Abstract
Copper is an essential heavy metal for diverse biological functions but toxic in excess. Consequently, a tightly regulated protein system is required to ensure adequate intracellular levels. In recent decades, several studies have explored the role of Cu+-ATPases in copper transport and homeostasis, revealing that these proteins are subject to kinase-mediated phosphorylation that significantly impacts their function. Techniques such as phosphoproteomic screening, site-directed mutagenesis, and artificial neural network tools demonstrated the regulatory effect of phosphorylation on these ATPases. Different protein kinases regulate Cu+-ATPases, modulating the active copper transport by affecting specific steps of the catalytic cycle, long-range intramolecular crosstalks, protein trafficking, gene expression, and protein stability. Therefore, the regulatory phosphorylation of Cu+-ATPases by kinases ultimately influences the intracellular copper distribution. This study aims to present a review of the scientific literature on the regulation of Cu+-ATPases by kinase-mediated phosphorylation as a crucial mechanism for copper homeostasis. This regulation offers new perspectives for developing therapies for disorders related to copper metabolism, such as Wilson and Menkes diseases, as well as cancer, diabetes mellitus, Parkinson’s, and Alzheimer’s diseases. These findings emphasize the need to further comprehend the signaling pathways involving protein kinases in the context of copper regulation.
1. Introduction
1.1. Copper
Copper is a metallic element with a density of 8.96 g/cm−3 at 20 °C, classifying it as a heavy metal due to its density being higher than 5 g/cm−3. This element has an atomic number of 29, an atomic weight of 63.5, and is classified in the periodic table as a transition element. Transition elements exhibit distinctive characteristics, notably the partial filling of d orbital across multiple compounds, and typically manifest multiple oxidation numbers. For instance, copper oxidation numbers include Cu+, Cu2+, Cu3+, and even Cu4+. Consequently, this element has the ability to mediate the transfer of electrons being highly active in oxidation-reduction reactions [1].
1.2. Copper in Biology
Copper is an essential trace element that plays a pivotal role in several physiological processes. Copper serves as a cofactor for enzymes involved in cellular respiration, antioxidant defense, connective tissue formation, neurotransmitter biosynthesis, iron transport, and more. Enzymes dependent on copper for their biosynthesis and catalytic functions are termed cuproenzymes (Table 1), and it is estimated that only a minor fraction has been described so far [2]. Additionally, copper contributes to chlorophyll formation, photosynthesis, oxidative stress protection, and metabolism in plants [3,4].

Table 1.
List of mammals cuproenzymes and their biological functions.
However, free Cu+ can be readily toxic to the cell since it participates in the production of hydroxyl radicals (OH●) in a Fenton-type reaction that could damage phospholipids and enzymes [13]. Another way to interfere with biological processes is through interaction with biomolecules like proteins. According to the “hard and soft acids and bases” concept [14], Cu+ is a “soft” cation and will interact with the “soft” thiolates of cysteine residues in proteins.
The biosynthetic incorporation of Cu+ into cuproenzymes occurs within the secretory pathway and is intricately regulated by copper-transporting ATPases, now referred to as Cu+-ATPases. As depicted in Figure 1, these enzymes display a typical perinuclear trans-Golgi network localization (TGN) and translocate to the plasma membrane through distinct pathways in response to altered intracellular Cu+ levels [15,16]. The native abundance of Cu+-ATPases is low, and they pump Cu+ across membranes using ATP energy, exhibiting tissue- and species-specific expression patterns, crucial for copper homeostasis and the vectorial displacement of bound ligands to target proteins, varying in mechanism and regulation [17].
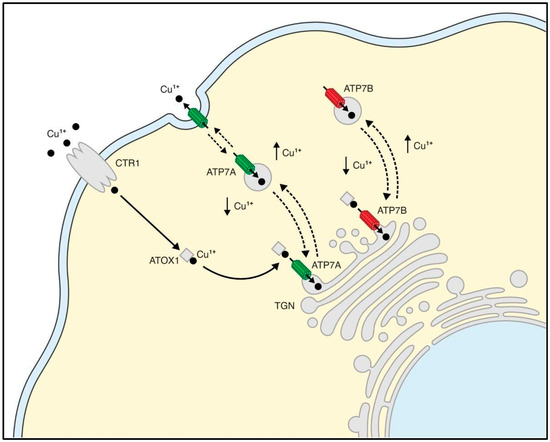
Figure 1.
Schematic representation of copper homeostasis in mammalian cells. Reduced Cu+ enters cells through Ctr1 at the plasma membrane. The chaperone ATOX1 carries Cu+ (black circles) to the trans-Golgi network (TGN) delivering it to the Cu+-ATPases ATP7A and ATP7B (solid arrows), where Cu+ is actively pumped into the Golgi. When intracellular Cu+ increases, Cu+-ATPases relocate to the plasma membrane (ATP7A) and to vesicles (in the case for ATP7B). This trafficking is reverted when the intracellular Cu+ concentration returns to basal levels (represented by dashlined arrows). Ctr1, high affinity Cu+ uptake protein 1; ATOX1, antioxidant protein 1; TGN, trans-Golgi network; ATP7A and ATP7B, Cu+-ATPases.
1.3. Cu+-ATPases
Cu+-ATPases have been identified across all life kingdoms, being well-characterized in bacteria, yeast, and mammals. This enzyme belongs to the P1-type ATPase family, corresponding to heavy-metal-transporting P-type ATPases [18]. P1-type ATPases share all structural attributes typical of P-type ATPases: DKTGT sequence, containing the aspartate residue transiently phosphorylated in the ATPase/transport cycle in the phosphorylation (P)-domain; TGES sequence, which corresponds to phosphatase activity in the actuator (A)-domain and the nucleotide-binding (N)-domain [19].
P-type ATPases are subject to two distinct types of phosphorylation: (i) in the conserved aspartate residue that undergoes autophosphorylation during the ATPase cycle; (ii) in non-mandatory serine or threonine residues phosphorylated by protein kinases that can modulate the enzyme activity, stability, or interaction with other proteins. These phosphorylations can be distinguished chemically. The former is alkali-labile phosphorylation, whereas the latter is alkali-resistant. P1-type ATPases share specific structural features such as eight putative transmembrane helices and one pair of membrane segments located in the C-terminal of the cytoplasmic ATP-binding domain [18]. Additionally, structural studies have provided insights into the mechanism of Cu+ transport by these ATPases, highlighting the importance of specific metal binding domains (MBD) in the N-terminal region. The number of MBD varies by species, from one to six GxMCxxC motifs, and the presence of two cysteines in the CxxC motif gives the enzyme a heavy-metal-binding site, where x indicates any amino acid [20]. At the time of initial investigations, there was a prevailing hypothesis suggesting that the transported ion by the ATPase was Cu2+. However, subsequent investigations revealed that it is the reduced Cu+ form that is, indeed, transported by Cu+-ATPases.
The first two genes encoding Cu+-ATPases in bacteria were described in Enterococcus hirae (CopA and CopB). CopA is responsible for Cu+ uptake in low Cu+ conditions, and CopB promotes Cu+ export at toxic levels [21]. The presence of Cu+-ATPases in bacteria underscores the significance of these proteins in maintaining copper balance and supporting essential cellular processes throughout evolution. In yeast, the Cu+-ATPase is encoded by the gene Ccc2p, and it was described to provide Cu+ to a cellular compartment rather than in the removal of excess Cu+ [22]. In that time, independent groups have cloned the two human Cu+-ATPases: ATP7A [23,24,25] and ATP7B [26,27,28]. Mutations in the genes encoding ATP7A and ATP7B are responsible for Cu+ deficiency or overload, associated with pathologic conditions such as Menkes (OMIM #309400) or Wilson disease (OMIM #277900), respectively. A diverse array of mutations has been screened for both genes, which have been documented in various published studies and collected by The Human Gene Mutation Database (HGMD®), available at http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 13 May 2024). Some mutations lead to aberrant intracellular targeting, protein stability, and Cu transport. In human health research, the homeostasis of copper has attracted increased attention due to its association with pathological conditions, such as diabetes mellitus (DM), Parkinson’s, Alzheimer’s, and cancer [29].
The post-translational modification of P-type ATPases through kinase-mediated phosphorylation and glycosylation is an extensively covered subject [30,31]. In the past two decades, the kinase-mediated modification of Cu+-ATPases has gained attention. A large number of phosphorylation sites were detected in phosphoproteomic screenings of both ATP7A and ATP7B [32,33], and the glycosylation of ATP7A was demonstrated in heterologous expression in COS-1 cells [34].
While some studies provide speculative indications of kinase action, others present more detailed molecular evidence for regulatory phosphorylation. This review comprehensively examines the ongoing knowledge on kinase-mediated phosphorylation of Cu+-ATPases.
2. Indirect Evidence for the Kinase Regulation of Cu+-ATPases
Even though the specific residues targeted for phosphorylation by protein kinases in Cu+-ATPases remained undetermined in some studies, the use of conventional pharmacological methodologies strongly implied their participation in regulatory mechanisms for copper homeostasis. cAMP-dependent protein kinase (PKA) and protein kinase C (PKC) are known regulators of the classical ATPases, such as Na+,K+-ATPase [30] and Ca2+-ATPase [35], and are related in pathological processes. Not surprisingly, those kinases were the first to be investigated in the modulation of different Cu+-ATPases.
A pioneering study on the regulation of ATP7A demonstrated that the normal trafficking of the pump is abrogated in the presence of H-89 and PKI(14–22), two potent PKA inhibitors [36]. PKA uniquely influences copper-dependent ATP7A trafficking, unlike ATP7B [37]. In HeLa cells, ATP7A’s copper-dependent trafficking to the plasma membrane is unaffected by Rho GTPases and PKD, known regulators of carrier vesicles in the TGN [38]. Although PKA activity was implicated in this process, the specific molecular mechanism governing the phosphorylation remained unclear. Regulation by PKA was also suggested for ATP7B as part of the hormonal response to insulin and glucagon in hepatocytes. In WIF-B9 hepatocyte-like cells, the treatment with insulin enhances ATP7B activity, promoting increased Cu+ sequestration in cells. In stark contrast, glucagon reduces ATP7B activity, leading to a diminished capacity of the transporter to manage Cu+ efficiently. Interestingly, when insulin and glucagon are administered together, they appear to counteract each other’s effects, indicating a balance controlled by these hormones to maintain Cu+ levels stable through the PKA-mediated signaling pathway. This hormonal response mechanism demonstrated the presence of a membrane-bound PKA and Akt activity, sensitive to their pharmacological inhibitors [39]. These findings provide a new perspective on copper homeostasis in metabolic disorders such as diabetes and obesity via the physiological significance of energy metabolism in regulating Golgi Cu+ availability.
The PKC-mediated signaling pathway was investigated in THP-1 monocytes. Treatment with phorbol 12-myristate 13-acetate (PMA), a classic activator of PKC, induced ATP7A expression and increased Cu+ egress. Cu+ transport by ATP7A regulates the expression of the vascular endothelial growth factor receptor 1 (VEGFR1). The proposed mechanism for a PMA-sensitive ATP7A-dependent copper metabolism in monocytes may contribute to advancements in the understanding of vascular function and potential cancer progression [40]. It is important to note that ATP7B is regulated by a specific PKC isoform in the porcine liver. However, the regulatory PKC-mediated phosphorylation did not alter ATP7B expression but does affect its catalytic cycle. This occurs after the dephosphorylation of the Cu+-ATPase, likely at the E2 → E1 transition during catalysis (Figure 1, step 1) [41]. The increased ATP7B activity by PKC opposes the inhibition attributed to PKA-mediated phosphorylation of the pump in the same model [39]. This raises the possibility that regulation of Cu+ transporters by these kinases could be a universal strategy for fine-tuning intracellular Cu+ levels in different cellular systems.
Another interesting scenario involves the connection between ATP7B regulation and the activities of MAP kinases such as p38 and c-Jun N terminal kinase (JNK). The specific genetic variation H1069Q found in Wilson disease disturbs the intracellular targeting of ATP7B, causing it to move to the endoplasmic reticulum (ER). This severely impairs the cells’ ability to export Cu+, which accumulates. The Cu+ accumulation due to H1069Q upregulates these kinases. There is a correlation between the expression of this particular mutant and the activation of p38 and JNK, which have been described as essential for the degradation of the H1069Q mutant [42]. The p38 and JNK signaling pathways respond to stress by becoming active following an increase in reactive oxygen species (ROS) [43]. The specific mechanism by which the inhibition of p38 and JNK works to restore the mutant ATP7B functionality and copper homeostasis is still unclear. However, these findings may underscore possible strategies for treating Wilson’s disease.
The effect of hormones like insulin and estrogen in the trafficking of ATP7A/B [44] and the involvement of Cu+ in the NMDA transmission pathway [45] hinted that post-translational modification could play a significant role in the regulation of ATP7A. Nevertheless, at least initially, the potential kinases involved in these phosphorylations were not determined.
However, some studies provided direct evidence of kinase-mediated phosphorylation in Cu+-ATPases, either through phosphoproteomic tools or site-directed mutagenesis, unraveling the molecular mechanisms triggered by this post-translational modification.
There are different ways to approach the modulation of Cu+-ATPases by kinases, whether through the effects caused by their phosphorylation, comparative analysis between Cu+-ATPases, or a sectorized analysis focusing on the involved kinases. Due to the wide variety of studies, we decided that it would be more enlightening to separate according to Cu+-ATPase, organizing the sessions according to the studies of the kinase-mediated phosphorylation of each transporter.
3. The Kinase Regulation of CCC2, the Yeast Cu+-ATPase
Only a few studies have aimed to elucidate the role of the kinase-mediated phosphorylation of CCC2, the Cu+-ATPase from Saccharomyces cerevisiae, with a particular interest in the modulation of its catalytic cycle (Figure 2).
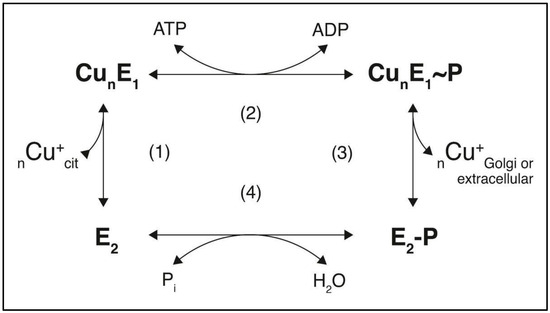
Figure 2.
Proposed catalytic cycle of Cu+-ATPases. In physiological Cu+ levels, the reaction actively transports Cu+ from the cytosol to acceptor proteins in the Golgi lumen or, in the overloaded Cu+ condition, to the extracellular compartment. Step 1: Cu+ binding to the pump; step 2: phosphorylation of the aspartate residue by ATP; step 3: conversion of the high-energy E~P form to the low-energy E-P form and Cu+ release; step 4: hydrolysis of the phosphorylated intermediate. The aspartyl-phosphate at the conserved DKTGT site in P-type ATPases’ high-energy state (E~P) is sensitive to ADP.
The use of artificial neural network analysis tools in silico resulted in potential targets for kinase phosphorylation in CCC2. Residues with higher predicted scores, serines 258 and 971 (Ser258 and Ser971, respectively), were replaced with alanines (S258A and S971A), and these mutations, both individually and in combination, were shown to impact CCC2 kinetic parameters and overall function. Ser258, located at the hinge of the CCC2 N-terminal domain, emerged as the primary target for PKA-mediated, Cu+-dependent phosphorylation. This amino acid would function as a master serine, and its phosphorylation would prime CCC2 for further kinase-mediated phosphorylations [46]. The importance of Ser258 in Cu+ handling was confirmed by in vivo essays accessing yeast growth in iron- and copper-deprived media [47].
The mutation of the conserved aspartate in the P-domain of the ATPase (D627A) significantly increases exogenous PKA-induced phosphorylation compared to wild-type CCC2. This suggests the existence of long-range negative feedback mechanisms linking Asp627 catalytic phosphorylation to Ser258 kinase-mediated phosphorylation in the N-terminal, modulating the catalytic cycle of CCC2. This intramolecular crosstalk extends to the dephosphorylation step of the catalytic cycle since the time constant of aspartyl–phosphate phosphoenzyme formation is delayed compared to the wild-type ATPase (Figure 2, step 4) [46]. A similar behavior occurs in ATP7B, where Cu+-dependent regulatory phosphorylations affect the catalytic turnover of the enzyme [48].
For CCC2, there is a gradual decrease in the proportion of ADP-sensitive aspartyl-phosphate intermediate enzymes over time, indicating a shift from a high-energy phosphorylated state (E1~P) to a low-energy (E2-P) one (Figure 2, step 3). The S258A mutant increases ADP sensitivity, leading to a sustained high-energy (E1~P) phosphorylated state. Conversely, disabling Ser971 impacts the breakdown of the ADP-insensitive E2-P intermediate associated with Cu+ release (Figure 2, steps 3 and 4). This body of evidence shows that PKA-mediated regulatory phosphorylation of CCC2 modulates the catalysis through long-range communication between Ser258, Ser971, and Asp627 [47]. It suggests a role for the kinase-mediated phosphorylation of yeast Cu+-ATPase, potentially mirroring the regulation of human Cu+ transporters.
4. The Kinase Regulation of ATP7A
The study of the kinase-mediated phosphorylation of ATP7A provided valuable information on different aspects of the Cu+-ATPase function beyond the catalytic cycle modulation observed for CCC2, such as trafficking and stability of the protein, evidencing the phosphorylation-driven modulation of ATP7A molecular dynamics, which is essential for Cu+ transport.
The first piece of evidence for the kinase regulation of ATP7A emerged from studies using CHO-K1 stably transfected with the wild-type ATP7A (wtATP7A) or mutants where all six metal-binding sites were disrupted (mMBS). This mutation impairs ATPase trafficking in response to increased Cu+. Phosphorylation assays performed in vivo and in vitro, followed by phosphopeptide analysis, revealed a Cu+-dependent phosphorylation of ATP7A [49]. The alkali-stable signal detected responded to even slight increases in Cu+ concentration, being 1 µM sufficient for provoking a hyperphosphorylated state. Since the mMBS mutant failed to hyperphosphorylate, the authors suggested that the traffic of ATP7A would require changes in protein dynamics arising from the binding of Cu+ at the MBDs. The conformational changes would allow a kinase-mediated phosphorylation to regulate ATP7A function. Even though no particular kinases were detected, ATP7A was phosphorylated in membrane preparations, suggesting the involvement of membrane-bound kinases [49].
Studies using a combination of immunoprecipitation, enrichment of phosphopeptides, and LC-MS/MS screening revealed that ATP7A was indeed subject to significant kinase-driven phosphorylation. These phosphorylation sites are mainly located in the N- and C-terminal cytosolic domains. Among the twenty-one observed phosphorylation sites, eight were sensitive to fluctuations in intracellular Cu+ levels. This indicates a system where the activity and targeting of ATP7A are finely adjusted based on the availability of intracellular Cu+ [50].
The phosphorylation of Ser1432 and Ser1469 of ATP7A explicitly affects its response to Cu+-induced trafficking. The former is phosphorylated regardless of Cu+, and S1432A leads to the misplacement of ATP7A in polarized cells in response to Cu+. Conversely, the phosphorylation at Ser1469 is sensitive to Cu+ levels, serving as a regulatory site that reacts specifically to fluctuations in intracellular Cu+. This specific phosphorylation site is pivotal for controlling the Cu+-sensitive trafficking of ATP7A towards the plasma membrane. Mutations that abrogate the phosphorylation at Ser1469 lead to improper localization patterns [50]. At the early stages of the investigations of Cu+-ATPases regulation by kinases, this was the first time specific phosphorylation sites were associated with functional outcomes in ATP7A trafficking. The phosphorylation at Ser1469 sits close to the 1459LL di-leucine motif in the C-terminal of ATP7A. This motif is crucial for ATP7A interaction with the AP-1 adaptor subunit necessary for the trafficking of ATP7A [51]. Adding a negative charge in the vicinity of 1459LL could favor this interaction and facilitate the trafficking of ATP7A [52].
Further investigation of the ATP7A phosphorylation profile revealed that the MBD3 plays a prominent role in sensing and regulating intracellular Cu+ levels. The phosphorylation of Ser339 and Thr327 influences the electrostatic properties of MBD3 and alters the dynamics in this domain. Results from dynamic simulations based on NMR structural analysis suggest that a constitutive phosphorylation of Ser339, independent of cellular Cu+, increases the backbone flexibility in the loop IV of MBD3. This phosphorylation facilitates the exposure of the CxxC Cu+-binding motif within MBD3. Cu+ binding primes the domain for a subsequent phosphorylation at Thr327. Adding a second phosphate reduces the flexibility in the loop when Cu+ is bound, impacting the efficiency of the Cu+ transport by ATP7A [50]. Therefore, this dual phosphorylation leads to a stable conformation of MBD3 in the presence of increased Cu+, potentially impacting the catalytic activity and trafficking of ATP7A.
Studies with type 2 diabetes (T2DM) in vascular smooth muscle cells (VSMCs) illustrate how phosphorylation-driven alterations in ATP7A’s molecular dynamics lead to functional changes in a physiopathological context. In VSMC activated by insulin, Akt2 phosphorylates the Ser1424, Ser1463, and Ser1466 of ATP7A. This process, impaired in T2DM, is linked to a stabilization of ATP7A since Akt2 phosphorylation prevents ATP7A proteasomal degradation. The pronounced downregulation of ATP7A observed in T2DM can be reverted by mutations that render Akt2 constitutively active, along with increased insulin [53]. A similar role in maintaining protein stability was also proposed for the regulatory phosphorylation of ATP7B [37]. The phosphorylation of Ser1424, Ser1463, and Ser1466 also controls the trafficking of ATP7A to the plasma membrane. This translocation is crucial for enabling ATP7A to transport Cu+ to superoxide dismutase 3 (SOD3), which assists VSMCs in protecting against superoxide radicals and endothelial dysfunction [53]. These findings suggest that intervening in the ATP7A-SOD3 pathway could be pivotal in developing strategies to alleviate complications associated with T2DM. The role of insulin in copper homeostasis extends beyond ATP7A, as suggested by indirect evidence associating insulin with reduced ATP7B activity in liver cells [39] and decreased ATP7B expression in placental cells [44].
5. The Kinase Regulation of ATP7B
5.1. Phosphorylation of the N-Terminal Trigger Intramolecular Interactions
Early studies proposing the regulatory phosphorylation of ATP7B relied on consistently detecting a stable phosphorylation signal at alkaline pH levels [54,55]. This post-translational modification was initially proposed to be Cu+-independent or Cu+-dependent phosphorylation. The Cu+-independent phosphorylation would be attributed to membrane-bound kinase activity, whereas soluble kinase would phosphorylate ATP7B in response to Cu+. The binding of Cu+ to the N-terminal domain triggers conformational changes that expose specific residues of ATP7B for kinase phosphorylation and allow for the trafficking of the Cu+-ATPase [33]. Analyzing phosphopeptides from wild-type ATP7B and a mutant lacking the N-terminal (N-ATP7B) reveals that Cu+ binding exposes the loop between MBD3 and MBD4, facilitating kinase-mediated phosphorylation. This indicates a hierarchical regulatory mechanism governing ATP7B post-translational modification and trafficking [56]. These structural changes resulting from this Cu+-dependent phosphorylation allow ATP7B to interact with clathrin vesicle adaptor proteins like AP-1, necessary for its trafficking [57], as described for ATP7A [51,57].
Site-directed mutagenesis analysis of the phosphorylation sites within the MBD3–4 loop yielded further insights into the protein functionality, particularly in vesicular trafficking. Mutations at Ser340/341 residues to Ala, Gly, Thr, or Asp result in a functionally active ATP7B with decreased protein phosphorylation, albeit with predominant localization in vesicles in a Cu+-independent process. The S340/341G mutation leads to reduced phosphorylation under basal conditions without being affected by Cu+, suggesting that phosphorylation would not be essential for sorting ATP7B from the TGN. The S340/341A mutation does not alter the overall structure of N-ATP7B but significantly reduces its interactions with the nucleotide-binding domain (N-domain). It mimics the effects of Cu+ binding to N-ATP7B, indicating structural changes, specifically in inter-domain contacts, as initiators of ATP7B relocation from the TGN. Moreover, these mutations may facilitate ATP7B vesicular exit by simulating the Cu+-saturated “trafficking-compatible” state of the protein, thereby suggesting that ATP7B exit from the TGN is governed primarily by its conformational state, which is influenced by Cu+ and disrupted by specific mutations in the regulatory N-terminal domain, particularly Ser340/341 [58].
Additionally, ATP7B phosphorylation dynamics revealed an intramolecular crosstalk between Cu+ binding in N-ATP7B and the phosphorylation in the C-terminal region. This conformational change within the ATP7B structure influences its function and Cu+ balance [33].
5.2. Multisite Phosphorylation of ATP7B Modulates the Catalytic Cycle
In a study aiming to investigate the phosphorylation dynamics and enzymatic activity of ATP7B, microsomal preparations of high-yield ATP7B and N-terminal mutants in COS-1 cells were utilized to identify key phosphorylation sites in the ATPase. Utilizing mass spectrometry for detailed phosphopeptide analysis evidenced Ser478 and Ser481 in the N-terminal metal binding domains (NMBD), Ser1121 in the N-domain, and Ser1453 in the C-terminal as the main phosphorylation sites. The ATP7B regulatory phosphorylation of those sites is Cu+-dependent and presumably affects the catalytic turnover of the enzyme. This phosphorylation was consistent across both ex vivo and in vitro samples, reinforcing the physiological relevance of these findings [48]. This activity modulation was demonstrated previously in the N-terminal of yeast’s CCC2, where Cu+-dependent kinase phosphorylations modulate the energy interconversion steps of its catalytic cycle [46]. A homologous mechanism was suggested for Ser971 in the C-terminal of CCC2, where the phosphorylation would trigger a long-range intramolecular crosstalk that affects the dephosphorylation step of the ATPase [47]. Another example comes from the modulation of ATP7B in hepatocytes, where phosphorylation by PKA decreased Cu+-ATPase activity [39].
5.3. Phosphorylation of ATP7B by PKD Increases Protein Stability
PKD-mediated phosphorylation of ATP7B plays a crucial role in protein stabilization. The ATP7B phosphopeptides obtained after treating COS-1 and HepG2 microsomes with [γ-32P] ATP were sensitive to inhibitors and activators of protein kinase D (PKD). Incubation with the PKD-specific inhibitor CID2011756 completely abolishes the phosphorylation of Ser478, Ser481, Ser1121, and Ser1453. Conversely, adding PMA has the opposite effect, increasing the detection of the phosphopeptides. PMA is a known activator of PKC, and PKD, in turn, is activated by PKC in a kinase cascade. The decreased ATP7B expression in cells treated with CID2011756 was reverted by the proteasome inhibitor MG132, pointing to the critical role of PKD-mediated phosphorylation for ATP7B protein stabilization. This phosphorylation in the mentioned Ser residues correlates to the trafficking of ATP7B from the TGN to cytosolic vesicles. The activity of PKD is known for regulating the fission of vesicular carriers departing the TGN, and the phosphorylation of ATP7B would then set the ATPase in a “trafficking-compatible” state [37].
Both Cu+-transporters are associated with increased resistance to drugs like cisplatin and carboplatin due to elevated Cu+-ATPases expression and alterations in their subcellular localization [59]. The inhibition of PKD-mediated phosphorylation of ATP7A/B was recently associated with increased susceptibility of cancer cells to platinum-based chemotherapy drugs involving proteasomal degradation [60]. Understanding the regulation of Cu+-ATPases by PKD and other kinases has proven crucial for integrating these transporters into broader biological processes, such as those associated with drug resistance.
5.4. The C-Terminal ATP7B Undergoes Multisite Kinase Phosphorylation
A Cu+-dependent ATP7B phosphorylation in the C-terminal region was previously suggested [54,61]. The most complete proteomic analysis of ATP7B identified twenty-four phosphorylatable Ser/Thr sites. Notably, the majority of these sites are at the N- and C-terminal regions of the pump, where multisite phosphorylations were detected in low and high Cu+, including the two C-terminal diphosphopeptides conserved in mammals: Ser1429/1432 and Ser1442/1453 [33]. Four residues are phosphorylated in the presence of added Cu+, Ser246, Ser1121, Ser1431, and Ser1442, three of which are in the C-terminal, emphasizing how each specific phosphorylation site responds to alterations triggered by Cu+. The proposed mechanism involves the binding of Cu+ to the N-ATP7B, resulting in structural changes that allow for phosphorylation at specific C-terminal sites [33].
5.5. Complex Regulation of ATP7B Trafficking and Phosphorylation by Kinases
The trafficking of ATP7B from the TGN to cytosolic vesicles was associated with PKD-mediated phosphorylations in response to Cu+ [37]. However, the Cu+-dependent multisite phosphorylation occurs even when ATP7B is restricted to the TGN, implying that the kinase(s) responsible would be found in this region when Cu+ is high. PKD is known for the constitutive regulation of carrier vesicle fission from the TGN, but its participation in the hyperphosphorylation of ATP7B remains to be further investigated [33]. Interestingly, in renal Hek293 cells, the movement of ATP7B to the apical membrane when stimulated by Cu+ does not always depend on phosphorylation [58]. Therefore, renal ATP7B is unlikely to mediate Cu+ export [62]. It suggests a complex connection between Cu+-induced phosphorylation and ATP7B trafficking. The substitution of amino acids at the C-terminal did not significantly impact how ATP7B moved in response to Cu+. While the N-ATP7B mutants display some degree of phosphorylation, they fail to hyperphosphorylate in response to Cu+ like wild-type ATP7B. It suggests that the N-ATP7B plays a significant role in Cu+-induced phosphorylation. In addition, disrupting all six MBDs at the N-ATP7B does not prevent apical trafficking triggered by Cu+ but does inhibit hyperphosphorylation in liver cells. One explanation would be the existence of other Cu+-binding sites in ATP7B. Cu+ binding could trigger conformational changes, exposing the C-terminal phosphorylation sites and the signal sequence for apical localization [33], but the molecular mechanism remains unclear.
6. Discussion
Copper plays a pivotal role in many biological processes essential for life. However, in excess, it leads to Cu+ toxicity via the generation of harmful ROS and interaction with proteins. Because of this dual role, the regulation of intracellular Cu+ levels should be strict. Cu+-ATPases are among the crucial enzymes responsible for Cu+ transport through biological membranes. The importance of these ATPases in human physiology is highlighted by genetic disorders like Menkes and Wilson diseases. The use of protein kinases such as p38 MAPK and JNK as new targets for treating Wilson disease has already been considered [42]. The molecular and cellular basis of Cu+ dysregulation can also exacerbate oxidative stress in any cell or tissue, which, in turn, could contribute to complications of other pathologies, for instance, diabetic kidney disease [63]. Understanding these copper-related diseases underscores the importance of maintaining proper copper homeostasis to prevent detrimental health outcomes.
More than one-third of all eukaryotic proteins undergo kinase-mediated phosphorylation, with every protein class being subject to this regulatory mechanism, affecting different aspects of protein function [64]. This review aimed to provide a detailed analysis of how Cu+-ATPases are regulated through phosphorylation by kinases. Studying the modulation of Cu+-ATPase and its function is essential for understanding copper homeostasis and developing therapies for diseases related to copper metabolism.
Although the literature on the modulation of Cu+-ATPase by kinases has increased over time, it is still limited and reveals a great complexity. The kinase-mediated phosphorylation of a P-type ATPase involves the presence of two different entities of phosphorylation: catalytic and regulatory. Conformational changes may mutually influence these events, thereby initiating long-range interactions among multiple domains of the protein [33,47,49,58].
Among all Cu+-ATPases, ATP7B was the most studied in kinase-mediated regulation. Different aspects were investigated, such as protein trafficking [33,56,58] and intramolecular dynamics characterized by long-range crosstalk between different domains of the pump [33,48,58]. ATP7B protein stability was also affected by kinase phosphorylation [37,60]. The work covering ATP7A focused on the Cu+-dependent phosphorylation impact on protein trafficking [49,50,52] and stability [53]. The modulation of the catalytic cycle of Cu+-ATPases by kinase-mediated phosphorylation was thoroughly described for CCC2 and ATP7B, with different protein kinases involved (PKA and PKD, respectively). Future experiments should be performed to clarify the enzymatic consequences of these phosphorylations.
Despite the knowledge that protein kinases modulate Cu+-ATPases, specific kinases involved have not been fully identified. So far, four kinases have been directly or indirectly involved with Cu+ transport regulation: PKA, PKC, PKD, and Akt. Other kinases are likely involved, so further research should focus on identifying and characterizing these additional kinases. Another aspect not considered is the direct influence of Cu+ on the activation of protein kinases since both Cu+ deficiency and excess can impact protein kinase activation [65,66]. The phosphorylation and activation of the MAPK Erk by MAPKK Mek1/2 requires Cu+ binding to the latter kinase [67]. This finding set the foreground for other studies, and the past decade has seen a rapid increase in reports describing the direct involvement of Cu+ with signaling pathways [68,69,70]. The direct correlation of Cu+ in protein kinase activation significantly impacts cell physiology. Fine adjustments in intracellular Cu+ levels by Cu+-ATPases may be crucial for these kinases’ regulation, showing a broader role for cellular copper homeostasis.
Recent cancer research has shown that disrupting cellular copper metabolism using Cu+ chelators can slow cell proliferation and tumor growth [71,72]. Cu+ is necessary for activating pro-oncogenic Erk1/2 and Akt signaling pathways, and using Cu+ chelators serves as an alternative or additional approach to traditional therapies that rely on pharmacological kinase inhibitors [67,68,73]. Although no gain-of-function mutations in Cu+-transporting proteins are associated with cancer, changes in the expression of Cu+ transporters directly affect cellular Cu+ levels. Thus, examining the expression profile of Cu+-ATPases, influenced by kinase-mediated phosphorylation [37], could provide valuable information about cancer clinical outcomes.
Considering the complexity of the pathogenesis of DM, it is vital to identify the factors that influence the onset of the disease. There has been a growing focus on studying the effect of Cu+ on the risk of diabetes [74,75]. It is important to note that Cu+ levels are not simply positively correlated with DM [63]. It involves various Cu+-related proteins, such as Cu+ chaperones, ATP7A [53], and ATP7B [76]. Additionally, copper chelators such as trientine have been researched in preclinical and clinical trials for diabetic kidney disease [77] and heart failure [78,79], respectively.
Cu+ ions play a crucial role in neuronal activation, synaptic transmission, myelination, and intracellular signaling in the central nervous system. Imbalanced copper levels in distinct regions of the human brain are related to dementia in Parkinson’s and Alzheimer’s diseases [80]; therefore, a copper-targeting strategy might be a potential therapeutic approach for these neurological disorders [81]. It is crucial to recognize that several challenges need to be addressed to develop safe and effective medications targeting copper. Understanding the active Cu+ transport by Cu+-ATPases and modulation by protein kinases should be a priority for advancing our understanding of cellular copper homeostasis.
In addition, imbalanced Cu+ levels lead to cell death through a novel mechanism called cuproptosis. In this process, demonstrated in livers from ATP7B−/− mice, excessive Cu+ binds and destabilizes Fe-S cluster proteins, causing the aggregation of lipoylated mitochondrial proteins that leads to cell death [82]. Protocols to induce cell death by increasing Cu+ levels and harnessing its therapeutic potential are still under development, and regulating Cu+-ATPases presents a promising alternative. Drugs that increase mitochondrial Cu+ and downregulate ATP7A [83] or Cu+-based nanocarriers containing ATP7A siRNA to promote Cu+ uptake and block Cu+ trafficking [84] induce cancer cell death via cuproptosis. Database screening and molecular docking analysis are significant approaches for small-molecule drug design that impact the cuproptosis. These examples highlight the clinical promise of targeting this mechanism [85,86].
7. Conclusions
In recent decades, significant progress has been made in understanding the regulation of Cu+-ATPase and copper homeostasis, revealing an intricate and sophisticated landscape. Kinase-mediated phosphorylation plays a key role in modulating various aspects of Cu+-ATPase functioning and is associated with physiological and pathological conditions in different cell types. These phosphorylations, performed by ubiquitous protein kinases, directly impact the management of intracellular Cu+ while being responsive to its availability, highlighting the dynamic nature of these processes.
A comprehensive understanding of the complex dynamics of kinases and Cu+-ATPases is critical for understanding the roles of copper in human physiology. It is essential for elucidating the mechanisms underlying various diseases associated with copper dysregulation, not only in Wilson and Menkes diseases but also in metabolic and neurological disorder such as DM, Parkinson’s, and Alzheimer’s.
Author Contributions
Conceptualization, J.L. and R.H.F.V.; Writing—original draft preparation, review and editing, J.L. and R.H.F.V.; Funding acquisition, J.L. and R.H.F.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), grant number E-26/210.147/2023.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge Stéphanie Chauvin for Figures illustration.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Malmström, B.G.; Leckner, J. The chemical biology of copper. Curr. Opin. Chem. Biol. 1998, 2, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Occurrence of copper proteins through the three domains of life: A bioinformatic approach. J. Proteome Res. 2008, 7, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Festa, R.A.; Thiele, D.J. Copper: An essential metal in biology. Curr. Biol. 2011, 21, R877–R883. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.; Liu, L.; Wang, Q.; Saleem, M.H.; Bashir, S.; Ullah, S.; Peng, D. Copper environmental toxicology, recent advances, and future outlook: A review. Environ. Sci. Pollut. Res. Int. 2019, 26, 18003–18016. [Google Scholar] [CrossRef]
- Holmberg, C.G.; Laurell, C.B. Investigations in serum copper. II. Isolation of the copper-containing protein, and a description of some of its properties. Acta Chem. Scand. 1948, 5, 476–478. [Google Scholar] [CrossRef]
- Vander Wende, C.; Wainio, W.W. The state of the copper in cytochrome c oxidase. J. Biol. Chem. 1960, 235, PC11–PC12. [Google Scholar] [CrossRef] [PubMed]
- Udenfriend, S.; Cooper, J.R. The enzymatic conversion of phenylalanine to tyrosine. J. Biol. Chem. 1952, 194, 503–511. [Google Scholar] [CrossRef]
- Harris, E.D.; Gonnerman, W.A.; Savage, J.E.; O’Dell, B.L. Connective tissue amine oxidase. II. Purification and partial characterization of lysyl oxidase from chick aorta. Biochim. Biophys. Acta 1974, 341, 332–344. [Google Scholar] [CrossRef]
- Eipper, B.A.; Milgram, S.L.; Husten, E.J.; Yun, H.Y.; Mains, R.E. Peptidylglycine α-amidating monooxygenase: A multifunctional protein with catalytic, processing, and routing domains. Protein Sci. 1993, 2, 489–497. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Marklund, S.L. Human copper-containing superoxide dismutase of high molecular weight. Proc. Natl. Acad. Sci. USA 1982, 79, 7634–7638. [Google Scholar] [CrossRef]
- Matoba, Y.; Kumagai, T.; Yamamoto, A.; Yoshitsu, H.; Sugiyama, M. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 2006, 281, 8981–8990. [Google Scholar] [CrossRef]
- Yoshida, Y.; Furuta, S.; Niki, E. Effects of metal chelating agents on the oxidation of lipids induced by copper and iron. Biochim. Biophys. Acta 1993, 1210, 81–88. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pase, L.; Voskoboinik, I.; Greenough, M.; Camakaris, J. Copper stimulates trafficking of a distinct pool of the Menkes copper ATPase (ATP7A) to the plasma membrane and diverts it into a rapid recycling pool. Biochem. J. 2004, 378, 1031–1037. [Google Scholar] [CrossRef]
- Nyasae, L.; Bustos, R.; Braiterman, L.; Eipper, B.; Hubbard, A. Dynamics of endogenous ATP7A (Menkes protein) in intestinal epithelial cells: Copper-dependent redistribution between two intracellular sites. Am. J. Phys. Gastr. Liver Physiol. 2007, 292, G1181–G1194. [Google Scholar] [CrossRef]
- Inesi, G. Calcium and copper transport ATPases: Analogies and diversities in transduction and signaling mechanisms. J. Cell Commun. Signal. 2011, 5, 227–237. [Google Scholar] [CrossRef]
- Lutsenko, S.; Kaplan, J.H. Organization of P-type ATPases: Significance of structural diversity. Biochemistry 1995, 34, 15607–15613. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Biology, structure and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295. [Google Scholar] [CrossRef]
- Silver, S.; Nucifora, G.; Phung, L.T. Human Menkes X-chromosome disease and the staphylococcal cadmium-resistance ATPase: A remarkable similarity in protein sequences. Mol. Microb. 1993, 10, 7–12. [Google Scholar] [CrossRef]
- Odermatt, A.; Suter, H.; Krapf, R.; Solioz, M. Primary structure of two P-type ATPases involved in copper homeostasis in Enterococcus hirae. J. Biol. Chem. 1993, 268, 12775–12779. [Google Scholar] [CrossRef]
- Fu, D.; Beeler, T.J.; Dunn, T.M. Sequence, mapping and disruption of CCC2, a gene that cross-complements the Ca2+-sensitive phenotype of csg1 mutants and encodes a P-type ATPase belonging to the Cu2+-ATPase subfamily. Yeast 1995, 11, 283–292. [Google Scholar] [CrossRef]
- Chelly, J.; Tümer, Z.; Tønnesen, T.; Petterson, A.; Ishikawa-Brush, Y.; Tommerup, N.; Horn, N.; Monaco, A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993, 3, 14–19. [Google Scholar] [CrossRef]
- Mercer, J.F.; Livingston, J.; Hall, B.; Paynter, J.A.; Begy, C.; Chandrasekharappa, S.; Lockhart, P.; Grimes, A.; Bhave, M.; Siemieniak, D. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet. 1993, 3, 20–25. [Google Scholar] [CrossRef]
- Vulpe, C.; Levinson, B.; Whitney, S.; Packman, S.; Gitschier, J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993, 3, 7–13. [Google Scholar] [CrossRef]
- Bull, P.C.; Thomas, G.R.; Rommens, J.M.; Forbes, J.R.; Cox, D.W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 1993, 5, 327–337. [Google Scholar] [CrossRef]
- Petrukhin, K.; Fischer, S.G.; Pirastu, M.; Tanzi, R.E.; Chernov, I.; Devoto, M.; Brzustowicz, L.M.; Cayans, E.; Vitale, E.; Russo, J.J.; et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat. Genet. 1993, 5, 338–343. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Parano, E.; Pavone, L.; Brzustowicz, L.M. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef]
- Maung, M.T.; Carlson, A.; Olea-Flores, M.; Elkhadragy, L.; Schachtschneider, K.M.; Navarro-Tito, N.; Padilla-Benavides, T. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB 2021, 35, e21810. [Google Scholar] [CrossRef]
- Poulsen, H.; Morth, P.; Egebjerg, J.; Nissen, P. Phosphorylation of the Na+,K+-ATPase and the H+,K+-ATPase. FEBS Lett. 2010, 584, 2589–2595. [Google Scholar] [CrossRef]
- Vagin, O.; Turdikulova, S.; Sachs, G. The H,K-ATPase beta subunit as a model to study the role of N-glycosylation in membrane trafficking and apical sorting. J. Biol. Chem. 2004, 279, 39026–39034. [Google Scholar] [CrossRef]
- Veldhuis, N.A.; Gaeth, A.P.; Pearson, R.B.; Gabriel, K.; Camakaris, J. The multi-layered regulation of copper translocating P-type ATPases. Biometals 2009, 22, 177–190. [Google Scholar] [CrossRef]
- Braiterman, L.T.; Gupta, A.; Chaerkady, R.; Cole, R.N.; Hubbard, A.L. Communication between the N and C termini is required for copper-stimulated Ser/Thr phosphorylation of Cu(I)-ATPase (ATP7B). J. Biol. Chem. 2015, 290, 8803–8819. [Google Scholar] [CrossRef]
- Liu, Y.; Pilankatta, R.; Hatori, Y.; Lewis, D.; Inesi, G. Comparative features of copper ATPases ATP7A and ATP7B heterologously expressed in COS-1 cells. Biochemistry 2010, 49, 10006–10012. [Google Scholar] [CrossRef]
- Di Leva, F.; Domi, T.; Fedrizzi, L.; Lim, D.; Carafoli, E. The plasma membrane Ca2+-ATPase of animal cells: Structure, function and regulation. Arch. Biochem. Biophys. 2008, 476, 65–74. [Google Scholar] [CrossRef]
- Cobbold, C.; Ponnambalam, S.; Francis, M.J.; Monaco, A.P. Novel membrane traffic steps regulate the exocytosis of the Menkes disease ATPase. Hum. Mol. Genet. 2002, 11, 2855–2866. [Google Scholar] [CrossRef]
- Pilankatta, R.; Lewis, D.; Inesi, G. Involvement of protein kinase D in expression and trafficking of ATP7B (copper ATPase). J. Biol. Chem. 2011, 286, 7389–7396. [Google Scholar] [CrossRef]
- Wakana, Y.; Campelo, F. The PKD-Dependent Biogenesis of TGN-to-Plasma Membrane Transport Carriers. Cells 2021, 10, 1618. [Google Scholar] [CrossRef]
- Hilário-Souza, E.; Cuillel, M.; Mintz, E.; Charbonnier, P.; Vieyra, A.; Cassio, D.; Lowe, J. Modulation of hepatic copper-ATPase activity by insulin and glucagon involves protein kinase A (PKA) signaling pathway. Biochim. Biophys. Acta 2016, 1862, 2086–2097. [Google Scholar] [CrossRef] [PubMed]
- Afton, S.E.; Caruso, J.A.; Britigan, B.E.; Qin, Z. Copper egress is induced by PMA in human THP-1 monocytic cell line. Biometals 2009, 22, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, L.H.; Britto-Borges, T.; Vieyra, A.; Lowe, J. ATP7B activity is stimulated by PKCe in porcine liver. Int. J. Biochem. Cell Biol. 2014, 54, 60–67. [Google Scholar] [CrossRef][Green Version]
- Chesi, G.; Hegde, R.N.; Iacobacci, S.; Concilli, M.; Parashuraman, S.; Festa, B.P.; Polishchuk, E.V.; Di Tullio, G.; Carissimo, A.; Montefusco, S.; et al. Identification of p38 MAPK and JNK as new targets for correction of Wilson disease-causing ATP7B mutants. Hepatology 2016, 63, 1842–1859. [Google Scholar] [CrossRef]
- Park, G.B.; Choi, Y.; Kim, Y.S.; Lee, H.K.; Kim, D.; Hur, D.Y. ROS-mediated JNK/p38-MAPK activation regulates Bax translocation in Sorafenib-induced apoptosis of EBV-transformed B cells. Int. J. Oncol. 2014, 44, 977–985. [Google Scholar] [CrossRef]
- Hardman, B.; Michalczyk, A.; Greenough, M.; Camakaris, J.; Mercer, J.F.; Ackland, M.L. Hormonal regulation of the Menkes and Wilson copper-transporting ATPases in human placental Jeg-3 cells. Biochem. J. 2007, 402, 241–250. [Google Scholar] [CrossRef]
- Schlief, M.L.; West, T.; Craig, A.M.; Holtzman, D.M.; Gitlin, J.D. Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc. Natl. Acad. Sci. USA 2006, 103, 14919–14924. [Google Scholar] [CrossRef]
- Valverde, R.H.; Morin, I.; Lowe, J.; Mintz, E.; Cuillel, M.; Vieyra, A. Cyclic AMP-dependent protein kinase controls energy interconversion during the catalytic cycle of the yeast copper-ATPase. FEBS Lett. 2008, 582, 891–895. [Google Scholar] [CrossRef]
- Valverde, R.H.; Britto-Borges, T.; Lowe, J.; Einicker-Lamas, M.; Mintz, E.; Cuillel, M.; Vieyra, A. Two serine residues control sequential steps during catalysis of the yeast copper ATPase through different mechanisms that involve kinase-mediated phosphorylations. J. Biol. Chem. 2011, 286, 6879–6889. [Google Scholar] [CrossRef]
- Pilankatta, R.; Lewis, D.; Adams, C.M.; Inesi, G. High yield heterologous expression of wild-type and mutant Cu+-ATPase (ATP7B, Wilson disease protein) for functional characterization of catalytic activity and serine residues undergoing copper-dependent phosphorylation. J. Biol. Chem. 2009, 284, 21307–21316. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Fernando, R.; Veldhuis, N.; Hannan, K.M.; Marmy-Conus, N.; Pearson, R.B.; Camakaris, J. Protein kinase-dependent phosphorylation of the Menkes copper P-type ATPase. Biochem. Biophys. Res. Commun. 2003, 303, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Veldhuis, N.A.; Kuiper, M.J.; Dobson, R.C.; Pearson, R.B.; Camakaris, J. In silico modeling of the Menkes copper-translocating P-type ATPase 3rd metal binding domain predicts that phosphorylation regulates copper-binding. Biomet. Int. J. Role Met. Ions Biol. Biochem. Med. 2011, 24, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Holloway, Z.G.; Velayos-Baeza, A.; Howell, G.J.; Levecque, C.; Ponnambalam, S.; Sztul, E.; Monaco, A.P. Trafficking of the Menkes copper transporter ATP7A is regulated by clathrin-, AP-2-, AP-1-, and Rab22-dependent steps. Mol. Biol. Cell 2013, 24, 1735–1748. [Google Scholar] [CrossRef]
- Zhu, S.; Shanbhag, V.; Hodgkinson, V.L.; Petris, M.J. Multiple di-leucines in the ATP7A copper transporter are required for retrograde trafficking to the trans-Golgi network. Metallomics 2016, 8, 993–1001. [Google Scholar] [CrossRef]
- Sudhahar, V.; Okur, M.N.; Bagi, Z.; O’Bryan, J.P.; Hay, N.; Makino, A.; Patel, V.S.; Phillips, S.A.; Stepp, D.; Ushio-Fukai, M.; et al. Akt2 (Protein Kinase B Beta) Stabilizes ATP7A, a Copper Transporter for Extracellular Superoxide Dismutase, in Vascular Smooth Muscle: Novel Mechanism to Limit Endothelial Dysfunction in Type 2 Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 529–541. [Google Scholar] [CrossRef]
- Vanderwerf, S.M.; Cooper, M.J.; Stetsenko, I.V.; Lutsenko, S. Copper specifically regulates intracellular phosphorylation of the Wilson’s disease protein, a human copper-transporting ATPase. J. Biol. Chem. 2001, 276, 36289–36294. [Google Scholar] [CrossRef]
- Tsivkovskii, R.; Eisses, J.F.; Kaplan, J.H.; Lutsenko, S. Functional properties of the copper-transporting ATPase ATP7B (the Wilson’s disease protein) expressed in insect cells. J. Biol. Chem. 2002, 277, 976–983. [Google Scholar] [CrossRef]
- Bartee, M.Y.; Ralle, M.; Lutsenko, S. The loop connecting metal-binding domains 3 and 4 of ATP7B is a target of a kinase-mediated phosphorylation. Biochemistry 2009, 48, 5573–5581. [Google Scholar] [CrossRef]
- Ruturaj Mishra, M.; Saha, S.; Maji, S.; Rodriguez-Boulan, E.; Schreiner, R.; Gupta, A. Regulation of the apico-basolateral trafficking polarity of the homologous copper-ATPases ATP7A and ATP7B. J. Cell Sci. 2024, 137, jcs261258. [Google Scholar] [CrossRef]
- Hasan, N.M.; Gupta, A.; Polishchuk, E.; Yu, C.H.; Polishchuk, R.; Dmitriev, O.Y.; Lutsenko, S. Molecular events initiating exit of a copper-transporting ATPase ATP7B from the trans-Golgi network. J. Biol. Chem. 2012, 287, 36041–36050. [Google Scholar] [CrossRef]
- Li, Y.Q.; Yin, J.Y.; Liu, Z.Q.; Li, X.P. Copper efflux transporters ATP7A and ATP7B: Novel biomarkers for platinum drug resistance and targets for therapy. IUBMB Life 2018, 70, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Janardhanan, P.; Somasundaran, A.K.; Balakrishnan, A.J.; Pilankatta, R. Sensitization of cancer cells towards Cisplatin and Carboplatin by protein kinase D inhibitors through modulation of ATP7A/B (copper transport ATPases). Cancer Treat. Res. Commun. 2022, 32, 100613. [Google Scholar] [CrossRef] [PubMed]
- Vanderwerf, S.M.; Lutsenko, S. The Wilson’s disease protein expressed in Sf9 cells is phosphorylated. Biochem. Soc. Trans. 2002, 30, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.; Bartee, M.Y.; Braiterman, L.; Gupta, A.; Ustiyan, V.; Zuzel, V.; Kaplan, J.H.; Hubbard, A.L.; Lutsenko, S. Cell-specific trafficking suggests a new role for renal ATP7B in the intracellular copper storage. Traffic 2009, 10, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Gembillo, G.; Labbozzetta, V.; Giuffrida, A.E.; Peritore, L.; Calabrese, V.; Spinella, C.; Stancanelli, M.R.; Spallino, E.; Visconti, L.; Santoro, D. Potential Role of Copper in Diabetes and Diabetic Kidney Disease. Metabolites 2023, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Plattner, F.; Bibb, J. Serine and Threonine Phosphorylation. In Basic Neurochemistry, 8th ed.; Brady, S.T., Siegel, G.J., Albers, R.W., Price, D.L., Eds.; Academic Press: Cambridge, MA, USA, 2012; pp. 467–492. ISBN 9780123749475. [Google Scholar] [CrossRef]
- Samet, J.M.; Graves, L.M.; Quay, J.; Dailey, L.A.; Devlin, R.B.; Ghio, A.J.; Wu, W.; Bromberg, P.A.; Reed, W. Activation of MAPKs in human bronchial epithelial cells exposed to metals. Am. J. Physiol. 1998, 275, L551–L558. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Ostrakhovitch, E.A.; Walter, P.L.; Kampkötter, A.; Klotz, L.O. Stimulation of phosphoinositide 3-kinase/Akt signaling by copper and zinc ions: Mechanisms and consequences. Arch. Biochem. Biophys. 2007, 463, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Cheng, J.; Zheng, N.; Zhang, X.; Dai, X.; Zhang, L.; Hu, C.; Wu, X.; Jiang, Q.; Wu, D.; et al. Copper Promotes Tumorigenesis by Activating the PDK1-AKT Oncogenic Pathway in a Copper Transporter 1 Dependent Manner. Adv. Sci. 2021, 8, e2004303. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, J.E.; Li, R.; Tsang, T.; Alfaran, F.H.; Dick, A.; Cocklin, S.; Brady, D.C.; Strochlic, T.I. Copper Modulates the Catalytic Activity of Protein Kinase CK2. Front. Mol. Biosci. 2022, 9, 878652. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.; Posimo, J.M.; Gudiel, A.A.; Cicchini, M.; Feldser, D.M.; Brady, D.C. Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma. Nat. Cell Biol. 2020, 22, 412–424. [Google Scholar] [CrossRef]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef]
- Brady, D.C.; Crowe, M.S.; Greenberg, D.N.; Counter, C.M. Copper Chelation Inhibits BRAFV600E-Driven Melanomagenesis and Counters Resistance to BRAFV600E and MEK1/2 Inhibitors. Cancer Res. 2017, 77, 6240–6252. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Casio, M.; Range, D.E.; Sosa, J.A.; Counter, C.M. Copper Chelation as Targeted Therapy in a Mouse Model of Oncogenic BRAF-Driven Papillary Thyroid Cancer. Clin. Cancer Res. 2018, 24, 4271–4281. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; Taveira-da-Silva, R.; Hilário-Souza, E. Dissecting copper homeostasis in diabetes mellitus. IUBMB Life 2017, 69, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Chen, L.; Kong, Y.; Zhuo, J.F.; Sun, Q.; Chang, J. The association between serum copper concentration and prevalence of diabetes among US adults with hypertension (NHANES 2011–2016). J. Cell Mol. Med. 2024, 28, e18270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, H.; Amarsingh, G.V.; Cheung, C.C.H.; Hogl, S.; Narayanan, U.; Zhang, L.; McHarg, S.; Xu, J.; Gong, D.; et al. Diabetic cardiomyopathy is associated with defective myocellular copper regulation and both defects are rectified by divalent copper chelation. Cardiovasc. Diabetol. 2014, 13, 100. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Lu, J.; Chen, X.; Reddy, S.; Crossman, D.J.; Glyn-Jones, S.; Choong, Y.S.; Kennedy, J.; Barry, B.; Zhang, S.; et al. A copper(II)-selective chelator ameliorates diabetes-evoked renal fibrosis and albuminuria, and suppresses pathogenic TGF-beta activation in the kidneys of rats used as a model of diabetes. Diabetologia 2008, 51, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Farrant, J.; Dodd, S.; Vaughan, C.; Reid, A.; Schmitt, M.; Garratt, C.; Akhtar, M.; Mahmod, M.; Neubauer, S.; Cooper, R.M.; et al. TEMPEST investigators. Rationale and design of a randomised trial of trientine in patients with hypertrophic cardiomyopathy. Heart 2023, 109, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Januzzi, J.; Butler, J.; Cleland, J.; Felker, M.; Mentz, R.; Wang, Y.; Zhang, Y.; Mou, H.; Yu, J.; Guo, L.; et al. Effect Of Trientine-hydrochloride In Heart Failure With Lower Left Ventricular Ejection Fraction: The TRACER-HF Trial. J. Card. Fail. 2024, 30, 314–315. [Google Scholar] [CrossRef]
- Scholefield, M.; Church, S.J.; Xu, J.; Patassini, S.; Roncaroli, F.; Hooper, N.M.; Unwin, R.D.; Cooper, G.J.S. Widespread Decreases in Cerebral Copper Are Common to Parkinson’s Disease Dementia and Alzheimer’s Disease Dementia. Front. Aging Neurosci. 2021, 13, 641222. [Google Scholar] [CrossRef]
- Patwa, J.; Flora, S.J.S. Copper: From enigma to therapeutic target for neurological disorder. Basic Clin. Pharmacol. Toxicol. 2024, 134, 778–791. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Huang, Z.; Duan, J.; Nice, E.C.; Lin, J.; Huang, C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol. Oncol. 2021, 15, 3527–3544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, M.; Zhang, J.; Abdalla, M.; Sun, P.; Yang, Z.; Zhang, C.; Liu, Y.; Chen, C.; Jiang, X. Syphilis mimetic nanoparticles for cuproptosis-based synergistic cancer therapy via reprogramming copper metabolism. Int. J. Pharm. 2023, 640, 123025. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Zhang, J.; Yang, Y.; Fleishman, J.S.; Wang, Y.; Wang, J.; Chen, J.; Li, Y.; Wang, H. Cuproptosis: A novel therapeutic target for overcoming cancer drug resistance. Drug Resist. Updates 2024, 72, 101018. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Huang, L.; Gan, Y.; Xia, Y.; Yu, W. The Molecular Mechanisms of Cuproptosis and Small-Molecule Drug Design in Diabetes Mellitus. Molecules 2024, 29, 2852. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).