
Opportunities and Difficulties in the Repurposing of HDAC Inhibitors as Antiparasitic Agents

 , and
, and
Abstract
:1. Introduction
2. Human Parasitic Illness
3. Classification and Biological Significance of Human Histone Deacetylases (HDACs)
Biological Significance of Human HDAC Isoforms
4. Functions and Roles of Human Parasite Histone Deacetylases (HDACs)
4.1. HDACs of Plasmodium Falciparum
4.2. HDACs of Toxoplasma
4.3. HDACs of Schistosoma
4.4. HDACs of Trypanosoma
4.5. HDACs of Leishmania
5. Opportunities of HDAC Inhibitors as Antiparasitic Agent
5.1. HDAC Inhibitors as Antimalarial Agent
5.1.1. Cyclictetrapeptide HDAC Inhibitors
5.1.2. Short-Chain Fatty Acid HDAC Inhibitors
5.1.3. Hydroxamate-Based HDAC Inhibitors
5.1.4. Thiol-Based HDAC Inhibitors
5.1.5. Ortho-Aminoanilides HDAC Inhibitors
5.2. Antimalarial Class-III HDAC (Sirtuin) Inhibitors
5.3. Antitrypanosomal HDAC Inhibitors
5.4. Antitrypanosomal Class-III HDAC (Sirtuin) Inhibitors
5.5. Antileishmanial HDAC Inhibitors
5.6. Antileishmanial Class-III HDAC (Sirtuin) Inhibitors
5.7. Antitoxoplasma HDAC Inhibitors
6. Challenges Regarding HDAC Inhibitors as Antiparasitic Agents
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- World Health Organization. World Malaria Report 2014: Summary; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Pigott, D.M.; Bhatt, S.; Golding, N.; Duda, K.A.; Battle, K.E.; Brady, O.J.; Messina, J.P.; Balard, Y.; Bastien, P.; Pratlong, F. Global distribution maps of the leishmaniases. eLife 2014, 3, e02851. [Google Scholar] [PubMed]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Nemati Zargaran, F.; Rostamian, M.; Kooti, S.; Madanchi, H.; Ghadiri, K. Co-infection of COVID-19 and parasitic diseases: A systematic review. Parasite Epidemiol. Control 2023, 21, e00299. [Google Scholar] [CrossRef] [PubMed]
- Gebrecherkos, T.; Gessesse, Z.; Kebede, Y.; Gebreegzabher, A.; Tasew, G.; Abdulkader, M.; Ebuy, H.; Desta, A.; Hailu, A.; Harris, V.; et al. Effect of co-infection with parasites on severity of COVID-19. EClinicalMedicine 2021, 39, 101054. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar]
- Labella, D. Design, Synthesis and Biological Evaluation of Novel Epigenetic Modulators; Sapienza University of Rome: Rome, Italy, 2014. [Google Scholar]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef]
- Berg, M.; García-Hernández, R.; Cuypers, B.; Vanaerschot, M.; Manzano, J.I.; Poveda, J.A.; Ferragut, J.A.; Castanys, S.; Dujardin, J.-C.; Gamarro, F. Experimental resistance to drug combinations in Leishmania donovani: Metabolic and phenotypic adaptations. Antimicrob. Agents Chemother. 2015, 59, 2242–2255. [Google Scholar] [CrossRef]
- Takala-Harrison, S.; Jacob, C.G.; Arze, C.; Cummings, M.P.; Silva, J.C.; Dondorp, A.M.; Fukuda, M.M.; Hien, T.T.; Mayxay, M.; Noedl, H. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J. Infect. Dis. 2015, 211, 670–679. [Google Scholar] [CrossRef]
- Andrews, K.T.; Haque, A.; Jones, M.K. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 2012, 90, 66–77. [Google Scholar] [CrossRef]
- Ay, F.; Bunnik, E.M.; Varoquaux, N.; Vert, J.P.; Noble, W.S.; Le Roch, K.G. Multiple dimensions of epigenetic gene regulation in the malaria parasite Plasmodium falciparum: Gene regulation via histone modifications, nucleosome positioning and nuclear architecture in P. falciparum. Bioessays 2015, 37, 182–194. [Google Scholar]
- Cheeseman, K.; Weitzman, J.B. Host–parasite interactions: An intimate epigenetic relationship. Cell. Microbiol. 2015, 17, 1121–1132. [Google Scholar] [PubMed]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [PubMed]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, M.; Sanchez-del-Campo, L.; Fernandez-Perez, M.; Saez-Ayala, M.; Cabezas-Herrera, J.; Rodriguez-Lopez, J. Targeting the epigenetic machinery of cancer cells. Oncogene 2015, 34, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Brien, G.L.; Valerio, D.G.; Armstrong, S.A. Exploiting the epigenome to control cancer-promoting gene-expression programs. Cancer Cell 2016, 29, 464–476. [Google Scholar] [CrossRef]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer therapy with HDAC inhibitors: Mechanism-based combination strategies and future perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef]
- Azzi, A.; Cosseau, C.; Grunau, C. Schistosoma mansoni: Developmental arrest of miracidia treated with histone deacetylase inhibitors. Exp. Parasitol. 2009, 121, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Coleman, B.I.; Skillman, K.M.; Jiang, R.H.; Childs, L.M.; Altenhofen, L.M.; Ganter, M.; Leung, Y.; Goldowitz, I.; Kafsack, B.F.; Marti, M. A Plasmodium falciparum histone deacetylase regulates antigenic variation and gametocyte conversion. Cell Host Microbe 2014, 16, 177–186. [Google Scholar] [CrossRef] [PubMed]
- T Andrews, K.; N Tran, T.; P Fairlie, D. Towards histone deacetylase inhibitors as new antimalarial drugs. Curr. Pharm. Des. 2012, 18, 3467–3479. [Google Scholar] [CrossRef]
- Marek, M.; Oliveira, G.; Pierce, R.J.; Jung, M.; Sippl, W.; Romier, C. Drugging the schistosome zinc-dependent HDACs: Current progress and future perspectives. Future Med. Chem. 2015, 7, 783–800. [Google Scholar] [PubMed]
- Melesina, J.; Robaa, D.; Pierce, R.J.; Romier, C.; Sippl, W. Homology modeling of parasite histone deacetylases to guide the structure-based design of selective inhibitors. J. Mol. Graph. Model. 2015, 62, 342–361. [Google Scholar] [CrossRef] [PubMed]
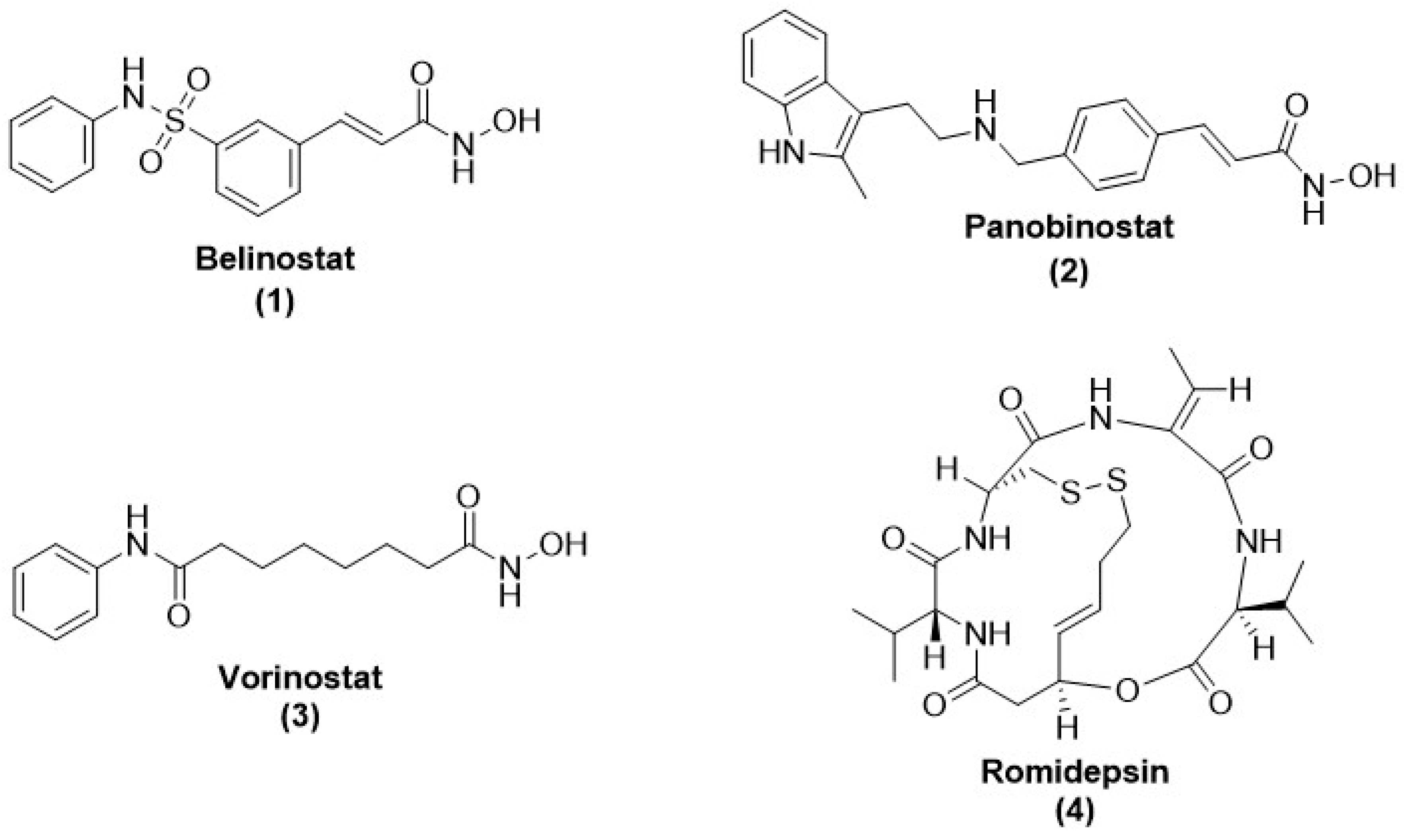
- Grant, S.; Easley, C.; Kirkpatrick, P. Vorinostat. Nat. Rev. Drug Discov. 2007, 6, 21–22. [Google Scholar] [CrossRef]
- Prince, H.M.; Dickinson, M. Romidepsin for Cutaneous T-cell Lymphoma. Clin. Cancer Res. 2012, 18, 3509–3515. [Google Scholar] [CrossRef]
- Thompson, C.A. Belinostat Approved for Use in Treating Rare Lymphoma; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704. [Google Scholar]
- Kogan, F.; Kogan, F. Malaria Burden. Remote Sensing for Malaria: Monitoring and Predicting Malaria from Operational Satellites; Springer Nature: Berlin, Germany, 2020; pp. 15–41. [Google Scholar]
- Monroe, A.; Williams, N.A.; Ogoma, S.; Karema, C.; Okumu, F. Reflections on the 2021 World Malaria Report and the Future of Malaria Control. Malar. J. 2022, 21, 154. [Google Scholar] [CrossRef]
- Nadeem, A.Y.; Shehzad, A.; Islam, S.U.; Al-Suhaimi, E.A.; Lee, Y.S. Mosquirix™ RTS, S/AS01 vaccine development, immunogenicity, and efficacy. Vaccines 2022, 10, 713. [Google Scholar]
- Brabin, B.J. An analysis of malaria in pregnancy in Africa. Bull. World Health Organ. 1983, 61, 1005. [Google Scholar]
- Musset, L.; Bouchaud, O.; Matheron, S.; Massias, L.; Le Bras, J. Clinical atovaquone-proguanil resistance of Plasmodium falciparum associated with cytochrome b codon 268 mutations. Microbes Infect. 2006, 8, 2599–2604. [Google Scholar] [CrossRef]
- Dondorp, A.M.; Yeung, S.; White, L.; Nguon, C.; Day, N.P.; Socheat, D.; Von Seidlein, L. Artemisinin resistance: Current status and scenarios for containment. Nat. Rev. Microbiol. 2010, 8, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.B.; Sande, M.A. Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. N. Engl. J. Med. 1992, 327, 1643–1648. [Google Scholar] [CrossRef]
- Carlier, Y.; Truyens, C.; Deloron, P.; Peyron, F. Congenital parasitic infections: A review. Acta Trop. 2012, 121, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.J.; Ross, A.G.; Li, Y.-S.; McManus, D.P. Diagnosis and management of schistosomiasis. BMJ 2011, 342, d2651. [Google Scholar] [PubMed]
- Hotez, P.J.; Kamath, A. Neglected tropical diseases in sub-Saharan Africa: Review of their prevalence, distribution, and disease burden. PLoS Neglected Trop. Dis. 2009, 3, e412. [Google Scholar] [CrossRef] [PubMed]
- WHO. Weekly Epidemiological Record Releve Epidemiologique Hebdomadaire; World Health Organization: Geneva, Switzerland, 2015; Volume 316, pp. 25–32. [Google Scholar]
- WHO. Weekly Epidemiological Record Releve Epidemiologique Hebdomadaire; World Health Organization: Geneva, Switzerland, 2016; Volume 91, pp. 53–60. [Google Scholar]
- Dömling, A.; Khoury, K. Praziquantel and schistosomiasis. ChemMedChem 2010, 5, 1420–1434. [Google Scholar] [CrossRef]
- WHO. Expert Committee on the Control of Schistosomiasisx. Bull. World Health Organ. 1993, 71, 657–662. [Google Scholar]
- King, C.H.; Dangerfield-Cha, M. The unacknowledged impact of chronic schistosomiasis. Chronic Illn. 2008, 4, 65–79. [Google Scholar] [CrossRef]
- King, C.H.; Dickman, K.; Tisch, D.J. Reassessment of the cost of chronic helmintic infection: A meta-analysis of disability-related outcomes in endemic schistosomiasis. Lancet 2005, 365, 1561–1569. [Google Scholar] [PubMed]
- Chitsulo, L.; Loverde, P.; Engels, D. Schistosomiasis. Nat. Rev. Microbiol. 2004, 2, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L. Praziquantel. Parasitol. Res. 2003, 90 (Supp. 1), S3–S9. [Google Scholar] [CrossRef]
- Doenhoff, M.J.; Cioli, D.; Utzinger, J. Praziquantel: Mechanisms of action, resistance and new derivatives for schistosomiasis. Curr. Opin. Infect. Dis. 2008, 21, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, C.R. Chemotherapy of schistosomiasis: Present and future. Curr. Opin. Chem. Biol. 2007, 11, 433–439. [Google Scholar] [CrossRef]
- Doenhoff, M.J.; Kusel, J.R.; Coles, G.C.; Cioli, D. Resistance of Schistosoma mansoni to praziquantel: Is there a problem? Trans. R. Soc. Trop. Med. Hyg. 2002, 96, 465–469. [Google Scholar] [CrossRef]
- Doenhoff, M.J.; Pica-Mattoccia, L. Praziquantel for the treatment of schistosomiasis: Its use for control in areas with endemic disease and prospects for drug resistance. Expert Rev. Anti-Infect. Ther. 2006, 4, 199–210. [Google Scholar] [CrossRef]
- Danso-Appiah, A.; De Vlas, S.J. Interpreting low praziquantel cure rates of Schistosoma mansoni infections in Senegal. Trends Parasitol. 2002, 18, 125–129. [Google Scholar] [CrossRef]
- Lawn, S.D.; Lucas, S.B.; Chiodini, P.L. Schistosoma mansoni infection: Failure of standard treatment with praziquantel in a returned traveller: A reply. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 720. [Google Scholar] [CrossRef]
- Bonesso-Sabadini, P.I.P.; Dias, L.C.d.S. Altered response of strain of Schistosoma mansoni to oxamniquine and praziquantel. Memórias Do Inst. Oswaldo Cruz 2002, 97, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Couto, F.F.; Coelho, P.M.Z.; Araújo, N.; Kusel, J.R.; Katz, N.; Jannotti-Passos, L.K.; Mattos, A.C.A. Schistosoma mansoni: A method for inducing resistance to praziquantel using infected Biomphalaria glabrata snails. Mem. Do Inst. Oswaldo Cruz 2011, 106, 153–157. [Google Scholar]
- Fallon, P.G.; Doenhoff, M.J. Drug-resistant schistosomiasis: Resistance to praziquantel and oxamniquine induced in Schistosoma mansoni in mice is drug specific. Am. J. Trop. Med. Hyg. 1994, 51, 83–88. [Google Scholar]
- Vaca, H.R.; Celentano, A.M.; Toscanini, M.A.; Heimburg, T.; Ghazy, E.; Zeyen, P.; Hauser, A.-T.; Oliveira, G.; Elissondo, M.C.; Jung, M. The potential for histone deacetylase (HDAC) inhibitors as cestocidal drugs. PLoS Negl. Trop. Dis. 2021, 15, e0009226. [Google Scholar] [CrossRef] [PubMed]
- Malik, L.H.; Singh, G.D.; Amsterdam, E.A. The epidemiology, clinical manifestations, and management of chagas heart disease. Clin. Cardiol. 2015, 38, 565–569. [Google Scholar] [CrossRef] [PubMed]
- WHO. Trypanosomiasis, Human African (Sleeping Sickness). 2016. Available online: http://www.who.int/mediacentre/factsheets/fs259/en/ (accessed on 10 June 2023).
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, randomised, phase III, non-inferiority trial. Lancet 2009, 374, 56–64. [Google Scholar] [CrossRef]
- Barrett, M.P.; Vincent, I.M.; Burchmore, R.J.; Kazibwe, A.J.; Matovu, E. Drug resistance in human African trypanosomiasis. Future Microbiol. 2011, 6, 1037–1047. [Google Scholar]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human african trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef]
- Matthews, K.R. The developmental cell biology of Trypanosoma brucei. J. Cell Sci. 2005, 118, 283–290. [Google Scholar]
- Maurice, J. New WHO plan targets the demise of sleeping sickness. Lancet 2013, 381, 13–14. [Google Scholar]
- WHO. Leishmaniasis. 2016. Available online: http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 10 June 2023).
- Pace, D. Leishmaniasis. J. Infect. 2014, 69 (Suppl. S1), S10–S18. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chakravarty, J. Leishmaniasis: An update of current pharmacotherapy. Expert Opin. Pharmacother. 2013, 14, 53–63. [Google Scholar]
- Mishra, J.; Saxena, A.; Singh, S. Chemotherapy of leishmaniasis: Past, present and future. Curr. Med. Chem. 2007, 14, 1153–1169. [Google Scholar] [CrossRef]
- Okwor, I.; Uzonna, J.E. The immunology of Leishmania/HIV co-infection. Immunol. Res. 2013, 56, 163–171. [Google Scholar] [PubMed]
- Cairns, B.R. Emerging roles for chromatin remodeling in cancer biology. Trends Cell Biol. 2001, 11, S15–S21. [Google Scholar] [CrossRef] [PubMed]
- H Beumer, J.; Tawbi, H. Role of histone deacetylases and their inhibitors in cancer biology and treatment. Curr. Clin. Pharmacol. 2010, 5, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Rotili, D.; Valente, S.; Kazantsev, A.G. Histone deacetylase inhibitors and neurodegenerative disorders: Holding the promise. Curr. Pharm. Des. 2009, 15, 3940–3957. [Google Scholar] [CrossRef]
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Tak, P.P.; Reedquist, K.A. Function of histone deacetylase inhibitors in inflammation. Crit. Rev. Immunol. 2011, 31, 233–263. [Google Scholar]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenetics 2016, 8, 61. [Google Scholar]
- Gregoretti, I.; Lee, Y.-M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [PubMed]
- Alcaín, F.J.; Villalba, J.M. Sirtuin inhibitors. Expert Opin. Ther. Pat. 2009, 19, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Alcaín, F.J.; Villalba, J.M. Sirtuin activators. Expert Opin. Ther. Pat. 2009, 19, 403–414. [Google Scholar] [CrossRef]
- Pasco, M.Y.; Rotili, D.; Altucci, L.; Farina, F.; Rouleau, G.A.; Mai, A.; Néri, C. Characterization of sirtuin inhibitors in nematodes expressing a muscular dystrophy protein reveals muscle cell and behavioral protection by specific sirtinol analogues. J. Med. Chem. 2010, 53, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.M.; Cockell, M.M. The molecular biology of the SIR proteins. Gene 2001, 279, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef]
- Rotili, D.; Simonetti, G.; Savarino, A.; Palamara, A.T.; Migliaccio, A.R.; Mai, A. Non-Cancer Uses of Histone Deacetylase Inhibitors: Effects on Infectious Diseases and β-Hemoglobinopathies. Curr. Top. Med. Chem. 2009, 9, 272–291. [Google Scholar]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S. PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37, D539–D543. [Google Scholar]
- Joshi, M.B.; Lin, D.T.; Chiang, P.H.; Goldman, N.D.; Fujioka, H.; Aikawa, M.; Syin, C. Molecular cloning and nuclear localization of a histone deacetylase homologue in Plasmodium falciparum. Mol. Biochem. Parasitol. 1999, 99, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Florens, L.; Washburn, M.P.; Raine, J.D.; Anthony, R.M.; Grainger, M.; Haynes, J.D.; Moch, J.K.; Muster, N.; Sacci, J.B.; Tabb, D.L. A proteomic view of the Plasmodium falciparum life cycle. Nature 2002, 419, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Pradhan, A.; Shah, F.; Tekwani, B.L.; Avery, M.A. Structural insights into the Plasmodium falciparum histone deacetylase 1 (PfHDAC-1): A novel target for the development of antimalarial therapy. Bioorganic Med. Chem. 2008, 16, 5254–5265. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.; Tran, T.; Lucke, A.; Kahnberg, P.; Le, G.; Boyle, G.; Gardiner, D.; Skinner-Adams, T.; Fairlie, D. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob. Agents Chemother. 2008, 52, 1454–1461. [Google Scholar] [CrossRef]
- Freitas-Junior, L.H.; Hernandez-Rivas, R.; Ralph, S.A.; Montiel-Condado, D.; Ruvalcaba-Salazar, O.K.; Rojas-Meza, A.P.; Mâncio-Silva, L.; Leal-Silvestre, R.J.; Gontijo, A.M.; Shorte, S. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 2005, 121, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Mazitschek, R.; Coleman, B.; Nguyen, C.; Urgaonkar, S.; Cortese, J.; Barker, R.H., Jr.; Greenberg, E.; Tang, W.; Bradner, J.E. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. J. Med. Chem. 2009, 52, 2185–2187. [Google Scholar] [CrossRef]
- Zhu, A.Y.; Zhou, Y.; Khan, S.; Deitsch, K.W.; Hao, Q.; Lin, H. Plasmodium falciparum Sir2A preferentially hydrolyzes medium and long chain fatty acyl lysine. ACS Chem. Biol. 2012, 7, 155–159. [Google Scholar] [CrossRef]
- Tonkin, C.J.; Carret, C.K.; Duraisingh, M.T.; Voss, T.S.; Ralph, S.A.; Hommel, M.; Duffy, M.F.; Silva, L.M.d.; Scherf, A.; Ivens, A. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 2009, 7, e1000084. [Google Scholar] [CrossRef]
- Duraisingh, M.T.; Voss, T.S.; Marty, A.J.; Duffy, M.F.; Good, R.T.; Thompson, J.K.; Freitas-Junior, L.H.; Scherf, A.; Crabb, B.S.; Cowman, A.F. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 2005, 121, 13–24. [Google Scholar] [CrossRef]
- Saksouk, N.; Bhatti, M.M.; Kieffer, S.; Smith, A.T.; Musset, K.; Garin, J.; Sullivan, W.J., Jr.; Cesbron-Delauw, M.-F.; Hakimi, M.-A. Histone-modifying complexes regulate gene expression pertinent to the differentiation of the protozoan parasite Toxoplasma gondii. Mol. Cell. Biol. 2005, 25, 10301–10314. [Google Scholar] [CrossRef]
- Oger, F.; Dubois, F.; Caby, S.; Noël, C.; Cornette, J.; Bertin, B.; Capron, M.; Pierce, R.J. The class I histone deacetylases of the platyhelminth parasite Schistosoma mansoni. Biochem. Biophys. Res. Commun. 2008, 377, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Caby, S.; Oger, F.; Cosseau, C.; Capron, M.; Grunau, C.; Dissous, C.; Pierce, R.J. Histone deacetylase inhibitors induce apoptosis, histone hyperacetylation and up-regulation of gene transcription in Schistosoma mansoni. Mol. Biochem. Parasitol. 2009, 168, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Theurkauf, W.E.; Baum, H.; Bo, J.; Wensink, P.C. Tissue-specific and constitutive alpha-tubulin genes of Drosophila melanogaster code for structurally distinct proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 8477–8481. [Google Scholar] [PubMed]
- Berriman, M.; Haas, B.J.; LoVerde, P.T.; Wilson, R.A.; Dillon, G.P.; Cerqueira, G.C.; Mashiyama, S.T.; Al-Lazikani, B.; Andrade, L.F.; Ashton, P.D. The genome of the blood fluke Schistosoma mansoni. Nature 2009, 460, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Ingram, A.K.; Horn, D. Histone deacetylases in Trypanosoma brucei: Two are essential and another is required for normal cell cycle progression. Mol. Microbiol. 2002, 45, 89–97. [Google Scholar] [CrossRef]
- Mandava, V.; Fernandez, J.P.; Deng, H.; Janzen, C.J.; Hake, S.B.; Cross, G.A. Histone modifications in Trypanosoma brucei. Mol. Biochem. Parasitol. 2007, 156, 41–50. [Google Scholar]
- Hertz-Fowler, C.; Figueiredo, L.M.; Quail, M.A.; Becker, M.; Jackson, A.; Bason, N.; Brooks, K.; Churcher, C.; Fahkro, S.; Goodhead, I. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS ONE 2008, 3, e3527. [Google Scholar]
- Wang, Q.P.; Kawahara, T.; Horn, D. Histone deacetylases play distinct roles in telomeric VSG expression site silencing in African trypanosomes. Mol. Microbiol. 2010, 77, 1237–1245. [Google Scholar] [CrossRef]
- Alsford, S.; Kawahara, T.; Isamah, C.; Horn, D. A sirtuin in the African trypanosome is involved in both DNA repair and telomeric gene silencing but is not required for antigenic variation. Mol. Microbiol. 2007, 63, 724–736. [Google Scholar] [CrossRef]
- García-Salcedo, J.A.; Gijón, P.; Nolan, D.P.; Tebabi, P.; Pays, E. A chromosomal SIR2 homologue with both histone NAD-dependent ADP-ribosyltransferase and deacetylase activities is involved in DNA repair in Trypanosoma brucei. EMBO J. 2003, 22, 5851–5862. [Google Scholar]
- Ritagliati, C.; Alonso, V.L.; Manarin, R.; Cribb, P.; Serra, E.C. Overexpression of Cytoplasmic Tc SIR2RP1 and Mitochondrial Tc SIR2RP3 Impacts on Trypanosoma cruzi Growth and Cell Invasion. PLoS Neglected Trop. Dis. 2015, 9, e0003725. [Google Scholar] [CrossRef] [PubMed]
- Moretti, N.S.; da Silva Augusto, L.; Clemente, T.M.; Antunes, R.P.P.; Yoshida, N.; Torrecilhas, A.C.; Cano, M.I.N.; Schenkman, S. Characterization of Trypanosoma cruzi sirtuins as possible drug targets for Chagas disease. Antimicrob. Agents Chemother. 2015, 59, 4669–4679. [Google Scholar]
- Saxena, A.; Lahav, T.; Holland, N.; Aggarwal, G.; Anupama, A.; Huang, Y.; Volpin, H.; Myler, P.; Zilberstein, D. Analysis of the Leishmania donovani transcriptome reveals an ordered progression of transient and permanent changes in gene expression during differentiation. Mol. Biochem. Parasitol. 2007, 152, 53–65. [Google Scholar] [CrossRef]
- Yahiaoui, B.; Taibi, A.; Ouaissi, A. A Leishmania major protein with extensive homology to silent information regulator 2 of Saccharomyces cerevisiae. Gene 1996, 169, 115–118. [Google Scholar] [PubMed]
- Vergnes, B.; Sereno, D.; Madjidian-Sereno, N.; Lemesre, J.-L.; Ouaissi, A. Cytoplasmic SIR2 homologue overexpression promotes survival of Leishmania parasites by preventing programmed cell death. Gene 2002, 296, 139–150. [Google Scholar] [CrossRef]
- Tavares, J.; Ouaissi, A.; Santarém, N.; Sereno, D.; Vergnes, B.; Sampaio, P.; Cordeiro-Da-Silva, A. The Leishmania infantum cytosolic SIR2-related protein 1 (LiSIR2RP1) is an NAD+-dependent deacetylase and ADP-ribosyltransferase. Biochem. J. 2008, 415, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, B.; Sereno, D.; Tavares, J.; Cordeiro-da-Silva, A.; Vanhille, L.; Madjidian-Sereno, N.; Depoix, D.; Monte-Alegre, A.; Ouaissi, A. Targeted disruption of cytosolic SIR2 deacetylase discloses its essential role in Leishmania survival and proliferation. Gene 2005, 363, 85–96. [Google Scholar]
- Adriano, M.-A.; Vergnes, B.; Poncet, J.; Mathieu-Daude, F.; da Silva, A.C.; Ouaissi, A.; Sereno, D. Proof of interaction between Leishmania SIR2RP1 deacetylase and chaperone HSP83. Parasitol. Res. 2007, 100, 811–818. [Google Scholar] [CrossRef]
- Silvestre, R.; Cordeiro-da-Silva, A.; Tavares, J.; Sereno, D.; Ouaissi, A. Leishmania cytosolic silent information regulatory protein 2 deacetylase induces murine B-cell differentiation and in vivo production of specific antibodies. Immunology 2006, 119, 529–540. [Google Scholar] [CrossRef]
- Silvestre, R.; Silva, A.M.; Cordeiro-da-Silva, A.; Ouaissi, A. The contribution of Toll-like receptor 2 to the innate recognition of a Leishmania infantum silent information regulator 2 protein. Immunology 2009, 128, 484–499. [Google Scholar] [CrossRef]
- Sodji, Q.; Patil, V.; Jain, S.; Kornacki, J.R.; Mrksich, M.; Tekwani, B.L.; Oyelere, A.K. The antileishmanial activity of isoforms 6-and 8-selective histone deacetylase inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 4826–4830. [Google Scholar] [CrossRef] [PubMed]
- Kadam, R.U.; Tavares, J.; Cordeiro, A.; Ouaissi, A.; Roy, N. Structure function analysis of Leishmania sirtuin: An ensemble of in silico and biochemical studies. Chem. Biol. Drug Des. 2008, 71, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Tavares, J.; Ouaissi, A.; Kong Thoo Lin, P.; Loureiro, I.; Kaur, S.; Roy, N.; Cordeiro-da-Silva, A. Bisnaphthalimidopropyl derivatives as inhibitors of Leishmania SIR2 related protein 1. ChemMedChem Chem. Enabling Drug Discov. 2010, 5, 140–147. [Google Scholar] [CrossRef]
- Darkin-Rattray, S.J.; Gurnett, A.M.; Myers, R.W.; Dulski, P.M.; Crumley, T.M.; Allocco, J.J.; Cannova, C.; Meinke, P.T.; Colletti, S.L.; Bednarek, M.A. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. USA 1996, 93, 13143–13147. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Cabrera, A.; Kono, M.; Mok, S.; Chaal, B.K.; Haase, S.; Engelberg, K.; Cheemadan, S.; Spielmann, T.; Preiser, P.R. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat. Biotechnol. 2010, 28, 91–98. [Google Scholar]
- Chaal, B.K.; Gupta, A.P.; Wastuwidyaningtyas, B.D.; Luah, Y.-H.; Bozdech, Z. Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLoS Pathog. 2010, 6, e1000737. [Google Scholar]
- Glaser, K.B.; Staver, M.J.; Waring, J.F.; Stender, J.; Ulrich, R.G.; Davidsen, S.K. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: Defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2003, 2, 151–163. [Google Scholar]
- Peart, M.J.; Smyth, G.K.; Van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702. [Google Scholar] [CrossRef]
- Meinke, P.T.; Colletti, S.L.; Doss, G.; Myers, R.W.; Gurnett, A.M.; Dulski, P.M.; Darkin-Rattray, S.J.; Allocco, J.J.; Galuska, S.; Schmatz, D.M. Synthesis of apicidin-derived quinolone derivatives: Parasite-selective histone deacetylase inhibitors and antiproliferative agents. J. Med. Chem. 2000, 43, 4919–4922. [Google Scholar]
- Colletti, S.L.; Myers, R.W.; Darkin-Rattray, S.J.; Gurnett, A.M.; Dulski, P.M.; Galuska, S.; Allocco, J.J.; Ayer, M.B.; Li, C.; Lim, J. Broad spectrum antiprotozoal agents that inhibit histone deacetylase: Structure–activity relationships of apicidin. Part 2. Bioorganic Med. Chem. Lett. 2001, 11, 113–117. [Google Scholar]
- Colletti, S.L.; Myers, R.W.; Darkin-Rattray, S.J.; Gurnett, A.M.; Dulski, P.M.; Galuska, S.; Allocco, J.J.; Ayer, M.B.; Li, C.; Lim, J. Broad spectrum antiprotozoal agents that inhibit histone deacetylase: Structure–activity relationships of apicidin. Part 1. Bioorganic Med. Chem. Lett. 2001, 11, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Kranz, M.; Ladlow, M.; Taylor, S.; Berst, F.; Holmes, A.B.; Keavey, K.N.; Jaxa-Chamiec, A.; Seale, P.W.; Stead, P. The synthesis of cyclic tetrapeptoid analogues of the antiprotozoal natural product apicidin. Bioorganic Med. Chem. Lett. 2001, 11, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Bougdour, A.; Maubon, D.; Baldacci, P.; Ortet, P.; Bastien, O.; Bouillon, A.; Barale, J.-C.; Pelloux, H.; Ménard, R.; Hakimi, M.-A. Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites. J. Exp. Med. 2009, 206, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 1–13. [Google Scholar] [CrossRef]
- Strobl, J.S.; Cassell, M.; Mitchell, S.M.; Reilly, C.M.; Lindsay, D.S. Scriptaid and suberoylanilide hydroxamic acid are histone deacetylase inhibitors with potent anti–Toxoplasma gondii activity in vitro. J. Parasitol. 2007, 93, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Jones-Brando, L.; Torrey, E.F.; Yolken, R. Drugs used in the treatment of schizophrenia and bipolar disorder inhibit the replication of Toxoplasma gondii. Schizophr. Res. 2003, 62, 237–244. [Google Scholar] [PubMed]
- Engel, J.A.; Jones, A.J.; Avery, V.M.; Sumanadasa, S.D.; Ng, S.S.; Fairlie, D.P.; Adams, T.S.; Andrews, K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 117–126. [Google Scholar] [CrossRef]
- Dow, G.S.; Chen, Y.; Andrews, K.T.; Caridha, D.; Gerena, L.; Gettayacamin, M.; Johnson, J.; Li, Q.; Melendez, V.; Obaldia III, N. Antimalarial activity of phenylthiazolyl-bearing hydroxamate-based histone deacetylase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 3467–3477. [Google Scholar] [CrossRef]
- Andrews, K.T.; Walduck, A.; Kelso, M.J.; Fairlie, D.P.; Saul, A.; Parsons, P.G. Anti-malarial effect of histone deacetylation inhibitors and mammalian tumour cytodifferentiating agents. Int. J. Parasitol. 2000, 30, 761–768. [Google Scholar] [CrossRef]
- Mai, A.; Cerbara, I.; Valente, S.; Massa, S.; Walker, L.A.; Tekwani, B.L. Antimalarial and antileishmanial activities of aroyl-pyrrolyl-hydroxyamides, a new class of histone deacetylase inhibitors. Antimicrob. Agents Chemother. 2004, 48, 1435. [Google Scholar] [CrossRef]
- Andrews, K.T.; Gupta, A.P.; Tran, T.N.; Fairlie, D.P.; Gobert, G.N.; Bozdech, Z. Comparative gene expression profiling of P. falciparum malaria parasites exposed to three different histone deacetylase inhibitors. PLoS ONE 2012, 7, e31847. [Google Scholar] [CrossRef] [PubMed]
- Marfurt, J.; Chalfein, F.; Prayoga, P.; Wabiser, F.; Kenangalem, E.; Piera, K.A.; Fairlie, D.P.; Tjitra, E.; Anstey, N.M.; Andrews, K.T. Ex vivo activity of histone deacetylase inhibitors against multidrug-resistant clinical isolates of Plasmodium falciparum and P. vivax. Antimicrob. Agents Chemother. 2011, 55, 961–966. [Google Scholar] [PubMed]
- Wheatley, N.C.; Andrews, K.T.; Tran, T.L.; Lucke, A.J.; Reid, R.C.; Fairlie, D.P. Antimalarial histone deacetylase inhibitors containing cinnamate or NSAID components. Bioorganic Med. Chem. Lett. 2010, 20, 7080–7084. [Google Scholar]
- Chen, Y.; Lopez-Sanchez, M.; Savoy, D.N.; Billadeau, D.D.; Dow, G.S.; Kozikowski, A.P. A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J. Med. Chem. 2008, 51, 3437–3448. [Google Scholar] [CrossRef] [PubMed]
- Agbor-Enoh, S.; Seudieu, C.; Davidson, E.; Dritschilo, A.; Jung, M. Novel inhibitor of Plasmodium histone deacetylase that cures P. berghei-infected mice. Antimicrob. Agents Chemother. 2009, 53, 1727–1734. [Google Scholar] [CrossRef]
- Patil, V.; Guerrant, W.; Chen, P.C.; Gryder, B.; Benicewicz, D.B.; Khan, S.I.; Tekwani, B.L.; Oyelere, A.K. Antimalarial and antileishmanial activities of histone deacetylase inhibitors with triazole-linked cap group. Bioorganic Med. Chem. 2010, 18, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Mwakwari, S.C.; Guerrant, W.; Patil, V.; Khan, S.I.; Tekwani, B.L.; Gurard-Levin, Z.A.; Mrksich, M.; Oyelere, A.K. Non-peptide macrocyclic histone deacetylase inhibitors derived from tricyclic ketolide skeleton. J. Med. Chem. 2010, 53, 6100–6111. [Google Scholar] [CrossRef]
- Hansen, F.K.; Skinner-Adams, T.S.; Duffy, S.; Marek, L.; Sumanadasa, S.D.; Kuna, K.; Held, J.; Avery, V.M.; Andrews, K.T.; Kurz, T. Synthesis, Antimalarial Properties, and SAR Studies of Alkoxyurea-Based HDAC Inhibitors. ChemMedChem 2014, 9, 665–670. [Google Scholar] [CrossRef]
- Trenholme, K.; Marek, L.; Duffy, S.; Pradel, G.; Fisher, G.; Hansen, F.K.; Skinner-Adams, T.S.; Butterworth, A.; Ngwa, C.J.; Moecking, J. Lysine acetylation in sexual stage malaria parasites is a target for antimalarial small molecules. Antimicrob. Agents Chemother. 2014, 58, 3666–3678. [Google Scholar]
- Giannini, G.; Battistuzzi, G.; Vignola, D. Hydroxamic acid based histone deacetylase inhibitors with confirmed activity against the malaria parasite. Bioorganic Med. Chem. Lett. 2015, 25, 459–461. [Google Scholar] [CrossRef]
- Itoh, Y.; Suzuki, T.; Kouketsu, A.; Suzuki, N.; Maeda, S.; Yoshida, M.; Nakagawa, H.; Miyata, N. Design, synthesis, structure−selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors. J. Med. Chem. 2007, 50, 5425–5438. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Altucci, L. Epi-drugs to fight cancer: From chemistry to cancer treatment, the road ahead. Int. J. Biochem. Cell Biol. 2009, 41, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Ontoria, J.M.; Paonessa, G.; Ponzi, S.; Ferrigno, F.; Nizi, E.; Biancofiore, I.; Malancona, S.; Graziani, R.; Roberts, D.; Willis, P. Discovery of a selective series of inhibitors of Plasmodium falciparum HDACs. ACS Med. Chem. Lett. 2016, 7, 454–459. [Google Scholar] [CrossRef]
- Verotta, L.; Appendino, G.; Bombardelli, E.; Brun, R. In vitro antimalarial activity of hyperforin, a prenylated acylphloroglucinol. A structure–activity study. Bioorganic Med. Chem. Lett. 2007, 17, 1544–1548. [Google Scholar]
- Prusty, D.; Mehra, P.; Srivastava, S.; Shivange, A.V.; Gupta, A.; Roy, N.; Dhar, S.K. Nicotinamide inhibits Plasmodium falciparum Sir2 activity in vitro and parasite growth. FEMS Microbiol. Lett. 2008, 282, 266–272. [Google Scholar]
- Guerrant, W.; Mwakwari, S.C.; Chen, P.C.; Khan, S.I.; Tekwani, B.L.; Oyelere, A.K. A structure–activity relationship study of the antimalarial and antileishmanial activities of nonpeptide macrocyclic histone deacetylase inhibitors. ChemMedChem 2010, 5, 1232–1235. [Google Scholar] [CrossRef]
- Sumanadasa, S.D.; Goodman, C.D.; Lucke, A.J.; Skinner-Adams, T.; Sahama, I.; Haque, A.; Do, T.A.; McFadden, G.I.; Fairlie, D.P.; Andrews, K.T. Antimalarial activity of the anticancer histone deacetylase inhibitor SB939. Antimicrob. Agents Chemother. 2012, 56, 3849–3856. [Google Scholar] [CrossRef]
- Hansen, F.K.; Sumanadasa, S.D.; Stenzel, K.; Duffy, S.; Meister, S.; Marek, L.; Schmetter, R.; Kuna, K.; Hamacher, A.; Mordmüller, B. Discovery of HDAC inhibitors with potent activity against multiple malaria parasite life cycle stages. Eur. J. Med. Chem. 2014, 82, 204–213. [Google Scholar] [CrossRef]
- Chakrabarty, S.P.; Saikumari, Y.K.; Bopanna, M.P.; Balaram, H. Biochemical characterization of Plasmodium falciparum Sir2, a NAD+-dependent deacetylase. Mol. Biochem. Parasitol. 2008, 158, 139–151. [Google Scholar] [CrossRef]
- Merrick, C.J.; Duraisingh, M.T. Plasmodium falciparum Sir2: An unusual sirtuin with dual histone deacetylase and ADP-ribosyltransferase activity. Eukaryot. Cell 2007, 6, 2081–2091. [Google Scholar] [CrossRef]
- Gey, C.; Kyrylenko, S.; Hennig, L.; Nguyen, L.H.D.; Büttner, A.; Pham, H.D.; Giannis, A. Phloroglucinol derivatives guttiferone G, aristoforin, and hyperforin: Inhibitors of human sirtuins SIRT1 and SIRT2. Angew. Chem. Int. Ed. 2007, 46, 5219–5222. [Google Scholar]
- Chakrabarty, S.P.; Ramapanicker, R.; Mishra, R.; Chandrasekaran, S.; Balaram, H. Development and characterization of lysine based tripeptide analogues as inhibitors of Sir2 activity. Bioorganic Med. Chem. 2009, 17, 8060–8072. [Google Scholar]
- Sheader, K.; te Vruchte, D.; Rudenko, G. Bloodstream form-specific up-regulation of silent vsg expression sites and procyclin in Trypanosoma brucei after inhibition of DNA synthesis or DNA damage. J. Biol. Chem. 2004, 279, 13363–13374. [Google Scholar] [PubMed]
- Respuela, P.; Ferella, M.; Rada-Iglesias, A.; Åslund, L. Histone acetylation and methylation at sites initiating divergent polycistronic transcription in Trypanosoma cruzi. J. Biol. Chem. 2008, 283, 15884–15892. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.M.; Taylor, M.C.; Horn, D.; Loza, E.; Kalvinsh, I.; Björkling, F. Inhibitors of human histone deacetylase with potent activity against the African trypanosome Trypanosoma brucei. Bioorganic Med. Chem. Lett. 2012, 22, 1886–1890. [Google Scholar]
- Carrillo, A.K.; Guiguemde, W.A.; Guy, R.K. Evaluation of histone deacetylase inhibitors (HDACi) as therapeutic leads for human African trypanosomiasis (HAT). Bioorganic Med. Chem. 2015, 23, 5151–5155. [Google Scholar] [CrossRef]
- Soares, M.B.; Silva, C.V.; Bastos, T.M.; Guimarães, E.T.; Figueira, C.P.; Smirlis, D.; Azevedo, W.F., Jr. Anti-Trypanosoma cruzi activity of nicotinamide. Acta Trop. 2012, 122, 224–229. [Google Scholar] [CrossRef]
- Lara, E.; Mai, A.; Calvanese, V.; Altucci, L.; Lopez-Nieva, P.; Martinez-Chantar, M.; Varela-Rey, M.; Rotili, D.; Nebbioso, A.; Ropero, S. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 2009, 28, 781–791. [Google Scholar] [CrossRef]
- Rotili, D.; Tarantino, D.; Nebbioso, A.; Paolini, C.; Huidobro, C.; Lara, E.; Mellini, P.; Lenoci, A.; Pezzi, R.; Botta, G. Discovery of salermide-related sirtuin inhibitors: Binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J. Med. Chem. 2012, 55, 10937–10947. [Google Scholar]
- Sacconnay, L.; Smirlis, D.; Queiroz, E.F.; Wolfender, J.L.; Soares, M.B.P.; Carrupt, P.-A.; Nurisso, A. Structural insights of SIR2rp3 proteins as promising biotargets to fight against Chagas disease and leishmaniasis. Mol. BioSyst. 2013, 9, 2223–2230. [Google Scholar] [CrossRef]
- Vergnes, B.; Vanhille, L.; Ouaissi, A.; Sereno, D. Stage-specific antileishmanial activity of an inhibitor of SIR2 histone deacetylase. Acta Trop. 2005, 94, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Sereno, D.; Alegre, A.M.; Silvestre, R.; Vergnes, B.; Ouaissi, A. In vitro antileishmanial activity of nicotinamide. Antimicrob. Agents Chemother. 2005, 49, 808–812. [Google Scholar] [CrossRef]
- Tavares, J.; Ouaissi, A.; Silva, A.M.; Lin, P.K.T.; Roy, N.; Cordeiro-da-Silva, A. Anti-leishmanial activity of the bisnaphthalimidopropyl derivatives. Parasitol. Int. 2012, 61, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Maubon, D.; Bougdour, A.; Wong, Y.-S.; Brenier-Pinchart, M.-P.; Curt, A.; Hakimi, M.-A.; Pelloux, H. Activity of the histone deacetylase inhibitor FR235222 on Toxoplasma gondii: Inhibition of stage conversion of the parasite cyst form and study of new derivative compounds. Antimicrob. Agents Chemother. 2010, 54, 4843–4850. [Google Scholar] [CrossRef] [PubMed]
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.-T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F. Structure-based design and synthesis of novel inhibitors targeting HDAC8 from Schistosoma mansoni for the treatment of schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar]
- Kozikowski, A.P.; Chen, Y.; Gaysin, A.; Chen, B.; D’Annibale, M.A.; Suto, C.M.; Langley, B.C. Functional differences in epigenetic modulators superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J. Med. Chem. 2007, 50, 3054–3061. [Google Scholar] [CrossRef]
- Ghazy, E.; Abdelsalam, M.; Robaa, D.; Pierce, R.J.; Sippl, W. Histone Deacetylase (HDAC) Inhibitors for the Treatment of Schistosomiasis. Pharmaceuticals 2022, 15, 80. [Google Scholar]
- Leung, D.; Abbenante, G.; Fairlie, D.P. Protease inhibitors: Current status and future prospects. J. Med. Chem. 2000, 43, 305–341. [Google Scholar] [CrossRef]
- Maolanon, A.R.; Madsen, A.S.; Olsen, C.A. Innovative strategies for selective inhibition of histone deacetylases. Cell Chem. Biol. 2016, 23, 759–768. [Google Scholar] [CrossRef]
- Kaur, S.; Shivange, A.V.; Roy, N. Structural analysis of trypanosomal sirtuin: An insight for selective drug design. Mol. Divers. 2010, 14, 169–178. [Google Scholar] [CrossRef]
- Zheng, W. Sirtuins as emerging anti-parasitic targets. Eur. J. Med. Chem. 2013, 59, 132–140. [Google Scholar] [PubMed]
- Kannan, S.; Melesina, J.; Hauser, A.-T.; Chakrabarti, A.; Heimburg, T.; Schmidtkunz, K.; Walter, A.; Marek, M.; Pierce, R.J.; Romier, C. Discovery of inhibitors of Schistosoma mansoni HDAC8 by combining homology modeling, virtual screening, and in vitro validation. J. Chem. Inf. Model. 2014, 54, 3005–3019. [Google Scholar] [PubMed]
- Schiedel, M.; Marek, M.; Lancelot, J.; Karaman, B.; Almlöf, I.; Schultz, J.; Sippl, W.; Pierce, R.J.; Romier, C.; Jung, M. Fluorescence-based screening assays for the NAD+-dependent histone deacetylase smSirt2 from Schistosoma mansoni. J. Biomol. Screen. 2015, 20, 112–121. [Google Scholar] [PubMed]
- Yamada, H.; Arakawa, Y.; Saito, S.; Agawa, M.; Kano, Y.; Horiguchi-Yamada, J. Depsipeptide-resistant KU812 cells show reversible P-glycoprotein expression, hyper-acetylated histones, and modulated gene expression profile. Leuk. Res. 2006, 30, 723–734. [Google Scholar] [CrossRef]
- Fedier, A.; Dedes, K.J.; Imesch, P.; Von Bueren, A.O.; Fink, D. The histone deacetylase inhibitors suberoylanilide hydroxamic (Vorinostat) and valproic acid induce irreversible and MDR1-independent resistance in human colon cancer cells. Int. J. Oncol. 2007, 31, 633–641. [Google Scholar] [CrossRef] [PubMed]
- John, C.C.; Kutamba, E.; Mugarura, K.; Opoka, R.O. Adjunctive therapy for cerebral malaria and other severe forms of Plasmodium falciparum malaria. Expert Rev. Anti-Infect. Ther. 2010, 8, 997–1008. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | Class/Inhibitor | Parasite Growth Inhibition (IC50) and Effective Concentration (EC50) | Mammalian Cell, Class-I hHDACs in HeLa Cells, and Sirutin Inhibition IC50 | References |
|---|---|---|---|---|
| 5 | Cyclic Tetrapeptide | P. falciparum IC50 ~0.200 μM | Mammalian Cell IC50 0.05–0.1 μM | [117,124] |
| 6 | Class-I/II/IV HDAC inhibitors | T. gondii IC50 ~10 nM | - | [132] |
| 7 | Short-Chain Fatty Acids | P. falciparum IC50 > 100 μM | Mammalian Cell IC50 ~1350 μM | [133] |
| 10 | Hydroxamic Acids | P. falciparum IC50 ~0.008–0.120 μM T. brucei IC50 ~7 μM T. gondi IC50 ~41 nM | Mammalian Cell IC50 0.2–0.3 μM | [86,92,128,131,134] |
| 3 | Hydroxamic Acids | P. falciparum IC50 0.025–2.2 μM T. gondi IC50 83 nM | Mammalian Cell IC50 0.26–20 μM | [128,130,133,135] |
| 11 | Hydroxamic Acids | P. falciparum IC50 0.8–2.3 μM T. gondi IC50 213 nM | Mammalian Cell IC50 50–300 μM | [128,131,139] |
| 12 | Hydroxamic Acids | P. falciparum IC50 0.9–1.1 μM | - | [131] |
| 13 | Hydroxamic Acids | P. falciparum IC50 1.2–4 μM | Mammalian Cell IC50 > 20 μM | [132] |
| 14 | Hydroxamic Acids | P. falciparum IC50 ~0.048 μM | Mammalian Cell IC50 ~0.6 μM | [93] |
| 15 | Hydroxamic Acids | P. falciparum IC50 0.015–0.039 μM | Mammalian Cell IC50 ~1.24 μM | [93,134,135] |
| 15 | Hydroxamic Acids | P. falciparum IC50 0.013–0.033 μM | Mammalian Cell IC50 ~0.34 μM | [93,135] |
| 17 | Hydroxamic Acids | P. falciparum IC50 ~0.013 μM | Mammalian Cell IC50 ~0.26 μM | [136] |
| 18 | Hydroxamic Acids | P. falciparum IC50 ~0.015 μM | - | [89] |
| 19 | Hydroxamic Acids | P. falciparum IC50 0.017–0.035 μM | Mammalian Cell IC50 0.02–0.8 μM | [137] |
| 20 | Hydroxamic Acids | P. falciparum IC50 0.0006–0.0016 μM | Mammalian Cell IC50 ~0.6 μM | [133,138] |
| 21 | Hydroxamic Acids | P. falciparum IC50 ~0.069 μM | - | [139] |
| 22 | Hydroxamic Acids | P. falciparum IC50 0.074–0.107 μM L. donovani IC50 ~32.1 μM | - | [139] |
| 23 | Hydroxamic Acids | P. falciparum IC50 0.144–0.148 μM | Mammalian Cell IC50 > 5.2 μM | [140] |
| 24 | Hydroxamic Acids | P. falciparum IC50 0.93–1.24 μM L. donovani IC50 ~5 μM | Mammalian Cell IC50 > 5 μM | [140] |
| 25 | Hydroxamic Acids | P. falciparum IC50 0.094–0.226 μM | - | [141] |
| 26 | Hydroxamic Acids | P. falciparum IC50 0.76–1.32 μM L. donovani IC50 3.2–4.7 μM | - | [141] |
| 27 | Hydroxamic Acids | P. falciparum IC50 0.08–0.15 μM | Mammalian Cell IC50 0.8–>100 μM | [142] |
| 28 | Hydroxamic Acids | P. falciparum IC50 ~0.09 μM | Mammalian Cell IC50 ~12.47 μM | [143] |
| 29 | Hydroxamic Acids | P. falciparum IC50 ~0.12 μM | Mammalian Cell IC50 ~3.24 μM | [143] |
| 30 | Hydroxamic Acids | P. falciparum IC50 ~0.17 μM | Mammalian Cell IC50 > 50 μM | [143] |
| 30 | Hydroxamic Acids | P. falciparum IC50 0.16 (3.45) μM | Mammalian Cell IC50 ~4.9 μM | [144] |
| 31 | Hydroxamic Acids | P. falciparum IC50 0.25–0.32 (2.12–2.25) μM | Mammalian Cell IC50 ~19.5 μM | [145] |
| 32 | Hydroxamic Acids | P. falciparum IC50 ~0.019 μM | Mammalian Cell IC50 ~75.1 μM | [146] |
| 33 | Cyclic Tetrapeptide | P. falciparum IC50 0.09–0.13 μM T. brucei IC50 35 nM | Mammalian Cell IC50 0.001–<0.005 μM | [130] |
| 34 | Hydroxamic Acids | P. falciparum IC50 0.06–0.13 μM T. brucei IC50 2.6 μM | Mammalian Cell IC50 1.4–2.4 μM | [130] |
| 35 | Hydroxamic Acids | P. falciparum IC50 0.01–0.03 μM T. brucei IC50 1.69 μM | Mammalian Cell IC50 0.07–0.18 μM | [130] |
| 37 | Thiols | P. falciparum IC50 15.2–19.9 μM | - | [23,26] |
| 38 | Ortho-Aminoanilide | P. falciparum IC50 7.8–8.3 μM | Mammalian Cell IC50 > 20 μM | [23,26,89] |
| 39 | N-Methyl Carboxamide | P. falciparum EC50 ~0.45 μM | Class-I hHDACs in HeLa cells IC50 ~16.6 μM | [147] |
| 40 | N-Methyl Carboxamide | P. falciparum EC50 ~0.90 μM | Class-I hHDACs in HeLa cells IC50 ~20.5 μM | [147] |
| 41 | N-Methyl Carboxamide | P. falciparum EC50 ~0.5 μM | Class-I hHDACs in HeLa cells IC50 > 25 μM | [147] |
| 42 | Class-I/II HDAC inhibitor | P. falciparum IC50 9–13 μM L. infantum IC50 30 μM | IC50 (PfSir2A) > 50μM IC50 (LiSir2rp1) 194μM IC50 (hSIRT1) = 245 μM | [89,148,149] |
| 43 | class-III HDAC (Sirutin) inhibitor | P. falciparum IC50 > 10 μM | IC50 (PfSir2A) > 400 μM | [89,148,149] |
| 44 | class-III HDAC (Sirutin) inhibitor | P. falciparum IC50 9 μM | IC50 (PfSir2A) 35 μM IC50 (hSIRT1) > 600 μM | [148] |
| 45 | class-III HDAC (Sirutin) inhibitor | P. falciparum IC50 1.5–2 μM | - | [150,151] |
| 46 | class-III HDAC (Sirutin) inhibitor | P. falciparum IC50 9.9 mM T. cruzi EC50 25.8 mM | IC50 (PfSir2A) 51 μM IC50 (hSIRT1) 88–250 μM | [148,149,152] |
| 47 | class-III HDAC (Sirutin) inhibitor | P. falciparum IC50 9.8 μM | IC50 (PfSir2A) 23 μM | [148] |
| 50 | class-III HDAC (Sirutin) inhibitor | T. brucei IC50 0.034 μM | - | [153] |
| 51 | class-III HDAC (Sirutin) inhibitor | T. brucei EC50 0.029 μM | - | [154] |
| 52 | Class-I/II HDAC and sirutin inhibitor | T. cruzi EC50 10.6 μM | IC50 (TcSir2rP3) 1 μM | [24] |
| 53 | Class-I/II HDAC and sirutin inhibitor | L. donovani IC50 4.4–8.8 μg/mL | IC50 (hHDAC6) 35 nM IC50 (hHDAC8) 854 nM | [114] |
| 54 | Class-I/II HDAC and sirutin inhibitor | L. donovani IC50 0.1–6.5 μg/mL | IC50 (hHDAC6) 457 nM IC50 (hHDAC8) 1272 nM | [114] |
| 55 | Class-I/II HDAC and sirutin inhibitor | L. donovani IC50 0.1–6.3 μg/mL | IC50 (hHDAC6) 847 nM IC50 (hHDAC8) 4283 nM | [114] |
| 56 | Class-I/II HDAC and sirutin inhibitor | L. infantum IC50 1.49 mM | - | [115] |
| 57 | Class-I/II HDAC and sirutin inhibitor | L. infantum IC50 6.03 μM | IC50 (LiSir2rp1) 5.7 μM IC50 (hSIRT1) 97.4 μM | [116] |
| 58 | class-I/II/IV HDAC inhibitors | T. gondi IC50 39 nM | - | [128] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohapatra, T.K.; Nayak, R.R.; Ganeshpurkar, A.; Tiwari, P.; Kumar, D. Opportunities and Difficulties in the Repurposing of HDAC Inhibitors as Antiparasitic Agents. Drugs Drug Candidates 2024, 3, 70-101. https://doi.org/10.3390/ddc3010006
Mohapatra TK, Nayak RR, Ganeshpurkar A, Tiwari P, Kumar D. Opportunities and Difficulties in the Repurposing of HDAC Inhibitors as Antiparasitic Agents. Drugs and Drug Candidates. 2024; 3(1):70-101. https://doi.org/10.3390/ddc3010006
Chicago/Turabian StyleMohapatra, Tapas Kumar, Reena Rani Nayak, Ankit Ganeshpurkar, Prashant Tiwari, and Dileep Kumar. 2024. "Opportunities and Difficulties in the Repurposing of HDAC Inhibitors as Antiparasitic Agents" Drugs and Drug Candidates 3, no. 1: 70-101. https://doi.org/10.3390/ddc3010006
APA StyleMohapatra, T. K., Nayak, R. R., Ganeshpurkar, A., Tiwari, P., & Kumar, D. (2024). Opportunities and Difficulties in the Repurposing of HDAC Inhibitors as Antiparasitic Agents. Drugs and Drug Candidates, 3(1), 70-101. https://doi.org/10.3390/ddc3010006