
Testolactone: The Rise and Fall of a Drug



Abstract
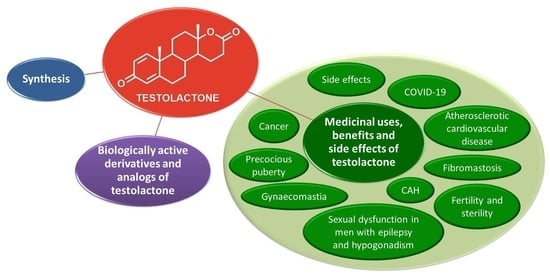
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
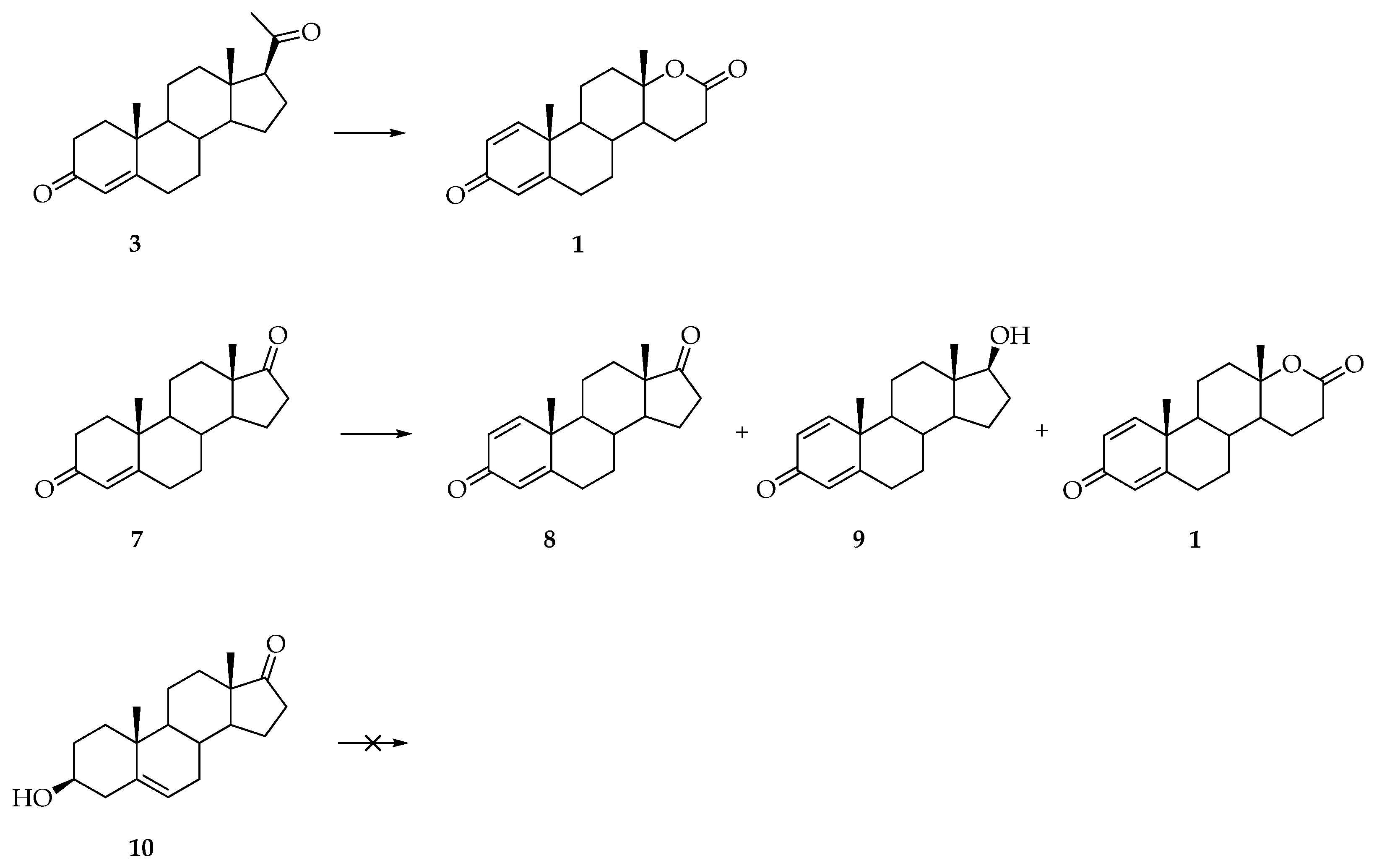{kind=link}
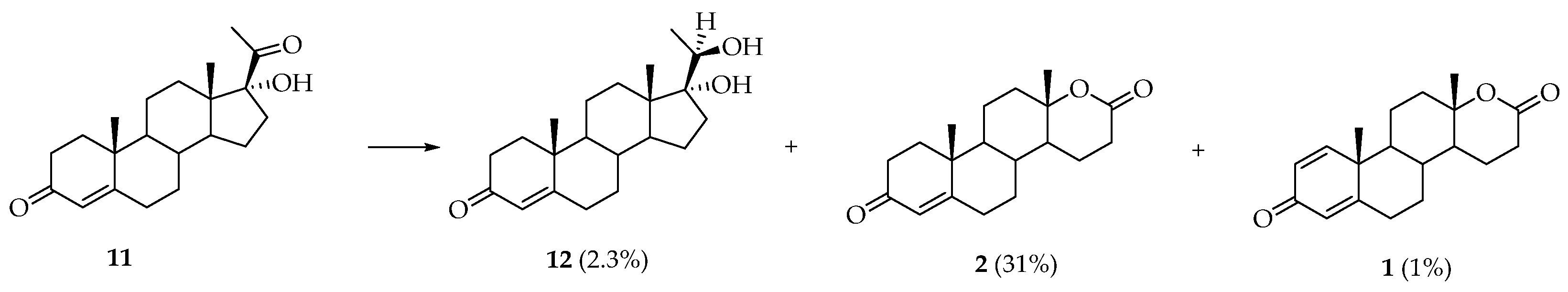{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
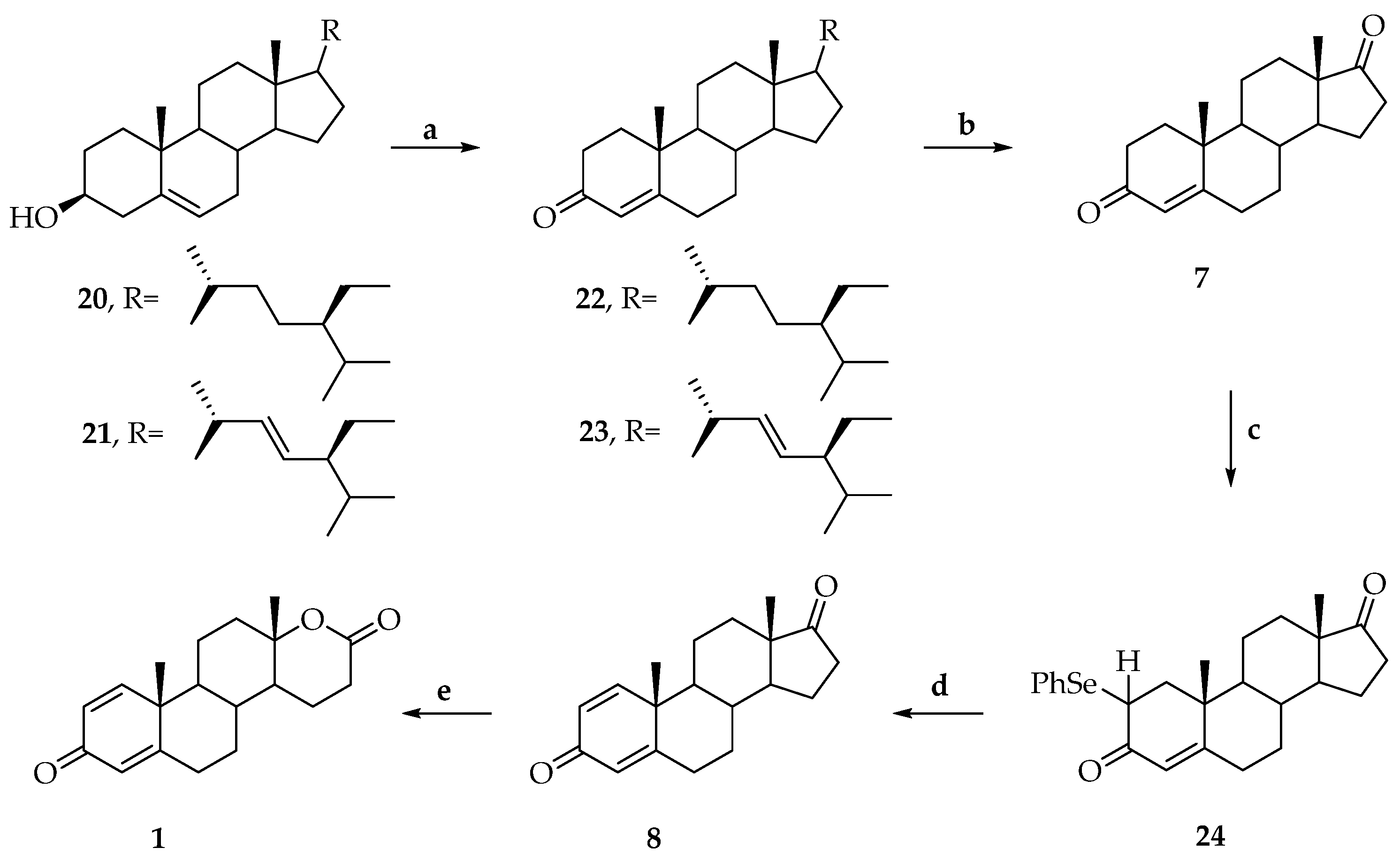{kind=link}
1. Introduction
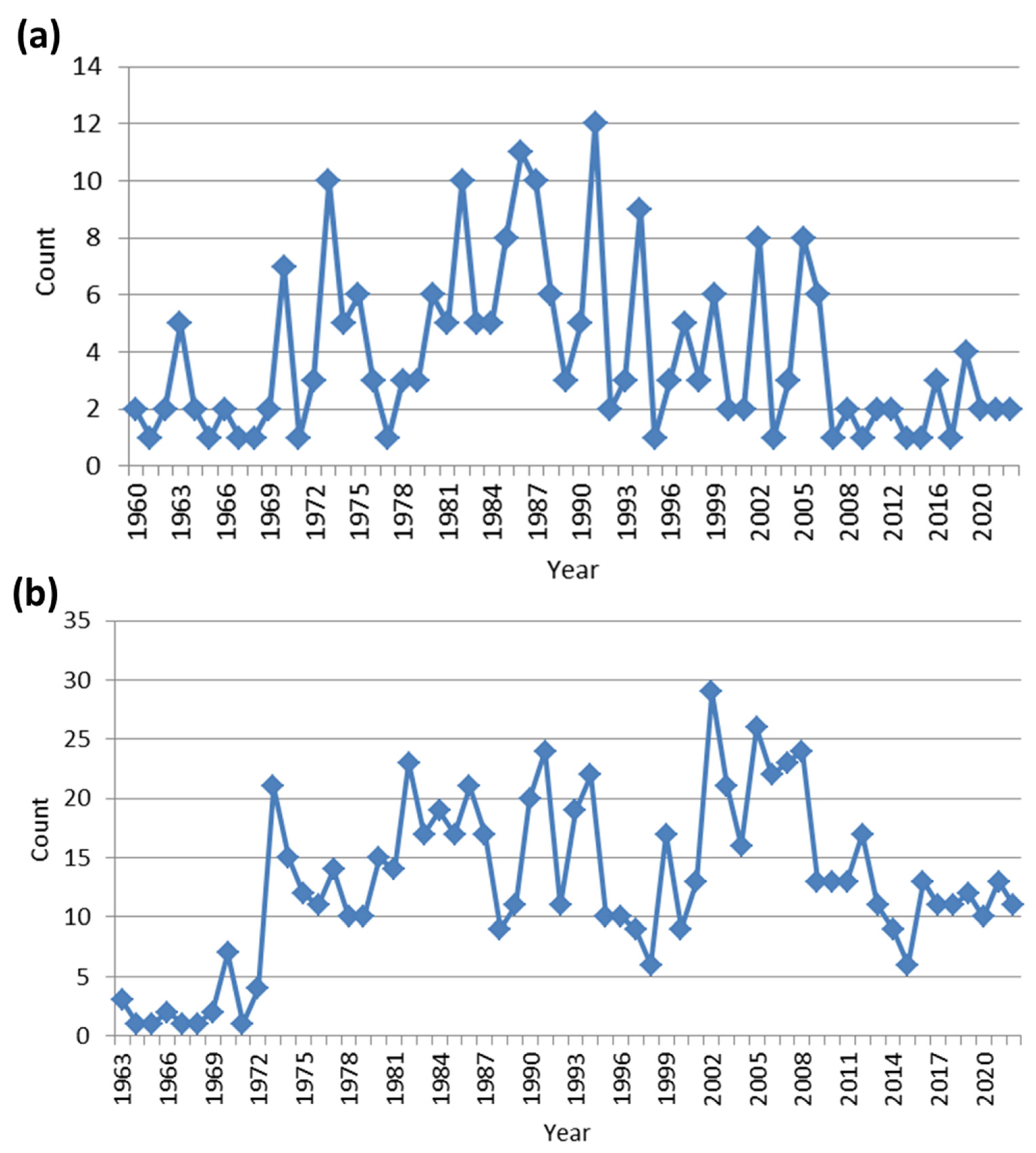
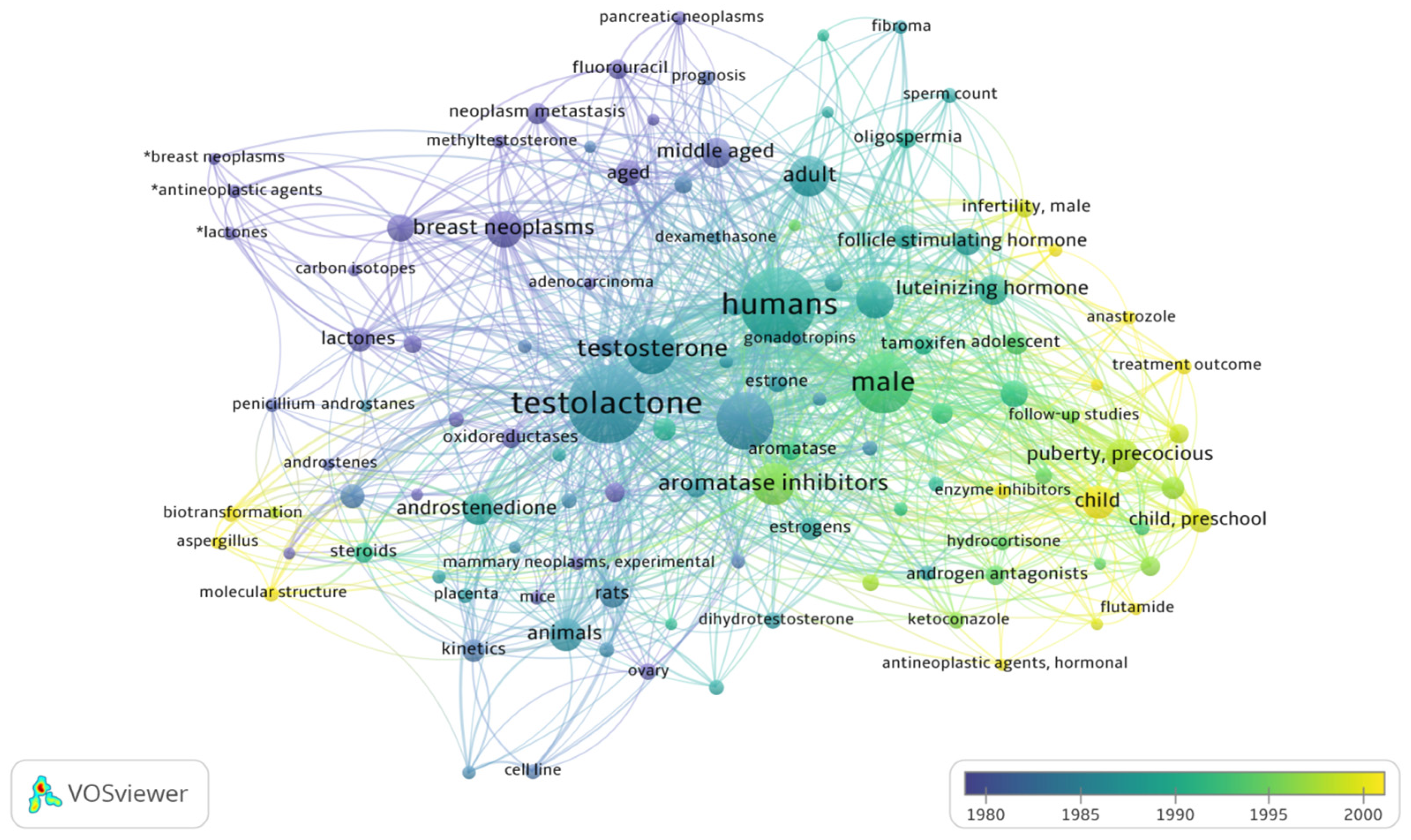
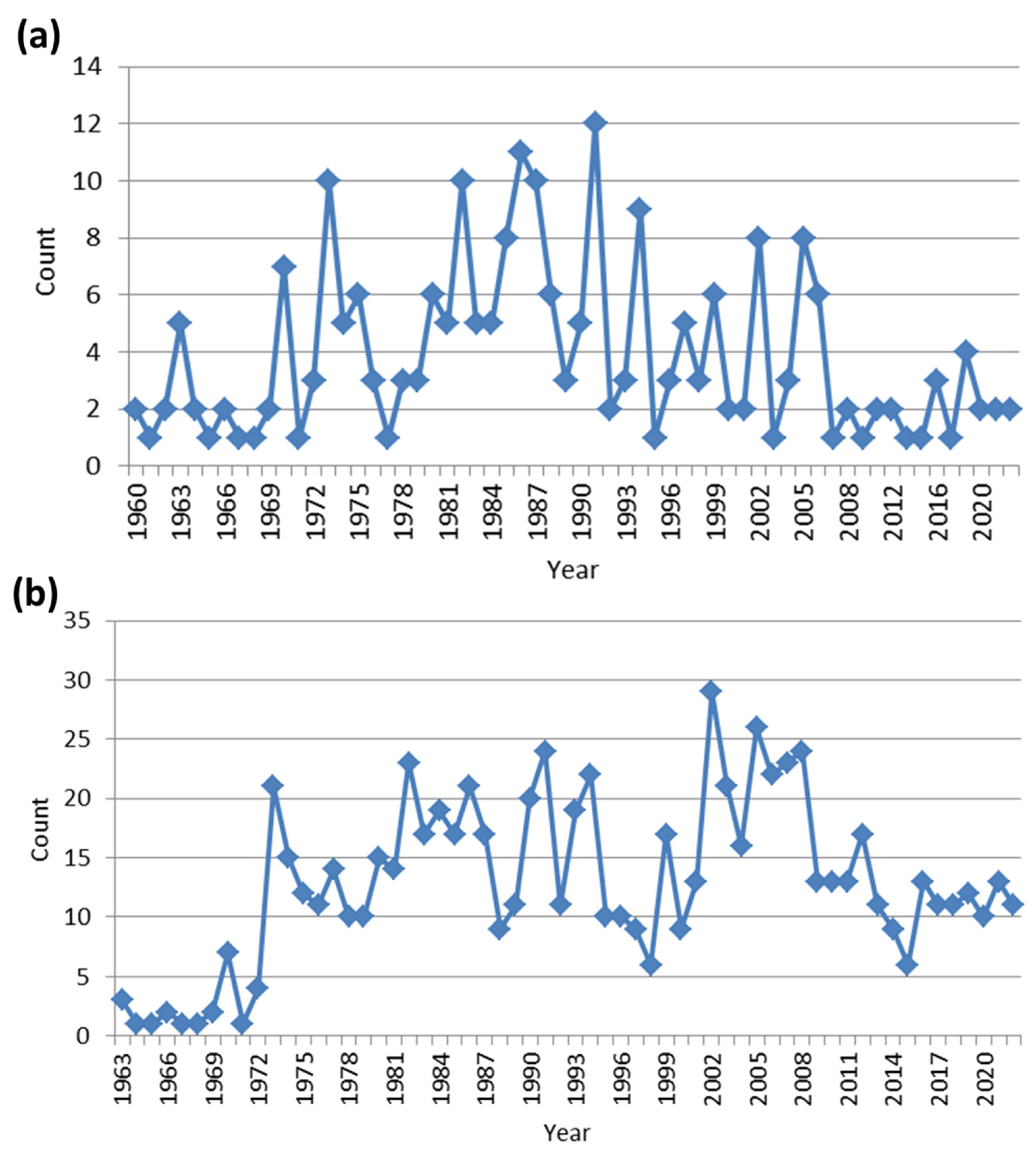
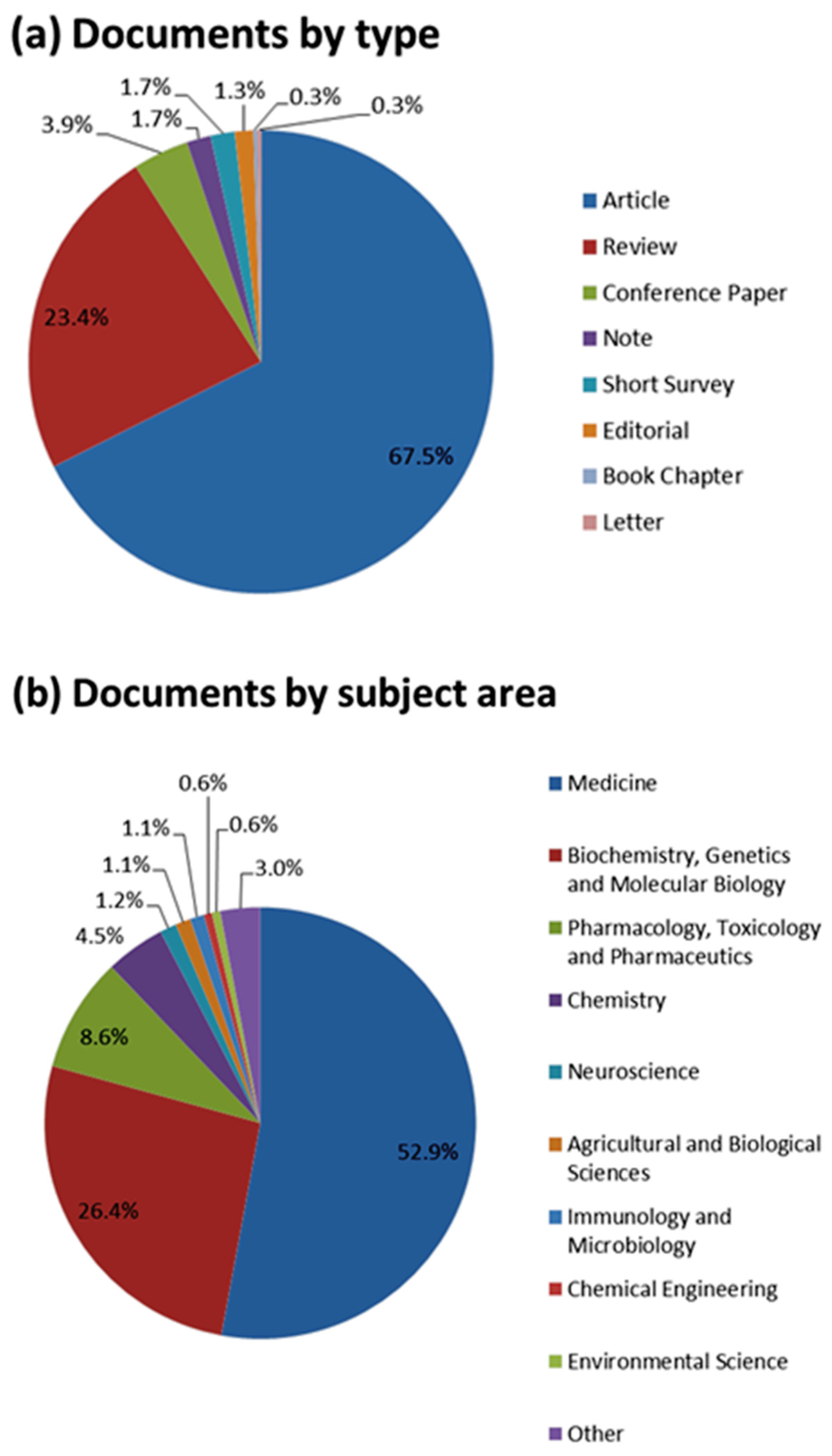
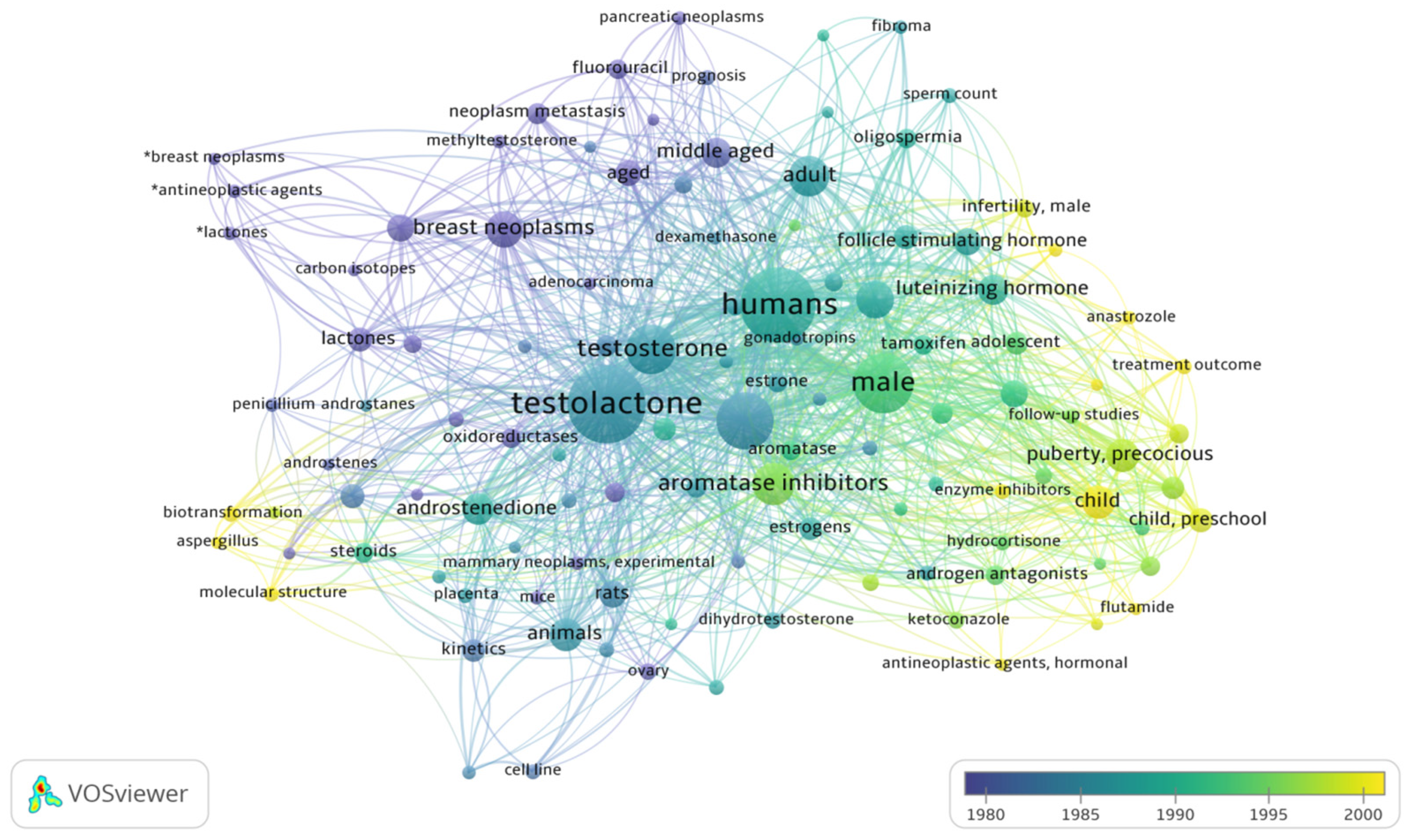
2. Literature Research Analysis
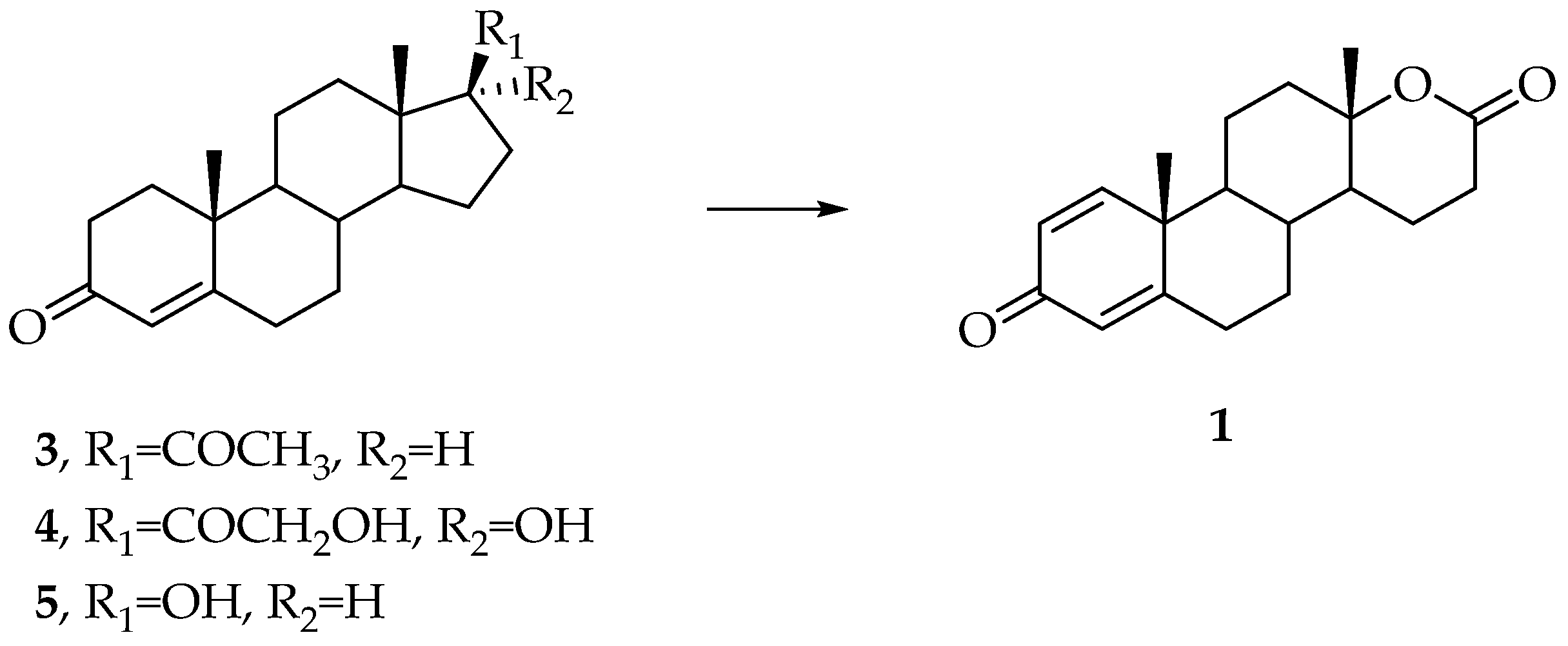
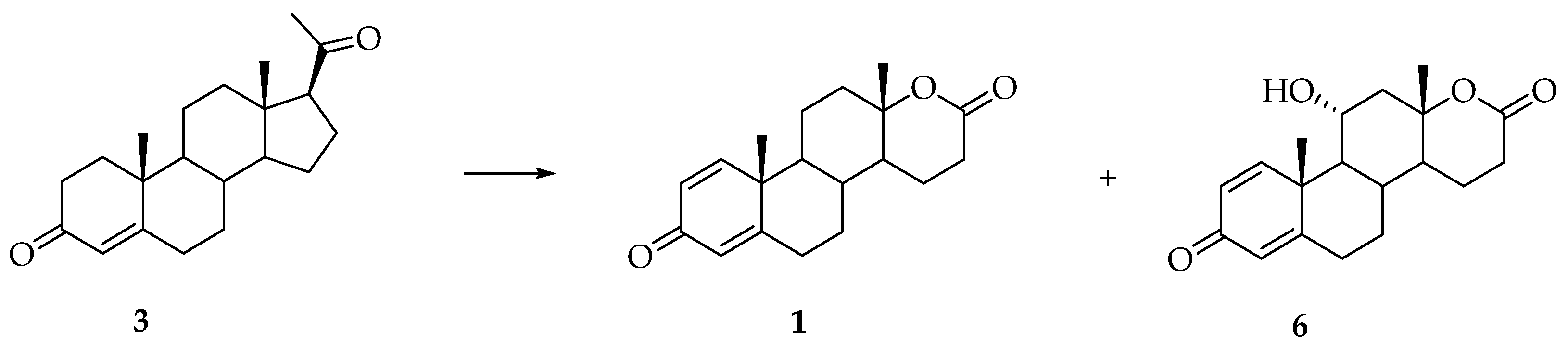
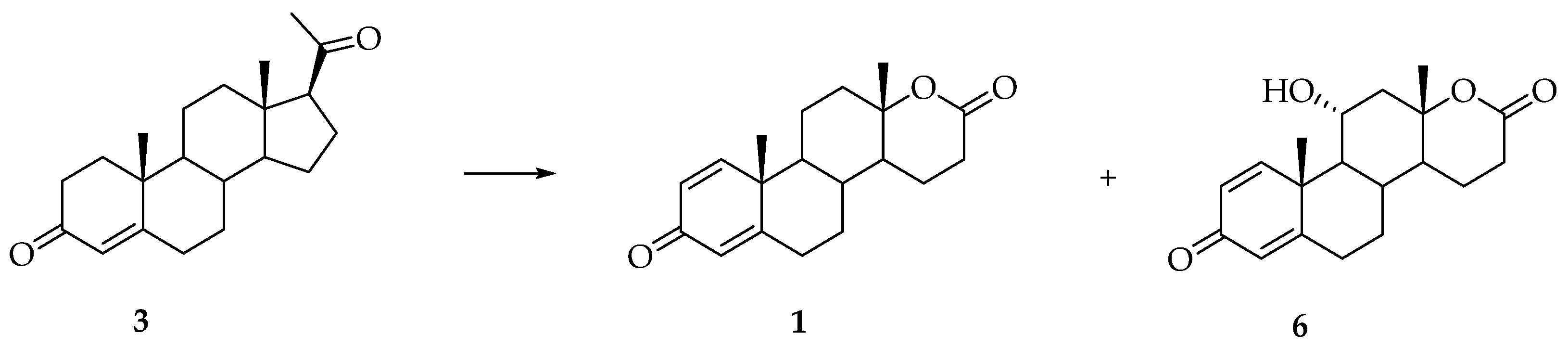
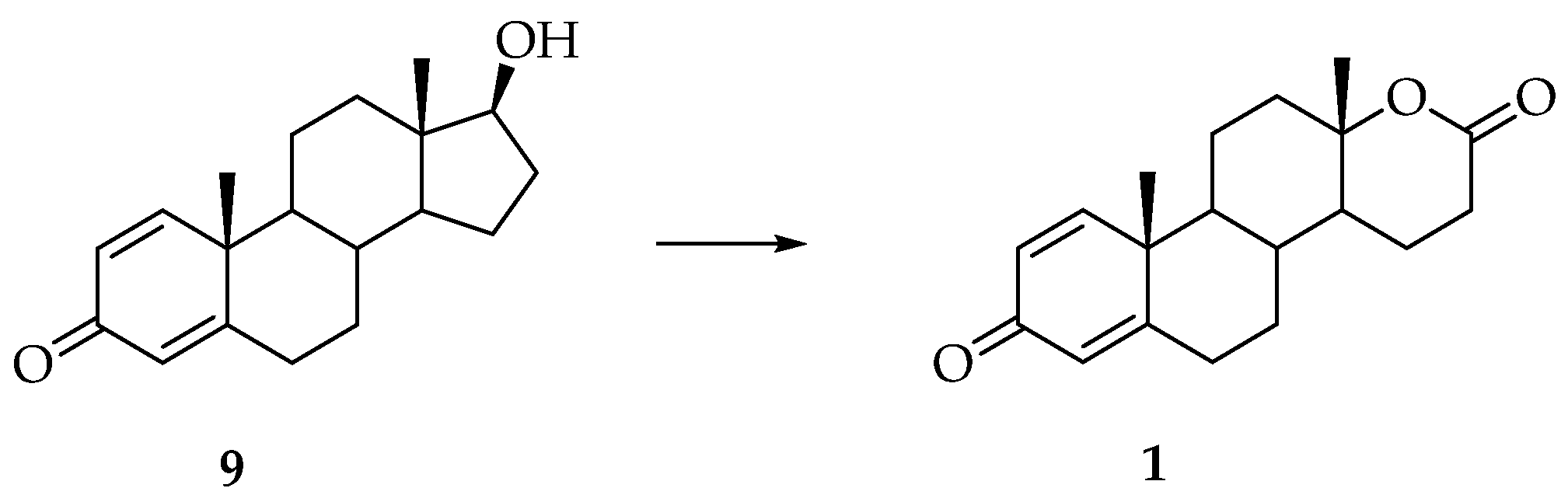
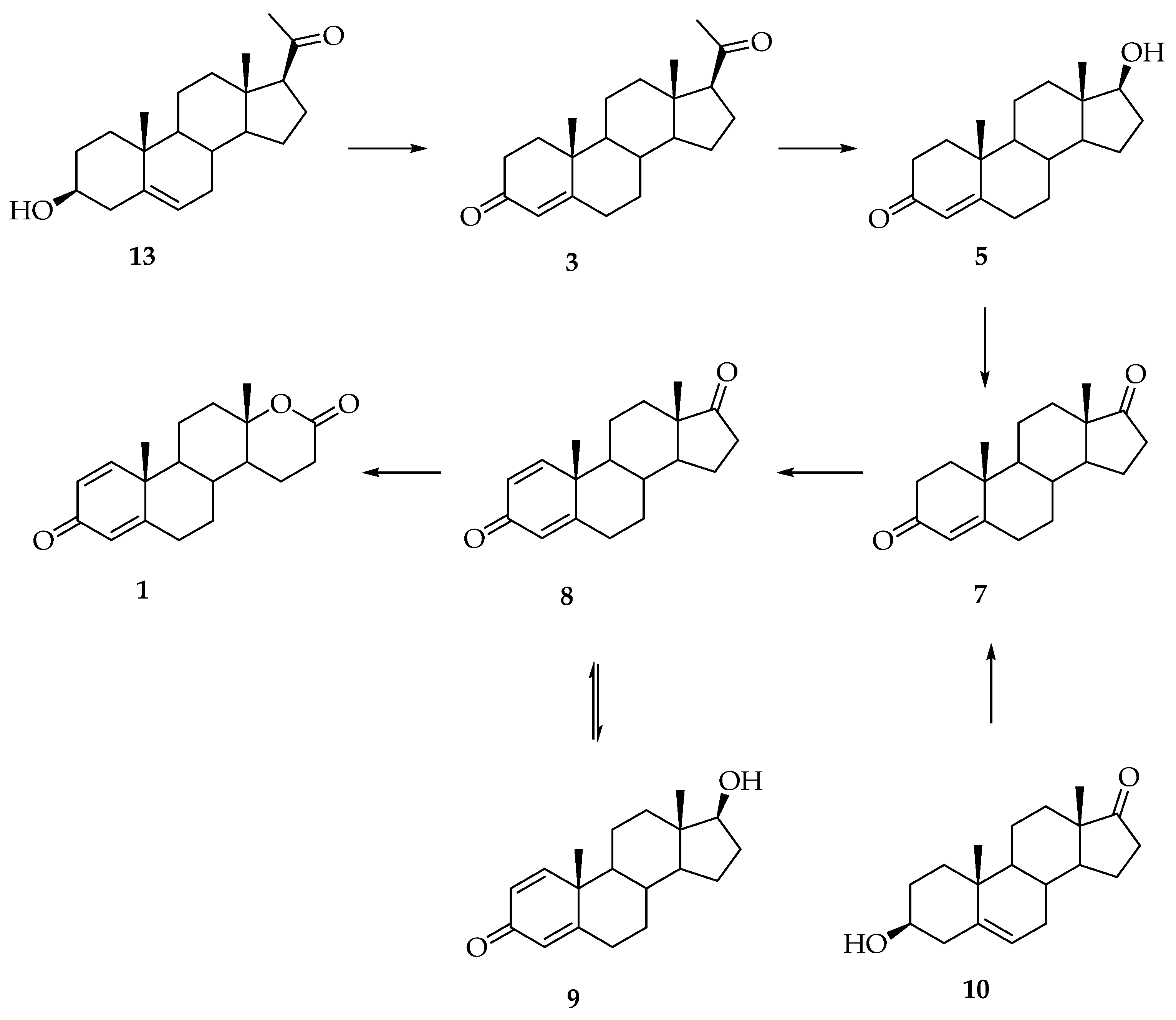
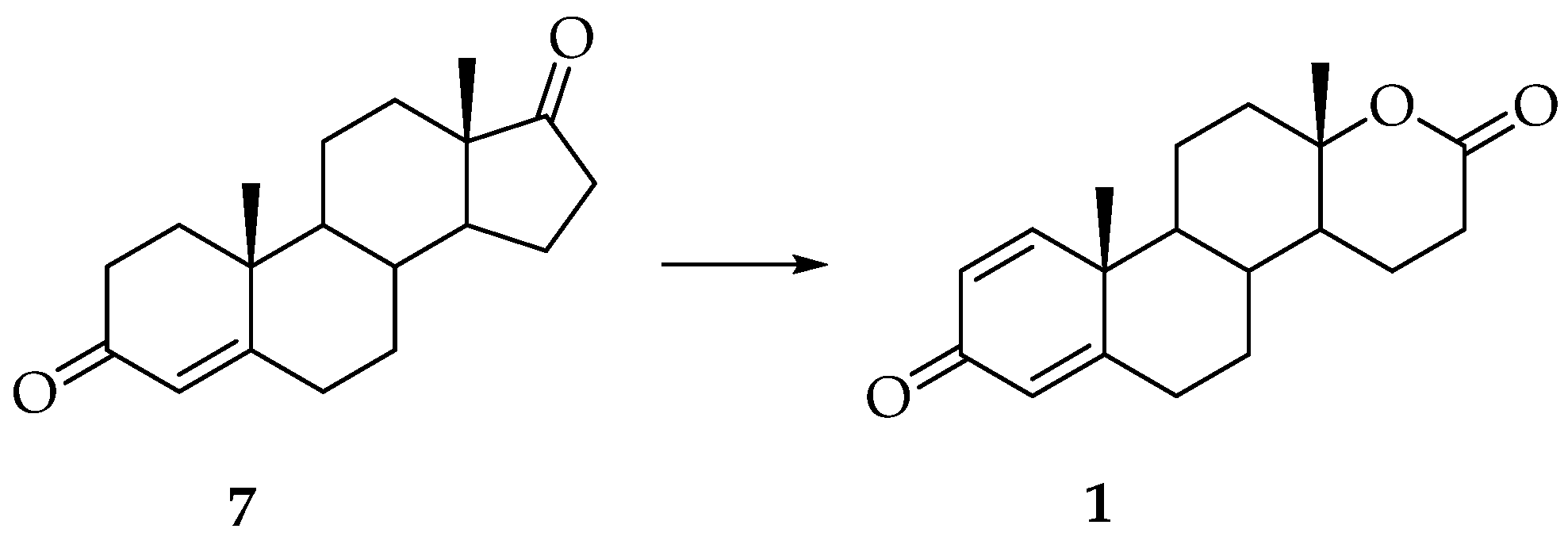
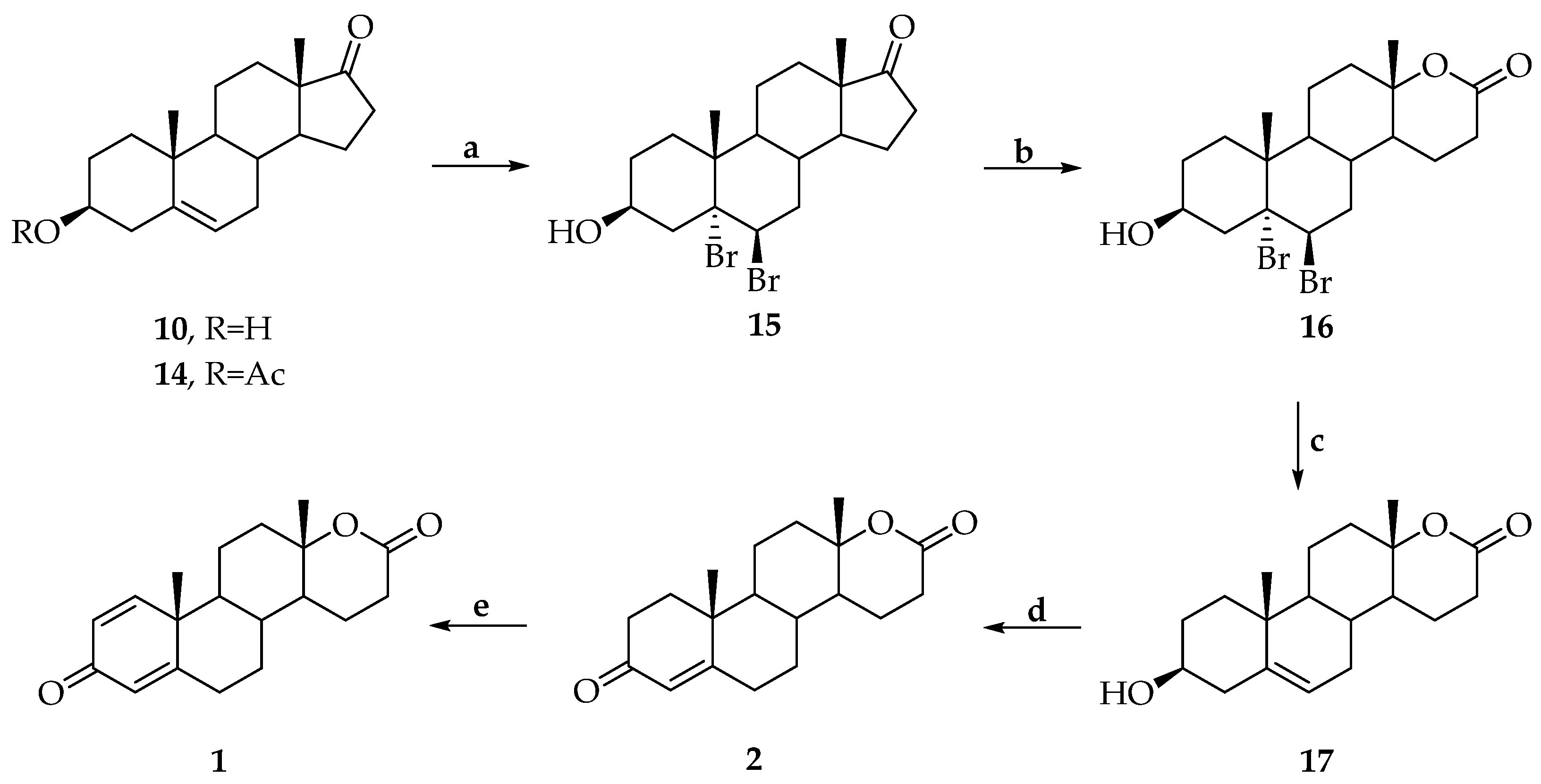
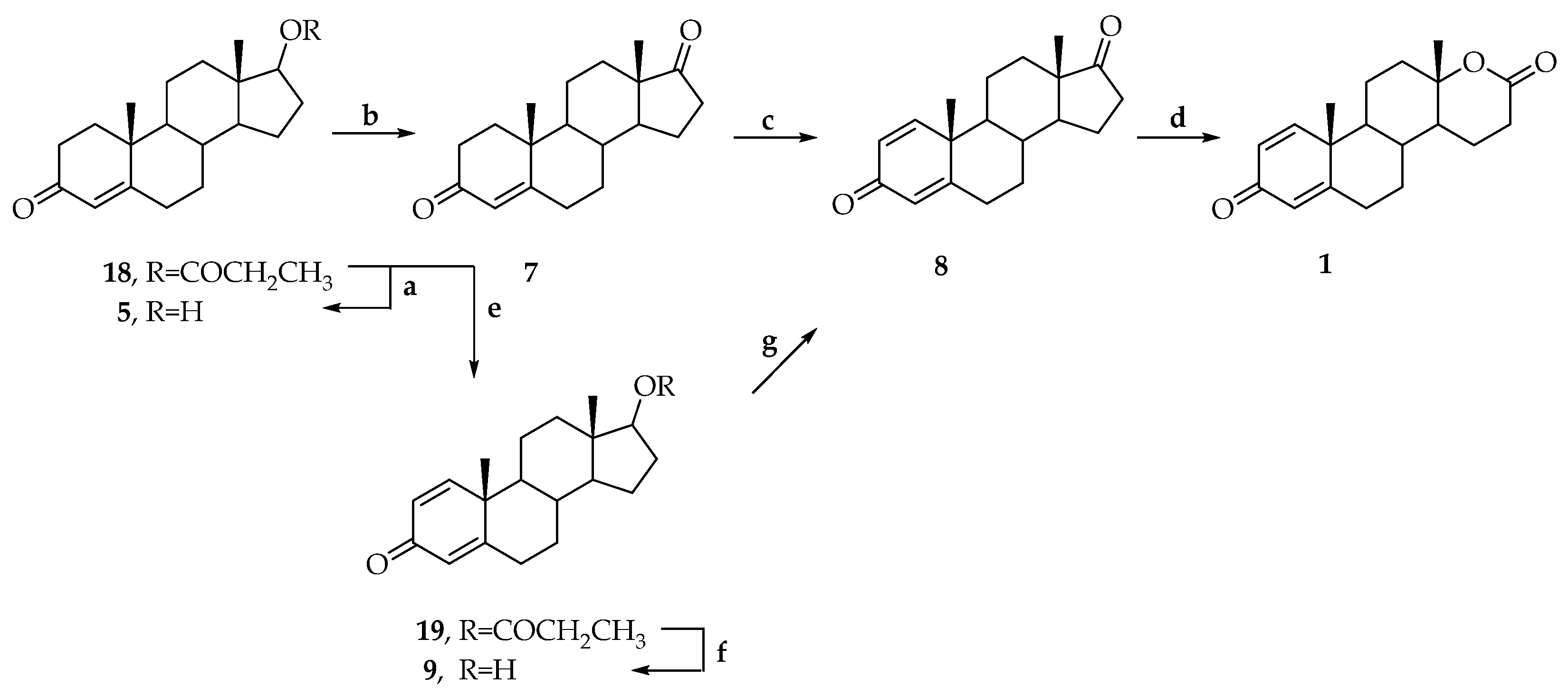
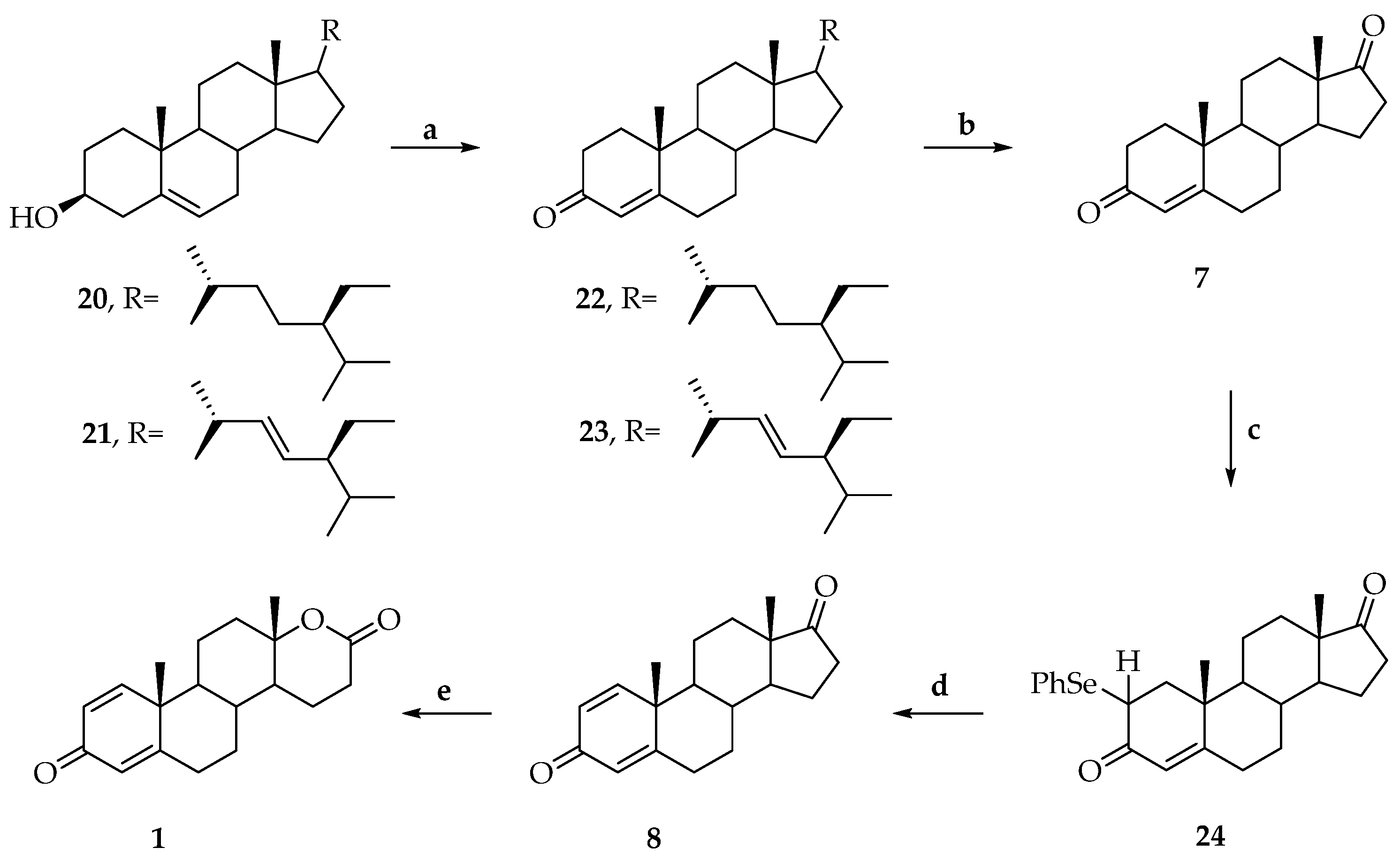
3. Synthesis of Testolactone
4. Medicinal Uses, Benefits and Side Effects of Testolactone
4.1. Cancer
4.1.1. Breast Cancer
4.1.2. Prostatic Carcinoma
4.1.3. Human Benign Prostatic Hyperplasia (BPH)
4.1.4. Desmoid Tumors
4.1.5. Carcinoma of the Pancreas
4.2. Precocious Puberty
4.3. Gynecomastia
4.4. Congenital Adrenal Hyperplasia (CAH)
4.5. Fertility and Sterility
4.6. Fibromatosis
4.7. Atherosclerotic Cardiovascular Disease
4.8. COVID-19
4.9. Side Effects
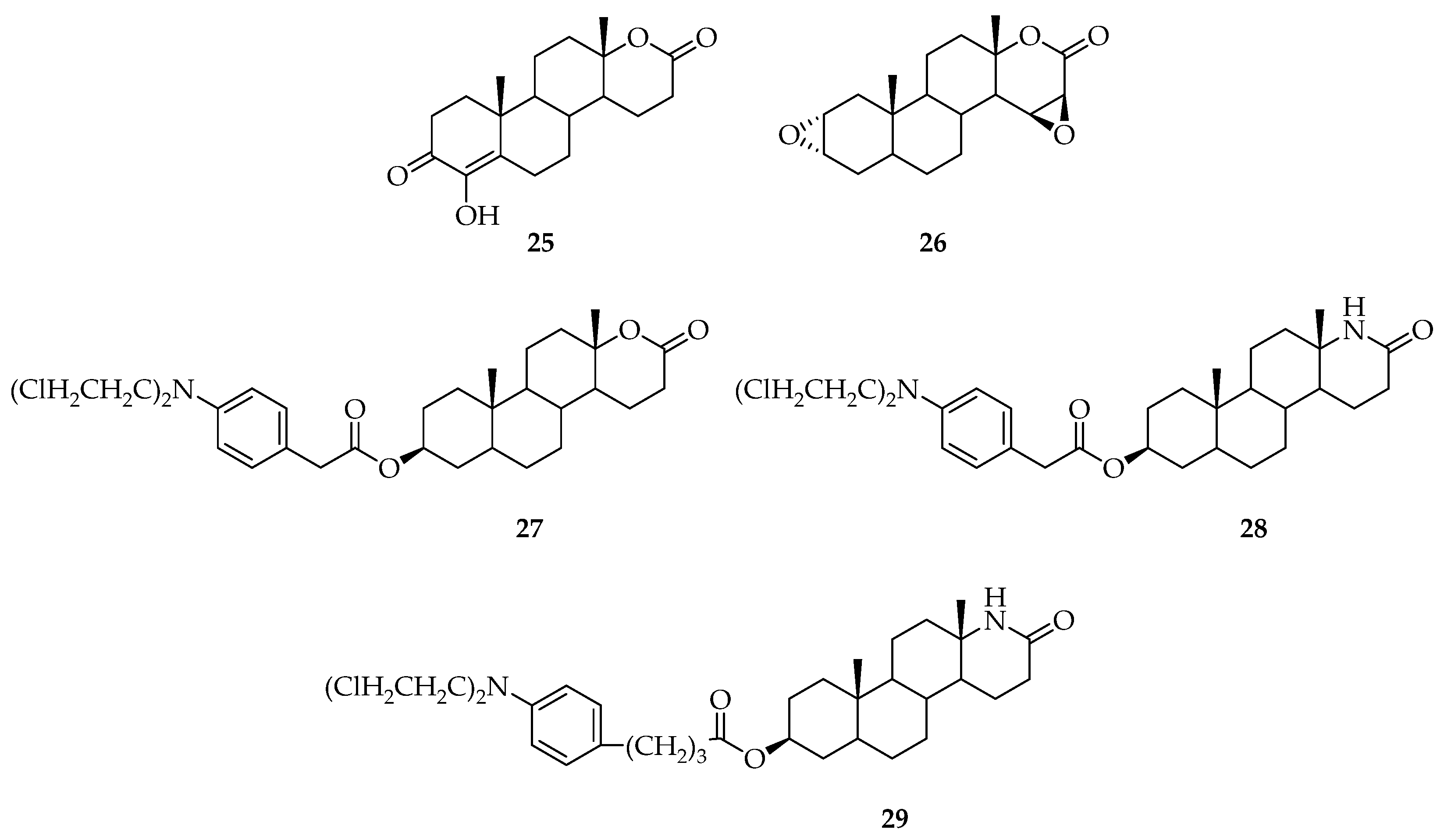
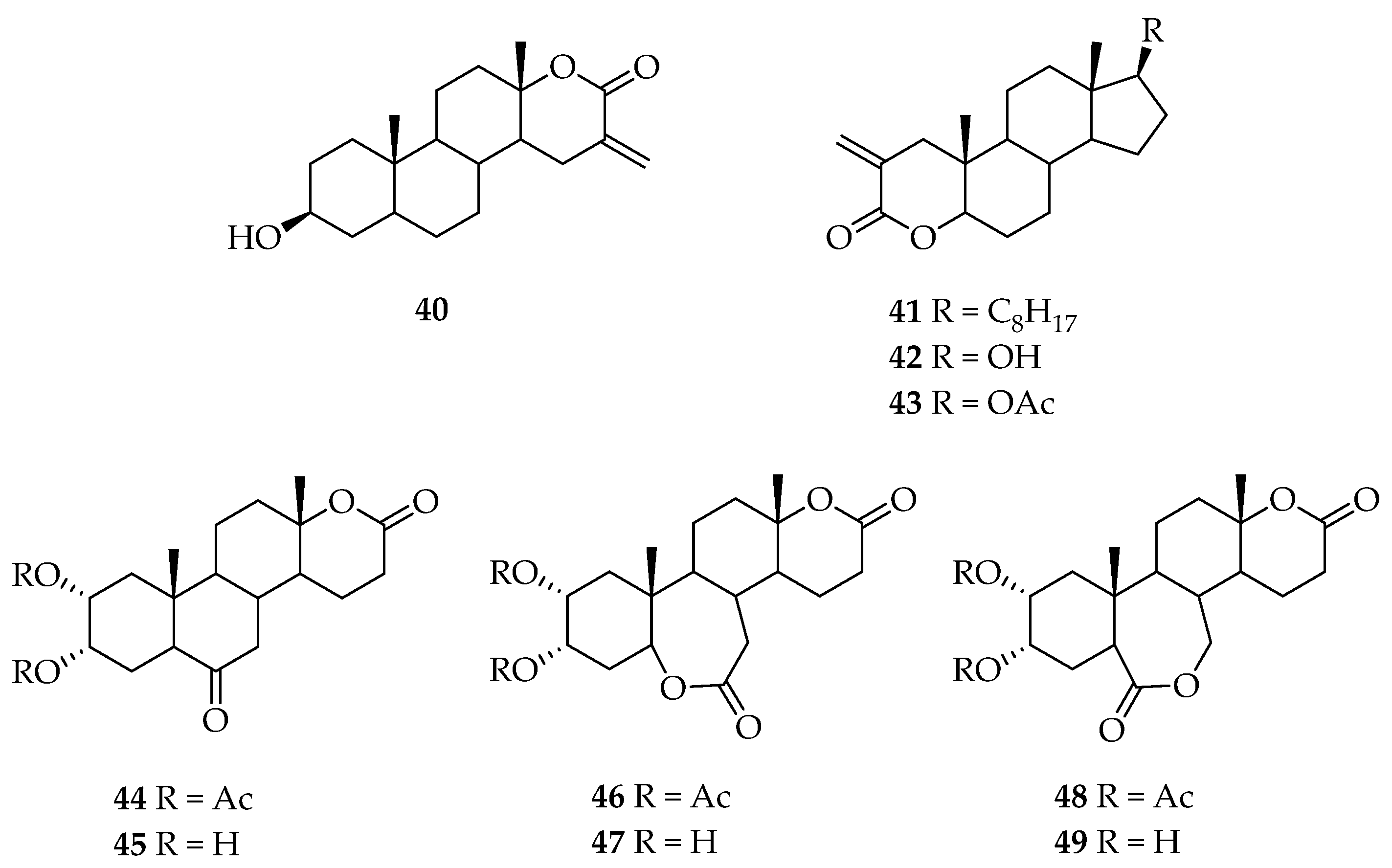
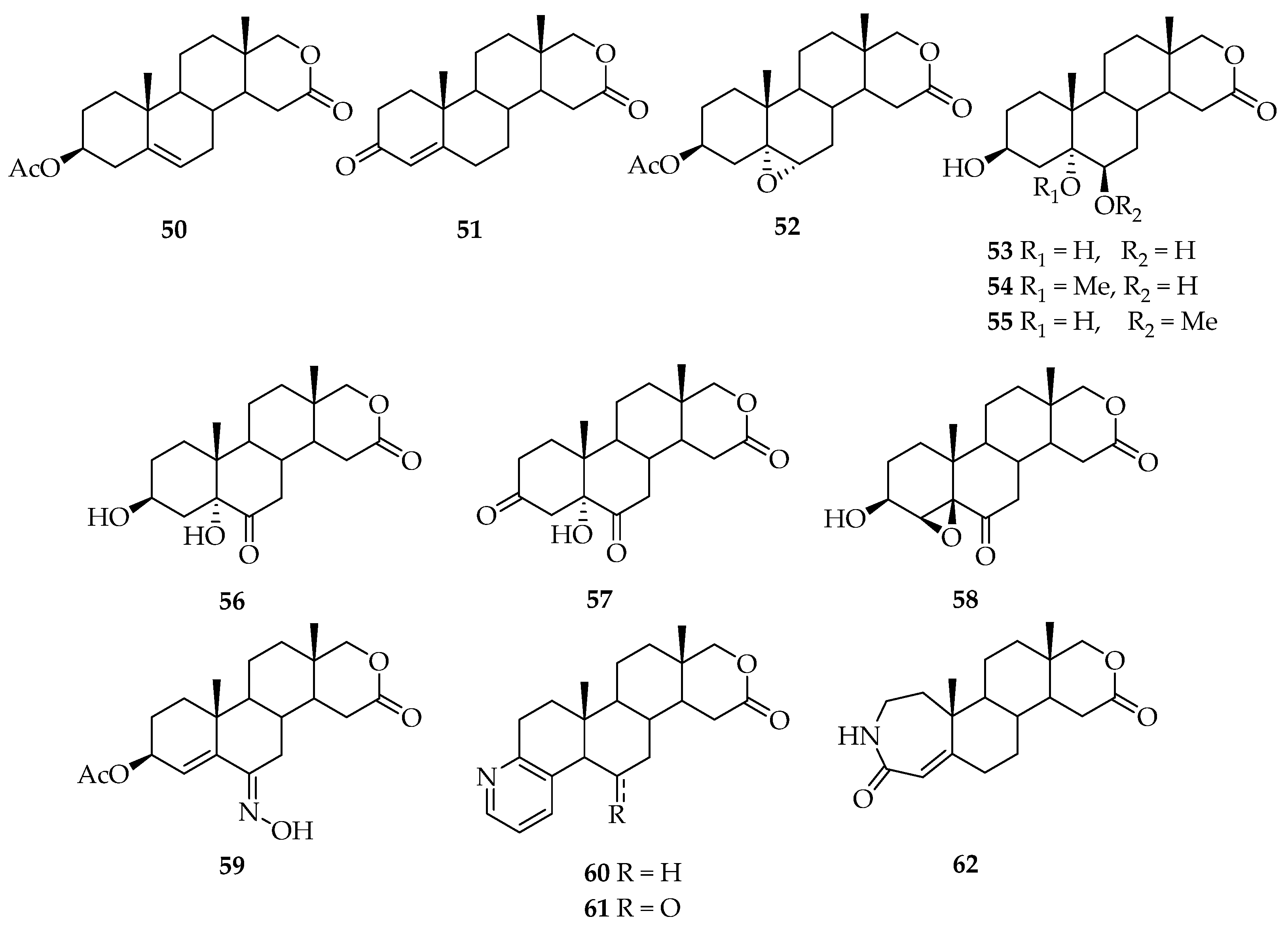
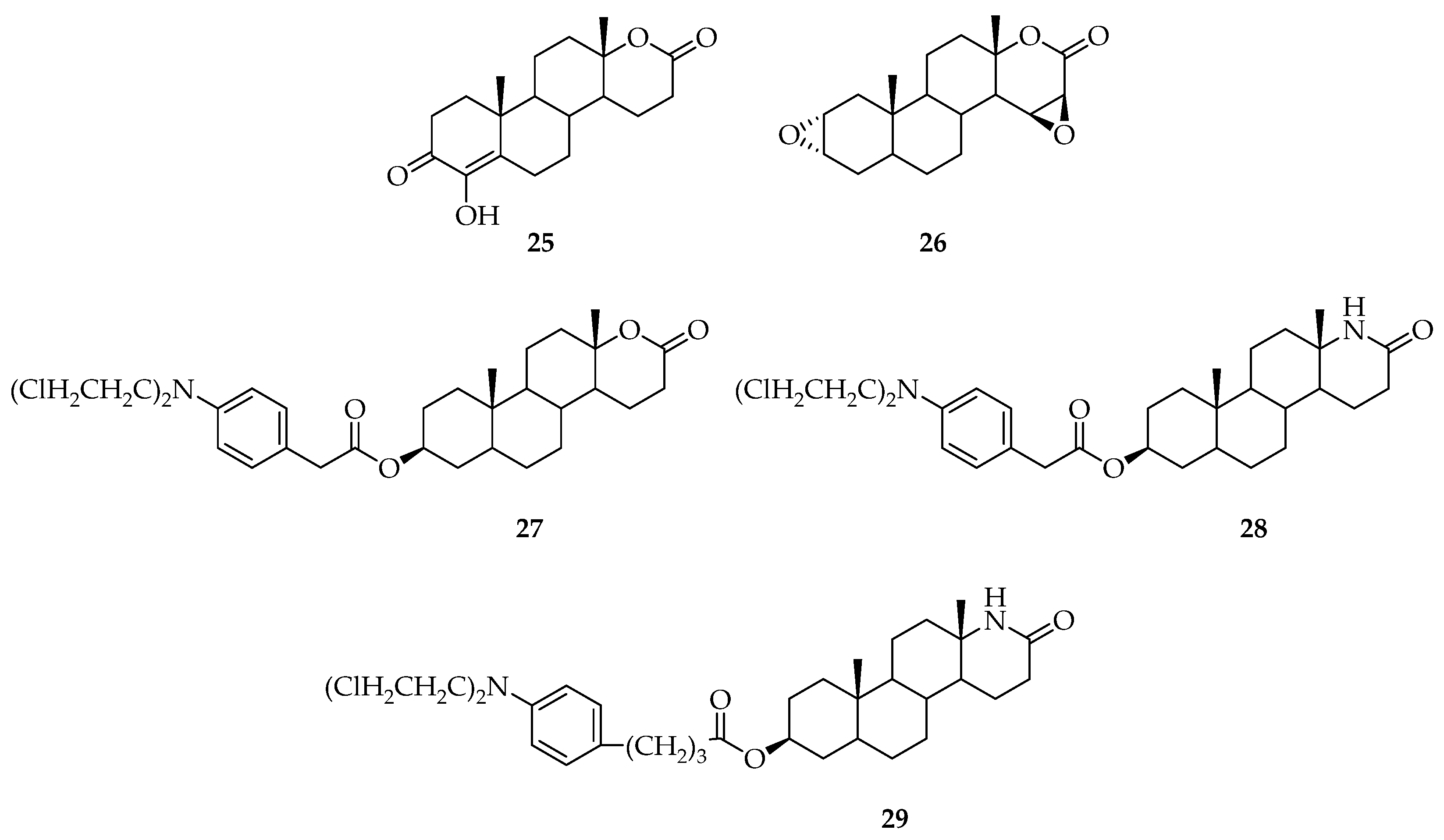
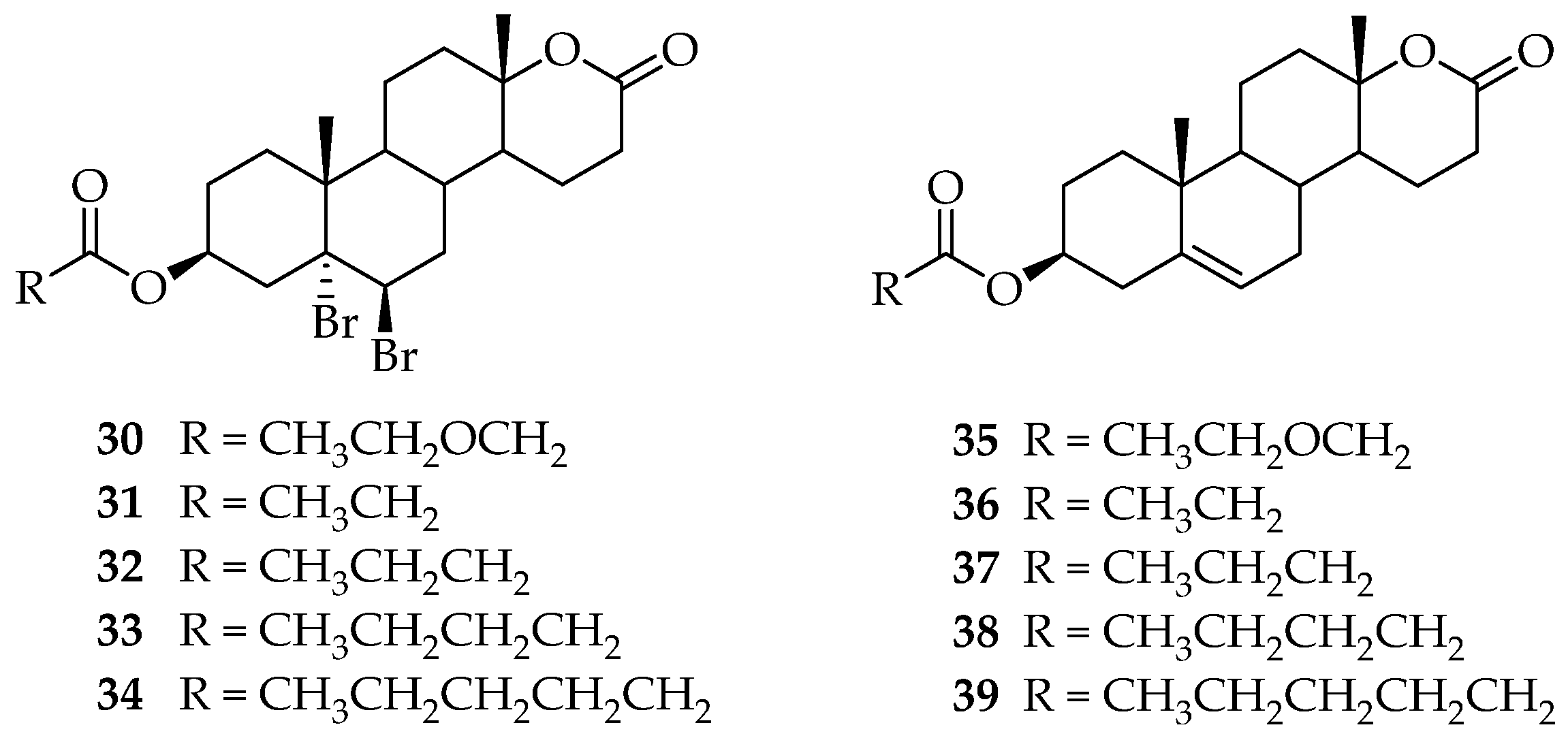
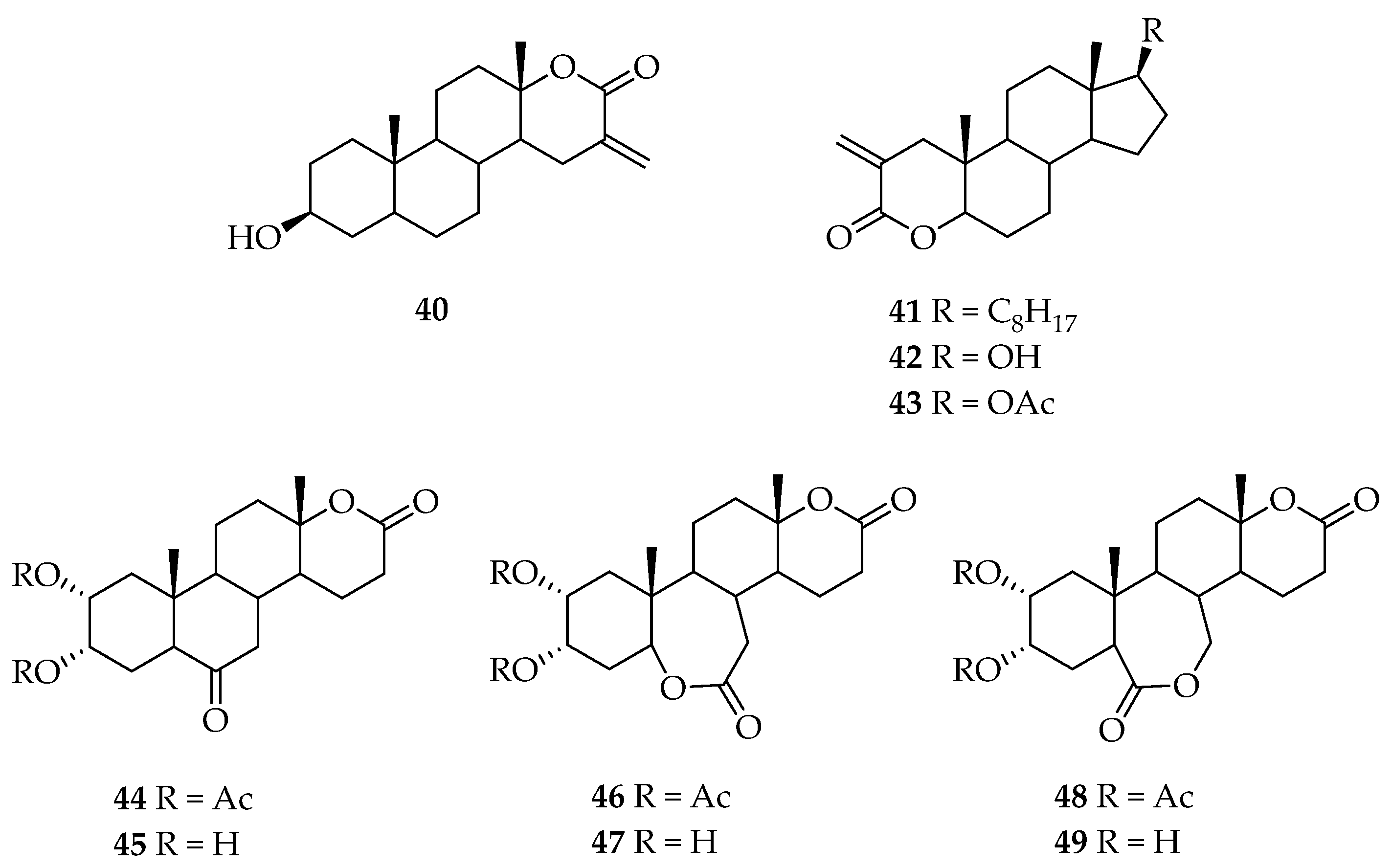
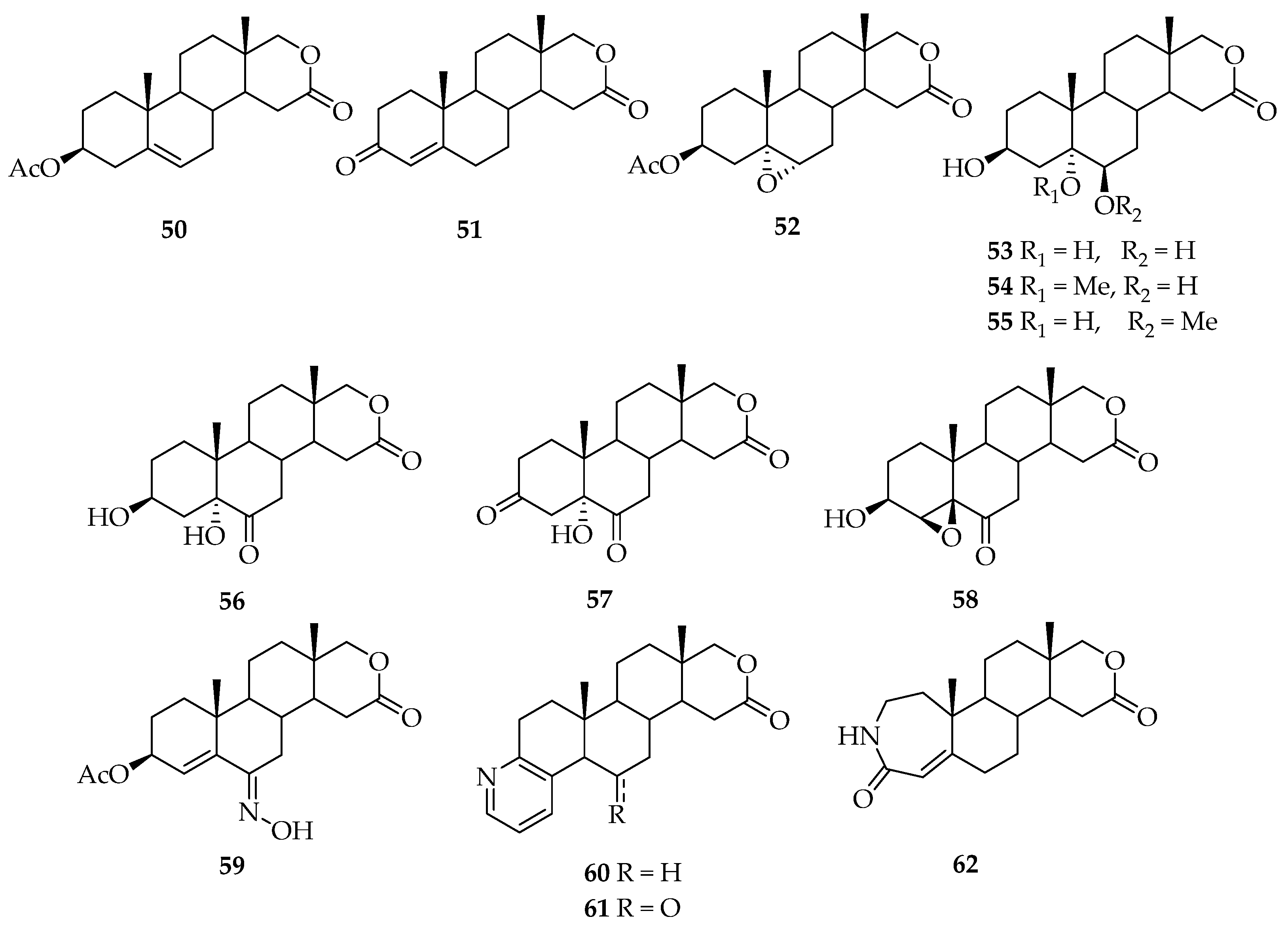
5. Biologically Active Derivatives and Analogs of Testolactone
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lemke, T.L.; Williams, D.A. Foye’s Principles of Medicinal Chemistry, 7th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; p. 1362. [Google Scholar]
- Siiteri, P.K.; Thompson, E.A. Studies of human placental aromatase. J. Steroid Biochem. 1975, 6, 317–322. [Google Scholar] [CrossRef]
- Florey, K. Testolactone. In Analytical Profiles of Drug Substances; Academic Press: Cambridge, MA, USA, 1976; Volume 5, pp. 533–553. [Google Scholar] [CrossRef]
- Dutta, U.; Pant, K. Aromatase inhibitors: Past, present and future in breast cancer therapy. Med. Oncol. 2008, 25, 113–124. [Google Scholar] [CrossRef]
- Dixon, J.M. Aromatase inhibitors in early breast cancer therapy: A variety of treatment strategies. Exp. Opin. Pharmacother. 2006, 7, 2465–2479. [Google Scholar] [CrossRef]
- Henderson, I.C.; Piccart-Gebhart, M.J. The evolving role of aromatase inhibitors in adjuvant breast cancer therapy. Clin. Breast Cancer 2005, 6, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, P.; Zheng, L. Clinical applications of aromatase inhibitors to treat male infertility. Hum. Reprod. Update 2022, 28, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.R.; Barmade, M.A.; Tamboli, R.S.; Murumkar, P.R. Developing steroidal aromatase inhibitors-an effective armament to win the battle against breast cancer. Eur. J. Med. Chem. 2015, 105, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Kharb, R.; Haider, K.; Neha, K.; Yar, M.S. Aromatase inhibitors: Role in postmenopausal breast cancer. Arch. Pharm. 2020, 353, e2000081. [Google Scholar] [CrossRef]
- Coney, P.; Yoshimura, Y.; Hosoi, Y.; Bongiovanni, A.; Wallach, E. Effect of aromatase inhibitors on the histology of the cycling rat ovary. Gynecol. Obstet. Investig. 1987, 23, 177–183. [Google Scholar] [CrossRef]
- Cocconi, G. First generation aromatase inhibitors—aminoglutethimide and testololactone. Breast Cancer Res. Treat. 1994, 30, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Lønning, P.E. Aromatase inhibitors and inactivators for breast cancer therapy. Drugs Aging 2002, 19, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Bartmanska, A.; Dmochowska-Gładysz, J.; Huszcza, E. Steroids’ transformations in Penicillium notatum culture. Steroids 2005, 70, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Li, H.; Shan, L.; Wu, J. Synthesis of steroidal lactone by penicillium citreo-viride. Steroids 2006, 71, 931–934. [Google Scholar] [CrossRef]
- Lønning, P.E. Additive endocrine therapy for advanced breast cancer—Back to the future. Acta Oncol. 2009, 48, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Tilson-Mallett, N.; Santner, S.J.; Feil, P.D.; Santen, R.J. Biological significance of aromatase activity in human breast tumors. J. Clin. Endocrinol. Metab. 1983, 57, 1125–1128. [Google Scholar] [CrossRef]
- Santner, S.J.; Rosen, H.; Osawa, Y.; Sanen, R.J. Additive effects of aminoglutethimide, testololactone, and 4-hydroxyandrostenedione as inhibitors of aromatase. J. Steroid Biochem. 1984, 20, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J.; Leszczynski, D.; Tilson-Mallet, N.; Feil, P.D.; Wright, C.; Manni, A.; Santner, S.J. Enzymatic control of estrogen production in human breast cancer: Relative significance of aromatase versus sulfatase pathways. Ann. N. Y. Acad. Sci. 1986, 464, 126–137. [Google Scholar] [CrossRef]
- Vigersky, R.A.; Mozingo, D.; Eil, C.; Purohit, V.; Bruton, J. The Antiandrogenic effects of Δ1-testolactone (Teslac) in vivo in rats and in vitro in human cultured fibroblasts, rat mammary carcinoma cells, and rat prostate cytosol. Endocrinology 1982, 110, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Fried, J.; Thoma, R.W.; Klingsberg, A. Oxidation of steroids by microörganisms. III. Side chain degradation, ring D-cleavage and dehydrogenation in ring A. J. Am. Chem. Soc. 1953, 75, 5764–5765. [Google Scholar] [CrossRef]
- Segaloff, A.; Weeth, J.B.; Rongone, E.L.; Murison, P.J.; Bowers, C.Y. Hormonal therapy in cancer of the breast. XVI. The effect of Δ1-testololactone on clinical course and hormonal excretion. Cancer 1960, 13, 1017–1020. [Google Scholar] [CrossRef]
- Lerner, L.J.; Bianchi, A.; Borman, A. Δ1-Testololactone, a nonandrogenic augmentor and inhibitor of androgens. Cancer 1960, 13, 1201–1205. [Google Scholar] [CrossRef]
- Peterson, D.H.; Eppstein, S.H.; Meister, P.D.; Murray, H.C.; Leigh, H.M.; Weintraub, A.; Reineke, L.M. Microbiological transformations of steroids. IX. Degradation of C21 steroids to C19 ketones and to testololactone. J. Am. Chem. Soc. 1953, 75, 5768–5769. [Google Scholar] [CrossRef]
- Holland, H.L. Organic Synthesis with Oxidative Enzymes, 1st ed.; Verlag Chemie: Weinheim, Germany, 1992; pp. 241–254. [Google Scholar]
- Fürst, M.J.L.J.; Gran-Scheuch, A.; Aalbers, F.S.; Fraaije, M.W. Baeyer–Villiger monooxygenases: Tunable oxidative biocatalysts. ACS Catal. 2019, 9, 11207–11241. [Google Scholar] [CrossRef]
- Mascotti, M.L.; Palazzolo, M.A.; Bisogno, F.R.; Kurina-Sanz, M. Biotransformation of dehydro-epi-androsterone by Aspergillus Parasiticus: Metabolic evidences of BVMO activity. Steroids 2016, 109, 44–49. [Google Scholar] [CrossRef]
- Haiilie, R.; TalalayP. Enzymatic formation of testololactone. Biochemistry 1963, 2, 203–208. [Google Scholar] [CrossRef]
- Brannon, D.R.; Martin, J.; Oehlschlager, A.C.; Durham, N.N.; Zalkow, L.H. Transformation of progesterone and related steroids by Aspergillus tamarii. J. Org. Chem. 1965, 30, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Panek, A.; Łyczko, P.; Swizdor, A. Microbial modifications of androstane and androstene steroids by Penicillium vinaceum. Molecules 2020, 25, 4226. [Google Scholar] [CrossRef] [PubMed]
- Świzdor, A.; Panek, A.; Milecka-Tronina, N. Microbial Baeyer-Villiger oxidation of 5α-steroids using Beauveria bassiana. A stereochemical requirement for the 11α-hydroxylation and the lactonization pathway. Steroids 2014, 82, 44–52. [Google Scholar] [CrossRef]
- Fried, J.; Brunswick, N.; Thoma, R.W.; Somerville, N.J. Synthesis of Steroids of the 1-Dehydrotestololactone Series. U.S. Patent 2,823,171, 11 February 1958. [Google Scholar]
- Gilbert, I.; White, M. Fermentation Method for the Preparation of Testolactone by Fusarium Species. U.S. Patent Application 10/573,636, 28 December 2006. [Google Scholar]
- Čapek, A.; Hanč, O.; Tadra, M. Microbial transformations of steroids. Folia Microbiol. 1963, 8, 120–124. [Google Scholar] [CrossRef]
- Hunter, A.C.; Carragher, N.E. Flexibility of the endogenous progesterone lactonisation pathway in Aspergillus tamarii KITA: Transformation of a series of cortical steroid analogues. J. Steroid Biochem. 2003, 87, 301–308. [Google Scholar] [CrossRef]
- Zhang, H.; Ren, J.; Wang, Y.; Sheng, C.; Wu, Q.; Diao, A.; Zhu, D. Effective multi-step functional biotransformations of steroids by a newly isolated Fusarium oxysporum SC1301. Tetrahedron 2013, 69, 184–189. [Google Scholar] [CrossRef]
- Lednicer, D.; Mitscher, L.A. The Organic Chemistry of Drug Synthesis; John-Wiley & Sons: New York, NY, USA, 1980; Volume 2, p. 160. ISBN 978-0-471-04392-8. [Google Scholar]
- Zinczuk, J.; Bacigaluppo, J.A.; Colombo, M.I.; Cravero, R.M.; González-Sierra, M.; Rúveda, E.A. An efficient and environmentally benign chemical synthesis of testolactone. J. Braz. Chem. Soc. 2003, 14, 970–974. [Google Scholar] [CrossRef]
- Lone, S.H.; Bhat, K.A. Phytosterols as precursors for the synthesis of aromatase inhibitors: Hemisynthesis of testololactone and testolactone. Steroids 2015, 96, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Testolactone aqueous suspension (Teslac). Clin. Pharmacol. Ther. 1970, 11, 302–306. [CrossRef]
- Sayyad, N.B.; Sabale, P.M.; Umare, M.D.; Bajaj, K.K. Aromatase inhibitors: Development and current perspectives. Indian J. Pharm. Educ. Res. 2022, 56, 311–320. [Google Scholar] [CrossRef]
- Manna, P.R.; Molehin, D.; Ahmed, A.U. Chapter eleven—Dysregulation of aromatase in breast, endometrial, and ovarian cancers: An overview of therapeutic strategies. Prog. Mol. Biol. Transl. 2016, 144, 487–537. [Google Scholar]
- Molehin, D.; Filleur, S.; Pruitt, K. Regulation of aromatase expression: Potential therapeutic insight into breast cancer treatment. Mol. Cell. Endocrinol. 2021, 531, 111321. [Google Scholar] [CrossRef]
- Covey, D.F.; Hood, W.F. A new hypothesis based on suicide substrate inhibitor studies for the mechanism of action of aromatase. Cancer Res. 1982, 42, 3327s–3333s. [Google Scholar]
- Klein, H.; Bartsch, W.; Niemand, A.; Stürenburg, H.; Voigt, K. Inhibition of human placental aromatase in a perfusion model. Comparison with kinetic, cell-free experiments. J. Steroid Biochem. 1988, 29, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Eil, C.; Edelson, S.K. The use of human skin fibroblasts to obtain potency estimates of drug binding. J. Clin. Endocrinol. Metab. 1984, 59, 51–55. [Google Scholar] [CrossRef]
- Segaloff, A.; Weeth, J.B.; Meyer, K.K.; Rongone, E.L.; Cuningham, M.E.G. Hormonal therapy in cancer of the breast. XIX. Effect of oral administration of Δ1-testololactone on clinical course and hormonal excretion. Cancer 1962, 15, 633–635. [Google Scholar] [CrossRef]
- Cantino, T.J. and Gorda, G.S. High dosage Δ1-testolactone therapy of disseminated carcinoma of the breast. Cancer 1967, 20, 458–461. [Google Scholar] [CrossRef]
- Goldenberg, I.S. Clinical trial of Δ1-testololactone (NSC 23759), medroxy progesterone acetate (NSC 26386) and oxylone acetate (NSC 47438) in advanced female mammary cancer: A report of the cooperative breast cancer group. Cancer 1969, 23, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.S.; Waters, M.N.; Ravdin, R.S.; Ansfield, F.J.; Segaloff, A. Androgenic therapy for advanced breast cancer in women: A report of the cooperative breast cancer group. JAMA 1973, 223, 1267–1268. [Google Scholar] [CrossRef]
- Goldenberg, I.S.; Sedransk, N.; Volk, H.; Segaloff, A.; Kelley, R.M.; Haines, C.R. Combined androgen and antimetabolite therapy of advanced female breast cancer: A report of the cooperative breast cancer group. Cancer 1975, 36, 308–310. [Google Scholar] [CrossRef]
- Barone, R.M.; Shamonki, I.M.; SiiteriI, P.K.; Judd, H.L. Inhibition of peripheral aromatization of androstenedione to estrone in postmenopausal women with breast cancer using Δ1-testololactone. J. Clin. Endocrinol. Metab. 1979, 49, 672–676. [Google Scholar] [CrossRef]
- Dao, T.L. Estrogen synthesis in human breast tumor and its inhibition by testololactone and bromoandrostenedione. Cancer Res. 1982, 42, 3338s–3341s. [Google Scholar]
- Budnick, R.M.; Dao, T.L. Inhibition of estrogen synthesis in human breast tumors by testololactone and bromoandrostenedione. Steroids 1980, 35, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Advanced breast cancer—Additive hormonal therapy. Cancer 1981, 47, 2398–2403. [Google Scholar] [CrossRef]
- Leinonen, P.; Bolton, N.J.; Kontturi, M.; Vihko, R. Rapid endocrine effects of tamoxifen and testolactone in prostatic carcinoma patients. Prostate 1982, 3, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Schweikert, H.; Tunn, U.W. Effects of the aromatase inhibitor testolactone on human benign prostatic hyperplasia, Steroids 1987, 50, 191–200. Steroids 1987, 50, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, W.; Klein, H.; Stürenburg, H.; Voigt, K. Metabolism of androgens in human benign prostatic hyperplasia; aromatase and its inhibition. J. Steroid Biochem. 1987, 27, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Church, J.M.; Jagelman, D.G.; Fazio, V.W.; McGannon, E.; George, C.R.; Schroeder, T.; Lavery, I.; Oakley, J. Noncytotoxic drug therapy for intra-abdominal desmoid tumor in patients with familial adenomatous polyposis. Dis. Colon Rectum 1992, 35, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Waddell, W.R. Treatment of intra-abdominal and abdominal wall desmoid tumors with drugs that affect the metabolism of cyclic 3′,5′-adenosine monophosphate. Ann. Surg. 1975, 181, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Waddell, W.R.; Kirsch, W.M. Testolactone, sulindac, warfarin, and vitamin K1 for unresectable desmoid tumors. Am. J. Surg. 1991, 161, 416–421. [Google Scholar] [CrossRef]
- Gansar, G.F.; Krementz, E.T. Desoid tumors: Experience with new modes of therapy. South Med. J. 1988, 81, 794–796. [Google Scholar] [CrossRef]
- Waddell, W.R. Chemotherapy for carcinoma of the pancreas. Surgery 1973, 74, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Engstrom, P.; Lavin, P.T.; Gelber, R.D.; Carbone, P.P. Chemotherapy of gastric and pancreatic carcinoma. Surgery 1979, 85, S09–S13. [Google Scholar]
- McCracken, J.D.; Ray, P.; Heilbrun, L.K.; Vaitkevicius, V.K.; Saiki, J.H.; Rivkin, S.E.; Rossof, A.H.; Moore, T.N. 5-Fluorouracil, methyl-CCNU, and radiotherapy with or without testolactone for localized adenocarcinoma of the exocrine pancreas. A Southwest Oncology Group Study. Cancer 1980, 46, 1518–1522. [Google Scholar] [CrossRef]
- Wheeler, M.D.; Styne, D.M. Diagnosis and management of precocious puberty. Pediatr. Clin. N. Am. 1990, 37, 1255–1271. [Google Scholar] [CrossRef]
- Wheeler, M.D.; Styne, D.M. Drug treatment in precocious puberty. Drugs 1991, 41, 717–728. [Google Scholar] [CrossRef]
- Gurnurkar, S.; DiLillo, E.; Carakushansky, M. A case of familial male-limited precocious puberty with a novel mutation. J. Clin. Res. Pediatr. Endocrinol. 2021, 13, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Eugster, E.A. Aromatase inhibitors in precocious puberty. Treat. Endocrinol. 2004, 3, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Laue, L.; Kenigsberg, D.; Pescovitz, O.H.; Hench, K.D.; Barnes, K.M.; Loriaux, D.L.; Cutler, G.B. Treatment of familial male precocious puberty with spironolactone and testolactone. N. Engl. J. Med. 1989, 320, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Laue, L.; Jones, J.; Barnes, K.M.; Cutler, G.B. Treatment of familial male precocious puberty with spironolactone, testolactone, and deslorelin. J. Clin. Endocrinol. Metab. 1993, 76, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Cummings, E.A.; Salisbury, S.R.; Givner, M.L.; Rittmaster, R.S. Testolactone-associated high androgen levels. J. Clin. Endocrinol. Metab. 1998, 83, 784–787. [Google Scholar] [CrossRef]
- Werber Leschek, E.; Jones, J.; Barnes, K.M.; Hill, S.C.; Cutler, G.B. Six-year results of spironolactone and testolactone. J. Clin. Endocrinol. Metab. 1999, 84, 175–178. [Google Scholar] [CrossRef]
- Werber Lescheck, E.; Flor, A.C.; Bryant, J.C.; Jones, J.; Barnes, K.M.; Cutler, G.B. Effect of antiandrogen, aromatase inhibitor, and gonadotropin-releasing hormone analog on adult height in familial male precocious puberty. J. Pediatr. 2017, 190, 229–235. [Google Scholar] [CrossRef]
- Foster, C.M.; Pescovitz, O.H.; Comite, F.; Feuillan, P.; Shawker, T.; Loriaux, D.L.; Cutler, G.B. Testolactone treatment of precocious puberty in McCune-Albright syndrome. Eur. J. Endocrinol. 1985, 109, 254–257. [Google Scholar] [CrossRef]
- Hauffa, B.P.; Havers, W.; Stolecke, H. Short-term effects of testolactone compared to other treatment modalities on longitudinal growth and ovarian activity in a girl with McCune-Albright syndrome. Helv. Paediat. Acta 1987, 42, 471–473. [Google Scholar] [CrossRef]
- Feuillan, P.P.; Foster, C.M.; Pescovitz, O.H.; Hench, K.D.; Shawker, T.; Dwyer, A.; Malley, J.D.; Barnes, K.; Loriaux, L.; Cutler, G.B. Treatment of precocious puberty in the McCune–Albright syndrome with the aromatase inhibitor testolactone. N. Engl. J. Med. 1986, 315, 1115–1119. [Google Scholar] [CrossRef]
- Feuillan, P.P.; Cutler, G.B.; Jones, J.; Oerter, K.E.; Manasco, P.K. Luteinizing hormone-releasing hormone (LHRH)-independent precocious puberty unresponsive to LHRH agonist therapy in two girls lacking features of the McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 1991, 73, 1370–1373. [Google Scholar] [CrossRef] [PubMed]
- Feuillan, P.P.; Jones, J.; Cutler, G.B. Long-term testolactone therapy for precocious puberty in girls with the McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 1993, 77, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Gryngarten, M.; Comar, H.; Arcari, A.; Boulgourdjian, E.; Escobar, M.E. McCune-Albright syndrome, a rare form of precocious puberty: Diagnosis, treatment, and follow-up. Arch. Argent. Pediatr. 2021, 119, e420–e427. [Google Scholar] [CrossRef]
- Papadopoulou, S.D.; Kitsios, K.; Kaltsas, K.; Kosta, K. A boy with McCune-Albright syndrome associated with GH associated with GH secreting pituitary microadenoma. Clinical findings and response to treatment. Hormones 2006, 5, 205–209. [Google Scholar] [CrossRef]
- Zacharin, M. Paediatric management of endocrine complications in McCune-Albright syndrome. J. Pediatr. Endocrinol. Metab. 2005, 18, 33–41. [Google Scholar] [CrossRef]
- Zachmann, M.; Eiholzer, U.; Muritano, M.; Werder, E.A.; Manella, B. Treatment of pubertal gynaecomastia with testolactone. Eur. J. Endocrinol. 1986, 113, S218–S226. [Google Scholar] [CrossRef] [PubMed]
- Binder, G.; Iliev, D.I.; Dufke, A.; Wabitsch, M.; Schweizer, R.; Ranke, M.B.; Schmidt, M. Dominant transmission of prepubertal gynecomastia due to serum estrone excess: Hormonal, biochemical, and genetic analysis in a large kindred. J. Clin. Endocrinol. Metab. 2005, 90, 484–492. [Google Scholar] [CrossRef]
- Auchus, R.J.; Lynch, S.C. Treatment of post-orchiectomy gynecomastia with testolactone. Endocrinologist 1994, 4, 429–432. [Google Scholar] [CrossRef]
- Kara, C.; Kutlu, A.O.; Tosun, M.S.; Apaydın, S.; Senel, F. Sertoli cell tumor causing prepubertal gynecomastia in a boy with Peutz-Jeghers syndrome: The outcome of 1-year treatment with the aromatase inhibitor testolactone. Horm. Res. Paediatr. 2005, 63, 252–256. [Google Scholar] [CrossRef]
- White, P.C.; Speiser, P.W. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr. Rev. 2000, 21, 245–291. [Google Scholar] [CrossRef]
- Charamandari, E.; Calis, K.A.; Keil, M.F.; Mohassel, M.R.; Remaley, A.; Merke, D.P. Flutamide decreases cortisol clearance in patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2002, 87, 3197–3200. [Google Scholar] [CrossRef] [PubMed]
- Merke, D.P.; Cutler, G.B. New approaches to the treatment of congenital adrenal hyperplasia. JAMA 1997, 227, 1073–1076. [Google Scholar] [CrossRef]
- Merke, D.P.; Keil, M.F.; Jones, J.V.; Fields, J.; Hill, S.; Cutler, G.B. Flutamide, testolactone, and reduced hydrocortisone dose maintain normal growth velocity and bone maturation despite elevated androgen levels in children with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2000, 85, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Laue, L.; Merke, D.P.; Jones, J.V.; Barnes, K.M.; Hill, S.; Cutler, G.B. A preliminary study of flutamide, testolactone, and reduced hydrocortisone dose in the treatment of congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 1996, 81, 3535–3539. [Google Scholar] [CrossRef]
- Vigersky, R.A.; Glass, A.R. Effects of Δ1-testolactone on the pituitary-testicular axis in oligospermic men. J. Clin. Endocrinol. Metab. 1981, 52, 897–902. [Google Scholar] [CrossRef]
- Dony, J.M.J.; Smals, A.G.H.; Rolland, R.; Fauser, B.C.J.M.; Thomas, C.M.G. Effect of aromatase inhibition by Δ1-testolactone on basal and luteinizing hormone-releasing hormone-stimulated pituitary and gonadal hormonal function in oligospermic men. Fertil. Steril. 1985, 43, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Dony, J.M.J.; Smals, A.G.H.; Rolland, R.; Fauser, B.C.J.M.; Thomas, C.M.G. Effect of chronic aromatase inhibition by Δ1-testolactone on pituitary-gonadal function in oligozoospermic men. Andrologia 1986, 18, 69–78. [Google Scholar] [CrossRef]
- Maier, U.; Hienert, G. Tamoxifen and testolactone in therapy of oligozoospermia: Results of a randomized study. Eur. Urol. 1988, 14, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Pavlovich, C.P.; King, P.; Goldstein, M.; Schlegel, P.N. Evidence of a treatable endocrinopathy in infertile men. J. Urol. 2001, 165, 837–841. [Google Scholar] [CrossRef]
- Dunaif, A.; Longcope, C.; Canick, J.; Badger, T.; Crowley, W.F. The effects of the aromatase inhibitor Δ1 -testolactone on gonadotropin release and steroid metabolism in polycystic ovarian disease. J. Clin. Endocrinol. Metab. 1985, 60, 773–780. [Google Scholar] [CrossRef]
- Martikainen, H.; Ruokonen, A.; Rönnberg, L.; Vihko, R. Short-term effects of testolactone on human testicular steroid production and on the response to human chorionic gonadotropin. Fertil. Steril. 1985, 43, 793–798. [Google Scholar] [CrossRef]
- Nagler, H.M.; deVere White, R.; Dyrenfurth, I.; Hembree, W.C. The effect of Δ1·testolactone on serum testosterone and estradiol in the adult male rat. Fertil. Steril. 1983, 40, 818–822. [Google Scholar] [CrossRef]
- Gooren, L.J.G.; van der Veen, E.A.; van Kessel, H.; Harmsen-Louman, W.; Wiegel, A.R. Prolactin secretion in the human male is increased by endogenous eostrogens and decreased by exogenous/endogenous androgen. Int. J. Androl. 1984, 7, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Paulson, R.J.; Francis, M.M.; Lobo, R.A. Modulation of prolactin responses to gonadotropin releasing hormone by acute testosterone infusions in normal women. Gynecol. Endocrinol. 1987, 1, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Marynick, S.P.; Loriaux, D.L.; Sherins, R.J.; Pita, J.C.; Lipsett, M.B. Evidence that testosterone can suppress pituitary gonadotropin secretion. J. Clin. Endocrinol. Metab. 1979, 49, 396–398. [Google Scholar] [CrossRef]
- Gooren, L.J.G.; Van der Veen, E.A.; Van Kessel, H.; Harmsen-Louman, W. Estrogens in the feedback regulation of gonadotropin secretion in men: Effects of administration of estrogen to agonadal subjects and the antiestrogen tamoxifen and the aromatase inhibitor Δ1-testolactone to eugonadal subjects. Andrologia 1984, 16, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Smals, A.G.H.; Dony, J.M.J.; Smals, A.E.M.; Pieters, G.F.F.M.; Hermus, A.R.M.M.; Boers, G.H.J.; Benraad, J.; Kloppenborg, P.W.C. Aromatase inhibition by Δ1-testolactone does not relieve the gonadotropin-induced late steroidogenic block in normal men. J. Clin. Endocrinol. Metab. 1985, 60, 1127–1131. [Google Scholar] [CrossRef]
- Zumoff, B.; Miller, L.K.; Strain, G.W. Reversal of the hypogonadotropic hypogonadism of obese men by administration of the aromatase inhibitor testolactone. Metabolism 2003, 52, 1126–1128. [Google Scholar] [CrossRef]
- Herzog, A.G.; Klein, P.; Jacobs, A.R. Reproductive endocrine considerations and hormonal therapy for men with epilepsy, Testosterone versus testosterone and testolactone in treating reproductive and sexual dysfunction in men with epilepsy and hypogonadism. Neurology 1998, 50, 782–784. [Google Scholar] [CrossRef]
- Sauven, P. Musculo-aponeurotic fibromatosis treated by surgery and testolactone. J. R. Soc. Med. 1982, 75, 281–283. [Google Scholar] [CrossRef]
- Zmuda, J.M.; Bausserman, L.L.; Maceroni, D.; Thompson, P.D. The effect of supraphysiologic doses of testosterone on fasting total homocysteine levels in normal men. Atherosclerosis 1997, 130, 199–202. [Google Scholar] [CrossRef]
- Mujwar, S. Computational repurposing of tamibarotene against triple mutant variant of SARS-CoV-2. Comput. Biol. Med. 2021, 136, 104748. [Google Scholar] [CrossRef]
- Volk, H.; Deupree, R.H.; Goldenberg, I.S.; Wilde, R.C.; Carabasi, R.A.; Escher, G.C. A dose response evaluation of delta-1-testololactone in advanced breast cancer. Cancer 1974, 33, 9–13. [Google Scholar] [CrossRef]
- Howard, R.P.; Furman, R.H. Metabolic and serum lipid effects of Δ1-testololactone. Exp. Biol. Med. 1962, 110, 227–229. [Google Scholar] [CrossRef]
- Friedl, K.E.; Hannan, C.J.; Jones, R.E.; Plymate, S.R. High-density lipoprotein cholesterol is not decreased. Metabolism 1990, 39, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Zmuda, J.M.; Fahrenbach, M.C.; Younkin, B.T.; Bausserman, L.L.; Terry, R.B.; Catlin, D.H.; Thompson, P.D. The effect of testosterone aromatization on high-density lipoprotein cholesterol. Metabolism 1993, 42, 446–450. [Google Scholar] [CrossRef]
- Sartori, S.K.; Nogueira Diaz, M.A.; Diaz-Munoz, G. Lactones: Classification, synthesis, biological activities, and industrial applications. Tetrahedron 2021, 84, 132001. [Google Scholar] [CrossRef]
- Ouellet, É.; Ayan, D.; Poirier, D. Synthesis and biological evaluation of estradiol-core derivatives bearing a fused γ-lactone as inhibitors of 17β-hydroxysteroid dehydrogenase type 1. Curr. Enzym. Inhib. 2014, 10, 39–52. [Google Scholar] [CrossRef]
- Kolodziejczyk-Czepas, J.; Stochmal, A. Bufadienolides of Kalanchoe species: An overview of chemical structure, biological activity and prospects for pharmacological use. Phytochem. Rev. 2017, 16, 1155–1171. [Google Scholar] [CrossRef]
- Huang, M.; He, J.X.; Xin Hu, H.; Zhang, K.; Wang, X.N.; Zhao, B.B.; Lou, H.X.; Ren, D.M.; She, T. Withanolides from the genus Physalis: A review on their phytochemical and pharmacological aspects. J. Pharm. Pharmacol. 2020, 72, 649–669. [Google Scholar] [CrossRef] [PubMed]
- Salaha, M.; Abdelsamieb, A.S.; Frotscher, M. Inhibitors of 17â-hydroxysteroid dehydrogenase type 1, 2 and 14: Structures, biological activities and future challenges. Mol. Cell Endocrinol. 2019, 489, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, K.; Joy, M.; Salas, S. First report of a lactonic disecosteroid from the buccinid gastropod Babylonia spirata. Steroids 2019, 143, 41–48. [Google Scholar] [CrossRef] [PubMed]
- D’Agata, R.; Aliffi, A.; Maugeri, G.; Mongioi, A.; Vicari, E.; Gulizia, S.; Polosa, P. Hydrotestolactone lowers serum oestradiol and PRL levels in normal men: Evidence of a role of oestradiol in PRL secretion. Clin. Endocrinol. 1982, 17, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Puck, T.T.; Webb, P.; Johnson, R. Cyclic AMP and the reverse transformation reaction. Ann. N. Y. Acad. Sci. 2002, 968, 122–138. [Google Scholar] [CrossRef]
- Cepa, M.M.D.S.; Tavares da Silva, E.J.; Correia-da-Silva, G.; Roleira, F.M.F.; Teixeira, N.A.A. Structure-activity relationships of new A,D-ring modified steroids as aromatase inhibitors: Design, synthesis, and biological activity evaluation. J. Med. Chem. 2005, 48, 6379–6385. [Google Scholar] [CrossRef]
- Clanton, N.A.; Hastings, S.D.; Foultz, G.B.; Contreras, J.A.; Yee, S.S.; Arman, H.D.; Risinger, A.L.; Frantz, D.E. Synthesis and biological evaluations of electrophilic steroids inspired by the Taccalonolides. ACS Med. Chem. Lett. 2020, 11, 2534–2543. [Google Scholar] [CrossRef]
- Mourelatos, D.; Petrou, C.; Boutis, L.; Papageorgiou, A.; Catsoulacos, P.; Dozi-Vassiliades, J. Induction of cytogenetic damage by modified steroidal derivatives of p-bis(2-chloroethyl)aminophenylacetic acid in human lymphocytes. Mutat. Res. Lett. 1987, 190, 205–210. [Google Scholar] [CrossRef]
- Geromichalos, G.D.; Geromichalou, E.; Camoutsis, C.; Kontos, M.; Dalezis, P.; Papageorgiou, A.; Grivas, A.A.; Tsigris, C.; Trafalis, D.T. In silico/in vitro study of hybrid D-modified steroidal alkylator anticancer activity using uridine phosphorylase as target protein. Anticancer Res. 2011, 31, 831–842. [Google Scholar]
- Ibrahim-Ouali, M.; Dumur, F. Steroidal lactams: A review. ARKIVOC 2022, 1, 262–284. [Google Scholar] [CrossRef]
- Garrido, M.; Bratoeff, E.; Bonilla, D.; Soriano, J.; Heuze, Y.; Cabeza, M. New steroidal lactones as 5α-reductase inhibitors and antagonists for the androgen receptor. J. Steroid Biochem. 2011, 127, 367–373. [Google Scholar] [CrossRef]
- Dehal, S.S.; Marples, B.A.; Stretton, R.J.; Traynor, J.R. Steroidal α-methylene δ-lactones as potential antitumor agents. J. Med. Chem. 1980, 23, 90–92. [Google Scholar] [CrossRef]
- Kohout, L.; Slavíková, B.; Strnad, M. 17a-Oxa-17a-homobrassinosteroid analogues. Collect. Czechoslov. Chem. Commun. 1998, 63, 646–654. [Google Scholar] [CrossRef]
- Kuzminac, I.; Klisuric, O.R.; Skoric, D.; Jakimov, D.; Sakac, M. Structural analysis and antitumor potential of novel 5,6-disubstituted-17a-homo-17-oxa-androstane derivatives. Struct. Chem. 2017, 28, 567–576. [Google Scholar] [CrossRef]
- Kulmany, A.E.; Edina Herman, B.; Zupko, I.; Sinreih, M.; Lanisnik Rizner, T.; Savic, M.; Okljesa, A.; Nikolic, A.; Nagy, V.; Ocsovszki, I.; et al. Heterocyclic androstane and estrane D-ring modified steroids: Microwave-assisted synthesis, steroid-converting enzyme inhibition, apoptosis induction, and effects on genes encoding estrogen inactivating enzymes. J. Steroid Biochem. 2021, 214, 105997. [Google Scholar] [CrossRef] [PubMed]
- Dzichenka, Y.; Shapira, M.; Yantsevich, A.; Cherkesova, T.; Grbovic, L.J.; Savic, M.; Usanov, S.; Jovanovic-Santa, S. Modified bile acids and androstanes—Novel promising inhibitors of human cytochrome P450 17A1. J. Steroid Biochem. 2021, 205, 105777. [Google Scholar] [CrossRef]
- Kovacevic, S.; Karadžic Banjac, M.; Anojcic, J.; Podunavac-Kuzmanovic, S.; Jevric, L.; Nikolic, A.; Savic, M.; Kuzminac, I. Chemometrics of anisotropic lipophilicity of anticancer androstane derivatives determined by reversed-phase ultra high performance liquid chromatography with polar aprotic and protic modifiers. J. Chromatogr. A 2022, 1673, 463197. [Google Scholar] [CrossRef]
- Kuzminac, I.Z.; Bekic, S.S.; Celic, A.S.; Jakimovic, D.S.; Sakac, M.N. Antitumor potential of novel 5α,6β-dibromo steroidal D-homo lactone. Steroids 2022, 188, 109118. [Google Scholar] [CrossRef]
- Sestic, T.L.J.; Ajdukovic, J.J.; Marinovic, M.A.; Petri, E.T.; Savic, M.P. In silico ADMET analysis of the A-, B- and D-modified androstane derivatives with potential anticancer effects. Steroids 2023, 189, 109147. [Google Scholar] [CrossRef]
- Savić, M.P.; Djurendić, E.A.; Petri, E.T.; Ćelić, A.; Klisurić, O.R.; Sakač, M.N.; Jakimov, D.S.; Kojić, V.V.; Penov Gaši, K.M. Synthesis, structural analysis and antiproliferative activity of some novel D-homo lactone androstane derivatives. RSC Adv. 2013, 3, 10385. [Google Scholar] [CrossRef]
- Savić, M.P.; Ajduković, J.J.; Plavša, J.J.; Bekić, S.S.; Ćelić, A.S.; Klisurić, O.R.; Jakimov, D.S.; Petri, E.T.; Djurendić, E.A. Evaluation of A-ring fused pyridine D-modified androstane derivatives for antiproliferative and aldo–keto reductase 1C3 inhibitory activity. MedChemComm 2018, 9, 969. [Google Scholar] [CrossRef] [PubMed]
- Savić, M.; Škorić, D.; Kuzminac, I.; Jakimov, D.; Kojić, V.; Rárová, L.; Strnad, M.; Djurendić, E. New A-homo lactam D-homo lactone androstane derivative: Synthesis and evaluation of cytotoxic and anti-inflammatory activities in vitro. Steroids 2020, 157, 108596. [Google Scholar] [CrossRef]
- Savić, M.; Kuzminac, I.; Škorić, D.; Jakimov, D.; Sakač, M.; Djurendić, E. Synthesis, NMR analysis and antiproliferative potential of some new oxygen-containing D-homo lactone androstane derivatives. J. Chem. Sci. 2020, 132, 98. [Google Scholar] [CrossRef]
- Kuzminac, I.Z.; Jakimov, D.S.; Bekić, S.S.; Ćelić, A.S.; Marinović, M.A.; Savić, M.P.; Raičević, V.N.; Kojić, V.V.; Sakač, M.N. Synthesis and anticancer potential of novel 5,6-oxygenated and/or halogenated steroidal D-homo lactones. Bioorg. Med. Chem. 2021, 30, 115935. [Google Scholar] [CrossRef] [PubMed]
- Kuzminac, I.Z.; Celic, A.S.; Bekic, S.S.; Kojic, V.; Savic, M.P.; Ignjatovic, N.L. Hormone receptor binding, selectivity and cytotoxicity of steroid D-homo lactone loaded chitosan nanoparticles for the treatment of breast and prostate cancer cells. Colloids Surf. B Biointerfaces 2022, 216, 112597. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savic, M.P.; Kuzminac, I.Z.; Nikolic, A.R. Testolactone: The Rise and Fall of a Drug. Drugs Drug Candidates 2023, 2, 69-94. https://doi.org/10.3390/ddc2010005
Savic MP, Kuzminac IZ, Nikolic AR. Testolactone: The Rise and Fall of a Drug. Drugs and Drug Candidates. 2023; 2(1):69-94. https://doi.org/10.3390/ddc2010005
Chicago/Turabian StyleSavic, Marina P., Ivana Z. Kuzminac, and Andrea R. Nikolic. 2023. "Testolactone: The Rise and Fall of a Drug" Drugs and Drug Candidates 2, no. 1: 69-94. https://doi.org/10.3390/ddc2010005
APA StyleSavic, M. P., Kuzminac, I. Z., & Nikolic, A. R. (2023). Testolactone: The Rise and Fall of a Drug. Drugs and Drug Candidates, 2(1), 69-94. https://doi.org/10.3390/ddc2010005