
Cyclic Peptides as Protein Kinase Modulators and Their Involvement in the Treatment of Diverse Human Diseases

 , ,
, , 
Abstract
1. Protein Kinases as Therapeutic Targets

1.1. Protein Kinase Inhibitors
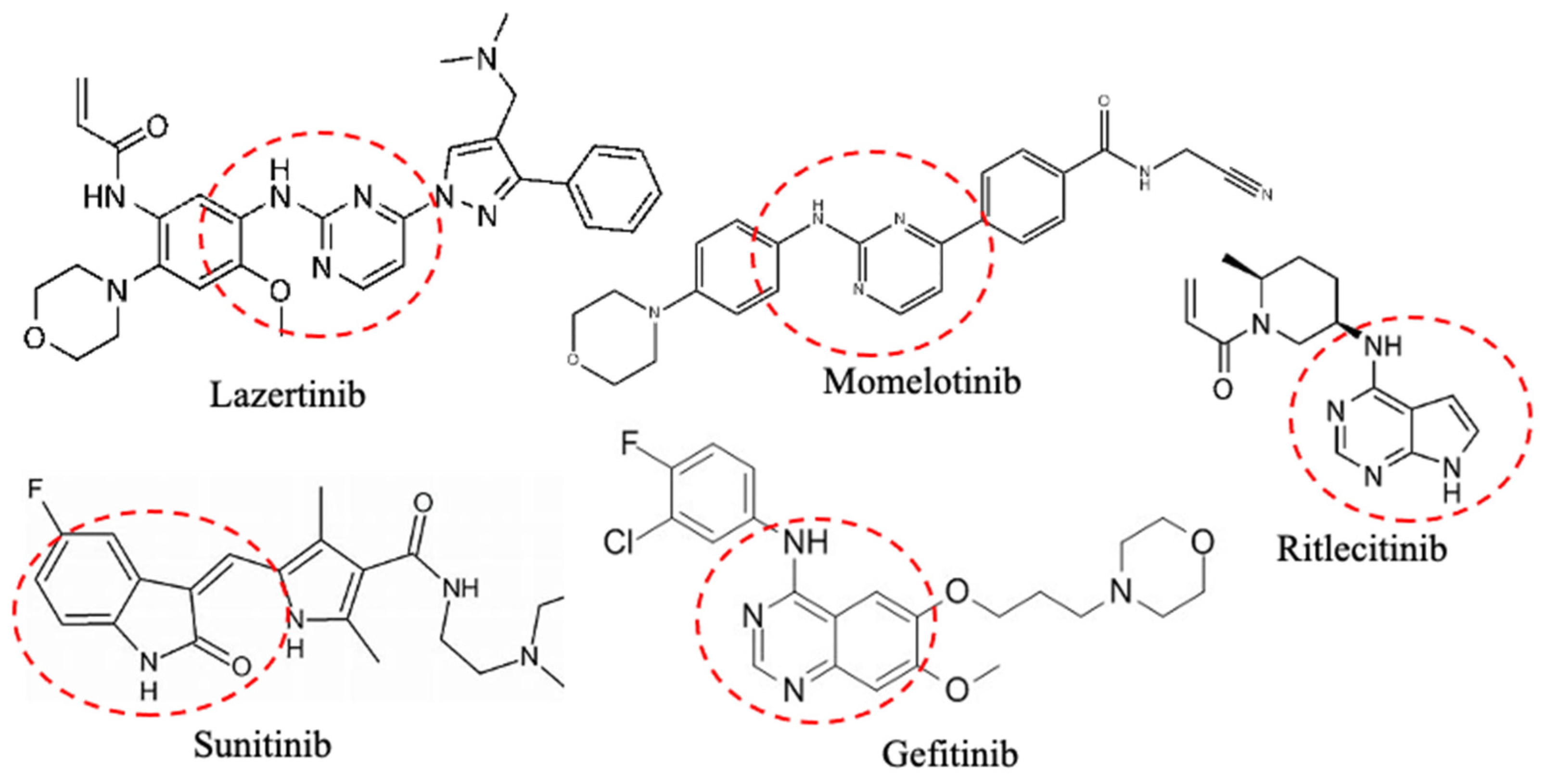
1.2. Small Protein Kinase Inhibitors Approved by FDA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Targets | Drug | Disease | References |
|---|---|---|---|
| CDK4/6 | Abemaciclib Palbociclib Ribociclib Trilaciclib | Breast cancer and myelosupression | [41,42,43,44] |
| AKT | Capivasertib | Breast cancer | [45] |
| HER2 | Lapatinib # Neratinib # Tucatinib # | Breast cancer | [46,47,48] |
| ErbB1/2/4 | Afatinib Lapatinib Neratinib Tucatinib | Non-small-cell lung cancer (NSCLC), breast cancer, and colon cancer | [47,49,50,51] |
| FKBP12/mTOR | Sirolimus Everolimus * | Breast cancer and * lymphangioleiomyomatosis | [52,53] |
| MEK1/2 | Binimetinib Cobimetinib Selumetinib | Melanoma and neurofibromatosis | [54,55,56] |
| BCR-Abl | Dasatinib # Imatinib # Nilotinib # Ponatinib # Asciminib Bosutinib # | Chronic myeloid leukemia (CML), and acute lymphocytic leukemia (ALL) | [57,58,59] |
| Flt3 | Gilteritinib Midostaurin # Quizatinib | Leukemia | [60,61,62] |
| BRAF | Dabrafenib Encorafenib Vemurafenib Tovorafenib ** | Melanoma | [63] |
| JAK1/2/3 | Abrocitinib * Momelotinib Pacritinib Baricitinib * Fedratinib Ritlecitinib * Tofacitinib * Upadacitibib * | * Atopic dermatitis, myelofibrosis, * rheumatoid arthritis, * alopecia areata, and * ulcerative colitis | [64,65,66,67,68,69] |
| ALK | Lorlatinib Alectinib Brigatinib # Ceritinib # Crizotinib # Entrectinib # | ALK-positive NSCLC, and inflammatory myofibroblastic tumor | [70,71] |
| PDGFR | Midostaurin # Ripretinib # Avapritinib # | Acute myeloid leukemia, and gastrointestinal stromal tumors | [72,73,74] |
| VEGFR 1/2/3 | Axitinib Cabozantinib # Fruquintinib Lenvatinib # Pazopanib # Regorafenib # Sorafenib # Sunitinib # Tivozanib # Vandetanib # | Advanced renal cell carcinoma, advanced medullary thyroid cancer, renal cell and hepatocellular carcinoma, metastatic colorectal cancer, differentiated thyroid cancer, gastrointestinal stromal, and pancreatic neuroendocrine tumors | [75,76,77,78,79] |
| ROCK2 | Belumosudil * | * Graft vs. host disease | [80] |
| Kit | Pexidartinib # Ripretinib # Avapritinib # | Tenosynovial giant cell tumors, and gastrointestinal stromal tumors | [81,82] |
| MET | Capmatinib Crizotinib # Tepotinib | NSCLC, anaplastic large cell lymphoma, inflammatory myofibroblastic tumor, and MET mutant NSCLC | [83] |
| EGFR | Dacomitinib # Erlotinib Gefitinib Mobcertinib Osimertinib Lazertinib ** | EGFR-mutant NSCLC, NSCLC, and pancreatic cancer | [84,85] |
| TYK2 | Deucravacitinib * | * Psoriasis | [86] |
| TRKA/B/C | Entrectinib Larotrectinib | Solid tumors with NTRK fusion proteins | [87] |
| ROS1 | Repotrectinib Entrectinib # | ROS1-positive NSCLC | [88] |
| FGFR1/2/3/4 | Erdafitinib Futibatinib Infigratinib Nintedanib Pemigatinib | Urothelial bladder cancer, bile duct cancer, and idiopathic pulmonary fibrosis | [89,90,91,92] |
| BTK | Ibrutinib Pirtobrutinib Zanubrutinib | Chronic lymphocytic leukemia (CLL), and small lymphocytic lymphoma | [93] |
| SYK | Fostamatinib | Chronic immune thrombocytopenia | [94] |
| T970M | Osimertinib # | NSCLC with exon 19 deletions or exon 21 substitutions | [95] |
| CSF1R | Pexidartinib # | Tenosynovial giant cell tumors | [96] |
| Axl | Sunitinib Cabozantinib | Renal cell carcinoma | [97,98] |
2. Cyclic Peptides as Small-Molecule Therapies Through Human Protein Kinase Inhibitors
Cyclic Peptides as Therapeutic Tools: Kinase Inhibition and Biomedical Applications
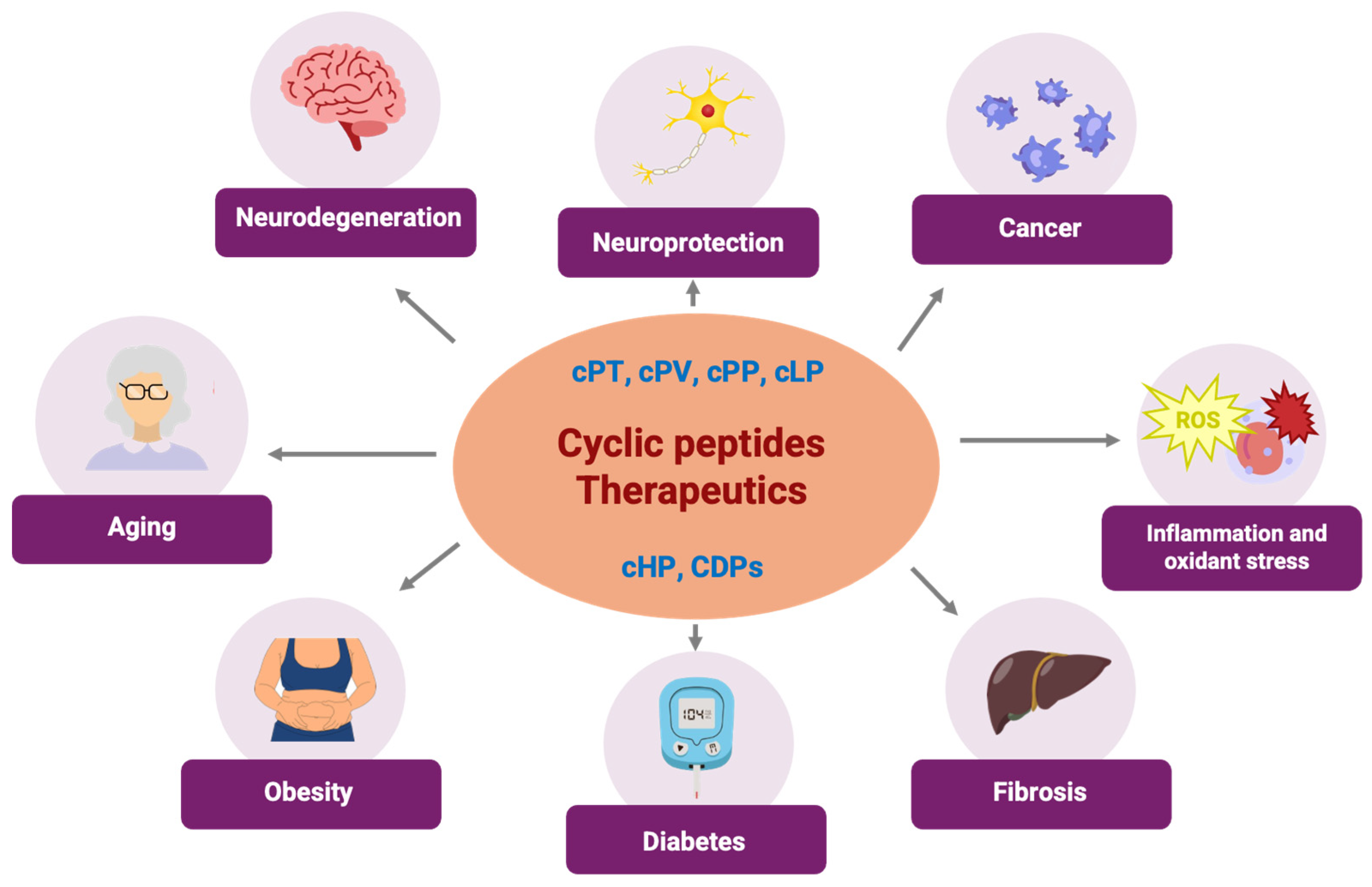
3. Cyclodipeptides, a Type of Cyclic Peptides with Potential Use in Human Diseases That Are Protein-Kinase-Associated
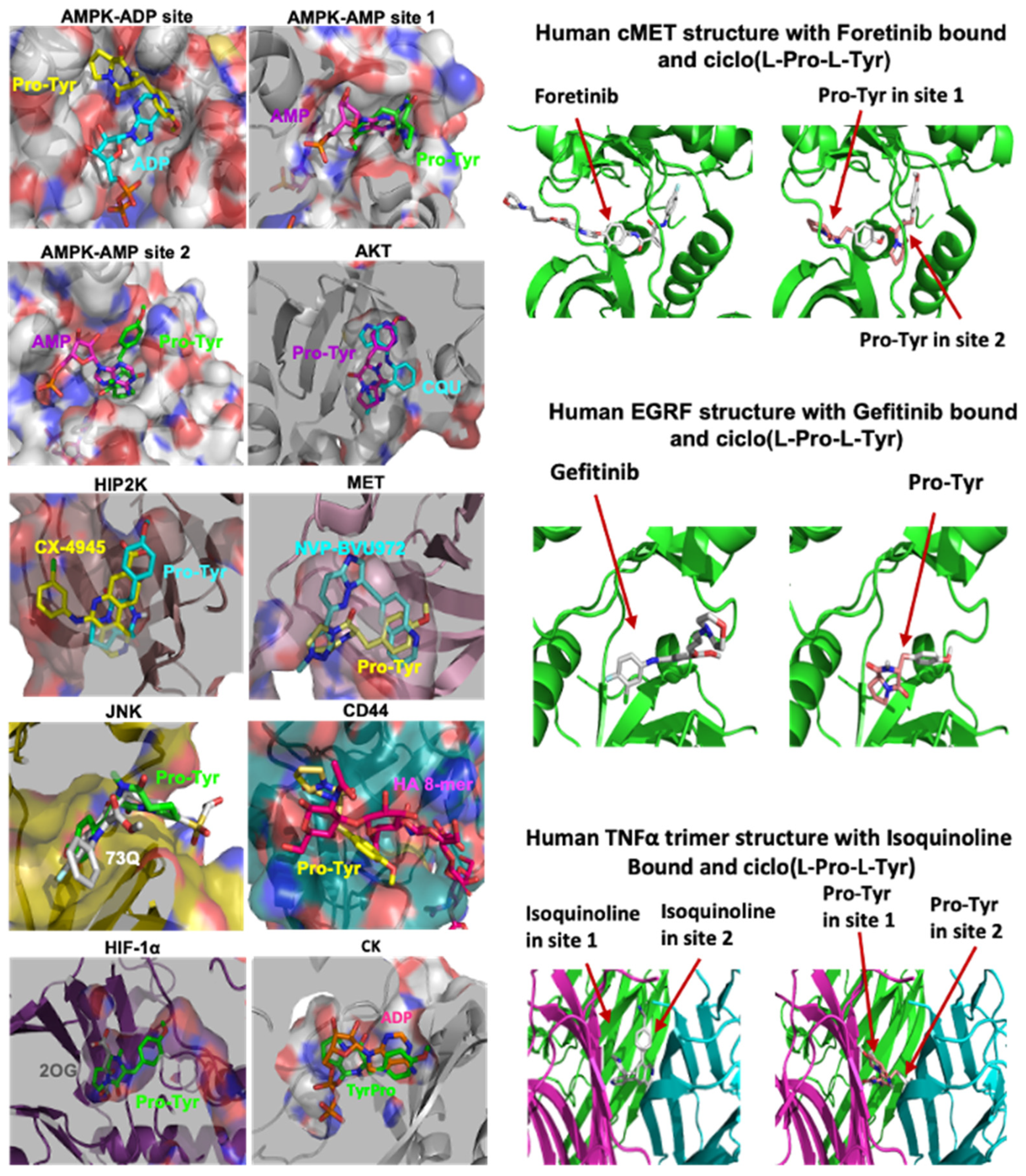
3.1. Cyclodipeptide and Protein Kinase Interaction in Cancer
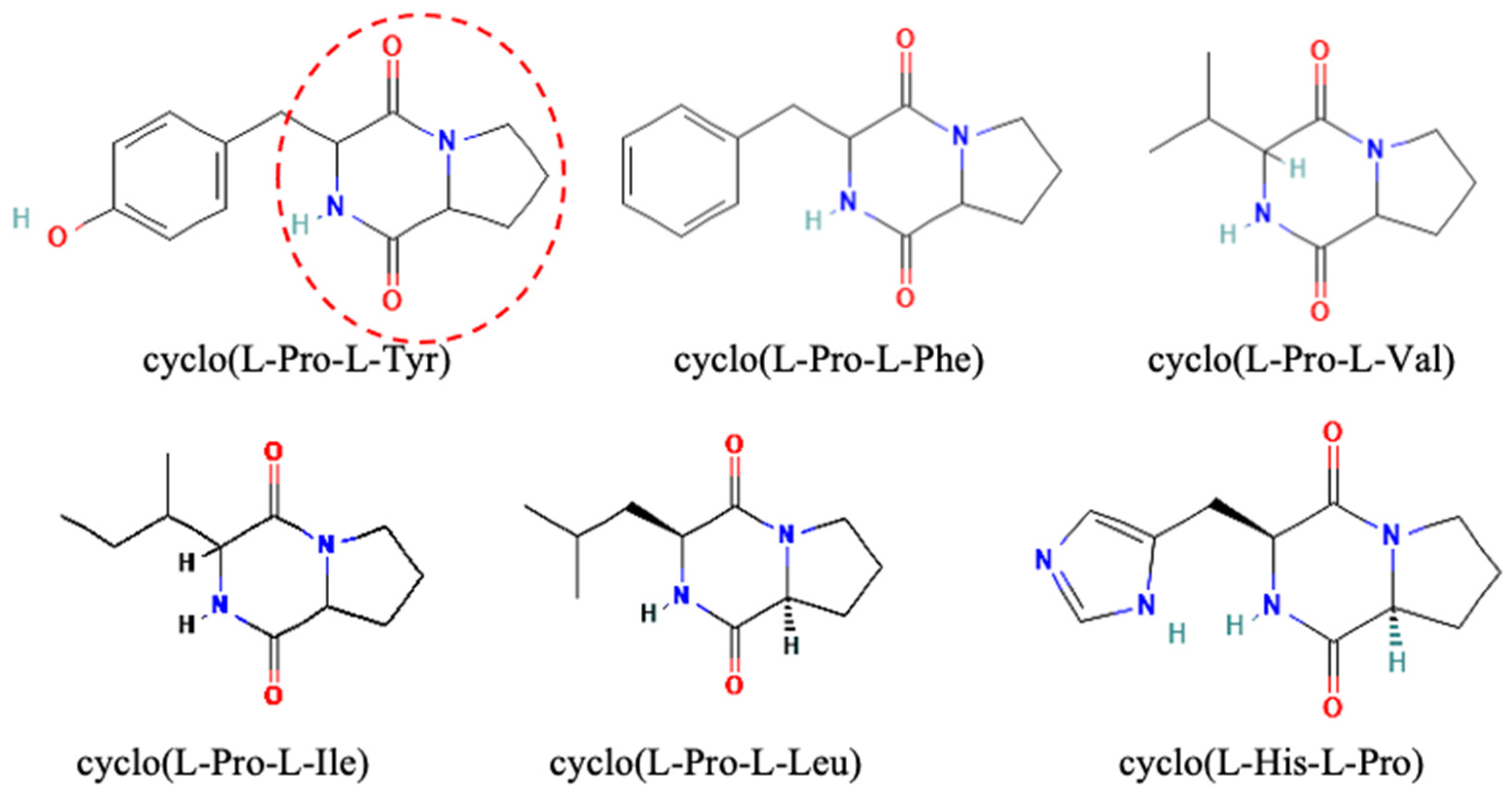
3.2. Cyclo(Pro-Tyr)
3.3. Cyclo(L-Leu-L-Pro)
3.4. Cyclodipeptide–Protein Kinase Interaction Related to Antioxidant and Anti-Inflammatory Effects
3.5. Cyclodipeptide–Protein Kinase Interaction Related to Neuroprotective Effects
3.6. Cyclodipeptide–Protein Kinase Interaction Related to Fibrosis
3.7. Cyclodipeptide–Protein Kinase Interaction Related to Aging
3.8. Cyclodipeptide–Protein Kinase Interaction Related to Diabetes
4. Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABL | Abelson kinase |
| AGEs | advanced glycation end products |
| AKT | protein kinase B |
| AMPK | AMP-activated protein kinase |
| ALK | anaplastic lymphoma kinase |
| ALL | acute lymphocytic leukemia |
| Arg | arginine |
| ATP | adenosine triphosphate |
| Axl | axl gene |
| Bax | BCL2-associated X protein |
| Bcl2 | B-cell lymphoma 2 protein |
| BCR | breakpoint cluster region |
| BCR-ABL | fusion protein of BCR gene and ABL gene |
| BDNF | brain derived neurotrophic factor |
| BRAF | v-raf murine sarcoma viral oncogene homolog B |
| BTK | Bruton’s tyrosine kinase |
| CCN2 | cellular communication network factor 2 |
| Cdc42 | cell division cycle 42 |
| CDK | Cyclin-dependent kinase |
| CDPs | cyclodipeptides |
| CDPs-PA | Pseudomonas aeruginosa cPT, cPV and cPP |
| cHP | cyclo(His-Pro) |
| CK | creatine kinase |
| CK2 | casein kinase 2 |
| CKD | chronic kidney disease |
| cLP | cyclo(L-Leu-L-Pro) |
| CML | carboxymethyl lysine |
| COL1A | collagen type I alpha 1 chain |
| CPP | cell-penetrating peptide |
| cPP | cyclo(L-Pro-L-Phe) |
| cPT | cyclo(L-Pro-L-Tyr) |
| cPV | cyclo(L-Pro-L-Val) |
| cRGDfK | cyclic-pentapeptide (-Arg-Gly-Asp-D-Phe-Lys) |
| cRGDyK | cyclic-pentapeptide (-Arg-Gly-Asp-D-Tyr-Lys) |
| CSC | cancer stem cells |
| CSF1R | colony-stimulating factor 1 receptor |
| cycloZ | cyclo(His-Pro) and zinc |
| DEN | N-diethylnitrosamine |
| DFG | Asp-Phe-Gly motif |
| dNa1 | d-b-naphthylalanine |
| EC50 | half maximal effective concentration |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial-mesenchymal transition |
| ePK | eucaryotic catalytic domain |
| ErbB | erb-b tyrosine kinase receptor |
| ERK1/2 | extracellular signal-regulated kinase |
| FAS | tumor necrosis factor receptor superfamily member 6 (TNFRSF6) |
| FDA | food and drug administration |
| FGF | Fibroblast Growth Factor |
| FGFR | fibroblast growth factor receptor |
| FKBP12 | FK506-binding protein 12 |
| Flt3 | fms related tyrosine kinase 3 |
| Fpa | 1-4-fluorophenylalanine |
| G12D | K-Ras gene |
| Gab1 | GRB2 associated binding protein 1 |
| GRTH | gonadotropin-regulated testicular RNA helicase |
| GSH | glutathione |
| HCC | hepatocellular carcinoma |
| HCD | Higher-energy collisional dissociation |
| hEGF-Hegfr | human EGF receptor bound ligand |
| HER2 | epidermal factor receptor 2 |
| HGF | hepatocyte growth factor |
| HIF-1 α | hypoxia-inducible factor 1-alpha |
| HiP-8 | HGF-inhibitory peptide-8 |
| HIP2K | homeodomain-interacting protein kinase 2 |
| HIV | human immunodeficiency virus |
| H-Ras | Harvey rat sarcoma viral oncogene homolog |
| IC50 | half-maximal inhibitory concentration |
| iNOS | inducible oxide nitric synthase |
| IRF-1 | interferon regulatory factor-1 |
| JAKs | Janus kinases |
| KD | dissociation constant |
| KIR | kinase inhibitory region |
| Kit | KIT proto-oncogene |
| KLIFS | kinase-ligand interaction fingerprint and structure |
| KLU | lytic peptide |
| K-Ras | Kirsten Ras oncogene homolog |
| LD50 | lethal dose 50 |
| LKB1 | liver kinase B1 |
| LMP | lysosomal membrane permeabilization |
| McoTI | Momordica cochinchinensis trypsin inhibitor |
| MEK | mitogen-activated protein kinase kinase 7 |
| MET | mesenchymal-epithelial transition factor |
| MF | intermediate-risk myelofibrosis |
| mRNAs | messenger ribonucleic acid |
| mTOR | mammalian target of rapamycin |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic fatty liver disease and steatohepatitis |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGF | nerve growth factor |
| NMDARs | N-methyl-D-aspartate receptors |
| nNOS | neuronal oxide synthase |
| Nrf2 | nuclear factor erythroid-related factor 2 |
| NSCLC | non-small cell lung cancer |
| OK2 | 7-mer cyclic peptide |
| P1GF | placental growth factor |
| P53 | Tumor Suppressor Protein p53 |
| PARP | Poly(ADP-ribose) polymerase |
| PBD | polo-box domain |
| PDGFR | platelet derived growth factor receptor |
| PDZ | binding domain PDZ |
| PGC-1alfa | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | phosphoinositide 3-kinase |
| PKA | protein kinase A |
| PKB | protein kinase B |
| PKC | protein kinase C |
| PKI | protein kinase inhibitors |
| Plk1 | polo like kinase |
| PPI | protein-protein interactions |
| PSD-95 | Postsynaptic density protein-95 |
| PTEN | phosphatase and tensin homolog |
| QR | VEGFR-targeting peptide VEGF125−136 |
| RA | rheumatoid arthritis |
| Rac1 | Rac family small GTPase 1 |
| RAF | RAF kinase |
| RAGE | receptor of advanced glycation end products |
| Ral-GDS | Ral guanine nucleotide dissociation stimulator |
| RBD | RAS binding domain |
| Ras-GDP | inactive GDP–bound state |
| Ras-GTP | active GTP-bound state |
| RGD | arginine-glycin-aspartic acid |
| ROCK | Rho kinase |
| ROS | reactive oxygen species |
| ROS1 | ROS proto-oncogene 1 receptor tyrosine kinase |
| ROSN | reactive oxygen-nitrogen species |
| RTK | receptor tyrosine kinase |
| SAPK | Stress-activated MAP kinases |
| SARS-CoV2 | severe acute respiratory syndrome coronavirus 2 |
| scHGF | single chain HGF |
| SFTI-1 | sunflower trypsin inhibitor |
| SH2 | Src homology 2 |
| SOCS | suppressors of cytokine signaling |
| Src | stored response chain kinase |
| STAT | signal transducer and activator of transcription |
| SYK | spleen tyrosine kinase |
| tcHGF | two chain HGF |
| TK | tyrosine kinase |
| TK-EGFR | tyrosine kinase domain of EGFR |
| TKI | tyrosine kinase inhibitor |
| TNBC | triple negative breast cancer |
| TRH | hypothalamic thyrotropin-releasing hormone |
| TrkA | Tropomyosin receptor kinase A |
| TRKs | tropomyosin receptor kinases |
| TyKs | tyrosin kinases |
| Val | valine |
| VEGFs | vascular endothelial growth factors |
| VEGFRs | vascular endothelial growth factor receptors |
| Wnt | Wingless-related integration site |
References
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase mutations in human disease: Interpreting genotype–phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Yang, J.; Zhang, J.; Yang, J.; Li, Q.; Zheng, T. Src-family protein tyrosine kinases: A promising target for treating cardiovascular diseases. Int. J. Med. Sci. 2021, 18, 1216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, Q.; Tan, Y.; Deng, M.; Zhang, L.; Cao, Y.; Guo, X. Mitogen-activated protein kinases MPK3 and MPK6 phosphorylate receptor-like cytoplasmic kinase CDL1 to regulate soybean basal immunity. Plant Cell 2024, 36, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Nikolic, I.; Leiva, M.; Sabio, G. The role of stress kinases in metabolic disease. Nat. Rev. Endocrinol. 2020, 16, 697–716. [Google Scholar] [CrossRef]
- Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.-H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Cheek, S.; Zhang, H.; Grishin, N.V. Sequence and structure classification of kinases. J. Mol. Biol. 2002, 320, 855–881. [Google Scholar] [CrossRef]
- Daly, N.L.; Wilson, D.T. Plant derived cyclic peptides. Biochem. Soc. Trans. 2021, 49, 1279–1285. [Google Scholar] [CrossRef]
- Buljan, M.; Ciuffa, R.; van Drogen, A.; Vichalkovski, A.; Mehnert, M.; Rosenberger, G.; Lee, S.; Varjosalo, M.; Pernas, L.E.; Spegg, V. Kinase interaction network expands functional and disease roles of human kinases. Mol. Cell 2020, 79, 504–520. e509. [Google Scholar] [CrossRef]
- Edelman, A.M.; Blumenthal, D.K.; Krebs, E.G. Protein serine/threonine kinases. Annu. Rev. Biochem. 1987, 56, 567–613. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2024 update. Pharmacol. Res. 2024, 107059. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fayçal, C.; Hatat, A.-S.; Gazzeri, S.; Eymin, B. Splice variants of the RTK family: Their role in tumour progression and response to targeted therapy. Int. J. Mol. Sci. 2017, 18, 383. [Google Scholar] [CrossRef]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFR v III): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Johnson, J.L.; Yaron, T.M.; Huntsman, E.M.; Kerelsky, A.; Song, J.; Regev, A.; Lin, T.-Y.; Liberatore, K.; Cizin, D.M.; Cohen, B.M. An atlas of substrate specificities for the human serine/threonine kinome. Nature 2023, 613, 759–766. [Google Scholar] [CrossRef]
- Oliveira, M.; Rugo, H.S.; Howell, S.J.; Dalenc, F.; Cortes, J.; Gomez, H.L.; Hu, X.; Toi, M.; Jhaveri, K.; Krivorotko, P. Capivasertib and fulvestrant for patients with hormone receptor-positive, HER2-negative advanced breast cancer (CAPItello-291): Patient-reported outcomes from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2024, 25, 1231–1244. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, X. Molecular Recognition of FDA-Approved Small Molecule Protein Kinase Drugs in Protein Kinases. Molecules 2022, 27, 7124. [Google Scholar] [CrossRef]
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S.; Bayliss, R. Structural features of the protein kinase domain and targeted binding by small-molecule inhibitors. J. Biol. Chem. 2022, 298. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Till, J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.; Rastelli, G. αC helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discov. Today 2013, 18, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peng, Y.-H.; Shiao, H.-Y.; Tu, C.-H.; Liu, P.-M.; Hsu, J.T.-A.; Amancha, P.K.; Wu, J.-S.; Coumar, M.S.; Chen, C.-H.; Wang, S.-Y. Protein kinase inhibitor design by targeting the Asp-Phe-Gly (DFG) motif: The role of the DFG motif in the design of epidermal growth factor receptor inhibitors. J. Med. Chem. 2013, 56, 3889–3903. [Google Scholar] [CrossRef] [PubMed]
- Kanev, G.K.; de Graaf, C.; Westerman, B.A.; de Esch, I.J.; Kooistra, A.J. KLIFS: An overhaul after the first 5 years of supporting kinase research. Nucleic Acids Res. 2021, 49, D562–D569. [Google Scholar] [CrossRef]
- Van Linden, O.P.; Kooistra, A.J.; Leurs, R.; De Esch, I.J.; De Graaf, C. KLIFS: A knowledge-based structural database to navigate kinase–ligand interaction space. J. Med. Chem. 2014, 57, 249–277. [Google Scholar] [CrossRef]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef]
- Vasquez, E.M. Sirolimus: A new agent for prevention of renal allograft rejection. Am. J. Health Syst. Pharm. 2000, 57, 437–448. [Google Scholar] [CrossRef]
- Fabbro, D.; Ruetz, S.; Buchdunger, E.; Cowan-Jacob, S.W.; Fendrich, G.; Liebetanz, J.; Mestan, J.; O’Reilly, T.; Traxler, P.; Chaudhuri, B. Protein kinases as targets for anticancer agents: From inhibitors to useful drugs. Pharmacol. Ther. 2002, 93, 79–98. [Google Scholar] [CrossRef]
- Ayala-Aguilera, C.C.; Valero, T.; Lorente-Macias, A.; Baillache, D.J.; Croke, S.; Unciti-Broceta, A. Small molecule kinase inhibitor drugs (1995–2021): Medical indication, pharmacology, and synthesis. J. Med. Chem. 2021, 65, 1047–1131. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, P.Y.; Yeoh, Y.; Low, T.Y. A recent update on small-molecule kinase inhibitors for targeted cancer therapy and their therapeutic insights from mass spectrometry-based proteomic analysis. FEBS J. 2023, 290, 2845–2864. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Essegian, D.; Khurana, R.; Stathias, V.; Schürer, S.C. The clinical kinase index: A method to prioritize understudied kinases as drug targets for the treatment of cancer. Cell Rep. Med. 2020, 1. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, Y.; Xiang, S.; Yang, F.; Lu, X. Targeting gatekeeper mutations for kinase drug discovery. J. Med. Chem. 2022, 65, 15540–15558. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H.; Cortes, J. Use of second-and third-generation tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia: An evolving treatment paradigm. Clin. Lymphoma Myeloma Leuk. 2015, 15, 323–334. [Google Scholar] [CrossRef]
- Cho, B.C.; Felip, E.; Hayashi, H.; Thomas, M.; Lu, S.; Besse, B.; Sun, T.; Martinez, M.; Sethi, S.N.; Shreeve, S.M. MARIPOSA: Phase 3 study of first-line amivantamab + lazertinib versus osimertinib in EGFR-mutant non-small-cell lung cancer. Future Oncol. 2022, 18, 639–647. [Google Scholar] [CrossRef]
- Isaak, A.J.; Clements, G.R.; Buenaventura, R.G.M.; Merlino, G.; Yu, Y. Development of Personalized Strategies for Precisely Battling Malignant Melanoma. Int. J. Mol. Sci. 2024, 25, 5023. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Derkach, A.; Masarova, L.; Pemmaraju, N.; Stein, E.M.; Mauro, M.; Rampal, R.K.; Bose, P. A Phase I Study of Ruxolitinib in Combination with Abemaciclib for Patients with Primary or Post-Polycythemia Vera/Essential Thrombocythemia Myelofibrosis. Blood 2023, 142, 6440. [Google Scholar] [CrossRef]
- Norman, H.; Lee, K.T.; Stearns, V.; Alcorn, S.R.; Mangini, N.S. Incidence and severity of myelosuppression with palbociclib after palliative bone radiation in advanced breast cancer: A single center experience and review of literature. Clin. Breast Cancer 2022, 22, e65–e73. [Google Scholar] [CrossRef] [PubMed]
- Eggersmann, T.K.; Degenhardt, T.; Gluz, O.; Wuerstlein, R.; Harbeck, N. CDK4/6 inhibitors expand the therapeutic options in breast cancer: Palbociclib, ribociclib and abemaciclib. BioDrugs 2019, 33, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.R.; Wright, G.S.; Thummala, A.R.; Danso, M.A.; Popovic, L.; Pluard, T.J.; Han, H.S.; Vojnović, Ž.; Vasev, N.; Ma, L. Trilaciclib prior to chemotherapy in patients with metastatic triple-negative breast cancer: Final efficacy and subgroup analysis from a randomized phase II study. Clin. Cancer Res. 2022, 28, 629–636. [Google Scholar] [CrossRef]
- Luboff, A.J.; DeRemer, D.L. Capivasertib: A novel AKT inhibitor approved for hormone-receptor-positive, HER-2-negative metastatic breast cancer. Ann. Pharmacother. 2024, 58, 10600280241241531. [Google Scholar] [CrossRef]
- Yuan, Y.; Liu, X.; Cai, Y.; Li, W. Lapatinib and lapatinib plus trastuzumab therapy versus trastuzumab therapy for HER2 positive breast cancer patients: An updated systematic review and meta-analysis. Syst. Rev. 2022, 11, 264. [Google Scholar] [CrossRef]
- Guo, L.; Shao, W.; Zhou, C.; Yang, H.; Yang, L.; Cai, Q.; Wang, J.; Shi, Y.; Huang, L.; Zhang, J. Neratinib for HER2-positive breast cancer with an overlooked option. Mol. Med. 2023, 29, 134. [Google Scholar] [CrossRef]
- Sirhan, Z.; Thyagarajan, A.; Sahu, R.P. The efficacy of tucatinib-based therapeutic approaches for HER2-positive breast cancer. Mil. Med. Res. 2022, 9, 39. [Google Scholar] [CrossRef]
- Zubair, T.; Bandyopadhyay, D. Small molecule EGFR inhibitors as anti-cancer agents: Discovery, mechanisms of action, and opportunities. Int. J. Mol. Sci. 2023, 24, 2651. [Google Scholar] [CrossRef]
- Normann, L.S. HER2-Positive Breast Cancer and Drug Response in Pre-Clinical Models. Ph.D. Thesis, University of Oslo, Oslo, Norway, 2023. [Google Scholar]
- Ippolitov, D.; Lin, Y.-H.; Spence, J.; Glogowska, A.; Thanasupawat, T.; Beiko, J.; Del Bigio, M.R.; Xu, X.; Wang, A.; Calvo, R. Overcoming brain-derived therapeutic resistance in HER2+ breast cancer brain metastasis. bioRxiv 2024. [Google Scholar]
- Lee, B.J.; Boyer, J.A.; Burnett, G.L.; Thottumkara, A.P.; Tibrewal, N.; Wilson, S.L.; Hsieh, T.; Marquez, A.; Lorenzana, E.G.; Evans, J.W. Selective inhibitors of mTORC1 activate 4EBP1 and suppress tumor growth. Nat. Chem. Biol. 2021, 17, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Artemenko, M.; Zhong, S.S.; To, S.K.; Wong, A.S. p70 S6 kinase as a therapeutic target in cancers: More than just an mTOR effector. Cancer Lett. 2022, 535, 215593. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.; Cohen, M.S. The discovery and development of binimetinib for the treatment of melanoma. Expert. Opin. Drug Discov. 2020, 15, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Mortensen, L.S.; Loeffler-Wirth, H.; Kosnopfel, C.; Krohn, K.; Binder, H.; Kunz, M. Single-cell trajectories of melanoma cell resistance to targeted treatment. Cancer Biol. Med. 2022, 19, 56. [Google Scholar] [CrossRef]
- O’Sullivan Coyne, G.H.; Gross, A.M.; Dombi, E.; Tibery, C.; Carbonell, A.; Takebe, N.; Derdak, J.; Pichard, D.; Srivastava, A.K.; Herrick, W. Phase II Trial of the MEK 1/2 Inhibitor Selumetinib (AZD6244, ARRY-142886 Hydrogen Sulfate) in Adults with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PN); American Society of Clinical Oncology: Alexandria, VA, USA, 2020. [Google Scholar]
- Wang, Z.; Wang, X.; Wang, Z.; Feng, Y.; Jia, Y.; Jiang, L.; Xia, Y.; Cao, J.; Liu, Y. Comparison of hepatotoxicity associated with new BCR-ABL tyrosine kinase inhibitors vs imatinib among patients with chronic myeloid leukemia: A systematic review and meta-analysis. JAMA Netw. Open 2021, 4, e2120165. [Google Scholar] [CrossRef]
- Choi, E.-J. Asciminib: The first-in-class allosteric inhibitor of BCR:: ABL1 kinase. Blood Res. 2023, 58, S29–S36. [Google Scholar] [CrossRef]
- van Outersterp, I.; Tasian, S.K.; Reichert, C.E.; Boeree, A.; de Groot-Kruseman, H.A.; Escherich, G.; Boer, J.M.; den Boer, M.L. Tyrosine kinase inhibitor response of ABL-class acute lymphoblastic leukemia: The role of kinase type and SH3 domain. Blood 2024, 143, 2178–2189. [Google Scholar] [CrossRef]
- Pulte, E.D.; Norsworthy, K.J.; Wang, Y.; Xu, Q.; Qosa, H.; Gudi, R.; Przepiorka, D.; Fu, W.; Okusanya, O.O.; Goldberg, K.B. FDA approval summary: Gilteritinib for relapsed or refractory acute myeloid leukemia with a FLT3 mutation. Clin. Cancer Res. 2021, 27, 3515–3521. [Google Scholar] [CrossRef]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood J. Am. Soc. Hematol. 2021, 137, 3093–3104. [Google Scholar] [CrossRef]
- Garcia-Horton, A.; Yee, K.W. Quizartinib for the treatment of acute myeloid leukemia. Expert Opin. Pharmacother. 2020, 21, 2077–2090. [Google Scholar] [CrossRef]
- Imani, S.; Roozitalab, G.; Emadi, M.; Moradi, A.; Behzadi, P.; Kaboli, P.J. The evolution of BRAF-targeted therapies in melanoma: Overcoming hurdles and unleashing novel strategies. Front. Oncol. 2024, 14, 1504142. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, T. Janus Kinases and Autoimmunity: Bridging Pathways to Therapy. Int. J. Drug Discov. Pharmacol. 2024, 3, 100007. [Google Scholar] [CrossRef]
- Duminuco, A.; Chifotides, H.T.; Giallongo, S.; Giallongo, C.; Tibullo, D.; Palumbo, G.A. ACVR1: A novel therapeutic target to treat anemia in myelofibrosis. Cancers 2023, 16, 154. [Google Scholar] [CrossRef] [PubMed]
- Sardana, K.; Bathula, S.; Khurana, A. Which is the Ideal JAK Inhibitor for Alopecia Areata–Baricitinib, Tofacitinib, Ritlecitinib or Ifidancitinib-Revisiting the Immunomechanisms of the JAK Pathway. Indian Dermatol. Online J. 2023, 14, 465–474. [Google Scholar] [CrossRef]
- Bu, Y.; Traore, M.D.M.; Zhang, L.; Wang, L.; Liu, Z.; Hu, H.; Wang, M.; Li, C.; Sun, D. A gastrointestinal locally activating Janus kinase inhibitor to treat ulcerative colitis. J. Biol. Chem. 2023, 299, 105467. [Google Scholar] [CrossRef]
- Talty, R.; Damsky, W.; King, B. Treatment of cutaneous sarcoidosis with tofacitinib: A case report and review of evidence for Janus kinase inhibition in sarcoidosis. JAAD Case Rep. 2021, 16, 62–64. [Google Scholar] [CrossRef]
- Ferreira, S.; Guttman-Yassky, E.; Torres, T. Selective JAK1 inhibitors for the treatment of atopic dermatitis: Focus on upadacitinib and abrocitinib. Am. J. Clin. Dermatol. 2020, 21, 783–798. [Google Scholar] [CrossRef]
- Chuang, C.-H.; Chen, H.-L.; Chang, H.-M.; Tsai, Y.-C.; Wu, K.-L.; Chen, I.-H.; Chen, K.-C.; Lee, J.-Y.; Chang, Y.-C.; Chen, C.-L. Systematic review and network meta-analysis of anaplastic lymphoma kinase (ALK) inhibitors for treatment-naïve ALK-positive lung cancer. Cancers 2021, 13, 1966. [Google Scholar] [CrossRef]
- Rolfo, C.; Ruiz, R.; Giovannetti, E.; Gil-Bazo, I.; Russo, A.; Passiglia, F.; Giallombardo, M.; Peeters, M.; Raez, L. Entrectinib: A potent new TRK, ROS1, and ALK inhibitor. Expert Opin. Investig. Drugs 2015, 24, 1493–1500. [Google Scholar] [CrossRef]
- Mathias, T.J.; Natarajan, K.; Shukla, S.; Doshi, K.A.; Singh, Z.N.; Ambudkar, S.V.; Baer, M.R. The FLT3 and PDGFR inhibitor crenolanib is a substrate of the multidrug resistance protein ABCB1 but does not inhibit transport function at pharmacologically relevant concentrations. Investig. New Drugs 2015, 33, 300–309. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Lu, W.-P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell 2019, 35, 738–751. e739. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.-K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Elgawish, M.S.; Abdeldayem, E. Vascular endothelial growth factor receptors (VEGFR/PDGFR) inhibitors. In Current Molecular Targets of Heterocyclic Compounds for Cancer Therapy; Elsevier: Amsterdam, The Netherlands, 2024; pp. 443–475. [Google Scholar]
- Zhang, Y.; Zou, J.-Y.; Wang, Z.; Wang, Y. Fruquintinib: A novel antivascular endothelial growth factor receptor tyrosine kinase inhibitor for the treatment of metastatic colorectal cancer. Cancer Manag. Res. 2019, 11, 7787–7803. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, M.; Papadimitriou, C.A. Antiangiogenic tyrosine kinase inhibitors in metastatic colorectal cancer: Focusing on regorafenib. Anticancer Res. 2021, 41, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Fountzilas, C.; Gupta, M.; Lee, S.; Krishnamurthi, S.; Estfan, B.; Wang, K.; Attwood, K.; Wilton, J.; Bies, R.; Bshara, W. A multicentre phase 1b/2 study of tivozanib in patients with advanced inoperable hepatocellular carcinoma. Br. J. Cancer 2020, 122, 963–970. [Google Scholar] [CrossRef]
- Cascone, T.; Sacks, R.; Subbiah, I.; Drobnitzky, N.; Piha-Paul, S.; Hong, D.; Hess, K.; Amini, B.; Bhatt, T.; Fu, S. Safety and activity of vandetanib in combination with everolimus in patients with advanced solid tumors: A phase I study. ESMO Open 2021, 6, 100079. [Google Scholar] [CrossRef]
- Jagasia, M.; Lazaryan, A.; Bachier, C.R.; Salhotra, A.; Weisdorf, D.J.; Zoghi, B.; Essell, J.; Green, L.; Schueller, O.; Patel, J. ROCK2 inhibition with belumosudil (KD025) for the treatment of chronic graft-versus-host disease. J. Clin. Oncol. 2021, 39, 1888–1898. [Google Scholar] [CrossRef]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.-Y.; Alcindor, T.; Ganjoo, K.; Martín-Broto, J.; Ryan, C.W. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Bauer, S.; George, S.; von Mehren, M.; Heinrich, M.C. Early and next-generation KIT/PDGFRA kinase inhibitors and the future of treatment for advanced gastrointestinal stromal tumor. Front. Oncol. 2021, 11, 672500. [Google Scholar] [CrossRef]
- Garon, E.B.; Brodrick, P. Targeted therapy approaches for MET abnormalities in non-small cell lung cancer. Drugs 2021, 81, 547–554. [Google Scholar] [CrossRef]
- Wang, J.; Lam, D.; Yang, J.; Hu, L. Discovery of mobocertinib, a new irreversible tyrosine kinase inhibitor indicated for the treatment of non-small-cell lung cancer harboring EGFR exon 20 insertion mutations. Med. Chem. Res. 2022, 31, 1647–1662. [Google Scholar] [CrossRef] [PubMed]
- Duggirala, K.B.; Lee, Y.; Lee, K. Chronicles of EGFR tyrosine kinase inhibitors: Targeting EGFR C797S containing triple mutations. Biomol. Ther. 2022, 30, 19. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.M.; Pathak, G.N.; Singal, A.; Taranto, V.; Rao, B.K. Deucravacitinib: The first FDA-approved oral TYK2 inhibitor for moderate to severe plaque psoriasis. Ann. Pharmacother. 2024, 58, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Harada, G.; Drilon, A. TRK inhibitor activity and resistance in TRK fusion-positive cancers in adults. Cancer Genet. 2022, 264, 33–39. [Google Scholar] [CrossRef]
- Barbato, M.I.; Bradford, D.; Ren, Y.; Aungst, S.L.; Miller, C.P.; Pan, L.; Zirkelbach, J.F.; Li, Y.; Bi, Y.; Fan, J. FDA Approval Summary: Repotrectinib for Locally Advanced or Metastatic ROS1-Positive Non–Small Cell Lung Cancer. Clin. Cancer Res. 2024, 30, 3364–3370. [Google Scholar] [CrossRef]
- Facchinetti, F.; Loriot, Y.; Brayé, F.; Vasseur, D.; Bahleda, R.; Bigot, L.; Barbé, R.; Nobre, C.; Combarel, D.; Michiels, S. Understanding and Overcoming Resistance to Selective FGFR inhibitors Across FGFR2-Driven Malignancies. Clin. Cancer Res. 2024, 30, 4943–4956. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Futibatinib, an investigational agent for the treatment of intrahepatic cholangiocarcinoma: Evidence to date and future perspectives. Expert Opin. Investig. Drugs 2021, 30, 317–324. [Google Scholar] [CrossRef]
- Repetto, M.; Crimini, E.; Giugliano, F.; Morganti, S.; Belli, C.; Curigliano, G. Selective FGFR/FGF pathway inhibitors: Inhibition strategies, clinical activities, resistance mutations, and future directions. Expert Rev. Clin. Pharmacol. 2021, 14, 1233–1252. [Google Scholar] [CrossRef]
- Morizumi, S.; Sato, S.; Koyama, K.; Okazaki, H.; Chen, Y.; Goto, H.; Kagawa, K.; Ogawa, H.; Nishimura, H.; Kawano, H. Blockade of pan-fibroblast growth factor receptors mediates bidirectional effects in lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 317–326. [Google Scholar] [CrossRef]
- Hatashima, A.; Shadman, M. BTK inhibitors: Moving the needle on the treatment of chronic lymphocytic leukemia. Expert Rev. Hematol. 2024, 17, 687–703. [Google Scholar] [CrossRef]
- Connell, N.T.; Berliner, N. Fostamatinib for the treatment of chronic immune thrombocytopenia. Blood J. Am. Soc. Hematol. 2019, 133, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N.; Scott, L.J. Osimertinib: A review in T790M-positive advanced non-small cell lung cancer. Target. Oncol. 2017, 12, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Monestime, S.; Lazaridis, D. Pexidartinib (TURALIO™): The first FDA-indicated systemic treatment for tenosynovial giant cell tumor. Drugs R&D 2020, 20, 189–195. [Google Scholar]
- Edwards, S.J.; Wakefield, V.; Cain, P.; Karner, C.; Kew, K.; Bacelar, M.; Masento, N.; Salih, F. Axitinib, cabozantinib, everolimus, nivolumab, sunitinib and best supportive care in previously treated renal cell carcinoma: A systematic review and economic evaluation. Health Technol. Assess. 2018, 22, 1–278. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shen, Y.; Chen, X.; He, J.; Liao, D.; Zu, X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018, 198, 99–111. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Carmena, M.; Earnshaw, W.C.; Glover, D.M. The dawn of aurora kinase research: From fly genetics to the clinic. Front. Cell Dev. Biol. 2015, 3, 73. [Google Scholar] [CrossRef]
- Loffet, A. Peptides as drugs: Is there a market? J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2002, 8, 1–7. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A Global Review on Short Peptides: Frontiers and Perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chan, K.H.; Xue, B.; Robinson, R.C.; Hauser, C.A.E. Systematic Moiety Variations of Ultrashort Peptides Produce Profound Effects on Self-Assembly, Nanostructure Formation, Hydrogelation, and Phase Transition. Sci. Rep. 2017, 7, 12897. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ji, X.; Nielsen, A.L.; Heinis, C. Cyclic peptides for drug development. Angew. Chem. Int. Ed. 2024, 63, e202308251. [Google Scholar] [CrossRef] [PubMed]
- White, T.R.; Renzelman, C.M.; Rand, A.C.; Rezai, T.; McEwen, C.M.; Gelev, V.M.; Turner, R.A.; Linington, R.G.; Leung, S.S.; Kalgutkar, A.S. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Villar, E.A.; Beglov, D.; Chennamadhavuni, S.; Porco Jr, J.A.; Kozakov, D.; Vajda, S.; Whitty, A. How proteins bind macrocycles. Nat. Chem. Biol. 2014, 10, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Jwad, R.; Weissberger, D.; Hunter, L. Strategies for Fine-Tuning the Conformations of Cyclic Peptides. Chem. Rev. 2020, 120, 9743–9789. [Google Scholar] [CrossRef]
- Akbarian, M.; Khani, A.; Eghbalpour, S.; Uversky, V.N. Bioactive peptides: Synthesis, sources, applications, and proposed mechanisms of action. Int. J. Mol. Sci. 2022, 23, 1445. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. 2012, 20, 19. [Google Scholar] [CrossRef]
- Alimbarashvili, E.; Samsonidze, N.; Grigolava, M.; Pirtskhalava, M. Small Natural Cyclic Peptides from DBAASP Database. Pharmaceuticals 2024, 17, 845. [Google Scholar] [CrossRef]
- Costa, L.; Sousa, E.; Fernandes, C. Cyclic peptides in pipeline: What future for these great molecules? Pharmaceuticals 2023, 16, 996. [Google Scholar] [CrossRef]
- Mustafa, K.; Kanwal, J.; Musaddiq, S.; Khakwani, S. Bioactive peptides and their natural sources. In Functional Foods and Nutraceuticals: Bioactive Components, Formulations and Innovations; Springer Nature: Berlin/Heidelberg, Germany, 2020; pp. 75–97. [Google Scholar]
- Robinson, S.D.; Undheim, E.A.; Ueberheide, B.; King, G.F. Venom peptides as therapeutics: Advances, challenges and the future of venom-peptide discovery. Expert Rev. Proteom. 2017, 14, 931–939. [Google Scholar] [CrossRef]
- Wang, X.; Lin, M.; Xu, D.; Lai, D.; Zhou, L. Structural diversity and biological activities of fungal cyclic peptides, excluding cyclodipeptides. Molecules 2017, 22, 2069. [Google Scholar] [CrossRef]
- Abdalla, M.A.; McGaw, L.J. Natural cyclic peptides as an attractive modality for therapeutics: A mini review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef] [PubMed]
- Merz, M.L.; Habeshian, S.; Li, B.; David, J.-A.G.; Nielsen, A.L.; Ji, X.; Il Khwildy, K.; Duany Benitez, M.M.; Phothirath, P.; Heinis, C. De novo development of small cyclic peptides that are orally bioavailable. Nat. Chem. Biol. 2024, 20, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Hosono, Y.; Uchida, S.; Shinkai, M.; Townsend, C.E.; Kelly, C.N.; Naylor, M.R.; Lee, H.-W.; Kanamitsu, K.; Ishii, M.; Ueki, R. Amide-to-ester substitution as a stable alternative to N-methylation for increasing membrane permeability in cyclic peptides. Nat. Commun. 2023, 14, 1416. [Google Scholar] [CrossRef]
- Nielsen, D.S.; Shepherd, N.E.; Xu, W.; Lucke, A.J.; Stoermer, M.J.; Fairlie, D.P. Orally absorbed cyclic peptides. Chem. Rev. 2017, 117, 8094–8128. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Northfield, S.E.; Colless, B.; Chaousis, S.; Hamernig, I.; Lohman, R.-J.; Nielsen, D.S.; Schroeder, C.I.; Liras, S.; Price, D.A. Rational design and synthesis of an orally bioavailable peptide guided by NMR amide temperature coefficients. Proc. Natl. Acad. Sci. USA 2014, 111, 17504–17509. [Google Scholar] [CrossRef]
- Sohrabi, C.; Foster, A.; Tavassoli, A. Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat. Rev. Chem. 2020, 4, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef]
- Plotkin, P.; Patel, K.; Uminski, A.; Marzella, N. Telavancin (vibativ), a new option for the treatment of gram-positive infections. Pharm. Ther. 2011, 36, 127. [Google Scholar]
- Steenbergen, J.N.; Alder, J.; Thorne, G.M.; Tally, F.P. Daptomycin: A lipopeptide antibiotic for the treatment of serious Gram-positive infections. J. Antimicrob. Chemother. 2005, 55, 283–288. [Google Scholar] [CrossRef]
- Brown, P.; Abdulle, O.; Boakes, S.; Duperchy, E.; Moss, S.; Simonovic, M.; Stanway, S.; Wilson, A.; Dawson, M.J. Direct modifications of the cyclic peptide Polymyxin B leading to analogues with enhanced in vitro antibacterial activity. Bioorganic Med. Chem. Lett. 2020, 30, 127163. [Google Scholar] [CrossRef]
- Davis, S.L.; Vazquez, J.A. Anidulafungin: An evidence-based review of its use in invasive fungal infections. Core Evid. 2007, 2, 241. [Google Scholar]
- Hoffmann, A.R.; Guha, S.; Wu, E.; Ghimire, J.; Wang, Y.; He, J.; Garry, R.F.; Wimley, W.C. Broad-spectrum antiviral entry inhibition by interfacially active peptides. J. Virol. 2020, 94, e01682-20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Norman, A.; Franck, C.; Christie, M.; Hawkins, P.M.; Patel, K.; Ashhurst, A.S.; Aggarwal, A.; Low, J.K.; Siddiquee, R.; Ashley, C.L. Discovery of cyclic peptide ligands to the SARS-CoV-2 spike protein using mRNA display. ACS Cent. Sci. 2021, 7, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Tavassoli, A.; Lu, Q.; Gam, J.; Pan, H.; Benkovic, S.J.; Cohen, S.N. Inhibition of HIV budding by a genetically selected cyclic peptide targeting the Gag−TSG101 interaction. ACS Chem. Biol. 2008, 3, 757–764. [Google Scholar] [CrossRef]
- Choi, J.-S.; Joo, S.H. Recent trends in cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. 2020, 28, 18. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Su, X.; Li, Z.; Deng, L.; Liu, X.; Feng, X.; Peng, J. HGF/c-MET pathway in cancer: From molecular characterization to clinical evidence. Oncogene 2021, 40, 4625–4651. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Umitsu, M.; De Silva, D.M.; Roy, A.; Bottaro, D.P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017, 108, 296–307. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sato, H.; Imamura, R.; Suga, H.; Matsumoto, K.; Sakai, K. Cyclic Peptide-Based Biologics Regulating HGF-MET. Int. J. Mol. Sci. 2020, 21, 7977. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Damalanka, V.C.; Voss, J.; Mahoney, M.W.; Primeau, T.; Li, S.; Klampfer, L.; Janetka, J.W. Macrocyclic Inhibitors of HGF-Activating Serine Proteases Overcome Resistance to Receptor Tyrosine Kinase Inhibitors and Block Lung Cancer Progression. J. Med. Chem. 2021, 64, 18158–18174. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chan, L.Y.; Du, J.; Craik, D.J. Tuning the Anti-Angiogenic Effect of the P15 Peptide Using Cyclic Trypsin Inhibitor Scaffolds. ACS Chem. Biol. 2021, 16, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef] [PubMed]
- Kunte, S.; Abraham, J.; Montero, A.J. Novel HER2–targeted therapies for HER2–positive metastatic breast cancer. Cancer 2020, 126, 4278–4288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zou, Y.; Cai, Y.; Chi, F.; Huang, W.; Shi, W.; Qian, H. A designed cyclic peptide based on Trastuzumab used to construct peptide-drug conjugates for its HER2-targeting ability. Bioorg Chem. 2021, 117, 105453. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A.; Sillje, H.H.; Nigg, E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, a potential target for cancer therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef]
- Ryu, S.; Park, J.E.; Ham, Y.J.; Lim, D.C.; Kwiatkowski, N.P.; Kim, D.H.; Bhunia, D.; Kim, N.D.; Yaffe, M.B.; Son, W.; et al. Novel Macrocyclic Peptidomimetics Targeting the Polo-Box Domain of Polo-Like Kinase 1. J. Med. Chem. 2022, 65, 1915–1932. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Park, K.Y.; Kim, J. Cyclic pentapeptide cRGDfK enhances the inhibitory effect of sunitinib on TGF-beta1-induced epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. PLoS ONE 2020, 15, e0232917. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Morlieras, J.; Dufort, S.; Sancey, L.; Truillet, C.; Mignot, A.; Rossetti, F.; Dentamaro, M.; Laurent, S.; Vander Elst, L.; Muller, R.N. Functionalization of small rigid platforms with cyclic RGD peptides for targeting tumors overexpressing αvβ3-integrins. Bioconjugate Chem. 2013, 24, 1584–1597. [Google Scholar] [CrossRef]
- Kulhari, H.; Pooja, D.; Kota, R.; Reddy, T.S.; Tabor, R.F.; Shukla, R.; Adams, D.J.; Sistla, R.; Bansal, V. Cyclic RGDfK peptide functionalized polymeric nanocarriers for targeting gemcitabine to ovarian cancer cells. Mol. Pharm. 2016, 13, 1491–1500. [Google Scholar] [CrossRef]
- Chen, K.; Chen, X. Integrin targeted delivery of chemotherapeutics. Theranostics 2011, 1, 189. [Google Scholar] [CrossRef] [PubMed]
- Roskoski Jr, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Bayliss, G.; Zhuang, S. Epidermal Growth Factor Receptor: A Potential Therapeutic Target for Diabetic Kidney Disease. Front. Pharmacol. 2020, 11, 598910. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jeon, H.-L.; Kwak, M.; Kim, S.; Yu, H.-Y.; Shin, J.-Y.; Jung, H.A. Comparative effectiveness of lazertinib in patients with EGFR T790M-positive non-small-cell lung cancer using a real-world external control. Sci. Rep. 2024, 14, 14659. [Google Scholar] [CrossRef]
- Jiwacharoenchai, N.; Tabtimmai, L.; Kiriwan, D.; Suwattanasophon, C.; Seetaha, S.; Sinthuvanich, C.; Choowongkomon, K. A novel cyclic NP1 reveals obstruction of EGFR kinase activity and attenuation of EGFR-driven cell lines. J. Cell Biochem. 2022, 123, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed] [PubMed Central]
- Sok, J.C.; Coppelli, F.M.; Thomas, S.M.; Lango, M.N.; Xi, S.; Hunt, J.L.; Freilino, M.L.; Graner, M.W.; Wikstrand, C.J.; Bigner, D.D.; et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin. Cancer Res. 2006, 12, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Li, C.; Zhou, M.; Jia, R.; Cai, G.; She, F.; Wei, L.; Wang, S.; Yu, J.; Wang, D. Discovery of cyclic peptidomimetic ligands targeting the extracellular domain of EGFR. J. Med. Chem. 2021, 64, 11219–11228. [Google Scholar] [CrossRef]
- Li, Q.; Li, B.; Wang, Q.; Wang, C.; Yu, M.; Xu, T. Marine-derived EGFR inhibitors: Novel compounds targeting breast cancer growth and drug resistance. Front. Pharmacol. 2024, 15, 1396605. [Google Scholar] [CrossRef]
- Liu, J.; Bai, Y.; Liu, X.; Zhou, B.; Sun, P.; Wang, Y.; Ju, S.; Zhou, C.; Wang, C.; Yao, W. Enhanced efficacy of combined VEGFR peptide–drug conjugate and anti-PD-1 antibody in treating hepatocellular carcinoma. Sci. Rep. 2024, 14, 21728. [Google Scholar] [CrossRef]
- Yamashita, N.; Kramann, R. Mechanisms of kidney fibrosis and routes towards therapy. Trends Endocrinol. Metab. 2024, 35, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Xiao, J.; Li, J.; Yu, H.; Zhao, Q.; Tang, Q.; Chen, H.; Liu, H.; Wu, K.; Lei, J. Discovery and design of novel cyclic peptides as specific inhibitors targeting CCN2 and disrupting CCN2/EGFR interaction for kidney fibrosis treatment. J. Med. Chem. 2023, 66, 8251–8266. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.E.; Jenna, S.; Beaulne, S.; Lee, E.H.; Bergeron, A.; Chauve, C.; Roby, P.; Rual, J.F.; Hill, D.E.; Vidal, M.; et al. Biochemical clustering of monomeric GTPases of the Ras superfamily. Mol. Cell Proteom. 2005, 4, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, X.; Upadhyaya, P.; Villalona-Calero, M.A.; Briesewitz, R.; Pei, D. Inhibition of Ras–effector interactions by cyclic peptides. Med. Chem. Comm. 2013, 4, 378–382. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Qian, Z.; Selner, N.G.; Clippinger, S.R.; Wu, Z.; Briesewitz, R.; Pei, D. Inhibition of Ras signaling by blocking Ras-effector interactions with cyclic peptides. Angew. Chem. Int. Ed. Engl. 2015, 54, 7602–7606. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takeuchi, K.; Misaki, I.; Tokunaga, Y.; Fujisaki, M.; Kamoshida, H.; Takizawa, T.; Hanzawa, H.; Shimada, I. Conformational Plasticity of Cyclic Ras-Inhibitor Peptides Defines Cell Permeabilization Activity. Angew. Chem. Int. Ed. Engl. 2021, 60, 6567–6572. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Masutani, T.; Hirokawa, T. Generation of KS-58 as the first K-Ras (G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 2020, 10, 21671. [Google Scholar] [CrossRef]
- Ismail, M.; Martin, S.R.; George, R.; Houghton, F.; Kelly, G.; Chaleil, R.A.; Anastasiou, P.; Wang, X.; O’Reilly, N.; Federico, S. Characterisation of a cyclic peptide that binds to the RAS binding domain of phosphoinositide 3-kinase p110α. Sci. Rep. 2023, 13, 1889. [Google Scholar] [CrossRef]
- Balboa, J.R.; Essig, D.J.; Ma, S.; Karer, N.; Clemmensen, L.S.; Pedersen, S.W.; Joerger, A.C.; Knapp, S.; Ostergaard, S.; Stromgaard, K. Development of a Potent Cyclic Peptide Inhibitor of the nNOS/PSD-95 Interaction. J. Med. Chem. 2023, 66, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Mihara, K.; Ueda, T.; Yamauchi, D.; Shimizu, M.; Ando, A.; Mayumi, K.; Nakata, Z.; Mikamiyama, H. Discovery and Hit to Lead Optimization of Macrocyclic Peptides as Novel Tropomyosin Receptor Kinase A Antagonists. J. Med. Chem. 2024, 67, 11197–11208. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Pardanani, A. Momelotinib for myelofibrosis: Our 14 years of experience with 100 clinical trial patients and recent FDA approval. Blood Cancer J. 2024, 14, 47. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yin, Y.; Shi, G.; Zhou, Y.; Shao, S.; Wei, Y.; Wu, L.; Zhang, D.; Sun, L.; Zhang, T. A highly selective JAK3 inhibitor is developed for treating rheumatoid arthritis by suppressing γc cytokine–related JAK-STAT signal. Sci. Adv. 2022, 8, eabo4363. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Lopez-Sanz, L.; Bernal, S.; Fortuna, S.; Mercurio, F.A.; Leone, M.; Gomez-Guerrero, C.; Marasco, D. Cyclic mimetics of kinase-inhibitory region of Suppressors of Cytokine Signaling 1: Progress toward novel anti-inflammatory therapeutics. Eur. J. Med. Chem. 2021, 221, 113547. [Google Scholar] [CrossRef]
- Raju, M.; Kavarthapu, R.; Anbazhagan, R.; Hassan, S.A.; Dufau, M.L. Blockade of GRTH/DDX25 Phosphorylation by Cyclic Peptides Provides an Avenue for Developing a Nonhormonal Male Contraceptive. J. Med. Chem. 2021, 64, 14715–14727. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sheng, Y.; Tsai-Morris, C.-H.; Gutti, R.; Maeda, Y.; Dufau, M.L. Gonadotropin-regulated testicular RNA helicase (GRTH/Ddx25) is a transport protein involved in gene-specific mRNA export and protein translation during spermatogenesis. J. Biol. Chem. 2006, 281, 35048–35056. [Google Scholar] [CrossRef]
- Anbazhagan, R.; Kavarthapu, R.; Coon, S.L.; Dufau, M.L. Role of Phosphorylated Gonadotropin-Regulated Testicular RNA Helicase (GRTH/DDX25) in the Regulation of Germ Cell Specific mRNAs in Chromatoid Bodies During Spermatogenesis. Front. Cell Dev. Biol. 2020, 8, 580019. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Buckton, L.K.; Rahimi, M.N.; McAlpine, S.R. Cyclic peptides as drugs for intracellular targets: The next frontier in peptide therapeutic development. Chem. Eur. J. 2021, 27, 1487–1513. [Google Scholar] [CrossRef]
- Imanishi, S.; Katoh, T.; Yin, Y.; Yamada, M.; Kawai, M.; Suga, H. In Vitro Selection of Macrocyclic d/l-Hybrid Peptides against Human EGFR. J. Am. Chem. Soc. 2021, 143, 5680–5684. [Google Scholar] [CrossRef] [PubMed]
- Appella, D.H.; Christianson, L.A.; Karle, I.L.; Powell, D.R.; Gellman, S.H. β-Peptide foldamers: Robust helix formation in a new family of β-amino acid oligomers. J. Am. Chem. Soc. 1996, 118, 13071–13072. [Google Scholar] [CrossRef]
- Cabrele, C.; Martinek, T.A.; Reiser, O.; Berlicki, L. Peptides containing beta-amino acid patterns: Challenges and successes in medicinal chemistry. J. Med. Chem. 2014, 57, 9718–9739. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, R.; Kawai, M.; Katoh, T.; Suga, H. In vitro selection of macrocyclic α/β3-peptides against human EGFR. J. Am. Chem. Soc. 2022, 144, 18504–18510. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Choi, J.; Choi, S.-J.; Baek, K.-H. Cyclodipeptides: An overview of their biosynthesis and biological activity. Molecules 2017, 22, 1796. [Google Scholar] [CrossRef] [PubMed]
- Widodo, W.S.; Billerbeck, S. Natural and engineered cyclodipeptides: Biosynthesis, chemical diversity, and engineering strategies for diversification and high-yield bioproduction. Eng. Microbiol. 2023, 3, 100067. [Google Scholar] [CrossRef]
- Mosetti, V.; Rosetti, B.; Pierri, G.; Bellotto, O.; Adorinni, S.; Bandiera, A.; Adami, G.; Tedesco, C.; Crosera, M.; Magnano, G.C. Cyclodipeptides: From their green synthesis to anti-age activity. Biomedicines 2022, 10, 2342. [Google Scholar] [CrossRef]
- Bojarska, J.; Mieczkowski, A.; Ziora, Z.M.; Skwarczynski, M.; Toth, I.; Shalash, A.O.; Parang, K.; El-Mowafi, S.A.; Mohammed, E.H.; Elnagdy, S. Cyclic dipeptides: The biological and structural landscape with special focus on the anti-cancer proline-based scaffold. Biomolecules 2021, 11, 1515. [Google Scholar] [CrossRef]
- Petrova, A.; Pancheva, M.; Kostova, K.; Zagranyarska, I.; Tavlinova-Kirilova, M.; Dimitrov, V. Stereoselective functionalization strategy of 2, 5-diketopiperazine derived from L-proline and glycine. Bulg. Chem. Commun. 2017, 49, 18–24. [Google Scholar]
- De Masi, A.; Zanou, N.; Strotjohann, K.; Lee, D.; Lima, T.I.; Li, X.; Jeon, J.; Place, N.; Jung, H.Y.; Auwerx, J. Cyclo His-Pro Attenuates Muscle Degeneration in Murine Myopathy Models. Adv. Sci. 2024, 2305927. [Google Scholar] [CrossRef]
- Durán-Maldonado, M.X.; Hernández-Padilla, L.; Gallardo-Pérez, J.C.; Díaz-Pérez, A.L.; Martínez-Alcantar, L.; Reyes De la Cruz, H.; Rodríguez-Zavala, J.S.; Pacheco-Rodríguez, G.; Moss, J.; Campos-García, J. Bacterial cyclodipeptides target signal pathways involved in malignant melanoma. Front. Oncol. 2020, 10, 1111. [Google Scholar] [CrossRef]
- Grottelli, S.; Bellezza, I.; Morozzi, G.; Peirce, M.J.; Marchetti, M.C.; Cacciatore, I.; Costanzi, E.; Minelli, A. Cyclo (His-Pro) Protects SOD1G93A Microglial Cells from Paraquat Induced Toxicity. J. Clin. Cell. Immunol. 2015, 6, 287. [Google Scholar] [CrossRef]
- Kim, J.E.; Han, D.; Kim, K.H.; Seo, A.; Moon, J.J.; Jeong, J.S.; Kim, J.H.; Kang, E.; Bae, E.; Kim, Y.C. Protective effect of Cyclo (His-Pro) on peritoneal fibrosis through regulation of HDAC3 expression. FASEB J. 2024, 38, e23819. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Jung, H.-Y.; Lee, D.; Jeon, J.; Kim, J.; Baek, S.; Lee, J.Y.; Kim, J.Y.; Kwon, H.J. Inhibition of chloride intracellular channel protein 1 (CLIC1) ameliorates liver fibrosis phenotype by activating the Ca2+-dependent Nrf2 pathway. Biomed. Pharmacother. 2023, 168, 115776. [Google Scholar] [CrossRef] [PubMed]
- Lazaro-Mixteco, P.E.; Gonzalez-Coronel, J.M.; Hernandez-Padilla, L.; Martinez-Alcantar, L.; Martinez-Carranza, E.; Lopez-Bucio, J.S.; Guevara-Garcia, A.A.; Campos-Garcia, J. Transcriptomics reveals the mevalonate and cholesterol pathways blocking as part of the bacterial cyclodipeptides cytotoxic effects in HeLa cells of human cervix adenocarcinoma. Front. Oncol. 2022, 12, 790537. [Google Scholar] [CrossRef]
- Hernández-Padilla, L.; Vázquez-Rivera, D.; Sánchez-Briones, L.A.; Díaz-Pérez, A.L.; Moreno-Rodríguez, J.; Moreno-Eutimio, M.A.; Meza-Carmen, V.; Reyes-De la Cruz, H.; Campos-García, J. The antiproliferative effect of cyclodipeptides from Pseudomonas aeruginosa PAO1 on HeLa cells involves inhibition of phosphorylation of Akt and S6k kinases. Molecules 2017, 22, 1024. [Google Scholar] [CrossRef]
- Vázquez-Rivera, D.; González, O.; Guzmán-Rodríguez, J.; Díaz-Pérez, A.L.; Ochoa-Zarzosa, A.; López-Bucio, J.; Meza-Carmen, V.; Campos-García, J. Cytotoxicity of cyclodipeptides from Pseudomonas aeruginosa PAO1 leads to apoptosis in human cancer cell lines. BioMed Res. Int. 2015, 2015, 197608. [Google Scholar] [CrossRef]
- Minelli, A.; Conte, C.; Grottelli, S.; Bellezza, M.; Cacciatore, I.; Bolaños, J.P. Cyclo (His-Pro) promotes cytoprotection by activating Nrf2-mediated up-regulation of antioxidant defence. J. Cell. Mol. Med. 2009, 13, 1149–1161. [Google Scholar] [CrossRef]
- Ortiz-Castro, R.; Díaz-Pérez, C.; Martínez-Trujillo, M.; del Río, R.E.; Campos-García, J.; López-Bucio, J. Transkingdom signaling based on bacterial cyclodipeptides with auxin activity in plants. Proc. Natl. Acad. Sci. USA 2011, 108, 7253–7258. [Google Scholar] [CrossRef]
- Hernández-Padilla, L.; Durán-Maldonado, M.X.; Martínez-Alcantar, L.; Rodríguez-Zavala, J.S.; Campos-Garcia, J. The HGF/Met Receptor Mediates Cytotoxic Effect of Bacterial Cyclodipeptides in Human Cervical Cancer Cells. Curr. Cancer Drug Targets 2024, 25, 230–243. [Google Scholar] [CrossRef]
- Hernández-Padilla, L.; Reyes de la Cruz, H.; Campos-García, J. Antiproliferative effect of bacterial cyclodipeptides in the HeLa line of human cervical cancer reveals multiple protein kinase targeting, including mTORC1/C2 complex inhibition in a TSC1/2-dependent manner. Apoptosis 2020, 25, 632–647. [Google Scholar] [CrossRef]
- Karanam, G.; Arumugam, M.K.; Sirpu Natesh, N. Anticancer effect of marine sponge-associated Bacillus pumilus AMK1 derived dipeptide Cyclo (-Pro-Tyr) in human liver cancer cell line through apoptosis and G2/M phase arrest. Int. J. Pept. Res. Ther. 2020, 26, 445–457. [Google Scholar] [CrossRef]
- Hong, S.-W.; Moon, B.-H.; Yong, Y.-J.; Shin, S.-Y.; Lee, Y.-H.; Lim, Y.-H. Inhibitory effect against Akt by cyclic dipeptides isolated from Bacillus sp. J. Microbiol. Biotechnol. 2008, 18, 682–685. [Google Scholar] [PubMed]
- Karanam, G.; Arumugam, M.K. Potential anticancer effects of cyclo (-pro-tyr) against n-diethyl nitrosamine induced hepatocellular carcinoma in mouse through pi3k/akt signaling. Environ. Toxicol. 2022, 37, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Karanam, G.; Arumugam, M.K. Reactive oxygen species generation and mitochondrial dysfunction for the initiation of apoptotic cell death in human hepatocellular carcinoma HepG2 cells by a cyclic dipeptide Cyclo (-Pro-Tyr). Mol. Biol. Rep. 2020, 47, 3347–3359. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.; Tariq, A.; Mustafa, G.; Javed, M.R.; Naheed, S.; Qamar, S.A. Cyclo (L-Leucyl-L-Prolyl) from Lactobacillus coryniformis BCH-4 inhibits the proliferation of Aspergillus flavus: An in vitro to in silico approach. Arch. Microbiol. 2022, 204, 267. [Google Scholar] [CrossRef]
- Lalitha, P.; Veena, V.; Vidhyapriya, P.; Lakshmi, P.; Krishna, R.; Sakthivel, N. Anticancer potential of pyrrole (1, 2, a) pyrazine 1, 4, dione, hexahydro 3-(2-methyl propyl)(PPDHMP) extracted from a new marine bacterium, Staphylococcus sp. strain MB30. Apoptosis 2016, 21, 566–577. [Google Scholar] [CrossRef]
- Deepak, K.; Kumari, S.; Shailender, G.; Malla, R.R. Marine natural compound cyclo (L-leucyl-L-prolyl) peptide inhibits migration of triple negative breast cancer cells by disrupting interaction of CD151 and EGFR signaling. Chem.-Biol. Interact. 2020, 315, 108872. [Google Scholar]
- Minelli, A.; Bellezza, I.; Grottelli, S.; Galli, F. Focus on cyclo (His-Pro): History and perspectives as antioxidant peptide. Amino Acids 2008, 35, 283–289. [Google Scholar] [CrossRef]
- Minelli, A.; Grottelli, S.; Mierla, A.; Pinnen, F.; Cacciatore, I.; Bellezza, I. Cyclo (His-Pro) exerts anti-inflammatory effects by modulating NF-κB and Nrf2 signalling. Int. J. Biochem. Cell Biol. 2012, 44, 525–535. [Google Scholar] [CrossRef]
- Mirzaei, S.; Mohammadi, A.T.; Gholami, M.H.; Hashemi, F.; Zarrabi, A.; Zabolian, A.; Hushmandi, K.; Makvandi, P.; Samec, M.; Liskova, A. Nrf2 signaling pathway in cisplatin chemotherapy: Potential involvement in organ protection and chemoresistance. Pharmacol. Res. 2021, 167, 105575. [Google Scholar] [CrossRef]
- Minelli, A.; Bellezza, I.; Grottelli, S.; Pinnen, F.; Brunetti, L.; Vacca, M. Phosphoproteomic analysis of the effect of cyclo-[His-Pro] dipeptide on PC12 cells. Peptides 2006, 27, 105–113. [Google Scholar] [CrossRef]
- Faden, A.I.; Fox, G.B.; Di, X.; Knoblach, S.M.; Cernak, I.; Mullins, P.; Nikolaeva, M.; Kozikowski, A.P. Neuroprotective and nootropic actions of a novel cyclized dipeptide after controlled cortical impact injury in mice. J. Cereb. Blood Flow. Metab. 2003, 23, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Movsesyan, V.A.; Knoblach, S.M.; Ahmed, F.; Cernak, I. Neuroprotective effects of novel small peptides in vitro and after brain injury. Neuropharmacology 2005, 49, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Prakash, K.; Tang, Y.; Kozikowski, A.P.; Flippen-Anderson, J.L.; Knoblach, S.M.; Faden, A.I. Synthesis and biological activity of novel neuroprotective diketopiperazines. Bioorganic Med. Chem. 2002, 10, 3043–3048. [Google Scholar] [CrossRef] [PubMed]
- De Masi, A.; Li, X.; Lee, D.; Jeon, J.; Wang, Q.; Baek, S.; Park, O.; Mottis, A.; Strotjohann, K.; Rapin, A. Cyclo (His-Pro): A further step in the management of steatohepatitis. Jhep Rep. 2023, 5, 100815. [Google Scholar] [CrossRef]
- Lucietto, F.; Milne, P.; Kilian, G.; Frost, C.; Van De Venter, M. The biological activity of the histidine-containing diketopiperazines cyclo (His-Ala) and cyclo (His-Gly). Peptides 2006, 27, 2706–2714. [Google Scholar] [CrossRef]
- André, A.; Touré, A.; Stien, D.; Eparvier, V. 2, 5-diketopiperazines mitigate the amount of advanced glycation end products accumulated with age in human dermal fibroblasts. Int. J. Cosmet. Sci. 2020, 42, 596–604. [Google Scholar] [CrossRef]
- Hwang, I.; Go, V.; Harris, D.; Yip, I.; Kang, K.; Song, M. Effects of cyclo (his–pro) plus zinc on glucose metabolism in genetically diabetic obese mice. Diabetes Obes. Metab. 2003, 5, 317–324. [Google Scholar] [CrossRef]
- Song, M.K.; Hwang, I.K.; Rosenthal, M.J.; Harris, D.M.; Yamaguchi, D.T.; Yip, I.; Go, V.L.W. Anti-hyperglycemic activity of zinc plus cyclo (his-pro) in genetically diabetic Goto-Kakizaki and aged rats. Exp. Biol. Med. 2003, 228, 1338–1345. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and obesity: Role and clinical implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Jeon, J.; Lee, D.; Kim, B.; Park, B.-Y.; Oh, C.J.; Kim, M.-J.; Jeon, J.-H.; Lee, I.-K.; Park, O.; Baek, S. CycloZ Improves Hyperglycemia and Lipid Metabolism by Modulating Lysine Acetylation in KK-Ay Mice. Diabetes Metab. J. 2023, 47, 653. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Gan, B.; Hu, J.; Jiang, S.; Liu, Y.; Sahin, E.; Zhuang, L.; Fletcher-Sananikone, E.; Colla, S.; Wang, Y.A.; Chin, L. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 2010, 468, 701–704. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Alcantar, L.; Hernández-Padilla, L.; Díaz-Pérez, A.L.; Villalón-Magallán, L.G.; Durán-Maldonado, M.X.; Díaz-Pérez, C.; Campos-Morales, M.E.; Figueroa-Guzmán, C.; Campos-García, J. Cyclic Peptides as Protein Kinase Modulators and Their Involvement in the Treatment of Diverse Human Diseases. Kinases Phosphatases 2024, 2, 346-378. https://doi.org/10.3390/kinasesphosphatases2040023
Martínez-Alcantar L, Hernández-Padilla L, Díaz-Pérez AL, Villalón-Magallán LG, Durán-Maldonado MX, Díaz-Pérez C, Campos-Morales ME, Figueroa-Guzmán C, Campos-García J. Cyclic Peptides as Protein Kinase Modulators and Their Involvement in the Treatment of Diverse Human Diseases. Kinases and Phosphatases. 2024; 2(4):346-378. https://doi.org/10.3390/kinasesphosphatases2040023
Chicago/Turabian StyleMartínez-Alcantar, Lorena, Laura Hernández-Padilla, Alma Laura Díaz-Pérez, Lizbeth Guadalupe Villalón-Magallán, Mayra Xóchitl Durán-Maldonado, César Díaz-Pérez, Marlene E. Campos-Morales, Citlali Figueroa-Guzmán, and Jesús Campos-García. 2024. "Cyclic Peptides as Protein Kinase Modulators and Their Involvement in the Treatment of Diverse Human Diseases" Kinases and Phosphatases 2, no. 4: 346-378. https://doi.org/10.3390/kinasesphosphatases2040023
APA StyleMartínez-Alcantar, L., Hernández-Padilla, L., Díaz-Pérez, A. L., Villalón-Magallán, L. G., Durán-Maldonado, M. X., Díaz-Pérez, C., Campos-Morales, M. E., Figueroa-Guzmán, C., & Campos-García, J. (2024). Cyclic Peptides as Protein Kinase Modulators and Their Involvement in the Treatment of Diverse Human Diseases. Kinases and Phosphatases, 2(4), 346-378. https://doi.org/10.3390/kinasesphosphatases2040023