
Organoid Models of Lymphoid Tissues


Abstract
:
1. Introduction
2. Bone Marrow
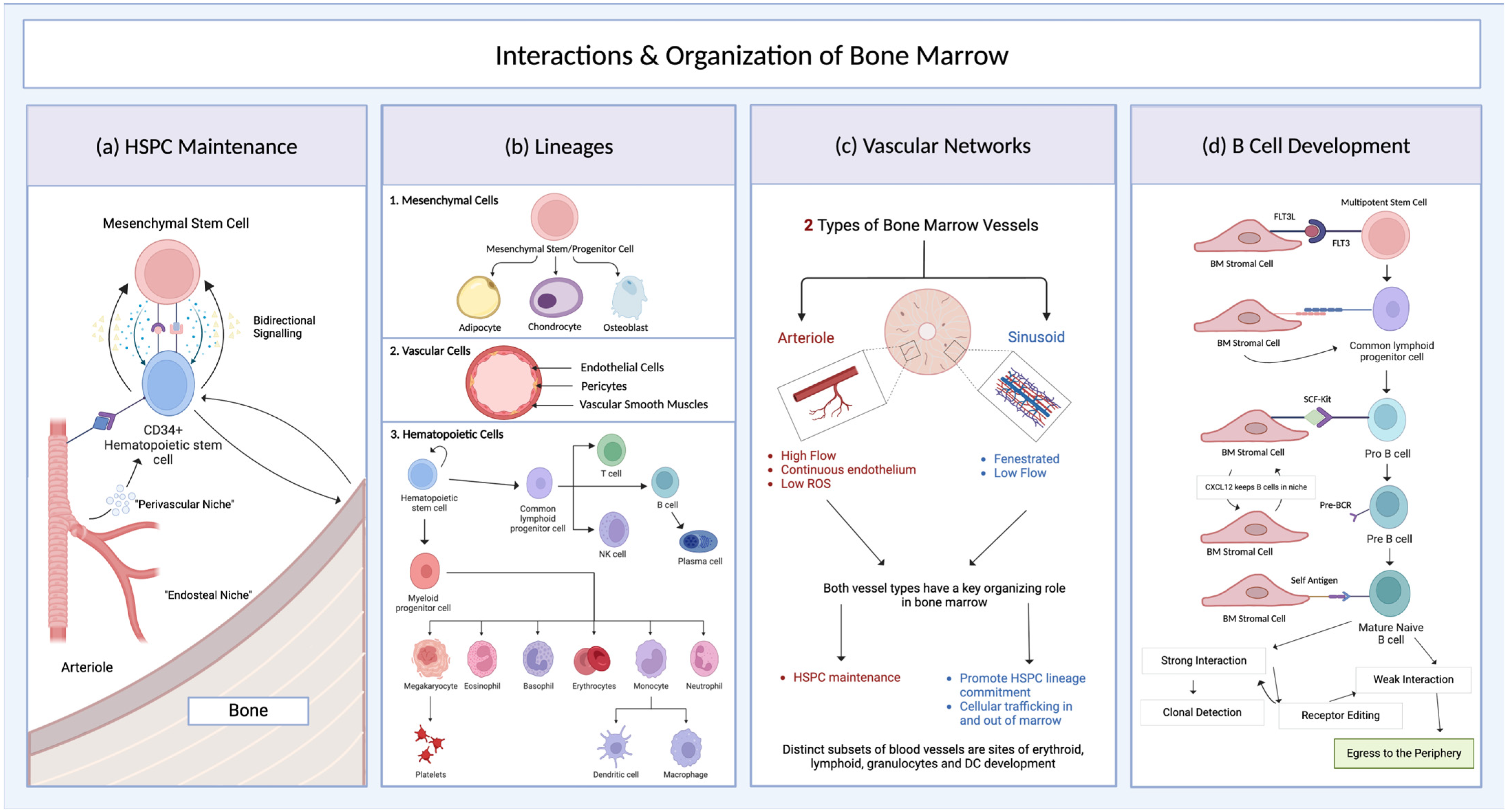
2.1. Bone Marrow Structure and Development
2.2. Mouse Models of Bone Marrow
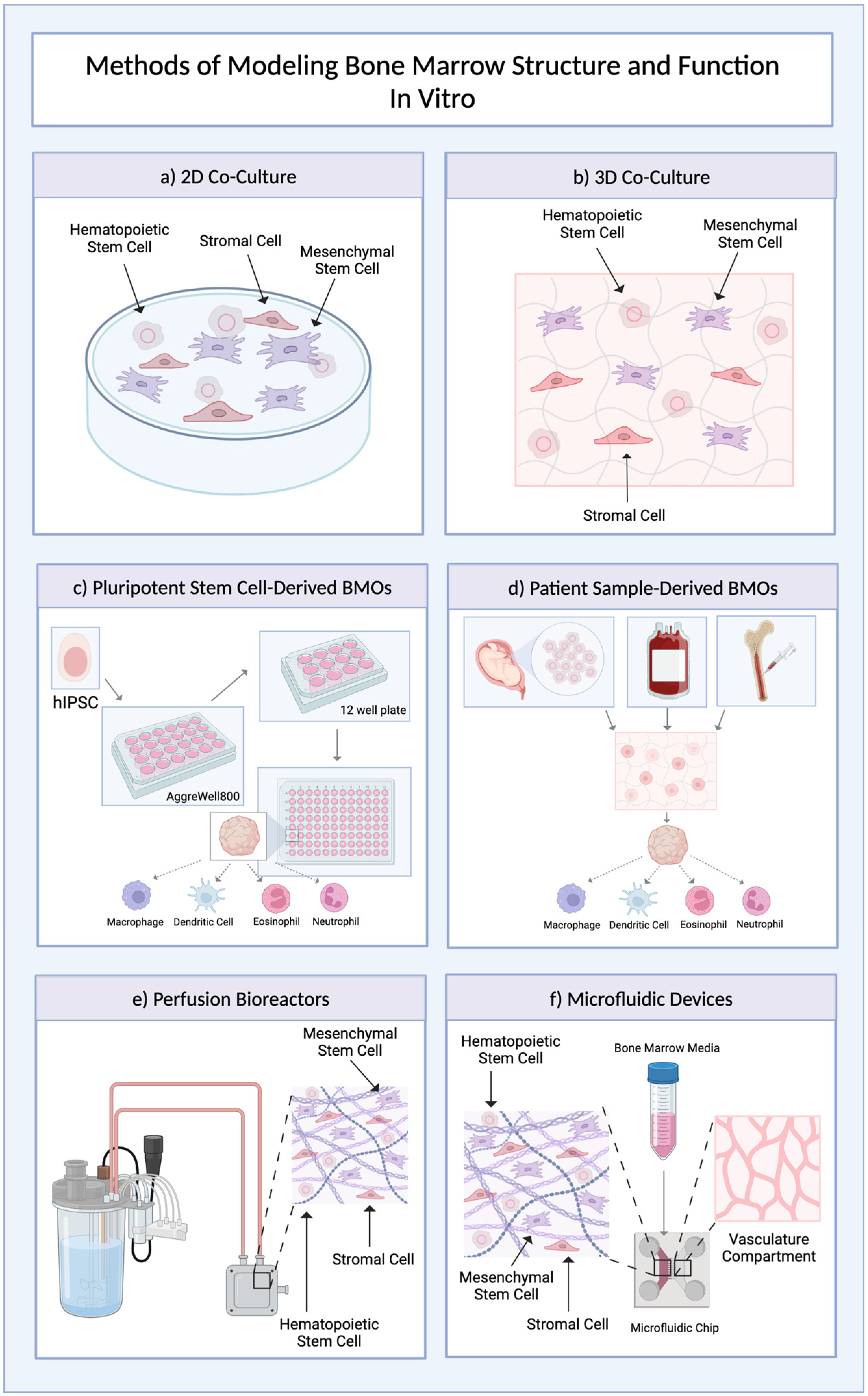
2.3. Bone Marrow Organoids
3. The Thymus
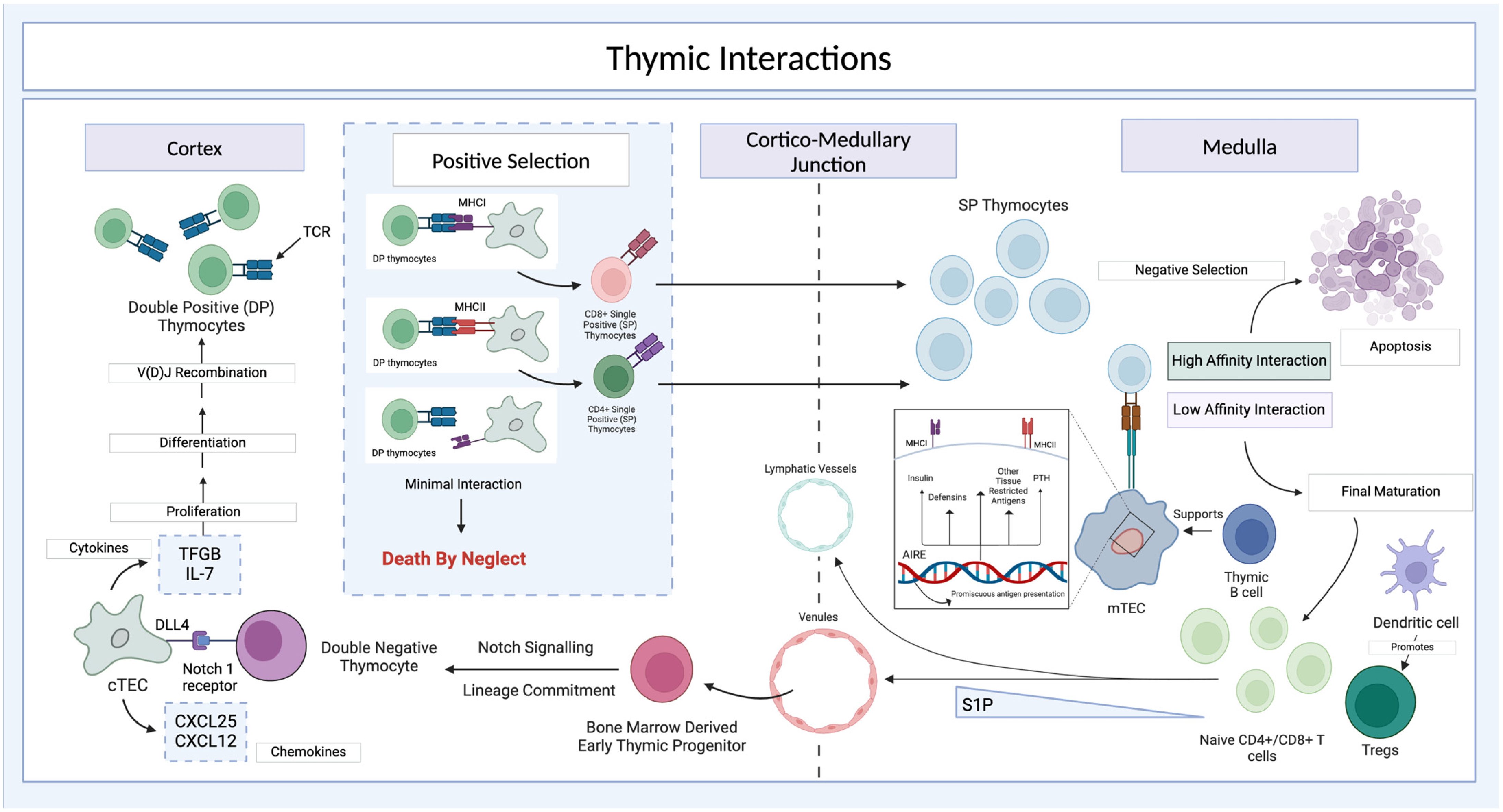
3.1. Thymic Development and Anatomy
3.2. Thymic Cortex and Medulla
3.3. Thymus Organoids
4. Lymph Nodes
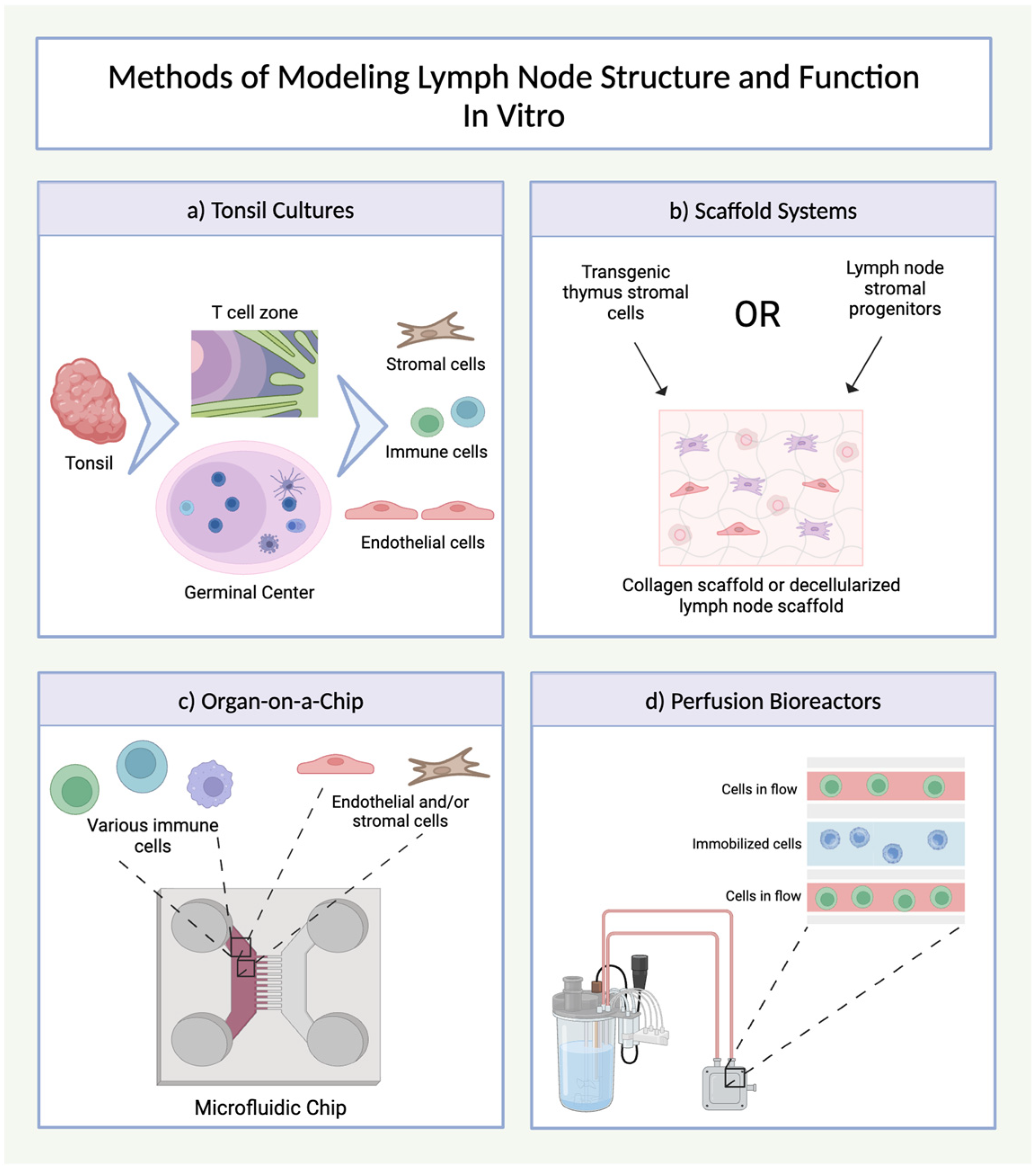
4.1. Lymph Node Structure and Function
4.2. Lymph Node Organoids
5. Spleen
5.1. Spleen Structure and Function
5.2. Spleen Organoid Models
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Carneiro, C.F.D.; Drude, N.; Hülsemann, M.; Collazo, A.; Toelch, U. Mapping Strategies towards Improved External Validity in Preclinical Translational Research. Expert Opin. Drug Discov. 2023, 18, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Pound, P.; Ritskes-Hoitinga, M. Is It Possible to Overcome Issues of External Validity in Preclinical Animal Research? Why Most Animal Models Are Bound to Fail. J. Transl. Med. 2018, 16, 304. [Google Scholar] [CrossRef] [PubMed]
- Contopoulos-Ioannidis, D.G.; Ntzani, E.E.; Ioannidis, J.P.A. Translation of Highly Promising Basic Science Research into Clinical Applications. Am. J. Med. 2003, 114, 477–484. [Google Scholar] [CrossRef]
- Fogel, D.B. Factors Associated with Clinical Trials That Fail and Opportunities for Improving the Likelihood of Success: A Review. Contemp. Clin. Trials Commun. 2018, 11, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Gore, S.D.; Vey, N.; Prebet, T. Lost in Translation? Ten Years of Development of Histone Deacetylase Inhibitors in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Expert Opin. Investig. Drugs 2016, 25, 307–317. [Google Scholar] [CrossRef]
- Witzig, T.E.; Tobinai, K.; Rigacci, L.; Ikeda, T.; Vanazzi, A.; Hino, M.; Shi, Y.; Mayer, J.; Costa, L.J.; Bermudez Silva, C.D.; et al. Adjuvant Everolimus in High-Risk Diffuse Large B-Cell Lymphoma: Final Results from the PILLAR-2 Randomized Phase III Trial. Ann. Oncol. 2018, 29, 707–714. [Google Scholar] [CrossRef]
- Xu, Z.-Z.; Wang, W.-F.; Fu, W.-B.; Wang, A.-H.; Liu, Z.-Y.; Chen, L.-Y.; Guo, P.; Li, J.-M. Combination of Rituximab and Mammalian Target of Rapamycin Inhibitor Everolimus (RAD001) in Diffuse Large B-Cell Lymphoma. Leuk. Lymphoma 2014, 55, 1151–1157. [Google Scholar] [CrossRef]
- Janning, M.; Fiedler, W. Volasertib for the Treatment of Acute Myeloid Leukemia: A Review of Preclinical and Clinical Development. Future Oncol. 2014, 10, 1157–1165. [Google Scholar] [CrossRef]
- Dohner, H.; Symeonidis, A.; Deeren, D.; Demeter, J.; Sanz, M.A.; Anagnostopoulos, A.; Esteve, J.; Fiedler, W.; Porkka, K.; Kim, H.J.; et al. Adjunctive Volasertib in Patients with Acute Myeloid Leukemia Not Eligible for Standard Induction Therapy: A Randomized, Phase 3 Trial. Hemasphere 2021, 5, e617. [Google Scholar] [CrossRef]
- An, Z.-Y.; Fu, H.; He, Y.; Zhu, X.; Huang, Q.-S.; Wu, J.; Liu, K.; Zhang, X. Projected Global Trends in Hematological Malignancies: Incidence, Mortality, and Disability-Adjusted Life Years from 2020 to 2030. Blood 2023, 142 (Suppl. 1), 3810. [Google Scholar] [CrossRef]
- Zhang, N.; Wu, J.; Wang, Q.; Liang, Y.; Li, X.; Chen, G.; Ma, L.; Liu, X.; Zhou, F. Global Burden of Hematologic Malignancies and Evolution Patterns over the Past 30 Years. Blood Cancer J. 2023, 13, 82. [Google Scholar] [CrossRef]
- Pulte, D.; Jansen, L.; Brenner, H. Changes in Long Term Survival after Diagnosis with Common Hematologic Malignancies in the Early 21st Century. Blood Cancer J. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Purton, L.E.; Scadden, D.T. The Hematopoietic Stem Cell Niche in Stembook; Harvard Stem Cell Institute: Cambridge, MA, USA, 2008; Available online: https://www.stembook.org/articles/the-hematopoietic-stem-cell-niche/ (accessed on 10 February 2025).
- Parekh, C.; Crooks, G.M. Critical Differences in Hematopoiesis and Lymphoid Development between Humans and Mice. J. Clin. Immunol. 2013, 33, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Nombela-Arrieta, C.; Manz, M.G. Quantification and Three-Dimensional Microanatomical Organization of the Bone Marrow. Blood Adv. 2017, 1, 407–416. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Takahashi, T. Organoids for Drug Discovery and Personalized Medicine. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 447–462. [Google Scholar] [CrossRef]
- Frenz-Wiessner, S.; Fairley, S.D.; Buser, M.; Goek, I.; Salewskij, K.; Jonsson, G.; Illig, D.; zu Putlitz, B.; Petersheim, D.; Li, Y.; et al. Generation of Complex Bone Marrow Organoids from Human Induced Pluripotent Stem Cells. Nat. Methods 2024, 21, 868–881. [Google Scholar] [CrossRef]
- Khan, A.O.; Rodriguez-Romera, A.; Reyat, J.S.; Olijnik, A.A.; Colombo, M.; Wang, G.; Wen, W.X.; Sousos, N.; Murphy, L.C.; Grygielska, B.; et al. Human Bone Marrow Organoids for Disease Modeling, Discovery, and Validation of Therapeutic Targets in Hematologic Malignancies. Cancer Discov. 2023, 13, 364–385. [Google Scholar] [CrossRef]
- Seet, C.S.; He, C.; Bethune, M.T.; Li, S.; Chick, B.; Gschweng, E.H.; Zhu, Y.; Kim, K.; Kohn, D.B.; Baltimore, D.; et al. Generation of Mature T Cells from Human Hematopoietic Stem and Progenitor Cells in Artificial Thymic Organoids. Nat. Methods 2017, 14, 521–530. [Google Scholar] [CrossRef]
- Montel-Hagen, A.; Seet, C.S.; Li, S.; Chick, B.; Zhu, Y.; Chang, P.; Tsai, S.; Sun, V.; Lopez, S.; Chen, H.C.; et al. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell 2019, 24, 376–389.e8. [Google Scholar] [CrossRef] [PubMed]
- Gee, K.; Isani, M.A.; Fode, A.; Maselli, K.M.; Zuber, S.M.; Fowler, K.L.; Squillaro, A.I.; Nucho, L.M.A.; Grikscheit, T.C. Spleen Organoid Units Generate Functional Human and Mouse Tissue-Engineered Spleen in a Murine Model. Tissue Eng. Part. A 2020, 26, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Wagar, L.E.; Salahudeen, A.; Constantz, C.M.; Wendel, B.S.; Lyons, M.M.; Mallajosyula, V.; Jatt, L.P.; Adamska, J.Z.; Blum, L.K.; Gupta, N.; et al. Modeling Human Adaptive Immune Responses with Tonsil Organoids. Nat. Med. 2021, 27, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef]
- Saglam-Metiner, P.; Devamoglu, U.; Filiz, Y.; Akbari, S.; Beceren, G.; Goker, B.; Yaldiz, B.; Yanasik, S.; Biray Avci, C.; Erdal, E.; et al. Spatio-Temporal Dynamics Enhance Cellular Diversity, Neuronal Function and Further Maturation of Human Cerebral Organoids. Commun. Biol. 2023, 6, 1–18. [Google Scholar] [CrossRef]
- Cho, Y.K.; Kim, H.K.; Kwon, S.S.; Jeon, S.H.; Cheong, J.W.; Nam, K.T.; Kim, H.S.; Kim, S.; Kim, H.O. In Vitro Erythrocyte Production Using Human-Induced Pluripotent Stem Cells: Determining the Best Hematopoietic Stem Cell Sources. Stem Cell Res. Ther. 2023, 14, 1–15. [Google Scholar] [CrossRef]
- Allenby, M.C.; Tahlawi, A.; Morais, J.C.F.; Li, K.; Panoskaltsis, N.; Mantalaris, A. Ceramic Hollow Fibre Constructs for Continuous Perfusion and Cell Harvest from 3D Hematopoietic Organoids. Stem Cells Int. 2018, 2018, 6230214. [Google Scholar] [CrossRef] [PubMed]
- Allenby, M.C.; Okutsu, N.; Brailey, K.; Guasch, J.; Zhang, Q.; Panoskaltsis, N.; Mantalaris, A. A Spatiotemporal Microenvironment Model to Improve Design of a Three-Dimensional Bioreactor for Red Cell Production. Tissue Eng. Part A 2022, 28, 38–53. [Google Scholar] [CrossRef]
- Torisawa, Y.S.; Spina, C.S.; Mammoto, T.; Mammoto, A.; Weaver, J.C.; Tat, T.; Collins, J.J.; Ingber, D.E. Bone Marrow–on–a–Chip Replicates Hematopoietic Niche Physiology in Vitro. Nat. Methods 2014, 11, 663–669. [Google Scholar] [CrossRef]
- Glaser, D.E.; Curtis, M.B.; Sariano, P.A.; Rollins, Z.A.; Shergill, B.S.; Anand, A.; Deely, A.M.; Shirure, V.S.; Anderson, L.; Lowen, J.M.; et al. Organ-on-a-Chip Model of Vascularized Human Bone Marrow Niches. Biomaterials 2022, 280, 121245. [Google Scholar] [CrossRef]
- Anderson, G.; Jenkinson, E.J. Investigating Central Tolerance with Reaggregate Thymus Organ Cultures. Methods Mol. Biol. 2007, 380, 185–196. [Google Scholar] [CrossRef]
- Chung, B.; Montel-Hagen, A.; Ge, S.; Blumberg, G.; Kim, K.; Klein, S.; Zhu, Y.; Parekh, C.; Balamurugan, A.; Yang, O.O.; et al. Engineering the Human Thymic Microenvironment to Support Thymopoiesis in Vivo. Stem Cells 2014, 32, 2386. [Google Scholar] [CrossRef] [PubMed]
- Bosticardo, M.; Notarangelo, L.D. Human Thymus in Health and Disease: Recent Advances in Diagnosis and Biology. Semin. Immunol. 2023, 66, 101732. [Google Scholar] [CrossRef] [PubMed]
- Ramos, S.A.; Armitage, L.H.; Morton, J.J.; Alzofon, N.; Handler, D.; Kelly, G.; Homann, D.; Jimeno, A.; Russ, H.A. Generation of Functional Thymic Organoids from Human Pluripotent Stem Cells. Stem Cell Rep. 2023, 18, 829–840. [Google Scholar] [CrossRef]
- Hübscher, T.; Lorenzo-Martín, L.F.; Barthlott, T.; Tillard, L.; Langer, J.J.; Rouse, P.; Blackburn, C.C.; Holländer, G.; Lutolf, M.P. Thymic Epithelial Organoids Mediate T-Cell Development. Development 2024, 151, dev202853. [Google Scholar] [CrossRef] [PubMed]
- Bogoslowski, A.; Butcher, E.C.; Kubes, P. Neutrophils Recruited through High Endothelial Venules of the Lymph Nodes via PNAd Intercept Disseminating Staphylococcus Aureus. Proc. Natl. Acad. Sci. USA 2018, 115, 2449–2454. [Google Scholar]
- Louie, D.A.P.; Liao, S. Lymp Node Subcapsular Sinus Macrophages as the Frontline of Lymphatic Immune Defense. Front. Immunol. 2019, 10, 438090. [Google Scholar] [CrossRef]
- Phan, T.G.; Green, J.A.; Xu, Y.; Cyster, J.G. Immune Complex Relay by Subcapsular Sinus Macrophages and Noncognate B Cells Drives Antibody Affinity Maturation. Nat. Immunol. 2009, 10, 786–793. [Google Scholar] [CrossRef]
- Skibinski, G.; Skibinska, A.; Deckers, M.; James, K. Tonsil Stromal-Cell Lines Expressing FDC-like Properties: Isolation, Characterization, and Interaction with B Lymphocytes. J. Immunol. Res. 1998, 6, 273–284. [Google Scholar] [CrossRef]
- Van Laar, J.M.; Melchers, M.; Teng, Y.K.O.; Van Der Zouwen, B.; Mohammadi, R.; Fischer, R.; Margolis, L.; Fitzgerald, W.; Grivel, J.C.; Breedveld, F.C.; et al. Sustained Secretion of Immunoglobulin by Long-Lived Human Tonsil Plasma Cells. Am. J. Pathol. 2007, 171, 917–927. [Google Scholar] [CrossRef]
- Moeller, T.D.; Shah, S.B.; Lai, K.; Lopez-Barbosa, N.; Desai, P.; Wang, W.; Zhong, Z.; Redmond, D.; Singh, A.; DeLisa, M.P. Profiling Germinal Center-like B Cell Responses to Conjugate Vaccines Using Synthetic Immune Organoids. ACS Cent. Sci. 2023, 9, 787–804. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, S.; Watanabe, T. Generation of a Synthetic Lymphoid Tissue–like Organoid in Mice. Nat. Biotechnol. 2004, 22, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Cuzzone, D.A.; Albano, N.J.; Aschen, S.Z.; Ghanta, S.; Mehrara, B.J. Decellularized Lymph Nodes as Scaffolds for Tissue Engineered Lymph Nodes. Lymphat. Res. Biol. 2015, 13, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Lenti, E.; Bianchessi, S.; Proulx, S.T.; Palano, M.T.; Genovese, L.; Raccosta, L.; Spinelli, A.; Drago, D.; Andolfo, A.; Alfano, M.; et al. Therapeutic Regeneration of Lymphatic and Immune Cell Functions upon Lympho-Organoid Transplantation. Stem Cell Rep. 2019, 12, 1260–1268. [Google Scholar] [CrossRef]
- Altamura, M.; Caradonna, L.; Amati, L.; Pellegrino, N.M.; Urgesi, G.; Miniello, S. SPLENECTOMY AND SEPSIS: THE ROLE OF THE SPLEEN IN THE IMMUNE-MEDIATED BACTERIAL CLEARANCE. Immunopharmacol. Immunotoxicol. 2001, 23, 153–161. [Google Scholar] [CrossRef]
- Golub, R.; Tan, J.; Watanabe, T.; Brendolan, A. Origin and Immunological Functions of Spleen Stromal Cells. Trends Immunol. 2018, 39, 503–514. [Google Scholar] [CrossRef]
- Schmidt, E.E.; MacDonald, I.C.; Groom, A.C. Comparative Aspects of Splenic Microcirculatory Pathways in Mammals: The Region Bordering the White Pulp. Scanning Microsc. 1993, 7, 17. [Google Scholar]
- Finetti, F.; Capitani, N.; Manganaro, N.; Tatangelo, V.; Libonati, F.; Panattoni, G.; Calaresu, I.; Ballerini, L.; Baldari, C.T.; Patrussi, L. Optimization of Organotypic Cultures of Mouse Spleen for Staining and Functional Assays. Front. Immunol. 2020. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct Bone Marrow Blood Vessels Differentially Regulate Haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Bessy, T.; Itkin, T.; Passaro, D. Bioengineering the Bone Marrow Vascular Niche. Front. Cell Dev. Biol. 2021, 9, 645496. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, J.; Kumar, S.; Shen, S.; Kincaid, M.; Johnson, C.B.; Zhang, Y.S.; Turcotte, R.; Alt, C.; Ito, K.; et al. Resilient Anatomy and Local Plasticity of Naive and Stress Haematopoiesis. Nature 2024, 627, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, Q.; Johnson, C.B.; Pham, G.; Kinder, J.M.; Olsson, A.; Slaughter, A.; May, M.; Weinhaus, B.; D’Alessandro, A.; et al. In Situ Mapping Identifies Distinct Vascular Niches for Myelopoiesis. Nature 2021, 590, 457–462. [Google Scholar] [CrossRef]
- Dennis, J.E.; Caplan, A.I. Bone Marrow Mesenchymal Stem Cells; Humana Press: Totowa, NJ, USA, 2004; pp. 107–117. [Google Scholar] [CrossRef]
- Lee, C.C.I.; Christensen, J.E.; Yoder, M.C.; Tarantal, A.F. Clonal Analysis and Hierarchy of Human Bone Marrow Mesenchymal Stem and Progenitor Cells. Exp. Hematol. 2010, 38, 46–54. [Google Scholar] [CrossRef]
- Hegner, B.; Lange, M.; Kusch, A.; Essin, K.; Sezer, O.; Schulze-Lohoff, E.; Luft, F.C.; Gollasch, M.; Dragun, D. MTOR Regulates Vascular Smooth Muscle Cell Differentiation from Human Bone Marrow-Derived Mesenchymal Progenitors. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Kolf, C.M.; Cho, E.; Tuan, R.S. Mesenchymal Stromal Cells. Biology of Adult Mesenchymal Stem Cells: Regulation of Niche, Self-Renewal and Differentiation. Arthritis Res. Ther. 2007, 9, 204. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, L.; Wang, S.; Huang, B.; Jing, Y.; Su, J. Bone Marrow Mesenchymal Stromal Cells: Identification, Classification, and Differentiation. Front. Cell Dev. Biol. 2022, 9, 787118. [Google Scholar] [CrossRef]
- Frenette, P.S.; Pinho, S.; Lucas, D.; Scheiermann, C. Mesenchymal Stem Cell: Keystone of the Hematopoietic Stem Cell Niche and a Stepping-Stone for Regenerative Medicine. Annu. Rev. Immunol. 2013, 31, 285–316. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.; Fonseca, A.V.; Alakel, N.; Fierro, F.A.; Muller, K.; Bornhauser, M.; Ehninger, G.; Corbeil, D.; Ordemann, R. Hematopoietic Stem Cells in Co-Culture with Mesenchymal Stromal Cells-Modeling the Niche Compartments In Vitro. Haematologica 2010, 95, 542–550. [Google Scholar] [CrossRef]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and Perivascular Cells Maintain Haematopoietic Stem Cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef]
- Poulos, M.G.; Guo, P.; Kofler, N.M.; Pinho, S.; Gutkin, M.C.; Tikhonova, A.; Aifantis, I.; Frenette, P.S.; Kitajewski, J.; Rafii, S.; et al. Endothelial Jagged-1 Is Necessary for Homeostatic and Regenerative Hematopoiesis. Cell Rep. 2013, 4, 1022–1034. [Google Scholar] [CrossRef]
- Wu, J.Y.; Scadden, D.T.; Kronenberg, H.M. Role of the Osteoblast Lineage in the Bone Marrow Hematopoietic Niches. J. Bone Miner. Res. 2009, 24, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Robles, H.; Park, S.J.; Joens, M.S.; Fitzpatrick, J.A.J.; Craft, C.S.; Scheller, E.L. Characterization of the Bone Marrow Adipocyte Niche with Three-Dimensional Electron Microscopy. Bone 2019, 118, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Cuminetti, V.; Arranz, L. Bone Marrow Adipocytes: The Enigmatic Components of the Hematopoietic Stem Cell Niche. J. Clin. Med. 2019, 8, 707. [Google Scholar] [CrossRef]
- Boulais, P.E.; Frenette, P.S. Making Sense of Hematopoietic Stem Cell Niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Belizario, J.E. Immunodeficient Mouse Models: An Overview. AACE Clin. Case Rep. 2021, 7, 79–85. [Google Scholar] [CrossRef]
- Fiedler, K.; Sindrilaru, A.; Terszowski, G.; Kokai, E.; Feyerabend, T.B.; Bullinger, L.; Rodewald, H.R.; Brunner, C. Neutrophil Development and Function Critically Depend on Bruton Tyrosine Kinase in a Mouse Model of X-Linked Agammaglobulinemia. Blood 2011, 117, 1329–1339. [Google Scholar] [CrossRef]
- Blundell, M.P.; Bouma, G.; Calle, Y.; Jones, G.E.; Kinnon, C.; Thrasher, A.J. Improvement of Migratory Defects in a Murine Model of Wiskott–Aldrich Syndrome Gene Therapy. Mol. Ther. 2008, 16, 836–844. [Google Scholar] [CrossRef]
- Peng, C.; Li, S. Chronic Myeloid Leukemia (CML) Mouse Model in Translational Research. Methods Mol. Biol. 2016, 1438, 225–243. [Google Scholar] [CrossRef]
- O’Neil, J.; Calvo, J.; McKenna, K.; Krishnamoorthy, V.; Aster, J.C.; Bassing, C.H.; Alt, F.W.; Kelliher, M.; Look, A.T. Activating Notch1 Mutations in Mouse Models of T-ALL. Blood 2006, 107, 781–785. [Google Scholar] [CrossRef]
- Deshpande, A.J.; Cusan, M.; Rawat, V.P.S.; Reuter, H.; Krause, A.; Pott, C.; Quintanilla-Martinez, L.; Kakadia, P.; Kuchenbauer, F.; Ahmed, F.; et al. Acute Myeloid Leukemia Is Propagated by a Leukemic Stem Cell with Lymphoid Characteristics in a Mouse Model of CALM/AF10-Positive Leukemia. Cancer Cell 2006, 10, 363–374. [Google Scholar] [CrossRef]
- Aydemir, N.; Bilaloǧlu, R. Genotoxicity of Two Anticancer Drugs, Gemcitabine and Topotecan, in Mouse Bone Marrow In Vivo. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2003, 537, 43–51. [Google Scholar] [CrossRef]
- Till, J.E.; Mcculloch, E.A. A Direct Measurement of the Radiation Sensitivity of Normal Mouse Bone Marrow Cells. Radiat. Res. 1961, 14, 213–222. [Google Scholar]
- Wolf, N.S.; Trentin, J.J. Hemopoietic colony studies: V. Effect of hemopoietic organ stroma on differentiation of pluripotent stem cells. J. Exp. Med. 1968, 127, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Negrin, R.; Hill, G.R. Mouse Models of Bone Marrow Transplantation. Biol. Blood Marrow Transplant. 2008, 14 (Suppl. 1), 129. [Google Scholar] [CrossRef]
- Almosailleakh, M.; Schwaller, J. Murine Models of Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 453. [Google Scholar] [CrossRef]
- Ilaria, R.L. Animal Models of Chronic Myelogenous Leukemia. Hematol. Oncol. Clin. N. Am. 2004, 18, 525–543. [Google Scholar] [CrossRef]
- Jacoby, E.; Chien, C.D.; Fry, T.J. Murine Models of Acute Leukemia: Important Tools in Current Pediatric Leukemia Research. Front. Oncol. 2014, 4, 95. [Google Scholar] [CrossRef]
- Simonetti, G.; Bertilaccio, M.T.S.; Ghia, P.; Klein, U. Mouse Models in the Study of Chronic Lymphocytic Leukemia Pathogenesis and Therapy. Blood 2014, 124, 1010–1019. [Google Scholar] [CrossRef]
- Rossi, M.; Botta, C.; Arbitrio, M.; Grembiale, R.D.; Tagliaferri, P.; Tassone, P. Mouse Models of Multiple Myeloma: Technologic Platforms and Perspectives. Oncotarget 2018, 9, 20119. [Google Scholar] [CrossRef]
- Beachy, S.H.; Aplan, P.D. Mouse Models of Myelodysplastic Syndromes. Hematol. Oncol. Clin. N. Am. 2010, 24, 361–375. [Google Scholar] [CrossRef]
- Jacquelin, S.; Kramer, F.; Mullally, A.; Lane, S.W. Murine Models of Myelofibrosis. Cancers 2020, 12, 2381. [Google Scholar] [CrossRef]
- Scheinberg, P.; Chen, J. Aplastic Anemia: What Have We Learned From Animal Models and From the Clinic. Semin. Hematol. 2013, 50, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Leblond, V.; Autran, B.; Cesbron, J.Y. The SCID Mouse Mutant: Definition and Potential Use as a Model for Immune and Hematological Disorders. Hematol. Cell Ther. 1997, 39, 213–221. [Google Scholar] [CrossRef]
- Blebea, J.S.; Houseni, M.; Torigian, D.A.; Fan, C.; Mavi, A.; Zhuge, Y.; Iwanaga, T.; Mishra, S.; Udupa, J.; Zhuang, J.; et al. Structural and Functional Imaging of Normal Bone Marrow and Evaluation of Its Age-Related Changes. Semin. Nucl. Med. 2007, 37, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Abarrategi, A.; Mian, S.A.; Passaro, D.; Rouault-Pierre, K.; Grey, W.; Bonnet, D. Modeling the Human Bone Marrow Niche in Mice: From Host Bone Marrow Engraftment to Bioengineering Approaches. J. Exp. Med. 2018, 215, 729–743. [Google Scholar] [CrossRef] [PubMed]
- Delorme, B.; Charbord, P. Culture and Characterization of Human Bone Marrow Mesenchymal Stem Cells. Methods Mol. Med. 2007, 140, 67–81. [Google Scholar] [CrossRef]
- Bonab, M.M.; Alimoghaddam, K.; Talebian, F.; Ghaffari, S.H.; Ghavamzadeh, A.; Nikbin, B. Aging of Mesenchymal Stem Cell in Vitro. BMC Cell Biol. 2006, 7, 14. [Google Scholar] [CrossRef]
- Frisch, B.J.; Calvi, L.M. Hematopoietic Stem Cell Cultures and Assays. Methods Mol. Biol. 2014, 1130, 315–324. [Google Scholar] [CrossRef]
- Audet, J.; Zandstra, P.W.; Eaves, C.J.; Piret, J.M. Advances in Hematopoietic Stem Cell Culture. Curr. Opin. Biotechnol. 1998, 9, 146–151. [Google Scholar] [CrossRef]
- Zhang, H.; Reilly, M.P. Human Induced Pluripotent Stem Cell? Derived Macrophages for Unraveling Human Macrophage Biology. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2000–2006. [Google Scholar] [CrossRef]
- Morishima, T.; Watanabe, K.; Niwa, A.; Fujino, H.; Matsubara, H.; Adachi, S.; Suemori, H.; Nakahata, T.; Heike, T. Neutrophil Differentiation from Human-Induced Pluripotent Stem Cells. J. Cell Physiol. 2011, 226, 1283–1291. [Google Scholar] [CrossRef]
- Nianias, A.; Themeli, M. Induced Pluripotent Stem Cell (IPSC)–Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S.; Tuan, R.S. Functional Comparison of Human-Induced Pluripotent Stem Cell-Derived Mesenchymal Cells and Bone Marrow-Derived Mesenchymal Stromal Cells from the Same Donor. Stem Cells Dev. 2014, 23, 1594–1610. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; James, W.; Moore, M.D. Human Induced Pluripotent Stem Cell-Derived Macrophages Share Ontogeny with MYB-Independent Tissue-Resident Macrophages. Stem Cell Rep. 2017, 8, 334–345. [Google Scholar] [CrossRef] [PubMed]
- de Janon, A.; Mantalaris, A.; Panoskaltsis, N. Three-Dimensional Human Bone Marrow Organoids for the Study and Application of Normal and Abnormal Hematoimmunopoiesis. J. Immunol. 2023, 210, 895–904. [Google Scholar] [CrossRef]
- Dexter, T.M.; Allen, T.D.; Lajtha, L.G. Conditions Controlling the Proliferation of Haemopoietic Stem Cells in Vitro. J. Cell Physiol. 1977, 91, 335–344. [Google Scholar] [CrossRef]
- Ogawa, M. Differentiation and Proliferation of Hematopoietic Stem Cells. Blood 1993, 81, 2844–2853. [Google Scholar] [CrossRef]
- Perdomo-Arciniegas, A.M.; Vernot, J.P. Co-Culture of Hematopoietic Stem Cells with Mesenchymal Stem Cells Increases VCAM-1-Dependent Migration of Primitive Hematopoietic Stem Cells. Int. J. Hematol. 2011, 94, 525–532. [Google Scholar] [CrossRef]
- Walenda, T.; Bork, S.; Horn, P.; Wein, F.; Saffrich, R.; Diehlmann, A.; Eckstein, V.; Ho, A.D.; Wagner, W. Co-Culture with Mesenchymal Stromal Cells Increases Proliferation and Maintenance of Haematopoietic Progenitor Cells. J. Cell Mol. Med. 2009, 14, 337–350. [Google Scholar] [CrossRef]
- Hatami, J.; Andrade, P.Z.; Alves de Matos, A.P.; Djokovic, D.; Lilaia, C.; Ferreira, F.C.; Cabral, J.M.S.; da Silva, C.L. Developing a Co-Culture System for Effective Megakaryo/Thrombopoiesis from Umbilical Cord Blood Hematopoietic Stem/Progenitor Cells. Cytotherapy 2015, 17, 428–442. [Google Scholar] [CrossRef]
- Gurel Pekozer, G.; Torun Kose, G.; Hasirci, V. Influence of Co-Culture on Osteogenesis and Angiogenesis of Bone Marrow Mesenchymal Stem Cells and Aortic Endothelial Cells. Microvasc. Res. 2016, 108, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Salati, S.; Lisignoli, G.; Manferdini, C.; Pennucci, V.; Zini, R.; Bianchi, E.; Norfo, R.; Facchini, A.; Ferrari, S.; Manfredini, R. Co-Culture of Hematopoietic Stem/Progenitor Cells with Human Osteblasts Favours Mono/Macrophage Differentiation at the Expense of the Erythroid Lineage. PLoS ONE 2013, 8, e53496. [Google Scholar] [CrossRef]
- Leisten, I.; Kramann, R.; Ventura Ferreira, M.S.; Bovi, M.; Neuss, S.; Ziegler, P.; Wagner, W.; Knüchel, R.; Schneider, R.K. 3D Co-Culture of Hematopoietic Stem and Progenitor Cells and Mesenchymal Stem Cells in Collagen Scaffolds as a Model of the Hematopoietic Niche. Biomaterials 2012, 33, 1736–1747. [Google Scholar] [CrossRef]
- Dhami, S.P.S.; Kappala, S.S.; Thompson, A.; Szegezdi, E. Three-Dimensional Ex Vivo Co-Culture Models of the Leukaemic Bone Marrow Niche for Functional Drug Testing. Drug Discov. Today 2016, 21, 1464–1471. [Google Scholar] [CrossRef]
- Huang, X.; Zhu, B.; Wang, X.; Xiao, R.; Wang, C. Three-Dimensional Co-Culture of Mesenchymal Stromal Cells and Differentiated Osteoblasts on Human Bio-Derived Bone Scaffolds Supports Active Multi-Lineage Hematopoiesis in Vitro: Functional Implication of the Biomimetic HSC Niche. Int. J. Mol. Med. 2016, 38, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Salman, Z.; Balandrán-Juárez, J.C.; Pelayo, R.; Guzman, M.L. A Novel Three-Dimensional Co-Culture System to Study Leukemia in the Bone Marrow Microenvironment. Blood 2015, 126, 1864. [Google Scholar] [CrossRef]
- Barbaglio, F.; Belloni, D.; Scarfò, L.; Sbrana, F.V.; Ponzoni, M.; Bongiovanni, L.; Pavesi, L.; Zambroni, D.; Stamatopoulos, K.; Caiolfa, V.R.; et al. Three-Dimensional Co-Culture Model of Chronic Lymphocytic Leukemia Bone Marrow Microenvironment Predicts Patient-Specific Response to Mobilizing Agents. Haematologica 2021, 106, 2334–2344. [Google Scholar] [CrossRef]
- Elizabeth, E.; Lewis, L. Modelling the Mesenchymal Stem Cell Niche In Vitro Using Magnetic Nanoparticles. Ph.D. Thesis, University of Glasgow, Glasgow, UK, 2016. [Google Scholar]
- Olijnik, A.A.; Rodriguez-Romera, A.; Wong, Z.C.; Shen, Y.; Reyat, J.S.; Jooss, N.J.; Rayes, J.; Psaila, B.; Khan, A.O. Generating Human Bone Marrow Organoids for Disease Modeling and Drug Discovery. Nat. Protoc. 2024, 19, 2117–2146. [Google Scholar] [CrossRef]
- Cuddihy, M.J.; Wang, Y.; MacHi, C.; Bahng, J.H.; Kotov, N.A. Replication of Bone Marrow Differentiation Niche: Comparative Evaluation of Different Three-Dimensional Matrices. Small 2013, 9, 1008–1015. [Google Scholar] [CrossRef]
- Ventura Ferreira, M.S.; Jahnen-Dechent, W.; Labude, N.; Bovi, M.; Hieronymus, T.; Zenke, M.; Schneider, R.K.; Neurs, S. Cord Blood-Hematopoietic Stem Cell Expansion in 3D Fibrin Scaffolds with Stromal Support. Biomaterials 2012, 33, 6987–6997. [Google Scholar] [CrossRef]
- de la Puente, P.; Muz, B.; Gilson, R.C.; Azab, F.; Luderer, M.; King, J.; Achilefu, S.; Vij, R.; Azab, A.K. 3D Tissue-Engineered Bone Marrow as a Novel Model to Study Pathophysiology and Drug Resistance in Multiple Myeloma. Biomaterials 2015, 73, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Belloni, D.; Heltai, S.; Ponzoni, M.; Villa, A.; Vergani, B.; Pecciarini, L.; Marcatti, M.; Girlanda, S.; Tonon, G.; Ciceri, F.; et al. Modeling Multiple Myeloma-Bone Marrow Interactions and Response to Drugs in a 3D Surrogate Microenvironment. Haematologica 2018, 103, 707–716. [Google Scholar] [CrossRef]
- Houshmand, M.; Soleimani, M.; Atashi, A.; Saglio, G.; Abdollahi, M.; Zarif, M.N. Mimicking the Acute Myeloid Leukemia Niche for Molecular Study and Drug Screening. Tissue Eng. Part. C Methods 2017, 23, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Isern, J.; Méndez-Ferrer, S. Stem Cell Interactions in a Bone Marrow Niche. Curr. Osteoporos. Rep. 2011, 9, 210–218. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 1–21. [Google Scholar] [CrossRef]
- Forsyth, R.; Ergün, S.; Rausch, M.; Iqbal, N.; Pathak, S.; Owston, H.E.; Ganguly, P. Organoid Models and Next-Generation Sequencing for Bone Marrow and Related Disorders. Organoids 2023, 2, 123–139. [Google Scholar] [CrossRef]
- Chen, K.; Li, Y.; Wu, X.; Tang, X.; Zhang, B.; Fan, T.; He, L.; Pei, X.; Li, Y. Establishment of Human Hematopoietic Organoids for Evaluation of Hematopoietic Injury and Regeneration Effect. Stem Cell Res. Ther. 2024, 15, 133. [Google Scholar] [CrossRef]
- Doherty-Boyd, W.S.; Donnelly, H.; Tsimbouri, M.P.; Dalby, M.J. Building Bones for Blood and beyond: The Growing Field of Bone Marrow Niche Model Development. Exp. Hematol. 2024, 135, 104232. [Google Scholar] [CrossRef]
- Isern, J.; Martín-Antonio, B.; Ghazanfari, R.; Martín, A.M.; López, J.A.; delToro, R.; Sánchez-Aguilera, A.; Arranz, L.; Martín-Pérez, D.; Suárez-Lledó, M.; et al. Self-Renewing Human Bone Marrow Mesenspheres Promote Hematopoietic Stem Cell Expansion. Cell Rep. 2013, 3, 1714–1724. [Google Scholar] [CrossRef]
- Giger, S.; Hofer, M.; Miljkovic-Licina, M.; Hoehnel, S.; Brandenberg, N.; Guiet, R.; Ehrbar, M.; Kleiner, E.; Gegenschatz-Schmid, K.; Matthes, T.; et al. Microarrayed Human Bone Marrow Organoids for Modeling Blood Stem Cell Dynamics. APL Bioeng. 2022, 6, 50. [Google Scholar] [CrossRef]
- Popova, G.; Soliman, S.S.; Kim, C.N.; Keefe, M.G.; Hennick, K.M.; Jain, S.; Li, T.; Tejera, D.; Shin, D.; Chhun, B.B.; et al. Human Microglia States Are Conserved across Experimental Models and Regulate Neural Stem Cell Responses in Chimeric Organoids. Cell Stem Cell 2021, 28, 2153–2166.e6. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Yang, X.; Liu, Y.; Deng, X.; Hayashi, Y.; Plummer, R.; Li, Q.; Luo, N.; Kasai, T.; Okumura, T.; et al. Integration of Kupffer Cells into Human IPSC-Derived Liver Organoids for Modeling Liver Dysfunction in Sepsis Ll Integration of Kupffer Cells into Human IPSC-Derived Liver Organoids for Modeling Liver Dysfunction in Sepsis. Cell Rep. 2024, 43, 113918. [Google Scholar] [CrossRef]
- Bogoslowski, A.; An, M.; Penninger, J.M. Incorporating Immune Cells into Organoid Models: Essential for Studying Human Disease. Organoids 2023, 2, 140–155. [Google Scholar] [CrossRef]
- Kunisaki, Y. Pericytes in Bone Marrow. Adv. Exp. Med. Biol. 2019, 1122, 101–114. [Google Scholar] [CrossRef]
- Piau, O.; Brunet-Manquat, M.; L’Homme, B.; Petit, L.; Birebent, B.; Linard, C.; Moeckes, L.; Zuliani, T.; Lapillonne, H.; Benderitter, M.; et al. Generation of Transgene-Free Hematopoietic Stem Cells from Human Induced Pluripotent Stem Cells. Cell Stem Cell 2023, 30, 1610–1623.e7. [Google Scholar] [CrossRef]
- Feyen, D.A.M.; McKeithan, W.L.; Bruyneel, A.A.N.; Spiering, S.; Hörmann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human IPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925. [Google Scholar] [CrossRef]
- Ho, R.; Sances, S.; Gowing, G.; Amoroso, M.W.; O’Rourke, J.G.; Sahabian, A.; Wichterle, H.; Baloh, R.H.; Sareen, D.; Svendsen, C.N. ALS Disrupts Spinal Motor Neuron Maturation and Aging Pathways within Gene Co-Expression Networks. Nat. Neurosci. 2016, 19, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gage, F.H.; Schafer, S.T. Transplantation Strategies to Enhance Maturity and Cellular Complexity in Brain Organoids. Biol. Psychiatry 2023, 93, 616–621. [Google Scholar] [CrossRef]
- Kolanowski, T.J.; Busek, M.; Schubert, M.; Dmitrieva, A.; Binnewerg, B.; Pöche, J.; Fisher, K.; Schmieder, F.; Grünzner, S.; Hansen, S.; et al. Enhanced Structural Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes under a Controlled Microenvironment in a Microfluidic System. Acta Biomater. 2020, 102, 273–286. [Google Scholar] [CrossRef]
- Allenby, M.C.; Panoskaltsis, N.; Tahlawi, A.; Dos Santos, S.B.; Mantalaris, A. Dynamic Human Erythropoiesis in a Three-Dimensional Perfusion Bone Marrow Biomimicry. Biomaterials 2019, 188, 24–37. [Google Scholar] [CrossRef]
- Bourgine, P.E.; Klein, T.; Paczulla, A.M.; Shimizu, T.; Kunz, L.; Kokkaliaris, K.D.; Coutu, D.L.; Lengerke, C.; Skoda, R.; Schroeder, T.; et al. In Vitro Biomimetic Engineering of a Human Hematopoietic Niche with Functional Properties. Proc. Natl. Acad. Sci. USA 2018, 115, E5688–E5695. [Google Scholar] [CrossRef] [PubMed]
- Born, G.; Nikolova, M.; Scherberich, A.; Treutlein, B.; García-García, A.; Martin, I. Engineering of Fully Humanized and Vascularized 3D Bone Marrow Niches Sustaining Undifferentiated Human Cord Blood Hematopoietic Stem and Progenitor Cells. J. Tissue Eng. 2021, 12, 20417314211044855. [Google Scholar] [CrossRef] [PubMed]
- Sieber, S.; Wirth, L.; Cavak, N.; Koenigsmark, M.; Marx, U.; Lauster, R.; Rosowski, M. Bone Marrow-on-a-Chip: Long-Term Culture of Human Haematopoietic Stem Cells in a Three-Dimensional Microfluidic Environment. J. Tissue Eng. Regen. Med. 2018, 12, 479–489. [Google Scholar] [CrossRef]
- Zhang, W.; Lee, W.Y.; Siegel, D.S.; Tolias, P.; Zilberberg, J. Patient-Specific 3D Microfluidic Tissue Model for Multiple Myeloma. Tissue Eng. Part. C Methods 2014, 20, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Witkowski, M.T.; Harris, J.; Dolgalev, I.; Sreeram, S.; Qian, W.; Tong, J.; Chen, X.; Aifantis, I.; Chen, W. Leukemia-on-a-Chip: Dissecting the Chemoresistance Mechanisms in B Cell Acute Lymphoblastic Leukemia Bone Marrow Niche. Sci. Adv. 2020, 6, eaba5536. [Google Scholar] [CrossRef]
- Chou, D.B.; Frismantas, V.; Milton, Y.; David, R.; Pop-Damkov, P.; Ferguson, D.; MacDonald, A.; Vargel Bölükbaşı, Ö.; Joyce, C.E.; Moreira Teixeira, L.S.; et al. On-Chip Recapitulation of Clinical Bone Marrow Toxicities and Patient-Specific Pathophysiology. Nat. Biomed. Eng. 2020, 4, 394–406. [Google Scholar] [CrossRef]
- Georgescu, A.; Oved, J.H.; Galarraga, J.H.; Cantrell, T.; Mehta, S.; Dulmovits, B.M.; Olson, T.S.; Fattahi, P.; Wang, A.; Candarlioglu, P.L.; et al. Self-Organization of the Hematopoietic Vascular Niche and Emergent Innate Immunity on a Chip. Cell Stem Cell 2024, 31, 1847–1864.e6. [Google Scholar] [CrossRef]
- Machiraju, P.; Greenway, S.C. Current Methods for the Maturation of Induced Pluripotent Stem Cell-Derived Cardiomyocytes. World J. Stem Cells 2019, 11, 33–43. [Google Scholar] [CrossRef]
- Jung, J.H.; Yang, S.R.; Kim, W.J.; Rhee, C.K.; Hong, S.H. Human Pluripotent Stem Cell-Derived Alveolar Organoids: Cellular Heterogeneity and Maturity. Tuberc. Respir. Dis. 2023, 87, 52–64. [Google Scholar] [CrossRef]
- Ergir, E.; Oliver-De La Cruz, J.; Fernandes, S.; Cassani, M.; Niro, F.; Pereira-Sousa, D.; Vrbský, J.; Vinarský, V.; Perestrelo, A.R.; Debellis, D.; et al. Generation and Maturation of Human IPSC-Derived 3D Organotypic Cardiac Microtissues in Long-Term Culture. Sci. Rep. 2022, 12, 17409. [Google Scholar] [CrossRef]
- Nishinakamura, R. Human Kidney Organoids: Progress and Remaining Challenges. Nat. Rev. Nephrol. 2019, 15, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Bogacheva, M.S.; Lou, Y.R. Challenges for the Applications of Human Pluripotent Stem Cell-Derived Liver Organoids. Front. Cell Dev. Biol. 2021, 9, 748576. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Q.; Jeong, H.W.; Koh, B.I.; Watson, E.C.; Xu, C.; Stehling, M.; Zhou, B.; Adams, R.H. A Specialized Bone Marrow Microenvironment for Fetal Haematopoiesis. Nat. Commun. 2022, 13, 1327. [Google Scholar] [CrossRef]
- Coşxkun, S.; Chao, H.; Vasavada, H.; Heydari, K.; Gonzales, N.; Zhou, X.; de Crombrugghe, B.; Hirschi, K.K. Development of the Fetal Bone Marrow Niche and Regulation of HSC Quiescence and Homing Ability by Emerging Osteolineage Cells. Cell Rep. 2014, 9, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Brady, K.; Dickinson, S.C.; Guillot, P.V.; Polak, J.; Blom, A.W.; Kafienah, W.; Hollander, A.P. Human Fetal and Adult Bone Marrow-Derived Mesenchymal Stem Cells Use Different Signaling Pathways for the Initiation of Chondrogenesis. Stem Cells Dev. 2014, 23, 541–554. [Google Scholar] [CrossRef]
- Newell, L.F.; Cook, R.J. Advances in Acute Myeloid Leukemia. BMJ 2021, 375, n2026. [Google Scholar] [CrossRef] [PubMed]
- Castagnetti, F.; Gugliotta, G.; Baccarani, M.; Breccia, M.; Specchia, G.; Levato, L.; Abruzzese, E.; Rossi, G.; Iurlo, A.; Martino, B.; et al. Differences among Young Adults, Adults and Elderly Chronic Myeloid Leukemia Patients. Ann. Oncol. 2015, 26, 185–192. [Google Scholar] [CrossRef]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic Lymphocytic Leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef]
- Trump, L.R.; Nayak, R.C.; Singh, A.K.; Emberesh, S.; Wellendorf, A.M.; Lutzko, C.M.; Cancelas, J.A. Neutrophils Derived from Genetically Modified Human Induced Pluripotent Stem Cells Circulate and Phagocytose Bacteria In Vivo. Stem Cells Transl. Med. 2019, 8, 557–567. [Google Scholar] [CrossRef]
- Gutbier, S.; Wanke, F.; Dahm, N.; Rümmelin, A.; Zimmermann, S.; Christensen, K.; Köchl, F.; Rautanen, A.; Hatje, K.; Geering, B.; et al. Large-Scale Production of Human IPSC-Derived Macrophages for Drug Screening. Int. J. Mol. Sci. 2020, 21, 4808. [Google Scholar] [CrossRef]
- Monkley, S.; Krishnaswamy, J.K.; Göransson, M.; Clausen, M.; Meuller, J.; Thörn, K.; Hicks, R.; Delaney, S.; Stjernborg, L. Optimised Generation of IPSC-Derived Macrophages and Dendritic Cells That Are Functionally and Transcriptionally Similar to Their Primary Counterparts. PLoS ONE 2020, 15, e0243807. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. IPSC-Derived NK Cells Maintain High Cytotoxicity and Enhance in Vivo Tumor Control in Concert with T Cells and Anti–PD-1 Therapy. Sci. Transl. Med. 2020, 12, eaaz5618. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Hogquist, K.A. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef]
- Petrie, H.T.; Zúñiga-Pflücker, J.C. Zoned out: Functional Mapping of Stromal Signaling Microenvironments in the Thymus. Annu. Rev. Immunol. 2007, 25, 649–679. [Google Scholar] [CrossRef]
- Zlotoff, D.A.; Bhandoola, A. Hematopoietic Progenitor Migration to the Adult Thymus. Ann. N. Y. Acad. Sci. 2011, 1217, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Pui, J.C.; Allman, D.; Xu, L.; DeRocco, S.; Karnell, F.G.; Bakkour, S.; Lee, J.Y.; Kadesch, T.; Hardy, R.R.; Aster, J.C.; et al. Notch1 Expression in Early Lymphopoiesis Influences B versus T Lineage Determination. Immunity 1999, 11, 299–308. [Google Scholar] [CrossRef]
- Radtke, F.; Fasnacht, N.; MacDonald, H.R. Notch Signaling in the Immune System. Immunity 2010, 32, 14–27. [Google Scholar] [CrossRef]
- Shortman, K.; Egerton, M.; Spangrude, G.J.; Scollay, R. The Generation and Fate of Thymocytes. Semin. Immunol. 1990, 2, 3–12. [Google Scholar]
- Gameiro, J.; Nagib, P.; Verinaud, L. The Thymus Microenvironment in Regulating Thymocyte Differentiation. Cell Adhes. Migr. 2010, 4, 382–390. [Google Scholar] [CrossRef]
- Han, J.; Zúñiga-Pflücker, J.C. A 2020 View of Thymus Stromal Cells in T Cell Development. J. Immunol. 2021, 206, 249–256. [Google Scholar] [CrossRef]
- Bleul, C.C.; Boehm, T. Chemokines Define Distinct Microenvironments in the Developing Thymus. Eur. J. Immunol. 2000, 30, 3371–3379. [Google Scholar] [CrossRef] [PubMed]
- Rodewald, H.R. Thymus Organogenesis. Annu. Rev. Immunol. 2008, 26, 355–388. [Google Scholar] [CrossRef] [PubMed]
- Su, D.M.; Navarre, S.; Oh, W.J.; Condie, B.G.; Manley, N.R. A Domain of Foxn1 Required for Crosstalk-Dependent Thymic Epithelial Cell Differentiation. Nat. Immunol. 2003, 4, 1128–1135. [Google Scholar] [CrossRef]
- Abramson, J.; Anderson, G. Thymic Epithelial Cells. Annu. Rev. Immunol. 2017, 35, 85–118. [Google Scholar] [CrossRef]
- Hozumi, K.; Mailhos, C.; Negishi, N.; Hirano, K.I.; Yahata, T.; Ando, K.; Zuklys, S.; Holländer, G.A.; Shima, D.T.; Habu, S. Delta-like 4 Is Indispensable in Thymic Environment Specific for T Cell Development. J. Exp. Med. 2008, 205, 2507. [Google Scholar] [CrossRef] [PubMed]
- Ohigashi, I.; Kozai, M.; Takahama, Y. Development and Developmental Potential of Cortical Thymic Epithelial Cells. Immunol. Rev. 2016, 271, 10–22. [Google Scholar] [CrossRef]
- Di Santo, J.P.; Aifantis, I.; Rosmaraki, E.; Garcia, C.; Feinberg, J.; Fehling, H.J.; Fischer, A.; Von Boehmer, H.; Rocha, B. The Common Cytokine Receptor γ Chain and the Pre-T Cell Receptor Provide Independent but Critically Overlapping Signals in Early α/β T Cell Development. J. Exp. Med. 1999, 189, 563. [Google Scholar] [CrossRef]
- Alves, N.L.; Goff, O.R.-L.; Huntington, N.D.; Sousa, A.P.; Ribeiro, V.S.G.; Bordack, A.; Vives, F.L.; Peduto, L.; Chidgey, A.; Cumano, A.; et al. Characterization of the Thymic IL-7 Niche in Vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 1512–1517. [Google Scholar] [CrossRef]
- Hewitt, S.L.; Chaumeil, J.; Skok, J.A. Chromosome Dynamics and the Regulation of V(D)J Recombination. Immunol. Rev. 2010, 237, 43–54. [Google Scholar] [CrossRef]
- Anderson, G.; Takahama, Y. Thymic Epithelial Cells: Working Class Heroes for T Cell Development and Repertoire Selection. Trends Immunol. 2012, 33, 256–263. [Google Scholar] [CrossRef]
- Starr, T.K.; Jameson, S.C.; Hogquist, K.A. Positive and Negative Selection of T Cells. Annu. Rev. Immunol. 2003, 21, 139–176. [Google Scholar] [CrossRef] [PubMed]
- Perniola, R. Twenty Years of AIRE. Front. Immunol. 2018, 9, 296687. [Google Scholar] [CrossRef]
- Perera, J.; Huang, H. The Development and Function of Thymic B Cells. Cell Mol. Life Sci. 2015, 72, 2657. [Google Scholar] [CrossRef]
- Yamano, T.; Steinert, M.; Klein, L. Thymic B Cells and Central T Cell Tolerance. Front. Immunol. 2015, 6, 155427. [Google Scholar] [CrossRef]
- Martín-Gayo, E.; Sierra-Filardi, E.; Corbí, A.L.; Toribio, M.L. Plasmacytoid Dendritic Cells Resident in Human Thymus Drive Natural Treg Cell Development. Blood 2010, 115, 5366–5375. [Google Scholar] [CrossRef] [PubMed]
- Proietto, A.I.; Van Dommelen, S.; Zhou, P.; Rizzitelli, A.; D’Amico, A.; Steptoe, R.J.; Naik, S.H.; Lahoud, M.H.; Liu, Y.; Zheng, P.; et al. Dendritic Cells in the Thymus Contribute to T-Regulatory Cell Induction. Proc. Natl. Acad. Sci. USA 2008, 105, 19869–19874. [Google Scholar] [CrossRef]
- Wang, H.; Zúñiga-Pflücker, J.C. Thymic Microenvironment: Interactions Between Innate Immune Cells and Developing Thymocytes. Front. Immunol. 2022, 13, 885280. [Google Scholar] [CrossRef]
- Kurd, N.S.; Lutes, L.K.; Yoon, J.; Chan, S.W.; Dzhagalov, I.L.; Hoover, A.R.; Robey, E.A. A Role for Phagocytosis in Inducing Cell Death during Thymocyte Negative Selection. Elife 2019, 8, e48097. [Google Scholar] [CrossRef]
- Hashimoto, D.; Gabriel, J.; Colet, R.; Murashima, A.; Fujimoto, K.; Ueda, Y.; Suzuki, K.; Hyuga, T.; Hemmi, H.; Kaisho, T.; et al. Radiation Inducible MafB Gene Is Required for Thymic Regeneration. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Holmes, R.; Zuñiga-Pflücker, J.C. The OP9-DL1 System: Generation of T-Lymphocytes from Embryonic or Hematopoietic Stem Cells In Vitro. Cold Spring Harb. Protoc. 2009, 2009, pdb.prot5156. [Google Scholar] [CrossRef]
- Fan, Y.; Tajima, A.; Goh, S.K.; Geng, X.; Gualtierotti, G.; Grupillo, M.; Coppola, A.; Bertera, S.; Rudert, W.A.; Banerjee, I.; et al. Bioengineering Thymus Organoids to Restore Thymic Function and Induce Donor-Specific Immune Tolerance to Allografts. Mol. Ther. 2015, 23, 1262–1277. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Schmidt, K.; Egle, S.; Stark, H.-J.; Boukamp, P.; Kyewski, B. An Organotypic Coculture Model Supporting Proliferation and Differentiation of Medullary Thymic Epithelial Cells and Promiscuous Gene Expression. J. Immunol. 2013, 190, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; McWilliams-Koeppen, H.P.; Reza, H.; Ostberg, J.R.; Chen, W.; Wang, X.; Huynh, C.; Vyas, V.; Chang, W.C.; Starr, R.; et al. 3D-Organoid Culture Supports Differentiation of Human CAR+ IPSCs into Highly Functional CAR T Cells. Cell Stem Cell 2022, 29, 515–527.e8. [Google Scholar] [CrossRef]
- Li, S.; Wang, C.S.; Montel-Hagen, A.; Chen, H.C.; Lopez, S.; Zhou, O.; Dai, K.; Tsai, S.; Satyadi, W.; Botero, C.; et al. Strength of CAR Signaling Determines T Cell versus ILC Differentiation from Pluripotent Stem Cells. Cell Rep. 2023, 42, 112241. [Google Scholar] [CrossRef] [PubMed]
- Pille, M.; Avila, J.; Sanchez, G.S.; Goetgeluk, G.; De Munter, S.; Jansen, H.; Billiet, L.; Weening, K.; Xue, H.; Bonte, S.; et al. The Wiskott–Aldrich Syndrome Protein Is Required for Positive Selection during T-Cell Lineage Differentiation. Front. Immunol. 2023, 14, 1188099. [Google Scholar] [CrossRef]
- Materna, M.; Delmonte, O.M.; Bosticardo, M.; Momenilandi, M.; Conrey, P.E.; Muylder, B.C.D.; Bravetti, C.; Bellworthy, R.; Cederholm, A.; Staels, F.; et al. The Immunopathological Landscape of Human Pre-TCRa Deficiency: From Rare to Common Variants. Science (1979) 2024, 383, eadh4059. [Google Scholar] [CrossRef]
- Brault, J.; Liu, T.; Bello, E.; Liu, S.; Sweeney, C.L.; Meis, R.J.; Koontz, S.; Corsino, C.; Choi, U.; Vayssiere, G.; et al. CRISPR-Targeted MAGT1 Insertion Restores XMEN Patient Hematopoietic Stem Cells and Lymphocytes. Blood 2021, 138, 2768–2780. [Google Scholar] [CrossRef]
- Brault, J.; Liu, T.; Liu, S.; Lawson, A.; Choi, U.; Kozhushko, N.; Bzhilyanskaya, V.; Pavel-Dinu, M.; Meis, R.J.; Eckhaus, M.A.; et al. CRISPR-Cas9-AAV versus Lentivector Transduction for Genome Modification of X-Linked Severe Combined Immunodeficiency Hematopoietic Stem Cells. Front. Immunol. 2023, 13, 1067417. [Google Scholar] [CrossRef]
- Castiello, M.C.; Brandas, C.; Ferrari, S.; Porcellini, S.; Sacchetti, N.; Canarutto, D.; Draghici, E.; Merelli, I.; Barcella, M.; Pelosi, G.; et al. Exonic Knockout and Knockin Gene Editing in Hematopoietic Stem and Progenitor Cells Rescues RAG1 Immunodeficiency. Sci. Transl. Med. 2024, 16, eadh8162. [Google Scholar] [CrossRef]
- Gardner, C.L.; Pavel-Dinu, M.; Dobbs, K.; Bosticardo, M.; Reardon, P.K.; Lack, J.; DeRavin, S.S.; Le, K.; Bello, E.; Pala, F.; et al. Gene Editing Rescues In Vitro T Cell Development of RAG2-Deficient Induced Pluripotent Stem Cells in an Artificial Thymic Organoid System. J. Clin. Immunol. 2021, 41, 852–862. [Google Scholar] [CrossRef]
- McAuley, G.E.; Yiu, G.; Chang, P.C.; Newby, G.A.; Campo-Fernandez, B.; Fitz-Gibbon, S.T.; Wu, X.; Kang, S.H.L.; Garibay, A.; Butler, J.; et al. Human T Cell Generation Is Restored in CD3δ Severe Combined Immunodeficiency through Adenine Base Editing. Cell 2023, 186, 1398–1416.e23. [Google Scholar] [CrossRef] [PubMed]
- Hubin, F.; Humblet, C.; Belaid, Z.; Lambert, C.; Boniver, J.; Thiry, A.; Defresne, M.-P. Murine Bone Marrow Stromal Cells Sustain In Vivo the Survival of Hematopoietic Stem Cells and the Granulopoietic Differentiation of More Mature Progenitors. Stem Cells 2005, 23, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Okabe, M.; Ito, S.; Nishio, N.; Tanaka, Y.; Isobe, K.I. Thymic Epithelial Cells Induced from Pluripotent Stem Cells by a Three-Dimensional Spheroid Culture System Regenerates Functional T Cells in Nude Mice. Cell. Reprogramming 2015, 17, 368–375. [Google Scholar] [CrossRef]
- Parent, A.V.; Russ, H.A.; Khan, I.S.; Laflam, T.N.; Metzger, T.C.; Anderson, M.S.; Hebrok, M. Generation of Functional Thymic Epithelium from Human Embryonic Stem Cells That Supports Host T Cell Development. Cell Stem Cell 2013, 13, 219–229. [Google Scholar] [CrossRef]
- Sun, X.; Xu, J.; Lu, H.; Liu, W.; Miao, Z.; Sui, X.; Liu, H.; Su, L.; Du, W.; He, Q.; et al. Directed Differentiation of Human Embryonic Stem Cells into Thymic Epithelial Progenitor-like Cells Reconstitutes the Thymic Microenvironment in Vivo. Cell Stem Cell 2013, 13, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Zeleniak, A.; Wiegand, C.; Liu, W.; McCormick, C.; Ravikumar, K.; Alavi, A.; Guan, H.; Bertera, S.; Lakomy, R.; Tajima, A.; et al. De Novo Construction of T Cell Compartment in Humanized Mice Engrafted with IPSC-Derived Thymus Organoids. Nat. Methods 2022, 19, 1306–1319. [Google Scholar] [CrossRef]
- Lim, S.; Van Son, G.J.F.; Luh Wisma, N.; Yanti, E.; Lee, H.-G.; Begthel, H. Derivation of Functional Thymic Epithelial Organoid Lines from Adult Murine Thymus. Cell Rep. 2024, 43, 114019. [Google Scholar] [CrossRef]
- Sultana, D.A.; Tomita, S.; Hamada, M.; Iwanaga, Y.; Kitahama, Y.; Van Khang, N.; Hirai, S.; Ohigashi, I.; Nitta, S.; Amagai, T.; et al. Gene Expression Profile of the Third Pharyngeal Pouch Reveals Role of Mesenchymal MafB in Embryonic Thymus Development. Blood 2009, 113, 2976. [Google Scholar] [CrossRef]
- James, K.D.; Jenkinson, W.E.; Anderson, G. Non-Epithelial Stromal Cells in Thymus Development and Function. Front. Immunol. 2021, 12, 634367. [Google Scholar] [CrossRef]
- Scimone, M.L.; Aifantis, I.; Apostolou, I.; Von Boehmer, H.; Von Andrian, U.H. A Multistep Adhesion Cascade for Lymphoid Progenitor Cell Homing to the Thymus. Proc. Natl. Acad. Sci. USA 2006, 103, 7006–7011. [Google Scholar] [CrossRef]
- Montel-Hagen, A.; Crooks, G.M. From Pluripotent Stem Cells to T Cells. Exp. Hematol. 2019, 71, 24–31. [Google Scholar] [CrossRef]
- Moore, J.E.; Bertram, C.D. Lymphatic System Flows. Annu. Rev. Fluid. Mech. 2018, 50, 459–482. [Google Scholar] [CrossRef] [PubMed]
- Margaris, K.N.; Black, R.A. Modelling the Lymphatic System: Challenges and Opportunities. J. R. Soc. Interface 2012, 9, 601–612. [Google Scholar] [CrossRef]
- Ansel, K.M.; Cyster, J.G. Chemokines in Lymphopoiesis and Lymphoid Organ Development. Curr. Opin. Immunol. 2001, 13, 172–179. [Google Scholar] [CrossRef]
- Von Andrian, U.H.; Mempel, T.R. Homing and Cellular Traffic in Lymph Nodes. Nat. Rev. Immunol. 2003, 3, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.L.; Acton, S.E.; Knoblich, K. Lymph Node Fibroblastic Reticular Cells in Health and Disease. Nat. Rev. Immunol. 2015, 15, 350–361. [Google Scholar] [CrossRef] [PubMed]
- León, B.; Lund, F.E. Compartmentalization of Dendritic Cell and T-Cell Interactions in the Lymph Node: Anatomy of T-Cell Fate Decisions. Immunol. Rev. 2019, 289, 84–100. [Google Scholar] [CrossRef]
- Victora, G.D.; Nussenzweig, M.C. Germinal Centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef]
- Nave, H.; Gebert, A.; Pabst, R. Morphology and Immunology of the Human Palatine Tonsil. Anat. Embryol. 2001, 204, 367–373. [Google Scholar] [CrossRef]
- Pipi, E.; Nayar, S.; Gardner, D.H.; Colafrancesco, S.; Smith, C.; Barone, F. Tertiary Lymphoid Structures: Autoimmunity Goes Local. Front. Immunol. 2018, 9, 363085. [Google Scholar] [CrossRef]
- Maher, D.M.; Zhang, Z.Q.; Schacker, T.W.; Southern, P.J. Ex Vivo Modeling of Oral HIV Transmission in Human Palatine Tonsil. J. Histochem. Cytochem. 2005, 53, 631–642. [Google Scholar] [CrossRef]
- Bonaiti, E.; Muraro, M.G.; Robert, P.A.; Jakscha, J.; Dirnhofer, S.; Martin, I.; Berger, C.T. Tonsil Explants as a Human in Vitro Model to Study Vaccine Responses. Front. Immunol. 2024, 15, 1425455. [Google Scholar] [CrossRef]
- Soto-Rivera, J.; Patterson, B.K.; Chen, Y.; Shen, C.; Ratner, D.; Ding, M.; Tumne, A.; Gupta, P. Study of HIV-1 Transmission across Cervical Mucosa to Tonsil Tissue Cells Using an Organ Culture. Am. J. Reprod. Immunol. 2013, 69, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Klinman, N.R. The Mechanism of Antigenic Stimulation of Primary and Secondary Clonal Precursor Cells. J. Exp. Med. 1972, 136, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Shanti, A.; Samara, B.; Abdullah, A.; Hallfors, N.; Accoto, D.; Sapudom, J.; Alatoom, A.; Teo, J.; Danti, S.; Stefanini, C. Multi-Compartment 3D-Cultured Organ-on-a-Chip: Towards a Biomimetic Lymph Node for Drug Development. Pharmaceutics 2020, 12, 464. [Google Scholar] [CrossRef]
- Birmingham, K.G.; O’Melia, M.J.; Bordy, S.; Reyes Aguilar, D.; El-Reyas, B.; Lesinski, G.; Thomas, S.N. Lymph Node Subcapsular Sinus Microenvironment-On-A-Chip Modeling Shear Flow Relevant to Lymphatic Metastasis and Immune Cell Homing. iScience 2020, 23, 101751. [Google Scholar] [CrossRef]
- Sonmez, U.M.; Wood, A.; Justus, K.; Jiang, W.; Syed-Picard, F.; LeDuc, P.R.; Kalinski, P.; Davidson, L.A. Chemotactic Responses of Jurkat Cells in Microfluidic Flow-Free Gradient Chambers. Micromachines 2020, 11, 384. [Google Scholar] [CrossRef] [PubMed]
- Ricart, B.G.; John, B.; Lee, D.; Hunter, C.A.; Hammer, D.A. Dendritic Cells Distinguish Individual Chemokine Signals through CCR7 and CXCR4. J. Immunol. 2011, 186, 53–61. [Google Scholar] [CrossRef]
- Haessler, U.; Kalinin, Y.; Swartz, M.A.; Wu, M. An Agarose-Based Microfluidic Platform with a Gradient Buffer for 3D Chemotaxis Studies. Biomed. Microdevices 2009. [Google Scholar] [CrossRef]
- Haessler, U.; Pisano, M.; Wu, M.; Swartz, M.A. Dendritic Cell Chemotaxis in 3D under Defined Chemokine Gradients Reveals Differential Response to Ligands CCL21 and CCL19. Proc. Natl. Acad. Sci. USA 2011, 108, 5614–5619. [Google Scholar] [CrossRef]
- Giese, C.; Lubitz, A.; Demmler, C.D.; Reuschel, J.; Bergner, K.; Marx, U. Immunological Substance Testing on Human Lymphatic Micro-Organoids In Vitro. J. Biotechnol. 2010, 148, 38–45. [Google Scholar] [CrossRef]
- Chadburn, A. The Spleen: Anatomy and Anatomical Function. Semin. Hematol. 2000, 37 (Suppl. 1), 13–21. [Google Scholar] [CrossRef] [PubMed]
- Luu, S.; Woolley, I.J.; Andrews, R.K. Platelet Phenotype and Function in the Absence of Splenic Sequestration (Review). Platelets 2021, 32, 47–52. [Google Scholar] [CrossRef]
- Udroiu, I. Storage of Blood in the Mammalian Spleen: An Evolutionary Perspective. J. Mamm. Evol. 2017, 24, 243–260. [Google Scholar] [CrossRef]
- Cesta, M.F. Normal Structure, Function, and Histology of the Spleen. Toxicol. Pathol. 2006, 34, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Mebius, R.E.; Kraal, G. Structure and Function of the Spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Picot, J.; Ndour, P.A.; Lefevre, S.D.; El Nemer, W.; Tawfik, H.; Galimand, J.; Da Costa, L.; Ribeil, J.A.; de Montalembert, M.; Brousse, V.; et al. A Biomimetic Microfluidic Chip to Study the Circulation and Mechanical Retention of Red Blood Cells in the Spleen. Am. J. Hematol. 2015. [Google Scholar] [CrossRef]
- Rigat-Brugarolas, L.G.; Elizalde-Torrent, A.; Bernabeu, M.; De Niz, M.; Martin-Jaular, L.; Fernandez-Becerra, C.; Homs-Corbera, A.; Samitier, J.; Del Portillo, H.A. A Functional Microengineered Model of the Human Splenon-on-a-Chip. Lab. Chip 2014, 14, 1715–1724. [Google Scholar] [CrossRef]
- Deniset, J.F.; Surewaard, B.G.; Lee, W.-Y.; Kubes, P. Splenic Ly6Ghigh Mature and Ly6Gint Immature Neutrophils Contribute to Eradication of S. Pneumoniae. J. Exp. Med. 2017, 214, 1333–1350. [Google Scholar]
- Liang, Z.; Dong, X.; Zhang, Z.; Zhang, Q.; Zhao, Y. Age-related Thymic Involution: Mechanisms and Functional Impact. Aging Cell 2022, 21, e13671. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lymphoid Organ | Model Description | Cell Source | Advantages | Limitations | Refs |
|---|---|---|---|---|---|
| Bone Marrow | iPSC-derived organoids | Human iPSCs |
|
| [19,20] |
| Perfusion-bioreactor-based organoids | Human umbilical cord blood |
|
| [26,27,28,29] | |
| Bone-marrow-on-a-chip | Human umbilical-cord-blood-derived endothelial cells and HSPCs, as well as human fetal osteoblast (hFOB) cells |
|
| [30,31] | |
| Thymus | Reaggregate thymus organ cultures | Murine thymic cells |
|
| [32,33] |
| Decellularized thymic scaffolds | Murine thymic tissue, TECs |
|
| [34] | |
| Artificial thymic organoids (ATOs) | Murine bone marrow stromal cells and human HSPC/iPSCs |
|
| [21,22] | |
| ATOs with iPSC-derived TECs | iPSC- or embryonic-tissue-derived TECs, HSPCs |
|
| [35,36] | |
| Lymph Nodes | Ex vivo tonsillar cultures | Human/mouse tonsil tissue |
|
| [37,38,39] |
| Tonsil-derived B cell follicle organoids | Human tonsil biopsy or lung lymph node samples |
|
| [24] | |
| Synthetic or decellularized scaffolds with stromal cells | Collagen scaffolds, thymic stromal cells |
|
| [40,41,42] | |
| Lymph-node-on-a-chip models | DCs, T cells, or Jurkat cells |
|
| [43,44,45] | |
| Spleen | Ex vivo spleen slice maintenance | Mouse spleen tissue |
|
| [46] |
| Donor-tissue-derived organoids | Human/mouse spleen |
|
| [24] | |
| Scaffold-based spleen organoids | Donor-spleen-derived cell clusters |
|
| [47] | |
| Microfluidic spleen models | Human erythrocytes |
|
| [48,49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogoslowski, A.; Ren, J.; Quintard, C.; Penninger, J.M. Organoid Models of Lymphoid Tissues. Organoids 2025, 4, 7. https://doi.org/10.3390/organoids4020007
Bogoslowski A, Ren J, Quintard C, Penninger JM. Organoid Models of Lymphoid Tissues. Organoids. 2025; 4(2):7. https://doi.org/10.3390/organoids4020007
Chicago/Turabian StyleBogoslowski, Ania, Joice Ren, Clément Quintard, and Josef M. Penninger. 2025. "Organoid Models of Lymphoid Tissues" Organoids 4, no. 2: 7. https://doi.org/10.3390/organoids4020007
APA StyleBogoslowski, A., Ren, J., Quintard, C., & Penninger, J. M. (2025). Organoid Models of Lymphoid Tissues. Organoids, 4(2), 7. https://doi.org/10.3390/organoids4020007