
Inhibitors of Farnesyl Diphosphate Synthase and Squalene Synthase: Potential Source for Anti-Trypanosomatidae Drug Discovery


Abstract
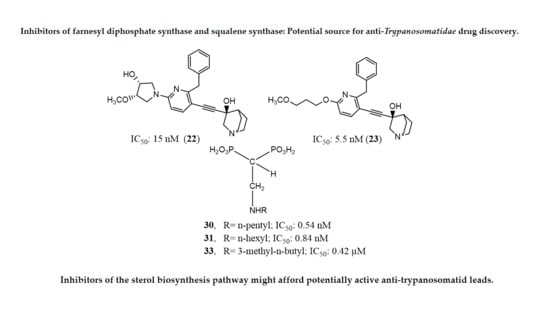
:
1. Introduction
2. Research Methods
2.1. Literature Search
2.2. Data Extraction and Synthesis
2.3. Results of the Literature Search
3. Human African Trypanosomiasis
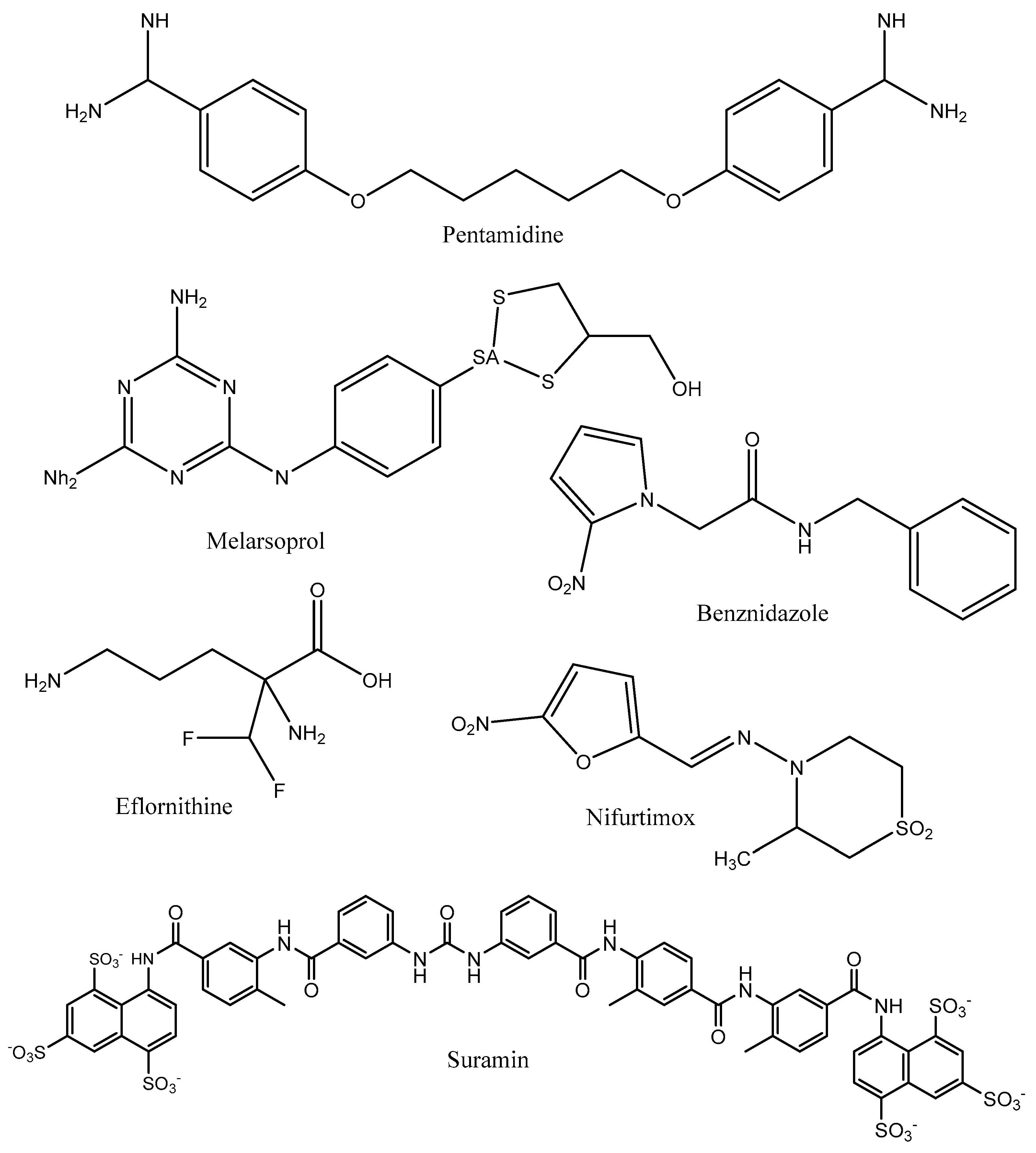
4. Human American Trypanosomiasis
5. Leishmaniases
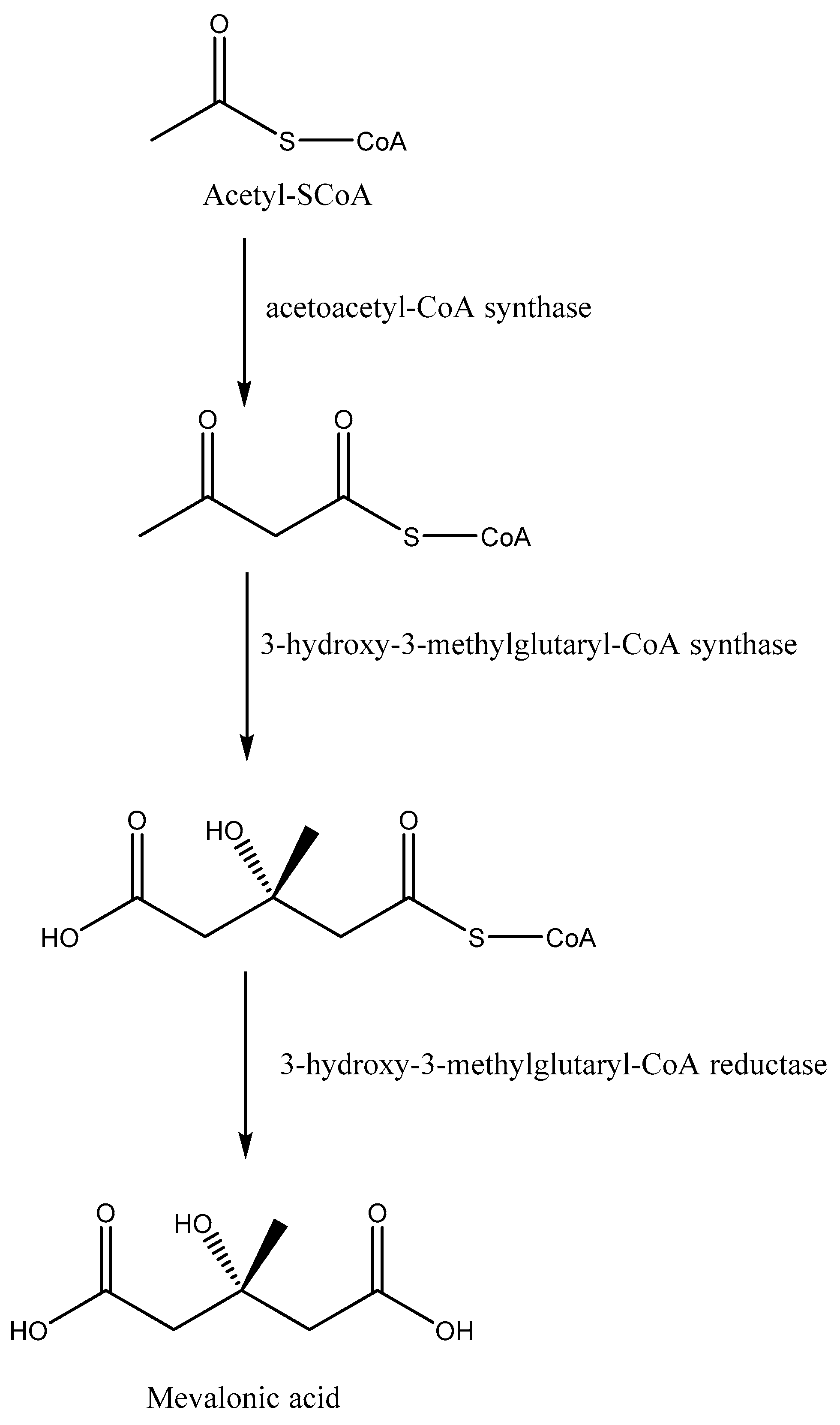
6. Squalene Synthase and Farnesyl Diphosphate Synthase: Targets for Anti-Trypanosomatidae Drug Discovery
7. Potential Inhibitors of Squalene Synthase
8. Potential Inhibitors of Farnesyl Diphosphate Synthase
9. Critical Assessment and Discussion
10. Authors’ Opinion on the Topic
11. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization (WHO). The Neglected Tropical Diseases. FactSheets. 2022. Available online: https://www.who.int/health-topics/neglected-tropical-diseases#tab=tab_1 (accessed on 26 April 2023).
- Ochola, E.A.; Karanja, D.M.S.; Elliott, S.J. The impact of Neglected Tropical Diseases (NTDs) on health and wellbeing in sub-Saharan Africa (SSA): A case study of Kenya. PLoS Negl. Trop. Dis. 2021, 15, e0009131. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J.; Kamath, A. Neglected tropical diseases in subsaharan Africa: Review of their prevalence, distribution, and disease burden. PLoS Negl. Trop. Dis. 2009, 3, e412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). The Fact Sheets. 2023. Available online: https://www.who.int/health-topics/chagas-disease#tab=tab_1 (accessed on 22 March 2023).
- World Health Organization (WHO). The Fact Sheets. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 22 March 2023).
- World Health Organization (WHO). The Fact Sheets. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 22 March 2023).
- Aronson, N.; Herwaldt, B.L.; Libman, M.; Pearson, R.; Lopez-Velez, R.; Weina, P.; Carvalho, E.; Ephros, M.; Jeronimo, S.; Magill, A. Diagnosis and treatment of leishmaniasis: Clinical practice guidelines by the infectious diseases society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Am. J. Trop. Med. Hyg. 2017, 96, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Basu, S.; Zhang, K. Farnesyl pyrophosphate synthase is essential for the promastigote and amastigote stages in Leishmania major. Mol. Biochem. Parasitol. 2019, 230, 8–15. [Google Scholar] [CrossRef]
- Rodrígues-Poveda, C.A.; González-Pacanowska, D.; Szajnman, S.H.; Rodríguez, J.B. 2-alkylaminoethyl-1,1-bisphosphonic acids are potent inhibitors of the enzymatic activity of Trypanosoma cruzi squalene synthase. Antimicrob. Agents Chemother. 2012, 56, 4483–4486. [Google Scholar] [CrossRef] [Green Version]
- Urbina, J.A.; Concepcion, J.L.; Rangel, S.; Visbal, G.; Lira, R. Squalene synthase as a chemotherapeutic target in Trypanosoma cruzi and Leishmania mexicana. Mol. Biochem. Parasitol. 2002, 125, 35–45. [Google Scholar] [CrossRef]
- Urbina, J.A.; Concepcion, J.L.; Caldera, A.; Payares, G.; Sanoja, C.; Otomo, T.; Hiyoshi, H. In vitro and in vivo activities of E5700 and ER-119884, two novel orally active squalene synthase inhibitors, against Trypanosoma cruzi. Antimicrob. Agents Chemother. 2004, 48, 2379–2387. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lindert, S.; Zhu, W.; Wang, K.; McCammon, J.A.; Oldfield, E. Taxodione and arenarone inhibit farnesyl diphosphate synthase by binding to the isopentenyl diphosphate site. Proc. Natl. Acad. Sci. USA 2014, 111, E2530–E2539. [Google Scholar] [CrossRef]
- Shang, N.; Li, Q.; Ko, T.P.; Chan, H.-C.; Li, J.; Zheng, Y.; Huang, C.-H.; Ren, F.; Chen, C.-C.; Zhu, Z.; et al. Squalene synthase as a target for Chagas disease therapeutics. PLoS Pathog. 2014, 10, e1004114. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; McKenna, C.E. Farnesyl pyrophosphate synthase modulators: A patent review (2006–2010). Expert Opin. Ther. Pat. 2011, 21, 1433–1451. [Google Scholar] [CrossRef]
- Elenga, V.A.; Lissom, A.; Elion, D.O.A.; Vouvoungui, J.C.; Djontu, J.C.; Boumpoutou, R.K.; Gabriel Ahombo, G.; Ntoumi, F. Risk factors and prevalence of human African trypanosomiasis in individuals living in remote areas of the republic of Congo. BMC Public Health 2022, 22, 2322. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Rodríguez, A.; Jin, B.K.; Radwanska, M.; Magez, S. Recent progress in diagnosis and treatment of Human African Trypanosomiasis has made the elimination of this disease a realistic target by 2030. Front. Med. 2022, 9, 1037094. [Google Scholar] [CrossRef] [PubMed]
- Bouteille, B.; Buguet, A. The detection and treatment of human African trypanosomiasis. Res. Rep. Trop. Med. 2012, 3, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Matthews, K.R. The developmental cell biology of Trypanosoma brucei. J. Cell Sci. 2005, 118 Pt 2, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Martinez, S.J.; Romano, P.S.; Engman, D.M. Precision health for Chagas disease: Integrating parasite and host factors to predict outcome of infection and response to therapy. Front. Cell Infect. Microbiol. 2020, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Balouz, V.; Agüero, F.; Buscaglia, C.A. Chagas disease diagnostic applications: Present knowledge and future steps. Adv. Parasitol. 2017, 97, 1–45. [Google Scholar] [PubMed] [Green Version]
- Bivona, A.E.; Alberti, A.S.; Cerny, N.; Trinitario, S.N.; Malchiodi, E.L. Chagas disease vaccine design: The search for an efficient Trypanosoma cruzi immune-mediated control. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165658. [Google Scholar] [CrossRef]
- Sosa-Estani, S.; Colantonio, L.; Segura, E.L. Therapy of Chagas disease: Implications for levels of prevention. J. Trop. Med. 2012, 2012, 292138. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhou, X.N. Preventing the transmission of American trypanosomiasis and its spread into nonendemic countries. Infect. Dis. Poverty 2015, 4, 60. [Google Scholar] [CrossRef] [Green Version]
- Madusanka, R.K.; Silva, H.; Karunaweera, N.D. Treatment of cutaneous leishmaniasis and insights into species-specific responses: A narrative review. Infect. Dis. Ther. 2022, 11, 695–711. [Google Scholar] [CrossRef]
- Arevalo, J.; Ramirez, L.; Adaui, V.; Zimic, M.; Tulliano, G.; Miranda-Verástegui, C.; Lazo, M.; Loayza-Muro, R.; De Doncker, S.; Maurer, A.; et al. Influence of Leishmania (Viannia) species on the response to antimonial treatment in patients with American tegumentary leishmaniasis. J. Infect. Dis. 2007, 195, 1846–1851. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Jaya, J. Liposomal amphotericin B and leishmaniasis: Dose and response. J. Glob. Infect. Dis. 2010, 2, 159–166. [Google Scholar] [CrossRef]
- Tahir, M.; Bashir, U.; Hafeez, J.; Ghafoor, R. Safety and efficacy of miltefosine in cutaneous leishmaniasis: An open label, noncomparative study from Balochistan. Pak. J. Med. Sci. 2019, 35, 495–499. [Google Scholar] [CrossRef] [Green Version]
- Asilian, A.; Davami, M. Comparison between the efficacy of photodynamic therapy and topical paromomycin in the treatment of Old World cutaneous leishmaniasis: A placebo-controlled, randomized clinical trial. Clin. Exp. Dermatol. 2006, 31, 634–637. [Google Scholar] [CrossRef]
- Galvão, E.L.; Rabello, A.; Cota, G.F. Efficacy of azole therapy for tegumentary leishmaniasis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0186117. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, D.E.; Benchimol, M.; Rodrigues, J.C.; Crepaldi, P.H.; Pimenta, P.F.; de Souza, W. The cell biology of Leishmania: How to teach using animations. PLoS Pathog. 2013, 9, e1003594. [Google Scholar] [CrossRef] [Green Version]
- Tansey, T.R.; Shechter, I. Squalene synthase: Structure and regulation. Prog. Nucleic Acid Res. Mol. Biol. 2001, 65, 157–195. [Google Scholar] [PubMed]
- Wadanambi, P.M.; Mannapperuma, U. Computational study to discover potent phytochemical inhibitors against drug target, squalene synthase from Leishmania donovani. Heliyon 2021, 7, e07178. [Google Scholar] [CrossRef] [PubMed]
- de Souza, W.; Rodrigues, J.C. Sterol biosynthesis pathway as target for anti-trypanosomatid drugs. Interdiscip. Perspect. Infect. Dis. 2009, 2009, 642502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrero-Lérida, J.; Pérez-Moreno, G.; Castillo-Acosta, V.M.; Ruiz-Pérez, L.M.; González-Pacanowskda, D. Intracellular location of the early steps of the isoprenoid biosynthetic pathway in the trypanosomatids Leishmania major and Trypanosoma brucei. Int. J. Parasitol. 2009, 39, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Gómez, A.; Jiménez, C.; Estévez, A.M.; Carrero-Lérida, J.; Ruiz-Pérez, L.M.; González-Pacanowska, D. Farnesyl diphosphate synthase is a cytosolic enzyme in Leishmania major promastigotes and its overexpression confers resistance to risedronate. Eukaryotic Cell. 2006, 5, 1057–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, T.; Kakuta, H.; Moritani, H.; Ugawa, T.; Yanagisawa, I. Synthesis and biological evaluation of novel propylamine derivatives as orally active squalene synthase inhibitors. Bioorg. Med. Chem. 2004, 12, 5899–5908. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Ohtsuka, M.; Ohki, H.; Ota, M.; Haginoya, N.; Itoh, M.; Shibata, Y.; Sugita, K.; Ishigai, Y.; Terayama, K.; et al. Discovery of DF-461, a potent squalene synthase inhibitor. ACS Med. Chem. Lett. 2013, 4, 932–936. [Google Scholar] [CrossRef] [Green Version]
- Pospiech, M.; Owens, S.E.; Miller, D.J.; Austin-Muttitt, K.; Mullins, J.G.L.; Cronin, J.G.; Allemann, R.K.; Sheldon, I.M. Bisphosphonate inhibitors of squalene synthase protect cells against cholesterol-dependent cytolysins. FASEB J. 2021, 35, e21640. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.; Fitzgerald, B.J.; Hutson, J.L.; McCarthy, A.D.; Motteram, J.M.; Ross, B.C.; Sapra, M.; Snowden, M.A.; Watson, N.S.; Williams, R.J.; et al. Squalestatin 1, a potent inhibitor of squalene synthase, which lowers serum cholesterol in vivo. J. Biol. Chem. 1992, 17, 11705–11708. [Google Scholar] [CrossRef]
- Wentzinger, L.F.; Bach, T.J.; Hartmann, M.A. Inhibition of squalene synthase and squalene epoxidase in tobacco cells triggers an upregulation of 3-hydroxy-3-methylglutaryl coenzyme a reductase. Plant Physiol. 2002, 130, 334–346. [Google Scholar] [CrossRef]
- Ward, W.H.; Holdgate, G.A.; Freeman, S.; McTaggart, F.; Girdwood, P.A.; Davidson, R.G.; Mallion, K.B.; Brown, G.R.; Eakin, M.A. Inhibition of squalene synthase in vitro by 3-(biphenyl-4-yl)-quinuclidine. Biochem. Pharmacol. 1996, 51, 1489–1501. [Google Scholar] [CrossRef]
- McTaggart, F.; Brown, G.R.; Davidson, R.G.; Freeman, S.; Holdgate, G.A.; Mallion, K.B.; Mirrlees, D.J.; Smith, G.J.; Ward, W.H. Inhibition of squalene synthase of rat liver by novel 3’ substituted quinuclidines. Biochem. Pharmacol. 1996, 51, 1477–1487. [Google Scholar] [CrossRef]
- Bergstrom, J.D.; Dufresne, C.; Bills, G.F.; Nallin-Omstead, M.; Byrne, K. Discovery, biosynthesis, and mechanism of action of the zaragozic acids: Potent inhibitors of squalene synthase. Annu. Rev. Microbiol. 1995, 49, 607–639. [Google Scholar] [CrossRef]
- Shechter, I.; Guan, G.; Boettcher, B.R. 2.09—Squalene synthase. Reference Module in Chemistry, Molecular Sciences and Chemical Engineering. Compr. Nat. Prod. Chem. 1999, 2, 245–266. [Google Scholar]
- Bedi, M.; Niesen, M.; Lopez, D. Inhibition of squalene synthase upregulates PCSK9 expression in rat liver. Arch. Biochem. Biophys. 2008, 470, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.; Rutledge, R.Z.; Needle, S.N.; Galczenski, H.F.; Neuenschwander, K.; Scotese, A.C.; Maguire, M.P.; Bush, R.C.; Hele, D.J.; Bilder, G.E.; et al. RPR 107393, a Potent Squalene Synthase Inhibitor and Orally Effective Cholesterol-Lowering Agent: Comparison with Inhibitors of HMG-CoA Reductase. J. Pharmacol. Exp. Ther. 1997, 281, 746–752. [Google Scholar] [PubMed]
- Ugawa, T.; Kakuta, H.; Moritani, H.; Matsuda, K.; Ishihara, T.; Yamaguchi, M.; Naganuma, S.; Lizumi, Y.; Shikama, H. A novel squalene synthase inhibitor, reduces plasma cholesterol and triglyceride levels in several animal species. Br. J. Pharmacol. 2000, 131, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiyoshi, H.; Yanagimachi, M.; Ito, M.; Ohtsuka, I.; Yoshida, I.; Saeki, T.; Tanaka, H. Effect of ER-27856, a novel squalene synthase inhibitor, on plasma cholesterol in rhesus monkeys: Comparison with 3-hydroxy-3-methylglutaryl-coa reductase inhibitors. J. Lipid Res. 2000, 41, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Altman, R.B.; Klein, T.E. Bisphosphonates pathway. Pharmacogenet. Genom. 2011, 21, 50–53. [Google Scholar] [CrossRef] [Green Version]
- Miki, T.; Kori, M.; Mabuchi, H.; Tozawa, R.; Nishimoto, T.; Sugiyama, Y.; Teshima, K.; Yukimasa, H. Synthesis of novel 4,1-benzoxazepine derivatives as squalene synthase inhibitors and their inhibition of cholesterol synthesis. J. Med. Chem. 2002, 45, 4571–4580. [Google Scholar] [CrossRef]
- Bailey, S.J.; Toth, M. Variability in the benzodiazepine response of serotonin 5-HT1A receptor null mice displaying anxiety-like phenotype: Evidence for genetic modifiers in the 5-HT-mediated regulation of GABAA receptors. J. Neurosci. 2004, 24, 6343–6351. [Google Scholar] [CrossRef]
- Jaka, O.; Iturria, I.; van der Toorn, M.; de Mendoza, J.H.; Latino, D.A.R.S.; Alzualde, A.; Peitsch, M.C.; Hoeng, J.; Koshibu, K. Effects of natural monoamine oxidase inhibitors on anxiety-like behavior in zebrafish. Front. Pharmacol. 2012, 12, 669370. [Google Scholar] [CrossRef]
- Scala, R.; Maqoud, F.; Antonacci, M.; Dibenedetto, J.R.; Perrone, M.G.; Scilimati, A.; Castillo, K.; Latorre, R.; Conte, D.; Bendahhou, S.; et al. Bisphosphonates Targeting Ion Channels and Musculoskeletal Effects. Front. Pharmacol. 2022, 13, 837534. [Google Scholar] [CrossRef]
- Ishihara, T.; Kakuta, H.; Moritani, H.; Ugawa, T.; Sakamoto, S.; Tsukamoto, S.; Yanagisawa, I. Syntheses and biological evaluation of novel quinuclidine derivatives as squalene synthase inhibitors. Bioorg. Med. Chem. 2003, 11, 2403–2414. [Google Scholar] [CrossRef]
- Ishihara, T.; Kakuta, H.; Moritani, H.; Ugawa, T.; Sakamoto, S.; Tsukamoto, S.; Yanagisawa, I. Syntheses of 3-ethylidenequinuclidine derivatives as squalene synthase inhibitors. Part 2: Enzyme inhibition and effects on plasma lipid levels. Bioorg. Med. Chem. 2003, 11, 3735–3745. [Google Scholar] [CrossRef] [PubMed]
- Sealey-Cardona, M.; Cammerer, S.; Jones, S.; Ruiz-Pérez, L.M.; Brun, R.; Gilbert, I.H.; Urbina, J.A.; González-Pacanowska, D. Kinetic characterization of squalene synthase from Trypanosoma cruzi: Selective inhibition by quinuclidine derivatives. Antimicrob. Agents Chemother. 2007, 51, 2123–2129. [Google Scholar] [CrossRef] [Green Version]
- Orenes Lorente, S.; Gómez, R.; Jiménez, C.; Cammerer, S.; Yardley, V.; de Luca-Fradley, K.; Croft, S.L.; Ruiz Perez, L.M.; Urbina, J.; Gonzalez Pacanowska, D.; et al. Biphenylquinuclidines as inhibitors of squalene synthase and growth of parasitic protozoa. Bioorg. Med. Chem. 2005, 13, 3519–3529. [Google Scholar] [CrossRef]
- Tavridou, A.; Kaklamanis, L.; Megaritis, G.; Kourounakis, A.P.; Papalois, A.; Roukounas, D.; Rekka, E.A.; Kourounakis, P.N.; Charalambous, A.; Manolopoulos, V.G. Pharmacological characterization in vitro of EP2306 and EP2302, potent inhibitors of squalene synthase and lipid biosynthesis. Eur. J. Pharmacol. 2006, 535, 34–42. [Google Scholar] [CrossRef]
- Fernandes Rodrigues, J.C.; Concepcion, J.L.; Rodrigues, C.; Caldera, A.; Urbina, J.A.; de Souza, W. In vitro activities of ER-119884 and E5700, two potent squalene synthase inhibitors, against Leishmania amazonensis: Antiproliferative, biochemical, and ultrastructural effects. Antimicrob. Agents Chemother. 2008, 52, 4098–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Chu, C.L.; Chen, L.; Shui, D. Evaluation of potential inhibitors of squalene synthase based on virtual screening and in vitro studies. Comput. Biol. Chem. 2019, 80, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Macías-Alonso, M.; Andrés, L.S.; Córdova-Guerrero, I.; Estolano-Cobián, A.; Díaz-Rubio, L.; Marrero, J.G. Inhibition of squalene synthase of rat liver by abietane diterpenes derivatives. Nat. Prod. Res. 2021, 35, 2972–2976. [Google Scholar] [CrossRef]
- Lage, O.M.; Ramos, M.C.; Calisto, R.; Almeida, E.; Vasconcelos, V.; Vicente, F. Current screening methodologies in drug discovery for selected human diseases. Mar. Drugs 2018, 16, 279. [Google Scholar] [CrossRef] [Green Version]
- Olías-Molero, A.I.; de la Fuente, C.; Cuquerella, M.; Torrado, J.J.; Alunda, J.M. Antileishmanial drug discovery and development: Time to reset the model? Microorganisms 2021, 9, 2500. [Google Scholar] [CrossRef]
- Yao, C.; Wilson, M.E. Dynamics of sterol synthesis during development of Leishmania spp. parasites to their virulent form. Parasit. Vectors 2016, 9, 200. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.È.; Nisole, A.; Béliveau, C.; Sen, S.; Barbar, A.; Shi, R.; Cusson, M. Structural characterization of a lepidopteran type-II farnesyl diphosphate synthase from the spruce budworm, Choristoneura fumiferana: Implications for inhibitor design. Insect Biochem. Mol. Biol. 2018, 92, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Szajnman, S.H.; Montalvetti, A.; Wang, Y.; Docampo, R.; Rodriguez, J.B. Bisphosphonates derived from fatty acids are potent inhibitors of Trypanosoma cruzi farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2003, 13, 3231–3235. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, E.; Paterson, A.H.; Powles, T.; Kanis, J.A. Clodronate. Bone 2021, 143, 115715. [Google Scholar] [CrossRef] [PubMed]
- Panagiotakou, A.; Yavropoulou, M.; Nasiri-Ansari, N.; Makras, P.; Basdra, E.K.; Papavassiliou, A.G.; Kassi, E.N. Extraskeletal effects of bisphosphonates. Metabolism 2020, 110, 154264. [Google Scholar] [CrossRef]
- Lehenkari, P.P.; Kellinsalmi, M.; Napankangas, J.P.; Ylitalo, K.V.; Monkkonen, J.; Rogers, M.J.; Azhayev, A.; Vaananen, K.; Hassinen, I.E. Further insight into mechanism of action of clodronate: Inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol. Pharmacol. 2002, 61, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Malwal, S.R.; O’Dowd, B.; Feng, X.; Turhanen, P.; Shin, C.; Yao, J.; Kim, B.K.; Baig, N.; Zhou, T.; Bansal, S.; et al. Bisphosphonate-generated ATP-analogs inhibit cell signaling pathways. J. Am. Chem. Soc. 2018, 140, 7568–7578. [Google Scholar] [CrossRef]
- Garzoni, L.R.; Caldera, A.; Meirelles, M.d.N.; de Castro, S.L.; Docampo, R.; Meints, G.A.; Oldfield, E.; Urbina, J.A. Selective in vitro effects of the farnesyl pyrophosphate synthase inhibitor risedronate on Trypanosoma cruzi. Int. J. Antimicrob. Agents 2004, 23, 273–285. [Google Scholar] [CrossRef]
- Szajnman, S.H.; Ravaschino, E.L.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 1-amino-1,1-bisphosphonates derived from fatty acids against Trypanosoma cruzi targeting farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2005, 15, 4685–4690. [Google Scholar] [CrossRef]
- Szajnman, S.H.; García Liñares, G.E.; Li, Z.H.; Jiang, C.; Galizzi, M.; Bontempi, E.J.; Ferella, M.; Moreno, S.N.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting farnesyl diphosphate synthase. Bioorg. Med. Chem. 2008, 16, 3283–3290. [Google Scholar] [CrossRef] [Green Version]
- Rosso, V.S.; Szajnman, S.H.; Malayil, L.; Galizzi, M.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of new 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting farnesyl diphosphate synthase. Bioorg. Med. Chem. 2011, 19, 2211–2217. [Google Scholar] [CrossRef] [Green Version]
- Aripirala, S.; Szajnman, S.H.; Jakoncic, J.; Rodriguez, J.B.; Docampo, R.; Gabelli, S.B.; Amzel, L.M. Design, synthesis, calorimetry, and crystallographic analysis of 2-alkylaminoethyl-1,1-bisphosphonates as inhibitors of Trypanosoma cruzi farnesyl diphosphate synthase. J. Med. Chem. 2012, 55, 6445–6454. [Google Scholar] [CrossRef] [Green Version]
- Recher, M.; Barboza, A.P.; Li, Z.H.; Galizzi, M.; Ferrer-Casal, M.; Szajnman, S.H.; Docampo, R.; Moreno, S.N.; Rodriguez, J.B. Design, synthesis and biological evaluation of sulfur-containing 1,1-bisphosphonic acids as antiparasitic agents. Eur. J. Med. Chem. 2013, 60, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindert, S.; Zhu, W.; Liu, Y.L.; Pang, R.; Oldfield, E.; McCammon, J.A. Farnesyl diphosphate synthase inhibitors from in silico screening. Chem. Biol. Drug Des. 2013, 81, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Aripirala, S.; Gonzalez-Pacanowska, D.; Oldfield, E.; Kaiser, M.; Amzel, L.M.; Gabelli, S.B. Structural and thermodynamic basis of the inhibition of Leishmania major farnesyl diphosphate synthase by nitrogen-containing bisphosphonates. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 802–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galaka, T.; Falcone, B.N.; Li, C.; Szajnman, S.H.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 1-alkylaminomethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii. Bioorg. Med. Chem. 2019, 27, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boran, A.D.; Iyengar, R. Systems approaches to polypharmacology and drug discovery. Curr. Opin. Drug Discov. Devel. 2010, 13, 297–309. [Google Scholar]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.J.; Gray, D.W.; Read, K.D.; Rycker, M.D.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Antitrypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial Number | Compound Name | Model | Significant Results | Reference |
|---|---|---|---|---|
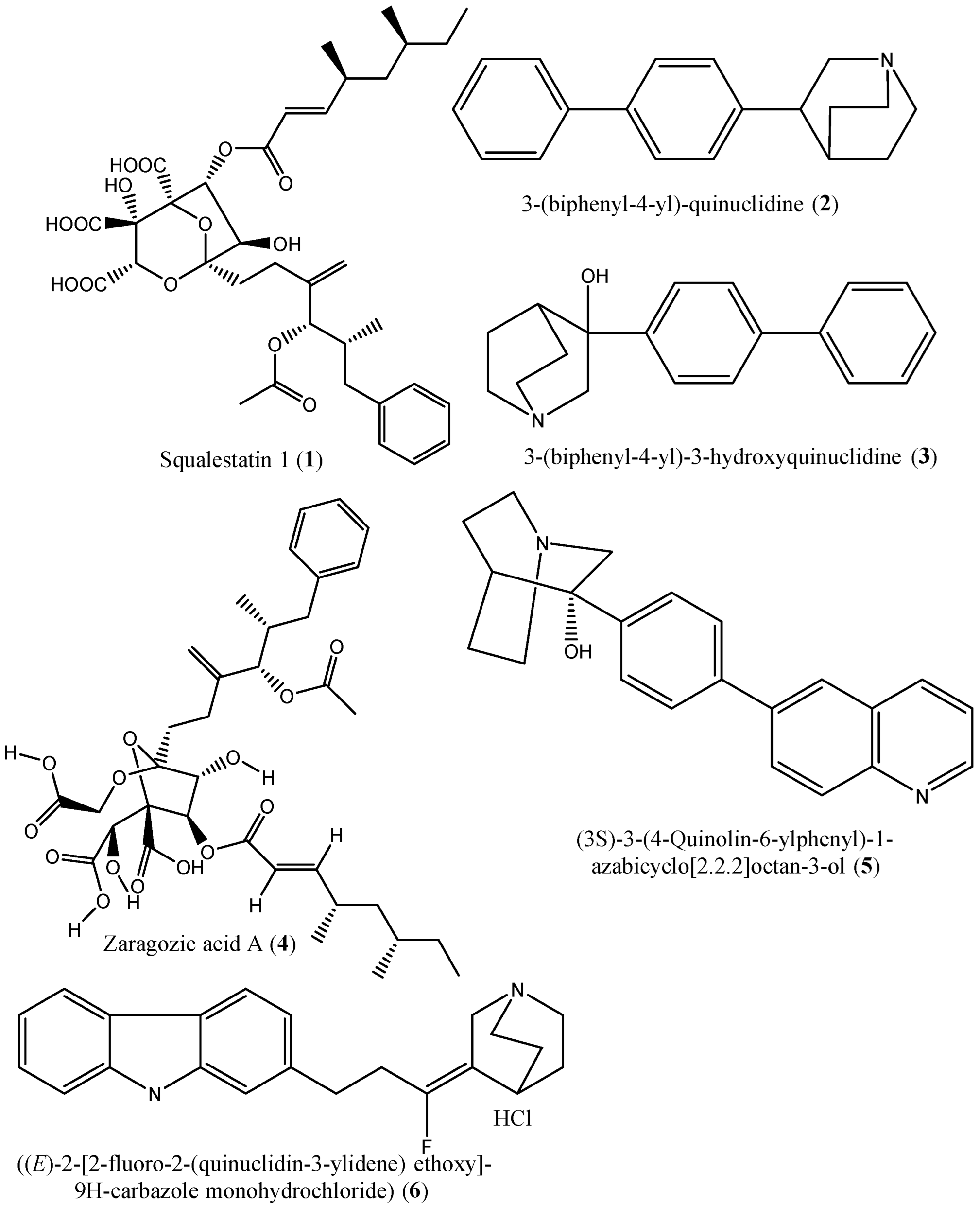
| 1 | Squalestatin-1 (1) | SQS in rat liver microsomes | IC50: 12 nM | [39] |
| 2 | 3-(biphenyl-4-yl)-quinuclidine (2) | Microsomal SQS from human or marmoset liver | Ki: 5 nM and 1300 nM in microsomes prepared from human and marmoset liver, respectively | [40] |
| 3 & 4 | 3-(biphenyl-4-yl) quinuclidine (BPQ) (2) and 3-(biphenyl-4-yl)-3-hydroxyquinuclidine (BPQ-OH) (3) | T. cruzi epimastigote’s squalene synthase; NS | Ki: 12 nM and 15 nM; Ki value: 48 nM | [10,42] |
| 5 | 3-hydroxy-3-[4-(quinolin-6-yl)phenyl]-1-azabicyclo[2-2-2]octane dihydrochloride (5) | Rat liver microsomal squalene synthase |
| [46] |
| 6 | ((E)-2-[2-uoro-2-(quinuclidin-3-ylidene) ethoxy]-9H-carbazole monohydrochloride) (6) | Hepatic microsomes of rats, Hamster, guinea pig, Rhesus monkey and HepG2 cells | In vitro studies: IC50: 90, 170, 46, 45, and 79 nM, respectively; In vivo studies: Reduction of cholesterol synthesis with ED50: 32 mg/kg | [47] |
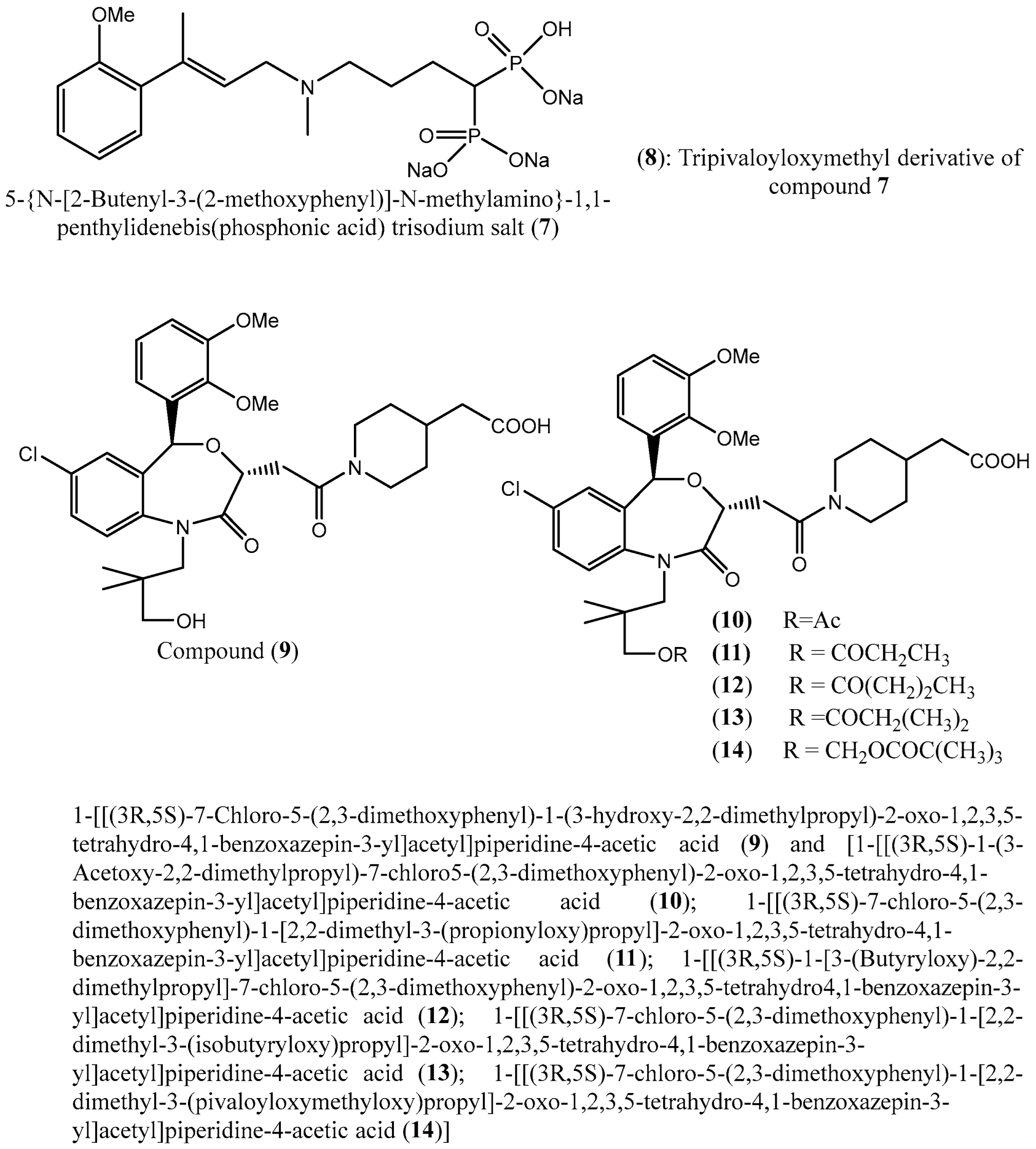
| 7 & 8 | 5-(N-[2-butenyl-3-(2-methoxyphenyl)]-N-methylamino)-1,1-penthylidenebis(phosphonic acid) trisodium salt (7) and tripivaloyloxymethyl derivative (8) | SQS in SD (Sprague Dawley) rat liver microsomes | IC50: 3.6 nM and 39 μM, respectively | [48] |
| 9–14 | 1-[[(3R,5S)-7-Chloro-5-(2,3-dimethoxyphenyl)-1-(3-hydroxy-2,2-dimethylpropyl)-2-oxo-1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl]acetyl]piperidine-4-acetic acid (9); 1-[[(3R,5S)-1-(3-Acetoxy-2,2-dimethylpropyl)-7-chloro5-(2,3-dimethoxyphenyl)-2-oxo-1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl]acetyl]piperidine-4-acetic acid (10); 1-[[(3R,5S)-7-chloro-5-(2,3-dimethoxyphenyl)-1-[2,2-dimethyl-3-(propionyloxy)propyl]-2-oxo-1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl]acetyl]piperidine-4-acetic acid (11); 1-[[(3R,5S)-1-[3-(Butyryloxy)-2,2-dimethylpropyl]-7-chloro-5-(2,3-dimethoxyphenyl)-2-oxo-1,2,3,5-tetrahydro4,1-benzoxazepin-3-yl]acetyl]piperidine-4-acetic acid (12); 1-[[(3R,5S)-7-chloro-5-(2,3-dimethoxyphenyl)-1-[2,2-dimethyl-3-(isobutyryloxy)propyl]-2-oxo-1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl]acetyl]piperidine-4-acetic acid (13); 1-[[(3R,5S)-7-chloro-5-(2,3-dimethoxyphenyl)-1-[2,2-dimethyl-3-(pivaloyloxymethyloxy)propyl]-2-oxo-1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl]acetyl]piperidine-4-acetic acid (14)] | Squalene synthase of rat liver | IC50 values: 45, 76, 87, 93, 89 and 471 nM, respectively. | [50] |
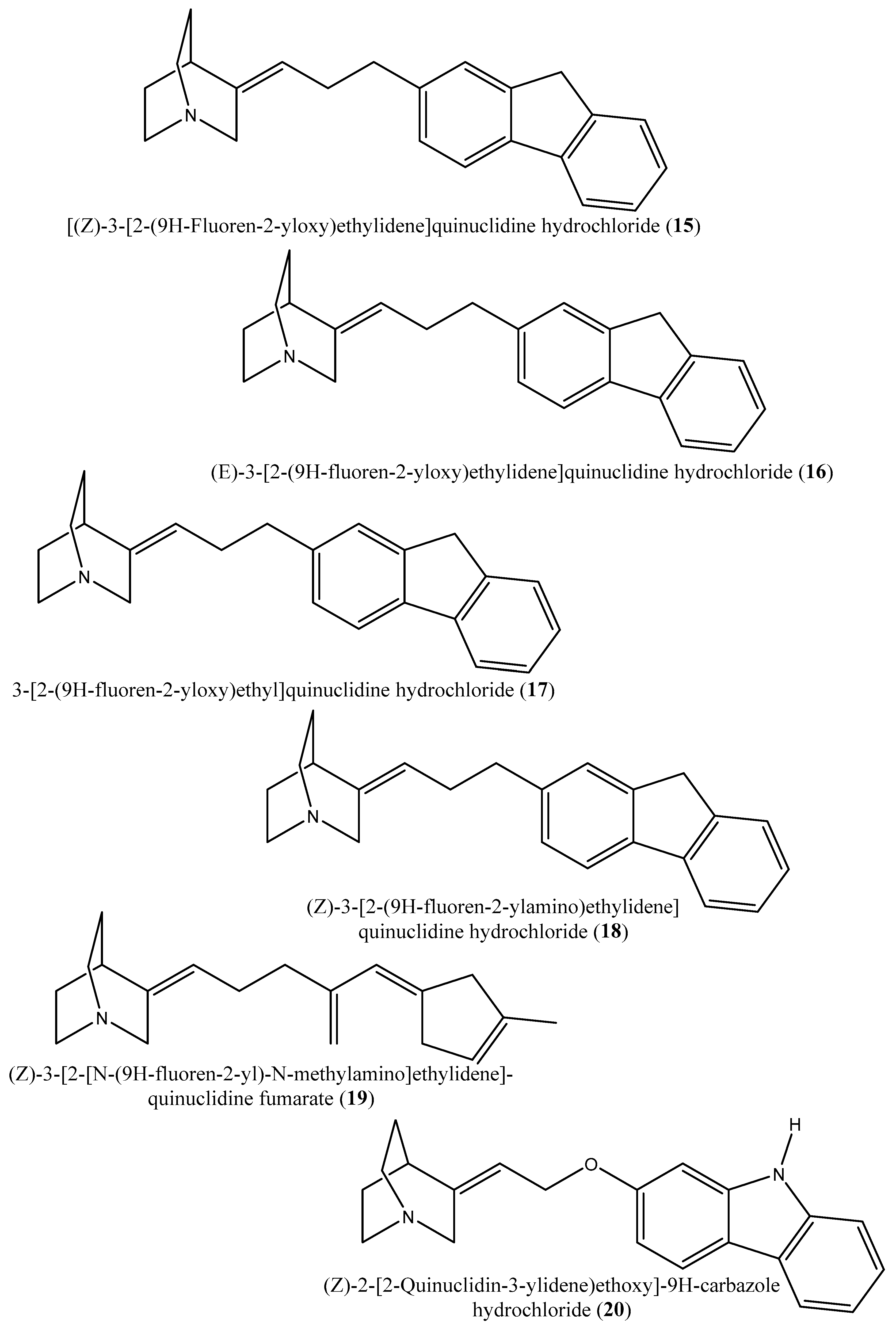
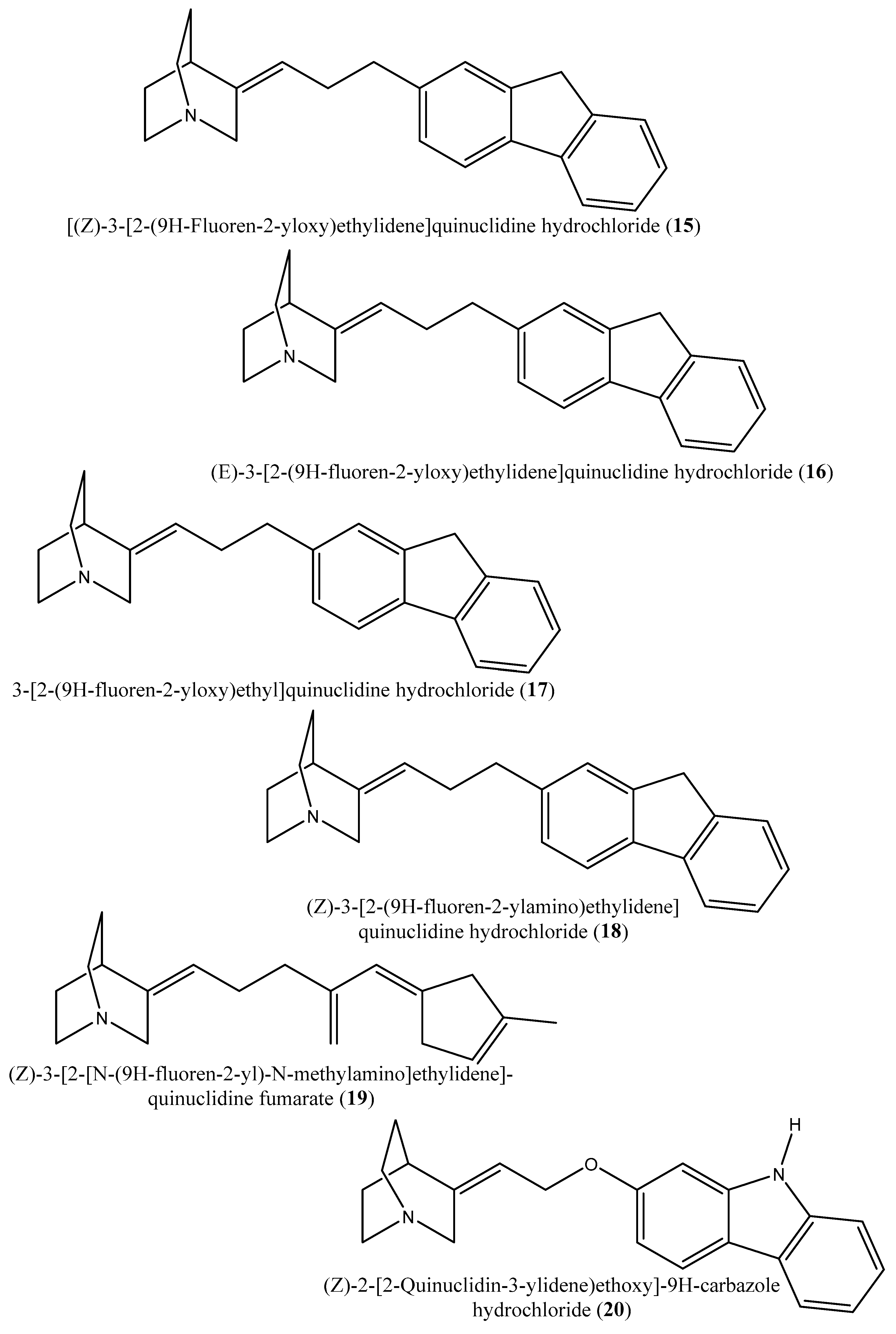
| 15–19 | [(Z)-3-[2-(9H-Fluoren-2-yloxy)ethylidene]quinuclidine hydrochloride (15); (E)-3-[2-(9H-fluoren-2-yloxy)ethylidene]quinuclidine hydrochloride (16); 3-[2-(9H-fluoren-2-yloxy)ethyl]quinuclidine hydrochloride (17); (Z)-3-[2-(9H-fluoren-2-ylamino)ethylidene]quinuclidine hydrochloride (18); (Z)-3-[2-[N-(9H-fluoren-2-yl)-N-methylamino]ethylidene] quinuclidine fumarate (19) | Squalene synthase from the hamster liver | IC50s: 0.076, 0.15, 0.25, 0.27 and 0.56 µM, for compounds 15–19, respectively | [54] |
| 20, 21 | (Z)-2-[2-(quinuclidin-3-ylidene)ethoxy]-9H-carbazole hydrochloride (20) and (E)-2-[2-fluoro-2-(quinuclidin-3-ylidene)ethoxy]-9H-carbazole hydrochloride (21) | Squalene synthase derived from human hepatoma cells | IC50s: 160 and 79 nM, respectively | [55] |
| 22, 23 | {(3R)-3-[[2-benzyl-6-[(3R,4S)-3-hydroxy-4-methoxypyrrolidin-1-yl]pyridin-3-yl]ethynyl]quinuclidin-3-ol monohydrate} (22) and {(3R)-3-[[2-benzyl-6-(3-methoxypropyloxy)pyridin-3-yl]ethynyl]quinuclidin-3-ol} (23) | Glycosomal and microsomal SQS of T. cruzi epimastigotes L. amazonensis squalene synthase |
| [11,56,59] |
| 24–26 | (3-(Biphenyl-4-yl)-3-hydroxyquinuclidine) (24); (3-(Biphenyl-4-yl)-2,3-dehydroquinuclidine); (25) and 5b (3-(Biphenyl-4-yl-40-hydroxy)-2,3-dehydroquinuclidine) (26) | L. major squalene synthase | IC50s: 0.013, 0.243 and 0.096 µM, for compounds 24–26, respectively; In vivo studies: Against L. donovani intracellular amastigotes, the following ED50 values were obtained: 29.0, 74.3 and >108 µM, respectively | [57] |
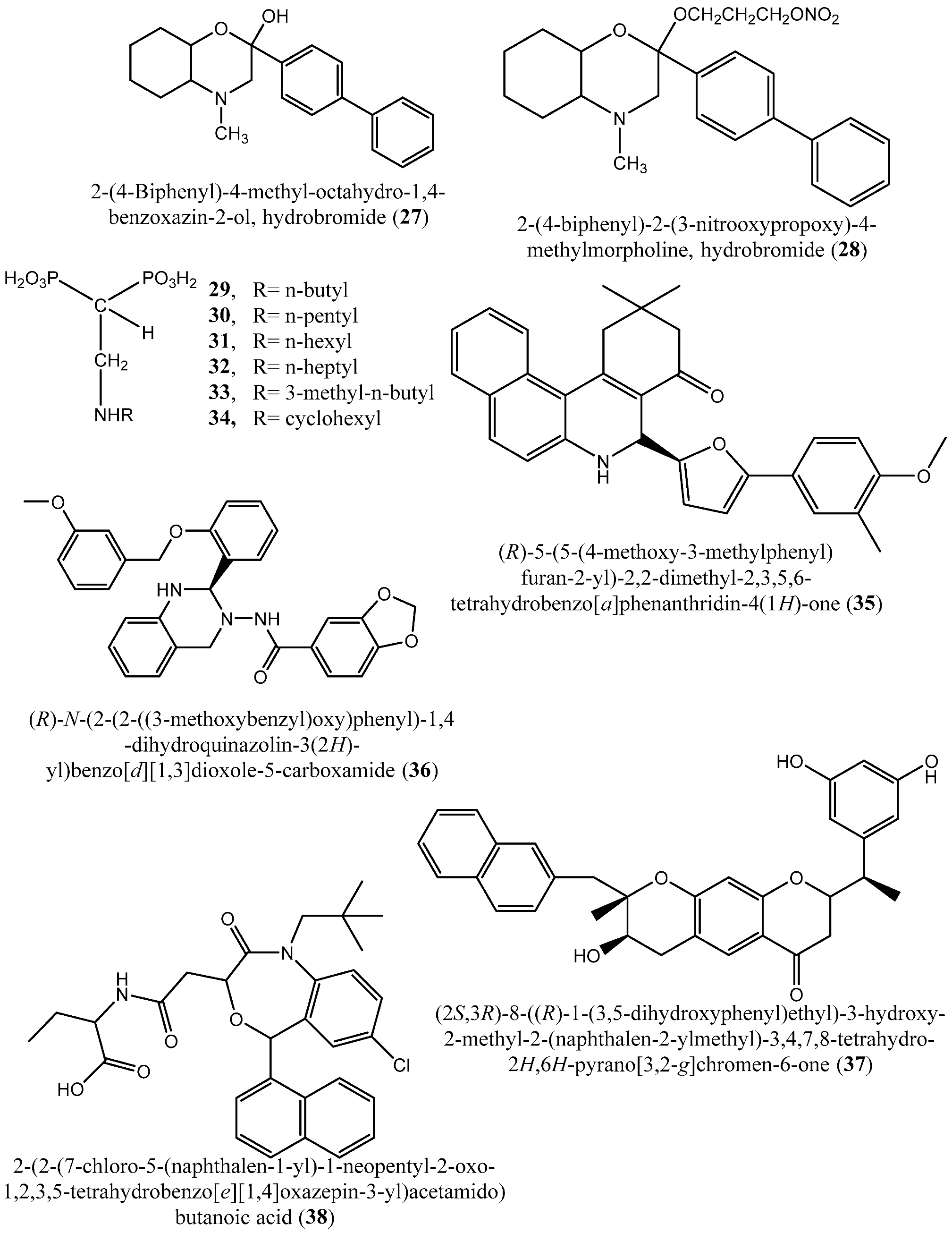
| 27–28 | 2-(4-Biphenyl)-4-methyl-octahydro-1,4-benzoxazin-2-ol, hydrobromide (27) and 2-(4-biphenyl)-2-(3-nitrooxypropoxy)-4-methylmorpholine, hydrobromide (28) | Squalene synthase in rabbit liver microsomes | IC50s: of 33 and 0.6 μM for compounds 27 and 28, respectively | [58] |
| 29–34 | Compounds 29–33 and 34 | T. cruzi squalene synthase | IC50s: 39.0, 5.0, 21.4, 11.9, 22.0 and 30.0 nM, respectively | [9] |
| 35–38 | (R)-5-(5-(4-methoxy-3-methylphenyl)furan-2-yl)-2,2-dimethyl-2,3,5,6-tetrahydrobenzo[a]phenanthridin-4(1H)-one (35); (R)-N-(2-(2-((3-methoxybenzyl)oxy)phenyl)-1,4-dihydroquinazolin-3(2H)-yl)benzo[d][1,3]dioxole-5-carboxamide (36); (2S,3R)-8-((R)-1-(3,5-dihydroxyphenyl)ethyl)-3-hydroxy-2-methyl-2-(naphthalen-2-ylmethyl)-3,4,7,8-tetrahydro-2H,6H-pyrano[3,2-g]chromen-6-one (37) and 2-(2-(7-chloro-5-(naphthalen-1-yl)-1-neopentyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-3-yl)acetamido)butanoic acid (38) | NS | IC50s: 1.7, 0.14, 191 and 63 nM for compounds 35–38, respectively | [60] |
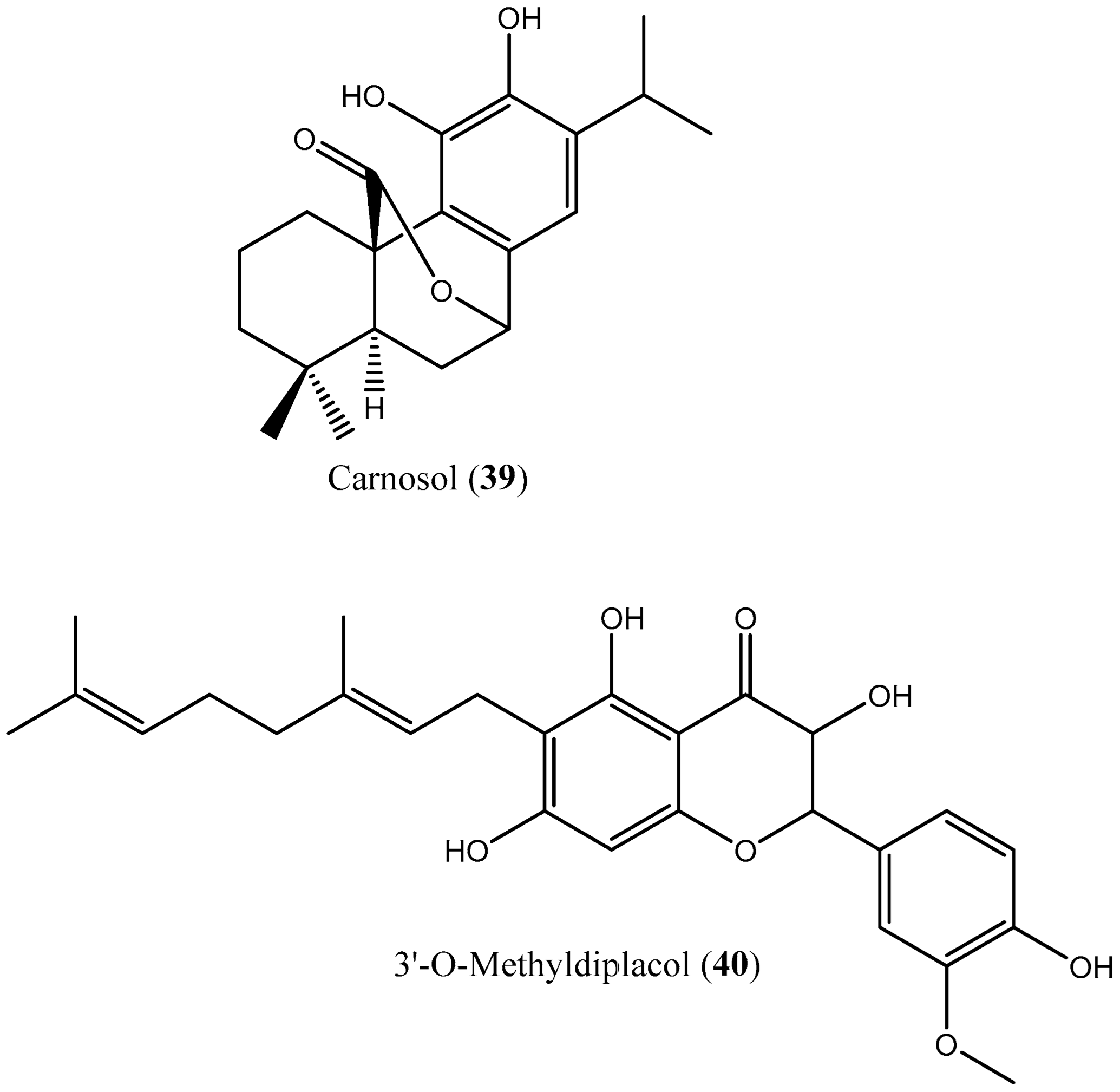
| 39 | Carnosol (39) | NS | IC50: 17.6 μM | [61] |
| Serial Number | Compound Name | Model | Significant Results | Reference |
|---|---|---|---|---|
| 41–50 | Compounds 41–45, 46, 47, 48, 49 and 50 | Trypanosoma cruzi farnesyl pyrophosphate synthase Trypanosoma brucei farnesyl pyrophosphate synthase | IC50 values: 42.83, 1.94, 2.37, 9.36, 8.45, 5.71, 5.67, 4.54, 19.73 and 4.25 µM for compounds 41–50, respectively. IC50 values: 3.12, 0.66, 3.57 and 4.54 µM, for compounds 41–45, respectively | [66] |
| 51 | Risedronate (51) | NS | IC50: 65.4 µM | [71] |
| 52–53 | Alkyl 1-amino-1,1-bisphosphonates (compounds 52 and 53) | T. cruzi farnesyl pyrophosphate synthase | IC50 values 0.382 and 3.57 µM, for compounds 10 and 11, respectively; compounds 52 and 53, respectively. | [72] |
| 54–55 | [1-[(n-But-1-ylamino)ethyl] 1,1-bisphosphonic acid (29); 1-[(n-Pent-1-ylamino)ethyl] 1,1-bisphosphonic acid (30); 1-[(n-Hex-1-ylamino)ethyl] 1,1-bisphosphonic acid (31); 1-[(n-Hept-1-ylamino)ethyl] 1,1-bisphosphonic acid (32); 1-[(n-Oct-1-ylamino)ethyl] 1,1-bisphosphonic acid (54); 1-[(3-Methyl-but-1-ylamino)ethyl] 1,1-bisphosphonic acid (33); and 1-[(tert-Butylamino)ethyl] 1,1-bisphosphonic acid (55)] | T. cruzi farnesyl diphosphate synthase | IC50s: 2.28, 1.84, 0.49, 0.058, 1.014, 0.42 and 1.21 µM, for compounds (29–32, 54, 33 and 55), respectively; | [73] |
| 56 | 1-[(n-dodec-1-ylamino)ethyl] 1,1-bisphosphonic acid (56) | T. cruzi farnesyl diphosphate synthase | IC50: 0.67 μM, vs. benznidazole (IC50: 2.77 μM) | [74] |
| 57 | 2-alkylaminoethyl-1,1-bisphosphonic acids (compounds (29–32, 55, 57 and 34)) | T. cruzi farnesyl pyrophosphate synthase | IC50 values: 4.8, 0.54, 0.84, 10.0, 10.0, 1.39 and >10 nM, for compounds 29–32, 55, 57 and 34, respectively | [9] |
| 58 | 2-(n-propylamino) ethane-1,1-diyl]bisphosphonic acid (58) and [2-(n-heptylamino)ethane-1,1-diyl]bisphosphonic acid (32) | T. cruzi farnesyl diphosphate synthase | IC50 values: 38.0 and 58.0 nM, for 58 and 32, respectively. | [75] |
| 59–61 | Compounds (59, 60 and 61) | Target enzyme TcFPPS | IC50 values: 6.4, 1.7, and 0.097 µM, for compounds 59–61, respectively | [76] |
| 73–74 | [1-(2-hydroxy-2,2-diphosphonoethyl)-3-phenylpyridinium (73) and 3-fluoro-1-(2-hydroxy-2,2-diphosphonoethyl)-pyridinium (74) | L. major farnesyl diphosphate synthase | Ki values: 9 and 50 µM for compounds 73 and 74, respectively, vs. zoledronate (11 µM) | [78] |
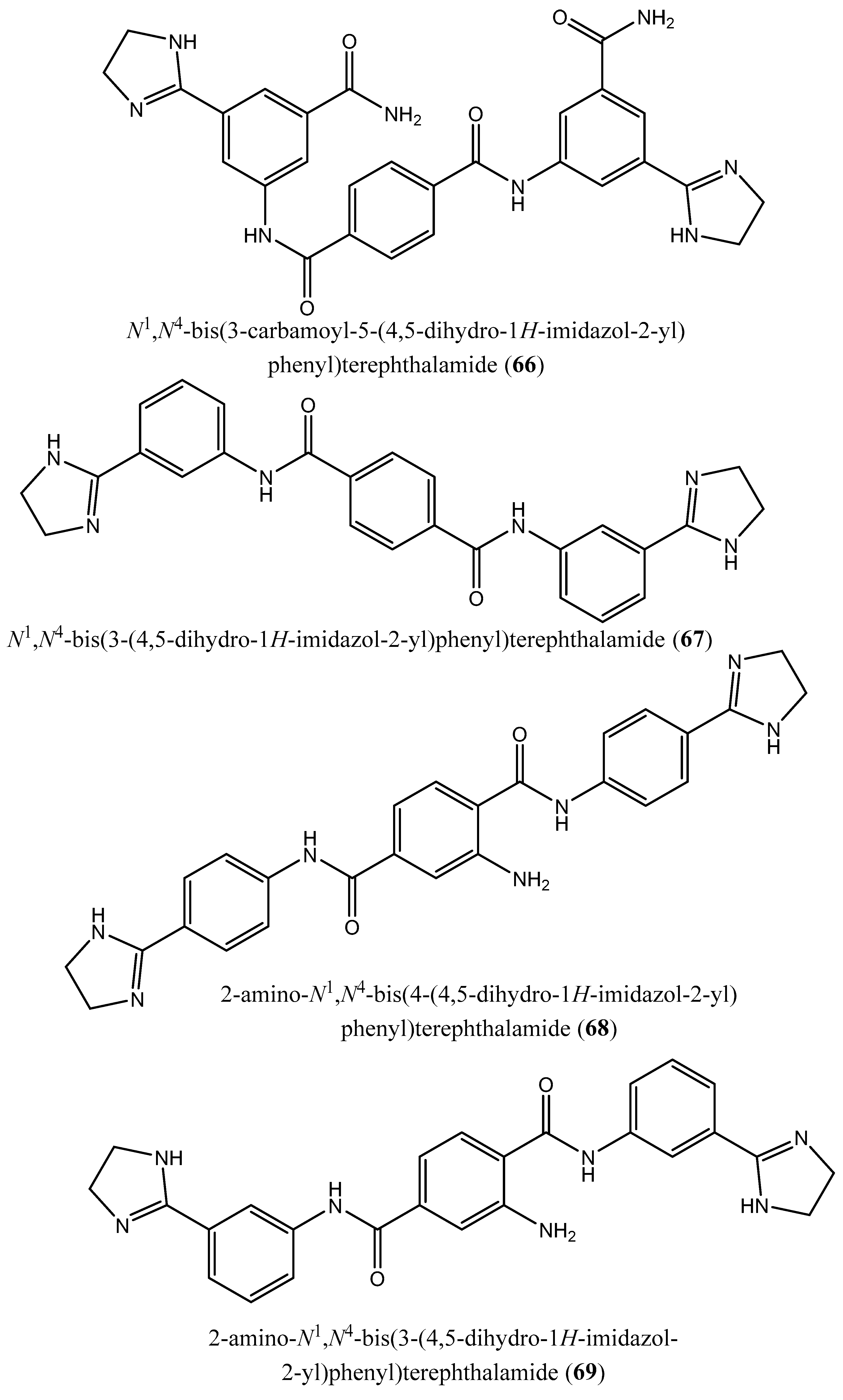
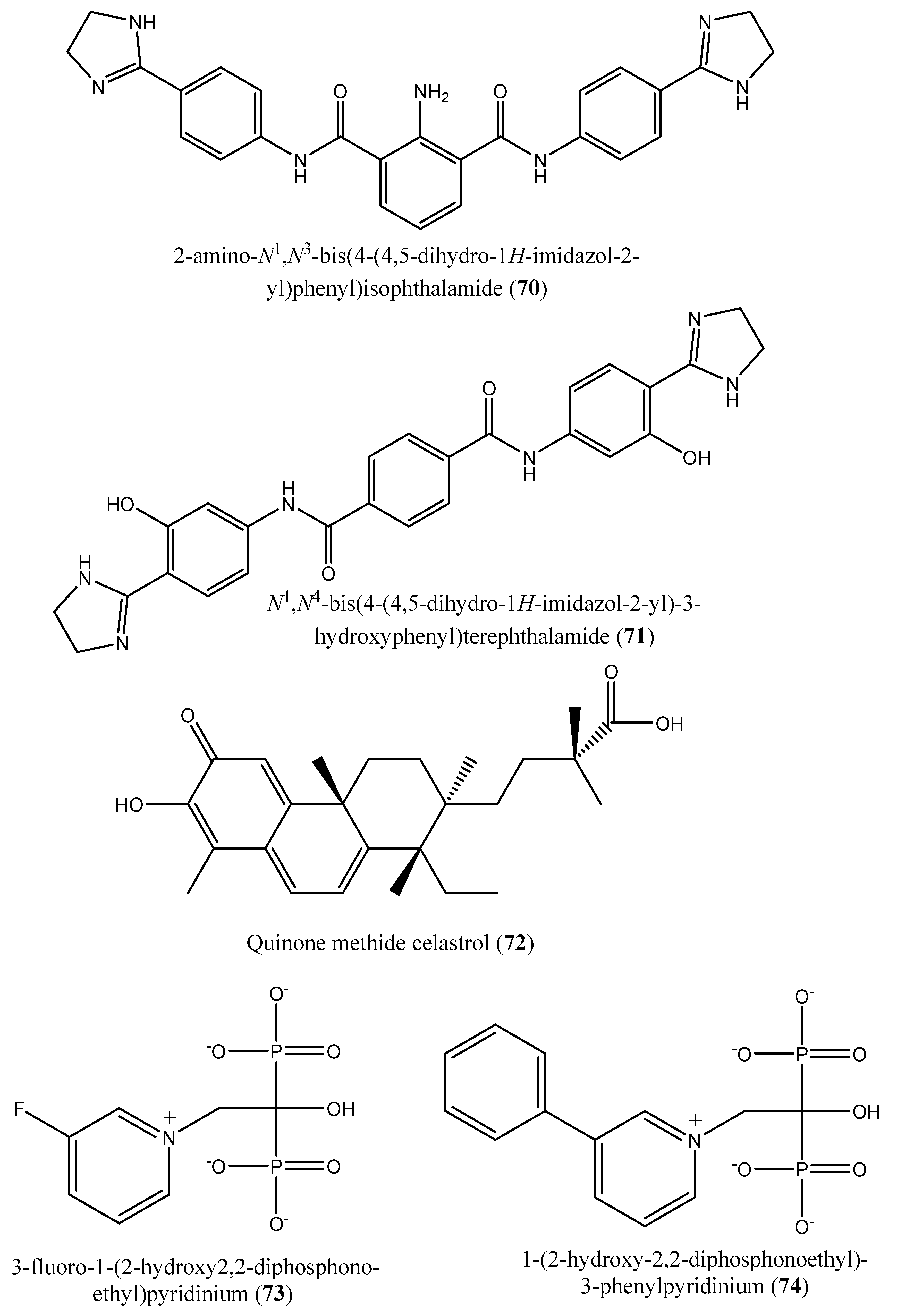
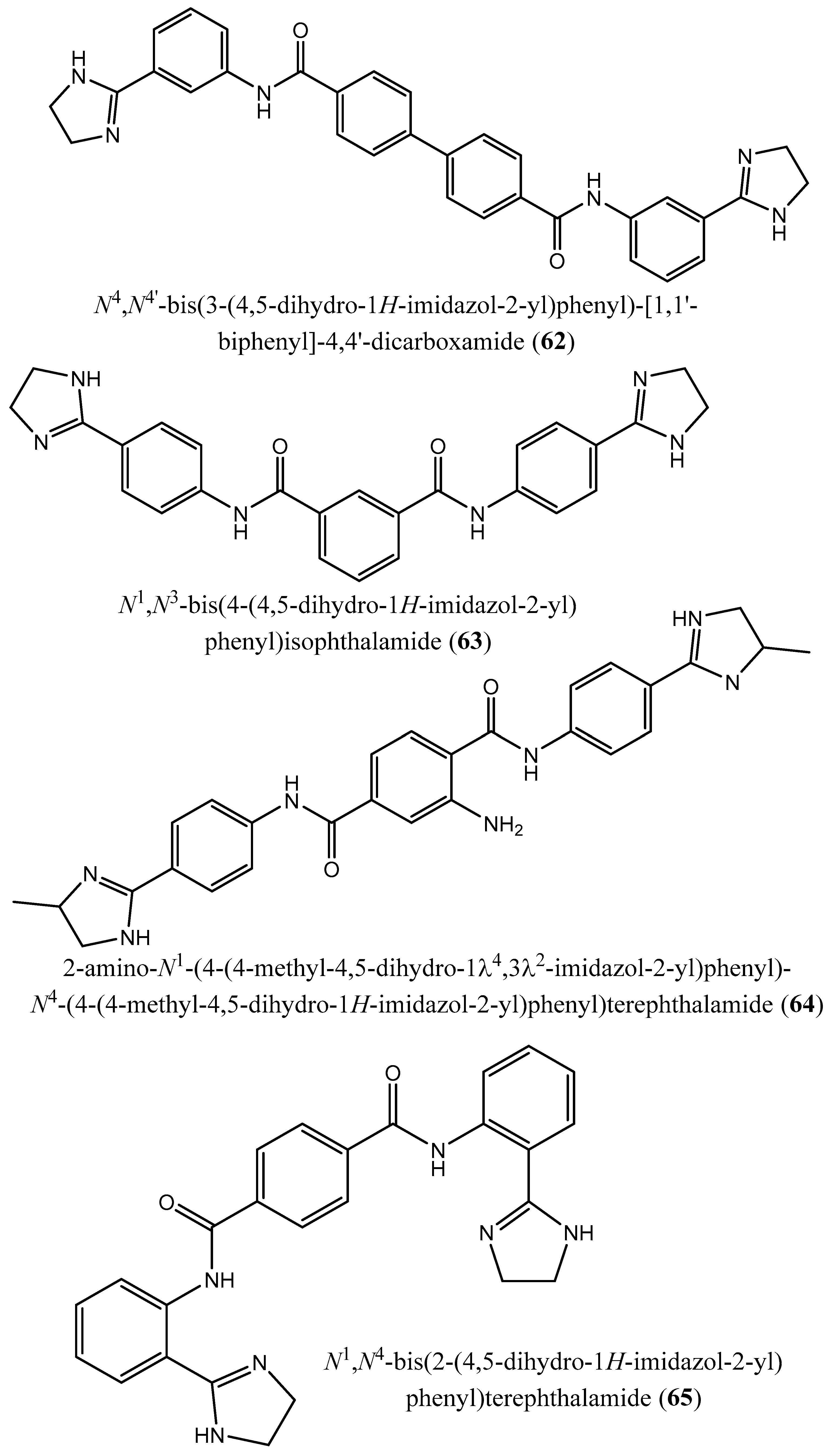
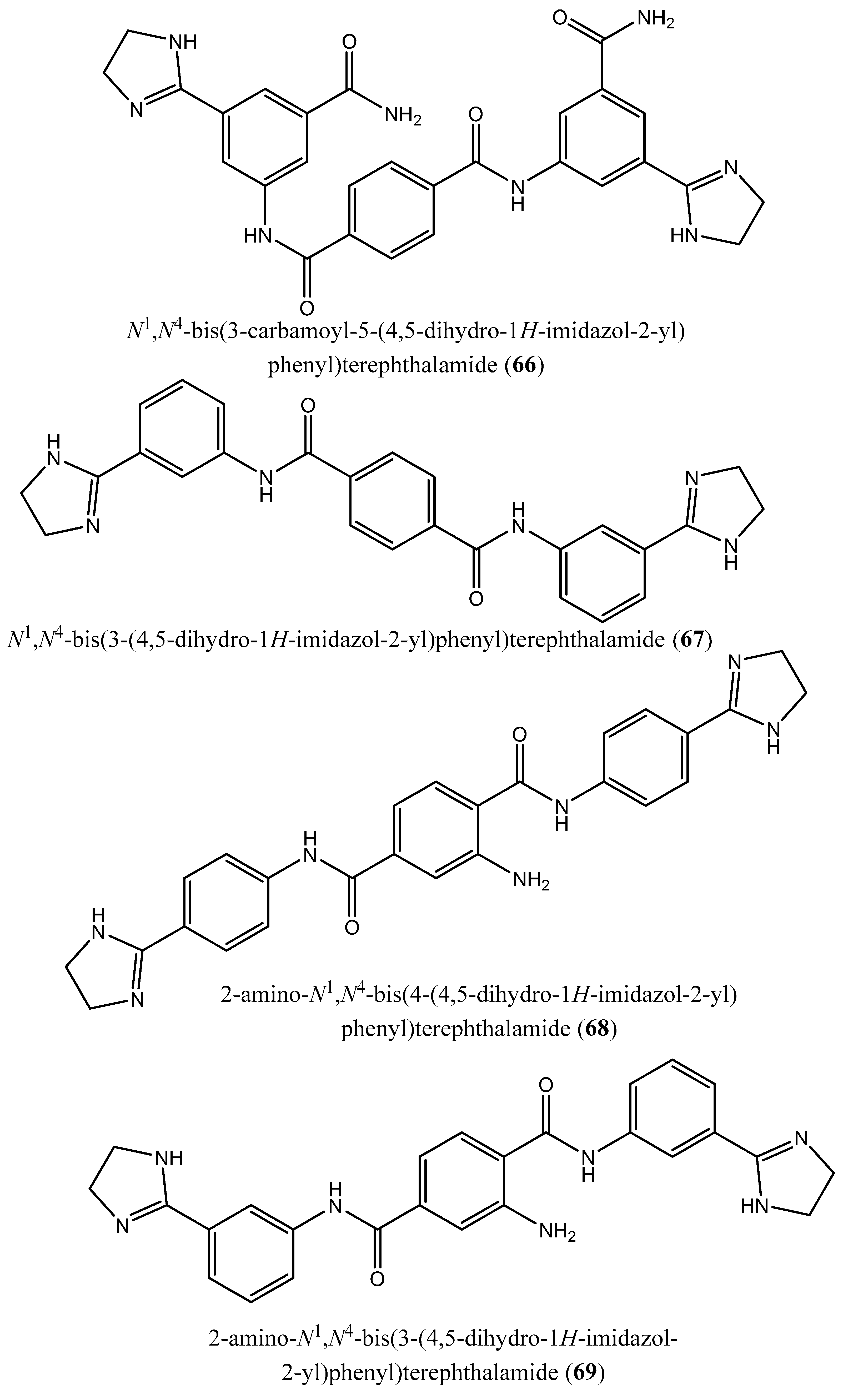
| 3 | N4,N4 ′-bis(3-(4,5-dihydro-1H-imidazol-2-yl)phenyl)-[1,1 ′-biphenyl]-4,4 ′-dicarboxamide (62); N1,N3-bis(4-(4,5-dihydro-1H-imidazol-2-yl)phenyl)isophthalamide (63); 2-amino-N1-(4-(4-methyl-4,5-dihydro-1l4,3l2-imidazol-2-yl)phenyl)-N4-(4-(4-methyl-4,5-dihydro-1H-imidazol-2-yl)phenyl)terephthalamide (64); N1,N4-bis(2-(4,5-dihydro-1H-imidazol-2-yl)phenyl)terephthalamide (65); N1,N4-bis(3-carbamoyl-5-(4,5-dihydro-1H-imidazol-2-yl)phenyl)terephthalamide (66); N1,N4-bis(3-(4,5-dihydro-1H-imidazol-2-yl)phenyl)terephthalamide (67); 2-amino-N1,N4-bis(4-(4,5-dihydro-1H-imidazol-2-yl)phenyl)terephthalamide (68); 2-amino-N1,N4-bis(3-(4,5-dihydro-1H-imidazol-2-yl)phenyl)terephthalamide (69); 2-amino-N1,N3-bis(4-(4,5-dihydro-1H-imidazol-2-yl)phenyl)isophthalamide (70); N1,N4-bis(4-(4,5-dihydro-1H-imidazol-2-yl)-3-hydroxyphenyl)terephthalamide (71). | Human FPPS | IC50: 1.8, 1.9, 2.5, 7.0, 10.7, 13.7, 20.3, 21.0, 22.3 and 35.0 µM, for compounds 62–71, respectively | [77] |
| 5 | Quinone methide celastrol (72) | Trypanosoma brucei farnesyl diphosphate synthase (FPPS) | IC50 value ∼20 µM | [12] |
| 11 | Compounds (31, 32 and 56) | Trypanosoma brucei farnesyl pyrophosphate synthase | IC50 values: 0.49, 0.058 and 0.81 µM for compounds (31, 32 and 56), respectively | [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamdem, B.P.; Boyom, F.F. Inhibitors of Farnesyl Diphosphate Synthase and Squalene Synthase: Potential Source for Anti-Trypanosomatidae Drug Discovery. Drugs Drug Candidates 2023, 2, 624-652. https://doi.org/10.3390/ddc2030032
Kamdem BP, Boyom FF. Inhibitors of Farnesyl Diphosphate Synthase and Squalene Synthase: Potential Source for Anti-Trypanosomatidae Drug Discovery. Drugs and Drug Candidates. 2023; 2(3):624-652. https://doi.org/10.3390/ddc2030032
Chicago/Turabian StyleKamdem, Boniface Pone, and Fabrice Fekam Boyom. 2023. "Inhibitors of Farnesyl Diphosphate Synthase and Squalene Synthase: Potential Source for Anti-Trypanosomatidae Drug Discovery" Drugs and Drug Candidates 2, no. 3: 624-652. https://doi.org/10.3390/ddc2030032
APA StyleKamdem, B. P., & Boyom, F. F. (2023). Inhibitors of Farnesyl Diphosphate Synthase and Squalene Synthase: Potential Source for Anti-Trypanosomatidae Drug Discovery. Drugs and Drug Candidates, 2(3), 624-652. https://doi.org/10.3390/ddc2030032