellular susceptibility to SARS-CoV-2 infections in human kidney organoids and patient cells. Cell Metab. 2022, 34, 857–873.e9. [Google Scholar] [CrossRef]
- Cassotta, M.; Forbes-Hernandez, T.Y.; Calderon Iglesias, R.; Ruiz, R.; Elexpuru Zabaleta, M.; Giampieri, F.; Battino, M. Links between Nutrition, Infectious Diseases, and Microbiota: Emerging Technologies and Opportunities for Human-Focused Research. Nutrients 2020, 12, 1827. [Google Scholar] [CrossRef]
- Rubert, J.; Schweiger, P.J.; Mattivi, F.; Tuohy, K.; Jensen, K.B.; Lunardi, A. Intestinal Organoids: A Tool for Modelling Diet-Microbiome-Host Interactions. Trends Endocrinol. Metab. 2020, 31, 848–858. [Google Scholar] [CrossRef]
- Nigro, G.; Hanson, M.; Fevre, C.; Lecuit, M.; Sansonetti, P.J. Intestinal Organoids as a Novel Tool to Study Microbes-Epithelium Interactions. Methods Mol. Biol. 2019, 1576, 183–194. [Google Scholar] [CrossRef]
- Fujii, M.; Matano, M.; Toshimitsu, K.; Takano, A.; Mikami, Y.; Nishikori, S.; Sugimoto, S.; Sato, T. Human Intestinal Organoids Maintain Self-Renewal Capacity and Cellular Diversity in Niche-Inspired Culture Condition. Cell Stem Cell 2018, 23, 787–793.e6. [Google Scholar] [CrossRef]
- Zietek, T.; Rath, E.; Haller, D.; Daniel, H. Intestinal organoids for assessing nutrient transport, sensing and incretin secretion. Sci. Rep. 2015, 5, 16831. [Google Scholar] [CrossRef]
- Zietek, T.; Giesbertz, P.; Ewers, M.; Reichart, F.; Weinmuller, M.; Urbauer, E.; Haller, D.; Demir, I.E.; Ceyhan, G.O.; Kessler, H.; et al. Organoids to Study Intestinal Nutrient Transport, Drug Uptake and Metabolism—Update to the Human Model and Expansion of Applications. Front. Bioeng. Biotechnol. 2020, 8, 577656. [Google Scholar] [CrossRef]
- Foulke-Abel, J.; In, J.; Yin, J.; Zachos, N.C.; Kovbasnjuk, O.; Estes, M.K.; de Jonge, H.; Donowitz, M. Human Enteroids as a Model of Upper Small Intestinal Ion Transport Physiology and Pathophysiology. Gastroenterology 2016, 150, 638–649.e8. [Google Scholar] [CrossRef]
- Jattan, J.; Rodia, C.; Li, D.; Diakhate, A.; Dong, H.; Bataille, A.; Shroyer, N.F.; Kohan, A.B. Using primary murine intestinal enteroids to study dietary TAG absorption, lipoprotein synthesis, and the role of apoC-III in the intestine. J. Lipid Res. 2017, 58, 853–865. [Google Scholar] [CrossRef]
- Cai, T.; Qi, Y.; Jergens, A.; Wannemuehler, M.; Barrett, T.A.; Wang, Q. Effects of six common dietary nutrients on murine intestinal organoid growth. PLoS ONE 2018, 13, e0191517. [Google Scholar] [CrossRef]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; de Vos, W.M.; Montijn, R.C.; Roeselers, G. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio 2014, 5, e01438-14. [Google Scholar] [CrossRef]
- Mathewson, N.D.; Jenq, R.; Mathew, A.V.; Koenigsknecht, M.; Hanash, A.; Toubai, T.; Oravecz-Wilson, K.; Wu, S.R.; Sun, Y.; Rossi, C.; et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat. Immunol. 2016, 17, 505–513. [Google Scholar] [CrossRef]
- Schilderink, R.; Verseijden, C.; Seppen, J.; Muncan, V.; van den Brink, G.R.; Lambers, T.T.; van Tol, E.A.; de Jonge, W.J. The SCFA butyrate stimulates the epithelial production of retinoic acid via inhibition of epithelial HDAC. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 310, G1138–G1146. [Google Scholar] [CrossRef] [PubMed]
- Bellono, N.W.; Bayrer, J.R.; Leitch, D.B.; Castro, J.; Zhang, C.; O’Donnell, T.A.; Brierley, S.M.; Ingraham, H.A.; Julius, D. Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell 2017, 170, 185–198.e16. [Google Scholar] [CrossRef] [PubMed]
- Mun, S.J.; Lee, J.; Chung, K.S.; Son, M.Y.; Son, M.J. Effect of Microbial Short-Chain Fatty Acids on CYP3A4-Mediated Metabolic Activation of Human Pluripotent Stem Cell-Derived Liver Organoids. Cells 2021, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Rosselot, A.E.; Park, M.; Kim, M.; Matsu-Ura, T.; Wu, G.; Flores, D.E.; Subramanian, K.R.; Lee, S.; Sundaram, N.; Broda, T.R.; et al. Ontogeny and function of the circadian clock in intestinal organoids. EMBO J. 2022, 41, e106973. [Google Scholar] [CrossRef]
- Yamada, R.G.; Ueda, H.R. The circadian clock ticks in organoids. EMBO J. 2022, 41, e110157. [Google Scholar] [CrossRef]
- Hou, Q.; Ye, L.; Liu, H.; Huang, L.; Yang, Q.; Turner, J.R.; Yu, Q. Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 2018, 25, 1657–1670. [Google Scholar] [CrossRef]
- Williamson, I.A.; Arnold, J.W.; Samsa, L.A.; Gaynor, L.; DiSalvo, M.; Cocchiaro, J.L.; Carroll, I.; Azcarate-Peril, M.A.; Rawls, J.F.; Allbritton, N.L.; et al. A High-Throughput Organoid Microinjection Platform to Study Gastrointestinal Microbiota and Luminal Physiology. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 301–319. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, 6487. [Google Scholar] [CrossRef]
- Goncalves, A.C.; Richiardone, E.; Jorge, J.; Polonia, B.; Xavier, C.P.R.; Salaroglio, I.C.; Riganti, C.; Vasconcelos, M.H.; Corbet, C.; Sarmento-Ribeiro, A.B. Impact of cancer metabolism on therapy resistance—Clinical implications. Drug Resist. Updat. 2021, 59, 100797. [Google Scholar] [CrossRef]
- Van den Bossche, V.; Zaryouh, H.; Vara-Messler, M.; Vignau, J.; Machiels, J.P.; Wouters, A.; Schmitz, S.; Corbet, C. Microenvironment-driven intratumoral heterogeneity in head and neck cancers: Clinical challenges and opportunities for precision medicine. Drug Resist. Updat. 2022, 60, 100806. [Google Scholar] [CrossRef]
- Fendt, S.M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Emerging roles of lipid metabolism in cancer progression. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 254–260. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 7–15. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Metabolic and mind shifts: From glucose to glutamine and acetate addictions in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 346–353. [Google Scholar] [CrossRef][Green Version]
- Vander Linden, C.; Corbet, C. Therapeutic Targeting of Cancer Stem Cells: Integrating and Exploiting the Acidic Niche. Front. Oncol. 2019, 9, 159. [Google Scholar] [CrossRef]
- Vander Linden, C.; Corbet, C.; Bastien, E.; Martherus, R.; Guilbaud, C.; Petit, L.; Wauthier, L.; Loriot, A.; De Smet, C.; Feron, O. Therapy-induced DNA methylation inactivates MCT1 and renders tumor cells vulnerable to MCT4 inhibition. Cell Rep. 2021, 35, 109202. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Driehuis, E.; Kolders, S.; Spelier, S.; Lohmussaar, K.; Willems, S.M.; Devriese, L.A.; de Bree, R.; de Ruiter, E.J.; Korving, J.; Begthel, H.; et al. Oral Mucosal Organoids as a Potential Platform for Personalized Cancer Therapy. Cancer Discov. 2019, 9, 852–871. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarro, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Norrie, J.L.; Nityanandam, A.; Lai, K.; Chen, X.; Wilson, M.; Stewart, E.; Griffiths, L.; Jin, H.; Wu, G.; Orr, B.; et al. Retinoblastoma from human stem cell-derived retinal organoids. Nat. Commun. 2021, 12, 4535. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat. Commun. 2019, 10, 4862. [Google Scholar] [CrossRef]
- Walsh, A.J.; Cook, R.S.; Sanders, M.E.; Aurisicchio, L.; Ciliberto, G.; Arteaga, C.L.; Skala, M.C. Quantitative optical imaging of primary tumor organoid metabolism predicts drug response in breast cancer. Cancer Res. 2014, 74, 5184–5194. [Google Scholar] [CrossRef]
- Sharick, J.T.; Walsh, C.M.; Sprackling, C.M.; Pasch, C.A.; Pham, D.L.; Esbona, K.; Choudhary, A.; Garcia-Valera, R.; Burkard, M.E.; McGregor, S.M.; et al. Metabolic Heterogeneity in Patient Tumor-Derived Organoids by Primary Site and Drug Treatment. Front. Oncol. 2020, 10, 553. [Google Scholar] [CrossRef]
- Sharick, J.T.; Jeffery, J.J.; Karim, M.R.; Walsh, C.M.; Esbona, K.; Cook, R.S.; Skala, M.C. Cellular Metabolic Heterogeneity In Vivo Is Recapitulated in Tumor Organoids. Neoplasia 2019, 21, 615–626. [Google Scholar] [CrossRef]
- Pasch, C.A.; Favreau, P.F.; Yueh, A.E.; Babiarz, C.P.; Gillette, A.A.; Sharick, J.T.; Karim, M.R.; Nickel, K.P.; DeZeeuw, A.K.; Sprackling, C.M.; et al. Patient-Derived Cancer Organoid Cultures to Predict Sensitivity to Chemotherapy and Radiation. Clin. Cancer Res. 2019, 25, 5376–5387. [Google Scholar] [CrossRef]
- Gillette, A.A.; Babiarz, C.P.; VanDommelen, A.R.; Pasch, C.A.; Clipson, L.; Matkowskyj, K.A.; Deming, D.A.; Skala, M.C. Autofluorescence Imaging of Treatment Response in Neuroendocrine Tumor Organoids. Cancers 2021, 13, 1873. [Google Scholar] [CrossRef]
- Shah, A.T.; Heaster, T.M.; Skala, M.C. Metabolic Imaging of Head and Neck Cancer Organoids. PLoS ONE 2017, 12, e0170415. [Google Scholar] [CrossRef]
- Walsh, A.J.; Castellanos, J.A.; Nagathihalli, N.S.; Merchant, N.B.; Skala, M.C. Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas 2016, 45, 863–869. [Google Scholar] [CrossRef]
- David, B.P.; Dubrovskyi, O.; Speltz, T.E.; Wolff, J.J.; Frasor, J.; Sanchez, L.M.; Moore, T.W. Using Tumor Explants for Imaging Mass Spectrometry Visualization of Unlabeled Peptides and Small Molecules. ACS Med. Chem. Lett. 2018, 9, 768–772. [Google Scholar] [CrossRef]
- Liu, X.; Flinders, C.; Mumenthaler, S.M.; Hummon, A.B. MALDI Mass Spectrometry Imaging for Evaluation of Therapeutics in Colorectal Tumor Organoids. J. Am. Soc. Mass. Spectrom. 2018, 29, 516–526. [Google Scholar] [CrossRef]
- Wang, Y.; Hummon, A.B. MS imaging of multicellular tumor spheroids and organoids as an emerging tool for personalized medicine and drug discovery. J. Biol. Chem. 2021, 297, 101139. [Google Scholar] [CrossRef]
- Vander Linden, C.; Corbet, C. Reconciling environment-mediated metabolic heterogeneity with the oncogene-driven cancer paradigm in precision oncology. Semin. Cell Dev. Biol. 2020, 98, 202–210. [Google Scholar] [CrossRef]
- Weygand, J.; Carter, S.E.; Salzillo, T.C.; Moussalli, M.; Dai, B.; Dutta, P.; Zuo, X.; Fleming, J.B.; Shureiqi, I.; Bhattacharya, P. Can an organoid recapitulate the metabolome of its parent tissue? A pilot NMR spectroscopy study. J. Cancer Prev. Curr. Res. 2017, 8, 425–429. [Google Scholar] [CrossRef][Green Version]
- Yoshizaki, H.; Ogiso, H.; Okazaki, T.; Kiyokawa, E. Comparative lipid analysis in the normal and cancerous organoids of MDCK cells. J. Biochem. 2016, 159, 573–584. [Google Scholar] [CrossRef]
- Feldman, A.; Mukha, D.; Maor, I.I.; Sedov, E.; Koren, E.; Yosefzon, Y.; Shlomi, T.; Fuchs, Y. Blimp1+ cells generate functional mouse sebaceous gland organoids in vitro. Nat. Commun. 2019, 10, 2348. [Google Scholar] [CrossRef]
- Lindeboom, R.G.; van Voorthuijsen, L.; Oost, K.C.; Rodriguez-Colman, M.J.; Luna-Velez, M.V.; Furlan, C.; Baraille, F.; Jansen, P.W.; Ribeiro, A.; Burgering, B.M.; et al. Integrative multi-omics analysis of intestinal organoid differentiation. Mol. Syst. Biol. 2018, 14, e8227. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Neef, S.K.; Janssen, N.; Winter, S.; Wallisch, S.K.; Hofmann, U.; Dahlke, M.H.; Schwab, M.; Murdter, T.E.; Haag, M. Metabolic Drug Response Phenotyping in Colorectal Cancer Organoids by LC-QTOF-MS. Metabolites 2020, 10, 494. [Google Scholar] [CrossRef]
- Hahn, S.; Nam, M.O.; Noh, J.H.; Lee, D.H.; Han, H.W.; Kim, D.H.; Hahm, K.B.; Hong, S.P.; Yoo, J.H.; Yoo, J. Organoid-based epithelial to mesenchymal transition (OEMT) model: From an intestinal fibrosis perspective. Sci. Rep. 2017, 7, 2435. [Google Scholar] [CrossRef]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schafer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bansal, A.; Dunbar, K.B.; Chang, Y.; Zhang, J.; Balaji, U.; Gu, J.; Zhang, X.; Podgaetz, E.; Pan, Z.; et al. A human Barrett’s esophagus organoid system reveals epithelial-mesenchymal plasticity induced by acid and bile salts. Am. J. Physiol.-Gastrointest. Liver Physiol. 2022, 322, G598–G614. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Powell, R.T.; Yuan, X.; Bae, G.; Roarty, K.P.; Stossi, F.; Strempfl, M.; Toneff, M.J.; Johnson, H.L.; Mani, S.A.; et al. Morphological screening of mesenchymal mammary tumor organoids to identify drugs that reverse epithelial-mesenchymal transition. Nat. Commun. 2021, 12, 4262. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Azambuja, A.P.; Simoes-Costa, M. Metabolic Reprogramming Promotes Neural Crest Migration via Yap/Tead Signaling. Dev. Cell 2020, 53, 199–211.e6. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Kaur, H.; Jung, K.H.; Yang, S.; Tripathi, S.; Galbraith, M.; Deng, Y.; Jolly, M.K.; Kaipparettu, B.A.; et al. Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br. J. Cancer 2021, 124, 1902–1911. [Google Scholar] [CrossRef]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef]
- Schutte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef]
- Zhao, H.; Jiang, E.; Shang, Z. 3D Co-culture of Cancer-Associated Fibroblast with Oral Cancer Organoids. J. Dent. Res. 2021, 100, 201–208. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.e6. [Google Scholar] [CrossRef]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor organoid-T-cell coculture systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Sivanand, S.; Lau, A.N.; Florek, L.V.; Barbeau, A.M.; Wyckoff, J.; Skala, M.C.; Vander Heiden, M.G. Interactions with stromal cells promote a more oxidized cancer cell redox state in pancreatic tumors. Sci. Adv. 2022, 8, eabg6383. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Perianez, R.; Molina-Privado, I.; Rojo, F.; Guijarro-Munoz, I.; Alonso-Camino, V.; Zazo, S.; Compte, M.; Alvarez-Cienfuegos, A.; Cuesta, A.M.; Sanchez-Martin, D.; et al. Basement membrane-rich organoids with functional human blood vessels are permissive niches for human breast cancer metastasis. PLoS ONE 2013, 8, e72957. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Goncalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
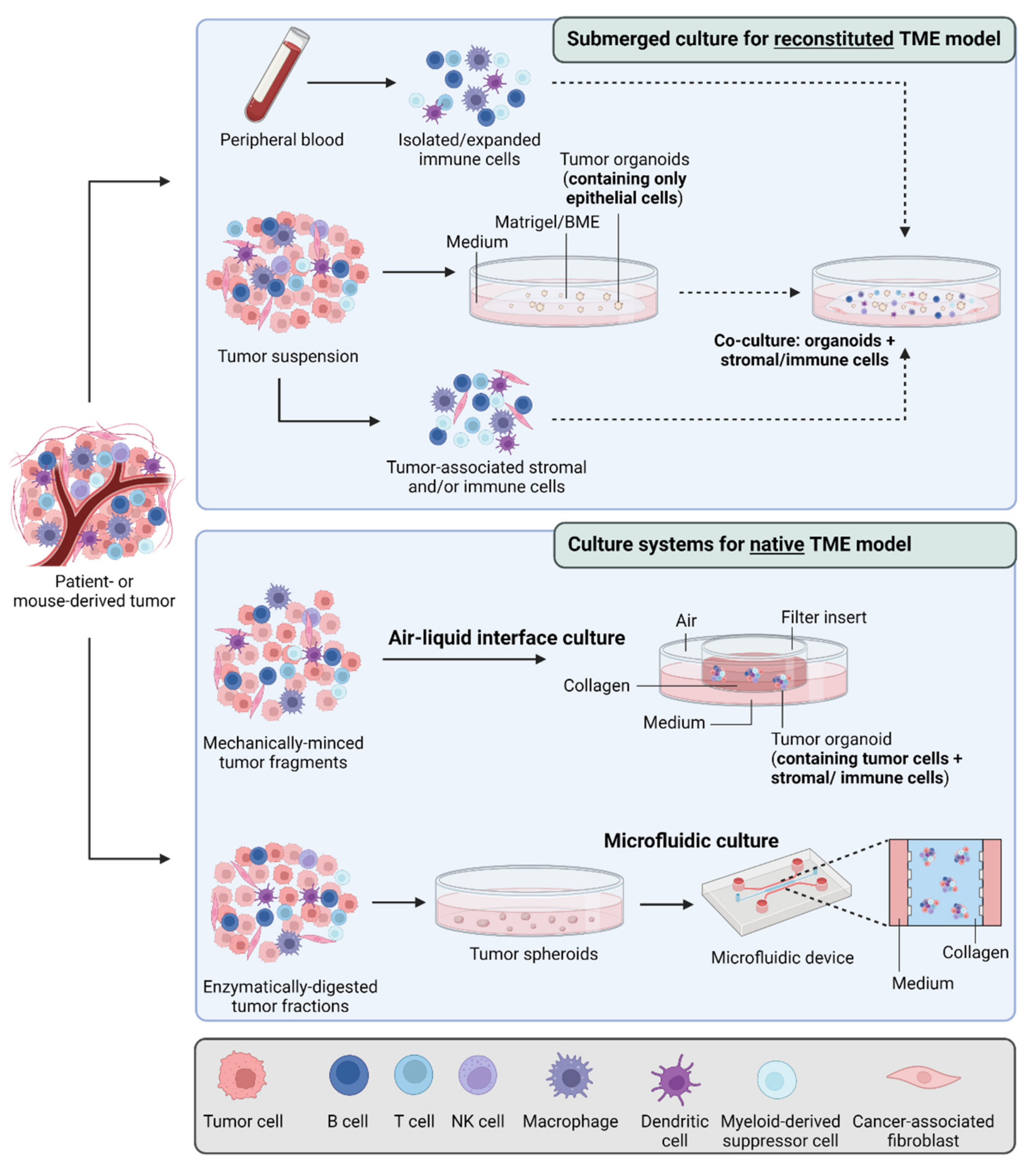{kind=link}
| Disease | Species | Organoid Source and Derivation | Refs |
|---|---|---|---|
| Kidney metabolic diseases | |||
| Fabry disease | Human | iPSCs (fibroblast-derived) | [44] |
| Liver metabolic diseases | |||
| Alpha-1 antitrypsin deficiency | Human | Adult tissue (surgical resection; liver transplantation; biopsy) | [45,46] |
| Citrullinemia type 1 | Human | iPSCs (fibroblast-derived) | [47] |
| Steatosis, steatohepatitis | Human Cat | iPSCs (fibroblast-derived) Adult tissue (post-mortem) | [48] [49,50] |
| Wilson’s disease | Dog | Adult tissue (surgical resection, needle biopsy, fine needle aspiration) | [51,52] |
| Wolman’s disease | Human | iPSCs (fibroblast-derived) | [48] |
| Methods | Applications | Advantages | Disadvantages | Refs |
|---|---|---|---|---|
| Extracellular flux analysis (Seahorse XF analyzer) | Evaluation of mitochondrial respiration (OCR) and glycolysis (ECAR) | Real-time and simultaneous measurements Up to 4 injections for nutritional and/or pharmacological modulation Label-free assay system, highly sensitive microplate format | No spatial resolution Technically challenging (e.g., need for accurate plating, optimal organoid density) Use of saturating concentrations of substrates and drugs | [16,17,18,19,20] |
| Fluorescence lifetime imaging microscopy (FLIM)/Optical metabolic imaging (OMI) | Live cell microscopy of endogenous (NAD(P)H, FAD) or exogenous chromophores (TMRM for mitochondrial membrane potential) | Non-invasive, cell-specific and direct analysis of metabolism within organoid models Compatible with other imaging methods (e.g., PLIM) for multiparametric quantitative analysis | Complex interpretation of fluorescence data (due to double or multi-exponential decays for most of fluorescent reporters) Shorter lifetimes than PLIM | [14,15,120,121,122,123,124,125,126,153] |
| Mass spectrometry-based metabolomics | Absolute or relative quantification of extra- and/or intracellular metabolites within organoids | Suitable for targeted and untargeted profiling of several metabolite classes (e.g., lipids, polar metabolites) | No spatial resolution No information on metabolic flux (steady-state conditions) Background signal from animal-derived extracellular matrix | [133,134,135] |
| Phosphorescence lifetime imaging microscopy (PLIM) | Live cell microscopy of oxygen (with dedicated cell-penetrating phosphorescent O2-sensitive probes) | Suitable for single-cell analysis of inter- and intra-organoid variability of oxygenation levels Direct, reversible and non-chemical process Compatible with other imaging methods (e.g., FLIM) for multiparametric quantitative analysis High sensitivity and high stability of the signal | Emission intensity dependent on the probe distribution within organoids Limited number of applications (depending on probe availability) | [12,13,14] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richiardone, E.; Van den Bossche, V.; Corbet, C. Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges. Organoids 2022, 1, 85-105. https://doi.org/10.3390/organoids1010008
Richiardone E, Van den Bossche V, Corbet C. Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges. Organoids. 2022; 1(1):85-105. https://doi.org/10.3390/organoids1010008
Chicago/Turabian StyleRichiardone, Elena, Valentin Van den Bossche, and Cyril Corbet. 2022. "Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges" Organoids 1, no. 1: 85-105. https://doi.org/10.3390/organoids1010008
APA StyleRichiardone, E., Van den Bossche, V., & Corbet, C. (2022). Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges. Organoids, 1(1), 85-105. https://doi.org/10.3390/organoids1010008