
SGLT2 Inhibitors and Uric Acid Homeostasis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Glucose and Uric Acid Handling in the Kidney
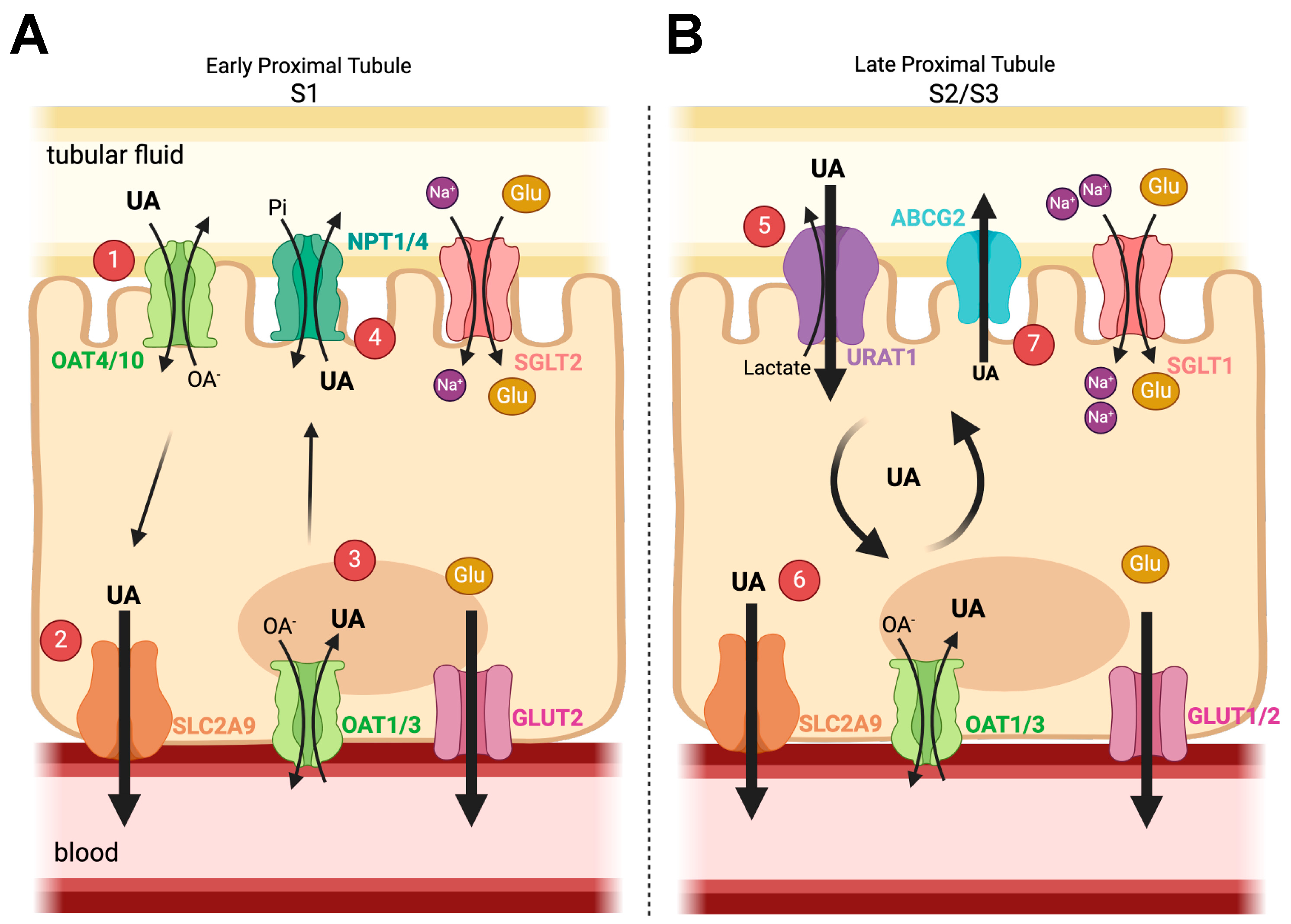
2.1. Uric Acid/Urate
2.2. Renal Glucose Physiology
3. Hyperglycemia, Type 2 Diabetes (T2D), and Urate
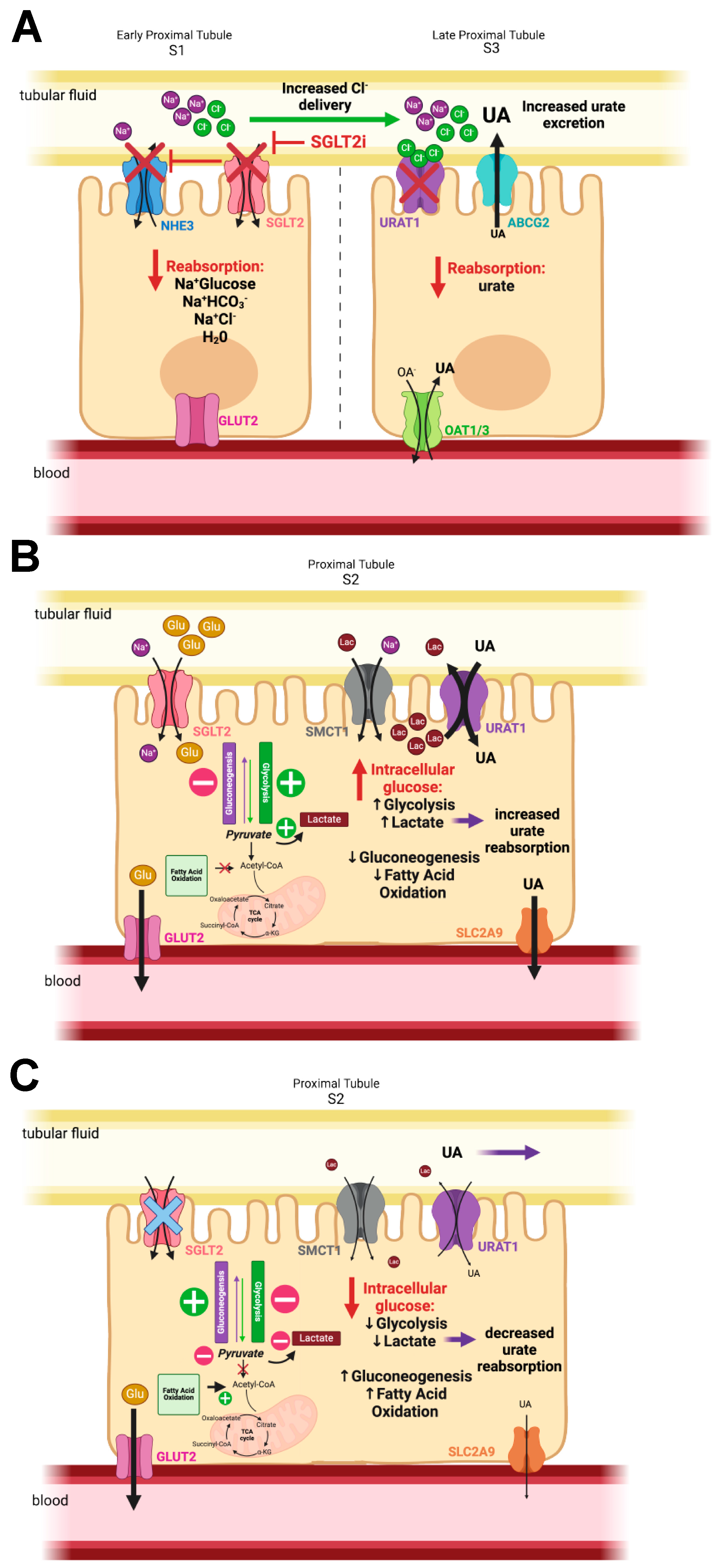
4. SGLT2 Inhibitors and Their Effects
Clinical Benefits
5. SGLT2 Inhibitors and Gout
6. How Do SGLT2 Inhibitors Lower Plasma Urate and Increase Uricosuria?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef]
- CDC. National Diabetes Statistics Report Website. Available online: https://www.cdc.gov/diabetes/data/statistics-report/index.html (accessed on 11 December 2023).
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef]
- Lingli, X.; Wenfang, X. Characteristics and molecular mechanisms through which SGLT2 inhibitors improve metabolic diseases: A mechanism review. Life Sci. 2022, 300, 120543. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef]
- Suijk, D.L.S.; van Baar, M.J.B.; van Bommel, E.J.M.; Iqbal, Z.; Krebber, M.M.; Vallon, V.; Touw, D.; Hoorn, E.J.; Nieuwdorp, M.; Kramer, M.M.H.; et al. SGLT2 Inhibition and Uric Acid Excretion in Patients with Type 2 Diabetes and Normal Kidney Function. Clin. J. Am. Soc. Nephrol. 2022, 17, 663–671. [Google Scholar] [CrossRef]
- Wei, J.; Choi, H.K.; Dalbeth, N.; Li, X.; Li, C.; Zeng, C.; Lei, G.; Zhang, Y. Gout Flares and Mortality After Sodium-Glucose Cotransporter-2 Inhibitor Treatment for Gout and Type 2 Diabetes. JAMA Netw. Open 2023, 6, e2330885. [Google Scholar] [CrossRef] [PubMed]
- Halperin Kuhns, V.L.; Woodward, O.M. Urate transport in health and disease. Best. Pract. Res. Clin. Rheumatol. 2021, 35, 101717. [Google Scholar] [CrossRef] [PubMed]
- Yokose, C.; McCormick, N.; Lu, N.; Tanikella, S.; Lin, K.; Joshi, A.D.; Raffield, L.M.; Warner, E.; Merriman, T.; Hsu, J.; et al. Trends in Prevalence of Gout Among US Asian Adults, 2011–2018. JAMA Netw. Open 2023, 6, e239501. [Google Scholar] [CrossRef]
- Somagutta, M.K.R.; Luvsannyam, E.; Jain, M.; Cuddapah, G.V.; Pelluru, S.; Mustafa, N.; Nasereldin, D.S.; Pendyala, S.K.; Jarapala, N.; Padamati, B. Sodium glucose co-transport 2 inhibitors for gout treatment. Discoveries 2022, 10, e152. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef]
- Reynolds, R.J.; Irvin, M.R.; Bridges, S.L.; Kim, H.; Merriman, T.R.; Arnett, D.K.; Singh, J.A.; Sumpter, N.A.; Lupi, A.S.; Vazquez, A.I. Genetic correlations between traits associated with hyperuricemia, gout, and comorbidities. Eur. J. Hum. Genet. 2021, 29, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L.; CARES Investigators. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N. Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Lanaspa, M.A.; Gaucher, E.A. Uric acid: A danger signal from the RNA world that may have a role in the epidemic of obesity, metabolic syndrome, and cardiorenal disease: Evolutionary considerations. Semin. Nephrol. 2011, 31, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Tin, A.; Marten, J.; Halperin Kuhns, V.L.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.B.; Qiu, C.; Gorski, M.; Yu, Z.; et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474. [Google Scholar] [CrossRef] [PubMed]
- Tin, A.; Li, Y.; Brody, J.A.; Nutile, T.; Chu, A.Y.; Huffman, J.E.; Yang, Q.; Chen, M.H.; Robinson-Cohen, C.; Macé, A.; et al. Large-scale whole-exome sequencing association studies identify rare functional variants influencing serum urate levels. Nat. Commun. 2018, 9, 4228. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S.; et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1, 5ra11. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.J.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P.; et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008, 5, e197. [Google Scholar] [CrossRef]
- Hoque, K.M.; Dixon, E.E.; Lewis, R.M.; Allan, J.; Gamble, G.D.; Phipps-Green, A.J.; Halperin Kuhns, V.L.; Horne, A.M.; Stamp, L.K.; Merriman, T.R.; et al. The ABCG2 Q141K hyperuricemia and gout associated variant illuminates the physiology of human urate excretion. Nat. Commun. 2020, 11, 2767. [Google Scholar] [CrossRef]
- Gutman, A.B.; Yu, T.F. A three-component system for regulation of renal excretion of uric acid in man. Trans. Assoc. Am. Physicians 1961, 74, 353–365. [Google Scholar]
- Maesaka, J.K.; Fishbane, S. Regulation of renal urate excretion: A critical review. Am. J. Kidney Dis. 1998, 32, 917–933. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takahashi, M.; Yan, K.; Sakurai, H. Expression of SLC2A9 isoforms in the kidney and their localization in polarized epithelial cells. PLoS ONE 2014, 9, e84996. [Google Scholar] [CrossRef] [PubMed]
- Bakhiya, A.; Bahn, A.; Burckhardt, G.; Wolff, N. Human organic anion transporter 3 (hOAT3) can operate as an exchanger and mediate secretory urate flux. Cell Physiol. Biochem. 2003, 13, 249–256. [Google Scholar] [CrossRef]
- Eraly, S.A.; Vallon, V.; Rieg, T.; Gangoiti, J.A.; Wikoff, W.R.; Siuzdak, G.; Barshop, B.A.; Nigam, S.K. Multiple organic anion transporters contribute to net renal excretion of uric acid. Physiol. Genom. 2008, 33, 180–192. [Google Scholar] [CrossRef]
- Ichida, K.; Hosoyamada, M.; Kimura, H.; Takeda, M.; Utsunomiya, Y.; Hosoya, T.; Endou, H. Urate transport via human PAH transporter hOAT1 and its gene structure. Kidney Int. 2003, 63, 143–155. [Google Scholar] [CrossRef]
- Wright, E.M.; Hirayama, B.A.; Loo, D.F. Active sugar transport in health and disease. J. Intern. Med. 2007, 261, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Vallon, V. Glucose transporters in the kidney in health and disease. Pflugers Arch. 2020, 472, 1345–1370. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Wilson-O’Brien, A.L.; Patron, N.; Rogers, S. Evolutionary ancestry and novel functions of the mammalian glucose transporter (GLUT) family. BMC Evol. Biol. 2010, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, A.M.; Weinbaum, S.; Duan, Y.; Du, Z.; Yan, Q.; Wang, T. Flow-dependent transport in a mathematical model of rat proximal tubule. Am. J. Physiol. Renal Physiol. 2007, 292, F1164–F1181. [Google Scholar] [CrossRef]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Renal Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, C.; Wright, E.M. Regulation of the human Na+-dependent glucose cotransporter hSGLT2. Am. J. Physiol. Cell Physiol. 2012, 303, C348–C354. [Google Scholar] [CrossRef]
- Toyoki, D.; Shibata, S.; Kuribayashi-Okuma, E.; Xu, N.; Ishizawa, K.; Hosoyamada, M.; Uchida, S. Insulin stimulates uric acid reabsorption via regulating urate transporter 1 and ATP-binding cassette subfamily G member 2. Am. J. Physiol. Renal Physiol. 2017, 313, F826–F834. [Google Scholar] [CrossRef]
- Mandal, A.K.; Leask, M.P.; Sumpter, N.A.; Choi, H.K.; Merriman, T.R.; Mount, D.B. Genetic and Physiological Effects of Insulin-Like Growth Factor-1 (IGF-1) on Human Urate Homeostasis. J. Am. Soc. Nephrol. 2023, 34, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Quiñones Galvan, A.; Natali, A.; Baldi, S.; Frascerra, S.; Sanna, G.; Ciociaro, D.; Ferrannini, E. Effect of insulin on uric acid excretion in humans. Am. J. Physiol. 1995, 268, E1–E5. [Google Scholar] [CrossRef] [PubMed]
- Guler, H.P.; Schmid, C.; Zapf, J.; Froesch, E.R. Effects of recombinant insulin-like growth factor I on insulin secretion and renal function in normal human subjects. Proc. Natl. Acad. Sci. USA 1989, 86, 2868–2872. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Ma, J.; Zhang, X. Higher Serum Uric Acid May Contribute to Cerebral Infarction in Patients with Type 2 Diabetes Mellitus: A Meta-Analysis. J. Mol. Neurosci. 2017, 61, 25–31. [Google Scholar] [CrossRef]
- Ji, P.; Zhu, J.; Feng, J.; Li, H.; Yu, Q.; Qin, H.; Wei, L.; Zhang, J. Serum uric acid levels and diabetic kidney disease in patients with type 2 diabetes mellitus: A dose-response meta-analysis. Prim. Care Diabetes 2022, 16, 457–465. [Google Scholar] [CrossRef]
- Lv, Q.; Meng, X.F.; He, F.F.; Chen, S.; Su, H.; Xiong, J.; Gao, P.; Tian, X.J.; Liu, J.S.; Zhu, Z.H.; et al. High serum uric acid and increased risk of type 2 diabetes: A systemic review and meta-analysis of prospective cohort studies. PLoS ONE 2013, 8, e56864. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, M.; Niwa, K.; Hisatome, I.; Nakagawa, T.; Roncal-Jimenez, C.A.; Andres-Hernando, A.; Bjornstad, P.; Jensen, T.; Sato, Y.; Milagres, T.; et al. Asymptomatic Hyperuricemia Without Comorbidities Predicts Cardiometabolic Diseases: Five-Year Japanese Cohort Study. Hypertension 2017, 69, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- van der Schaft, N.; Brahimaj, A.; Wen, K.X.; Franco, O.H.; Dehghan, A. The association between serum uric acid and the incidence of prediabetes and type 2 diabetes mellitus: The Rotterdam Study. PLoS ONE 2017, 12, e0179482. [Google Scholar] [CrossRef] [PubMed]
- Moleda, P.; Fronczyk, A.; Safranow, K.; Majkowska, L. Is Uric Acid a Missing Link between Previous Gestational Diabetes Mellitus and the Development of Type 2 Diabetes at a Later Time of Life? PLoS ONE 2016, 11, e0154921. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.C.; Wang, A.P.; Li, L.X.; Li, T.T.; Chen, M.Y.; Zhu, Y.; Yu, T.P.; Bao, Y.Q.; Jia, W.P. Urine uric acid excretion is associated with nonalcoholic fatty liver disease in patients with type 2 diabetes. J. Diabetes Complicat. 2016, 30, 1074–1080. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, J.; Shi, H.; Hu, W.; Song, L.; Zhao, Q. Serum uric acid is positively associated with the prevalence of nonalcoholic fatty liver in non-obese type 2 diabetes patients in a Chinese population. J. Diabetes Complicat. 2021, 35, 107874. [Google Scholar] [CrossRef] [PubMed]
- Sekula, P.; Del Greco, M.F.; Pattaro, C.; Köttgen, A. Mendelian Randomization as an Approach to Assess Causality Using Observational Data. J. Am. Soc. Nephrol. 2016, 27, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Keerman, M.; Yang, F.; Hu, H.; Wang, J.; Wang, F.; Li, Z.; Yuan, J.; Yao, P.; Zhang, X.; Guo, H.; et al. Mendelian randomization study of serum uric acid levels and diabetes risk: Evidence from the Dongfeng-Tongji cohort. BMJ Open Diabetes Res. Care 2020, 8, e000834. [Google Scholar] [CrossRef] [PubMed]
- Sluijs, I.; Holmes, M.V.; van der Schouw, Y.T.; Beulens, J.W.; Asselbergs, F.W.; Huerta, J.M.; Palmer, T.M.; Arriola, L.; Balkau, B.; Barricarte, A.; et al. A Mendelian Randomization Study of Circulating Uric Acid and Type 2 Diabetes. Diabetes 2015, 64, 3028–3036. [Google Scholar] [CrossRef]
- Hu, X.; Rong, S.; Wang, Q.; Sun, T.; Bao, W.; Chen, L.; Liu, L. Association between plasma uric acid and insulin resistance in type 2 diabetes: A Mendelian randomization analysis. Diabetes Res. Clin. Pract. 2021, 171, 108542. [Google Scholar] [CrossRef]
- McCormick, N.; O’Connor, M.J.; Yokose, C.; Merriman, T.R.; Mount, D.B.; Leong, A.; Choi, H.K. Assessing the Causal Relationships Between Insulin Resistance and Hyperuricemia and Gout Using Bidirectional Mendelian Randomization. Arthritis Rheumatol. 2021, 73, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; McCormick, N.; Yokose, C. Excess comorbidities in gout: The causal paradigm and pleiotropic approaches to care. Nat. Rev. Rheumatol. 2022, 18, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Afkarian, M.; Polsky, S.; Parsa, A.; Aronson, R.; Caramori, M.L.; Cherney, D.Z.; Crandall, J.P.; de Boer, I.H.; Elliott, T.G.; Galecki, A.T.; et al. Preventing Early Renal Loss in Diabetes (PERL) Study: A Randomized Double-Blinded Trial of Allopurinol-Rationale, Design, and Baseline Data. Diabetes Care 2019, 42, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.V.; Pascoe, E.M.; Tiku, A.; Boudville, N.; Brown, F.G.; Cass, A.; Clarke, P.; Dalbeth, N.; Day, R.O.; de Zoysa, J.R.; et al. Effects of Allopurinol on the Progression of Chronic Kidney Disease. N. Engl. J. Med. 2020, 382, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, M.; de Vinuesa, S.G.; Verdalles, U.; Ruiz-Caro, C.; Ampuero, J.; Rincón, A.; Arroyo, D.; Luño, J. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin. J. Am. Soc. Nephrol. 2010, 5, 1388–1393. [Google Scholar] [CrossRef]
- Kanbay, M.; Huddam, B.; Azak, A.; Solak, Y.; Kadioglu, G.K.; Kirbas, I.; Duranay, M.; Covic, A.; Johnson, R.J. A randomized study of allopurinol on endothelial function and estimated glomular filtration rate in asymptomatic hyperuricemic subjects with normal renal function. Clin. J. Am. Soc. Nephrol. 2011, 6, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chen, Y.; Wang, B.; Zhang, F.; Wang, D.; Wang, Y. Allopurinol treatment improves renal function in patients with type 2 diabetes and asymptomatic hyperuricemia: 3-year randomized parallel-controlled study. Clin. Endocrinol. 2015, 83, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hu, Y.; Huang, T.; Zhang, Y.; Li, Z.; Luo, C.; Luo, Y.; Yuan, H.; Hisatome, I.; Yamamoto, T.; et al. High uric acid directly inhibits insulin signalling and induces insulin resistance. Biochem. Biophys. Res. Commun. 2014, 447, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Halperin Kuhns, V.L.; Woodward, O.M. Sex Differences in Urate Handling. Int. J. Mol. Sci. 2020, 21, 4269. [Google Scholar] [CrossRef]
- Woodward, O.M.; Kottgen, A.; Kottgen, M. ABCG transporters and disease. FEBS J. 2011, 278, 3215–3225. [Google Scholar] [CrossRef]
- Lu, J.; Dalbeth, N.; Yin, H.; Li, C.; Merriman, T.R.; Wei, W.H. Mouse models for human hyperuricaemia: A critical review. Nat. Rev. Rheumatol. 2019, 15, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; Norton, L.; DeFronzo, R.A. Renal sodium-glucose cotransporter inhibition in the management of type 2 diabetes mellitus. Am. J. Physiol. Renal Physiol. 2015, 309, F889–F900. [Google Scholar] [CrossRef] [PubMed]
- Beitelshees, A.L.; Leslie, B.R.; Taylor, S.I. Sodium-Glucose Cotransporter 2 Inhibitors: A Case Study in Translational Research. Diabetes 2019, 68, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Vick, H.; Diedrich, D.F.; Baumann, K. Reevaluation of renal tubular glucose transport inhibition by phlorizin analogs. Am. J. Physiol. 1973, 224, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, L.; Smith, D.; Shulman, G.I.; Papachristou, D.; DeFronzo, R.A. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J. Clin. Investig. 1987, 79, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, L.; Shulman, G.I.; Zawalich, W.; DeFronzo, R.A. Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J. Clin. Investig. 1987, 80, 1037–1044. [Google Scholar] [CrossRef]
- Nomura, S. Renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors for new anti-diabetic agent. Curr. Top. Med. Chem. 2010, 10, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Thomson, S.C.; Rieg, T.; Miracle, C.; Mansoury, H.; Whaley, J.; Vallon, V.; Singh, P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R75–R83. [Google Scholar] [CrossRef]
- Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; Ng, S.Y.A.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Trujillo, A.; Vijapurkar, U.; Damaraju, C.V.; Meininger, G. Effect of canagliflozin on serum uric acid in patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2015, 17, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Furtado, R.H.M.; Raz, I.; Goodrich, E.L.; Murphy, S.A.; Bhatt, D.L.; Leiter, L.A.; McGuire, D.K.; Wilding, J.P.H.; Aylward, P.; Dalby, A.J.; et al. Efficacy and Safety of Dapagliflozin in Type 2 Diabetes According to Baseline Blood Pressure: Observations From DECLARE-TIMI 58 Trial. Circulation 2022, 145, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yang, Y.; Jia, X.; Liu, H.; Wei, M.; Lyu, Z. Effects of sodium-glucose cotransporter 2 inhibitors on serum uric acid in patients with type 2 diabetes mellitus: A systematic review and network meta-analysis. Diabetes Obes. Metab. 2022, 24, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.K.; Mercado, A.; Foster, A.; Zandi-Nejad, K.; Mount, D.B. Uricosuric targets of tranilast. Pharmacol. Res. Perspect. 2017, 5, e00291. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2018, 20, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Notsu, S.; Kawamura, Y.; Mitani, W.; Tamai, S.; Morimoto, M.; Yamato, M. The effect of dapagliflozin on uric acid excretion and serum uric acid level in advanced CKD. Sci. Rep. 2023, 13, 4849. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.P.; Inzucchi, S.E.; Mattheus, M.; Meinicke, T.; Steubl, D.; Wanner, C.; Zinman, B. Empagliflozin and uric acid metabolism in diabetes: A post hoc analysis of the EMPA-REG OUTCOME trial. Diabetes Obes. Metab. 2022, 24, 135–141. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kasahara, M.; Ueshima, K.; Uemura, S.; Kashihara, N.; Kimura, K.; Konta, T.; Shoji, T.; Mima, A.; Mukoyama, M.; et al. Multicenter randomized controlled trial of intensive uric acid lowering therapy for CKD patients with hyperuricemia: TARGET-UA. Clin. Exp. Nephrol. 2024. [Google Scholar] [CrossRef]
- Mohammed, E.; Browne, L.D.; Kumar, A.U.A.; Adeeb, F.; Fraser, A.D.; Stack, A.G. Prevalence and treatment of gout among patients with chronic kidney disease in the Irish health system: A national study. PLoS ONE 2019, 14, e0210487. [Google Scholar] [CrossRef]
- Fralick, M.; Chen, S.K.; Patorno, E.; Kim, S.C. Assessing the Risk for Gout With Sodium-Glucose Cotransporter-2 Inhibitors in Patients With Type 2 Diabetes: A Population-Based Cohort Study. Ann. Intern. Med. 2020, 172, 186–194. [Google Scholar] [CrossRef]
- Yokose, C.; McCormick, N.; Abhishek, A.; Dalbeth, N.; Pascart, T.; Lioté, F.; Gaffo, A.; FitzGerald, J.; Terkeltaub, R.; Sise, M.E.; et al. The clinical benefits of sodium-glucose cotransporter type 2 inhibitors in people with gout. Nat. Rev. Rheumatol. 2024, 20, 216–231. [Google Scholar] [CrossRef]
- Skeith, M.D.; Healey, L.A.; Cutler, R.E. Effect of phloridzin on uric acid excretion in man. Am. J. Physiol. 1970, 219, 1080–1082. [Google Scholar] [CrossRef]
- Novikov, A.; Fu, Y.; Huang, W.; Freeman, B.; Patel, R.; van Ginkel, C.; Koepsell, H.; Busslinger, M.; Onishi, A.; Nespoux, J.; et al. SGLT2 inhibition and renal urate excretion: Role of luminal glucose, GLUT9, and URAT1. Am. J. Physiol. Renal Physiol. 2019, 316, F173–F185. [Google Scholar] [CrossRef]
- Santer, R.; Calado, J. Familial renal glucosuria and SGLT2: From a mendelian trait to a therapeutic target. Clin. J. Am. Soc. Nephrol. 2010, 5, 133–141. [Google Scholar] [CrossRef]
- Aires, I.; Santos, A.R.; Pratas, J.; Nolasco, F.; Calado, J. Hypouricaemia and hyperuricosuria in familial renal glucosuria. Clin. Kidney J. 2013, 6, 523–525. [Google Scholar] [CrossRef]
- Calado, J.; Santer, R.; Rueff, J. Effect of kidney disease on glucose handling (including genetic defects). Kidney Int. Suppl. 2011, 79, S7–S13. [Google Scholar] [CrossRef]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for tubular Na. Am. J. Physiol. Renal Physiol. 2020, 319, F712–F728. [Google Scholar] [CrossRef]
- Steele, T.H.; Manuel, M.A.; Boner, G. Diuretics, urate excretion and sodium reabsorption: Effect of acetazolamide and urinary alkalinization. Nephron 1975, 14, 49–61. [Google Scholar] [CrossRef]
- Anzai, N.; Ichida, K.; Jutabha, P.; Kimura, T.; Babu, E.; Jin, C.J.; Srivastava, S.; Kitamura, K.; Hisatome, I.; Endou, H.; et al. Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans. J. Biol. Chem. 2008, 283, 26834–26838. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Uchimura, K.; Donnelly, E.L.; Kirita, Y.; Morris, S.A.; Humphreys, B.D. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell 2018, 23, 869–881.e868. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Malone, A.F.; Donnelly, E.L.; Kirita, Y.; Uchimura, K.; Ramakrishnan, S.M.; Gaut, J.P.; Humphreys, B.D. Single-Cell Transcriptomics of a Human Kidney Allograft Biopsy Specimen Defines a Diverse Inflammatory Response. J. Am. Soc. Nephrol. 2018, 29, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Wilcox, C.S.; Testani, J.M. Critical Analysis of the Effects of SGLT2 Inhibitors on Renal Tubular Sodium, Water and Chloride Homeostasis and Their Role in Influencing Heart Failure Outcomes. Circulation 2023, 148, 354–372. [Google Scholar] [CrossRef] [PubMed]
- Tin, A.; Woodward, O.M.; Kao, W.H.; Liu, C.T.; Lu, X.; Nalls, M.A.; Shriner, D.; Semmo, M.; Akylbekova, E.L.; Wyatt, S.B.; et al. Genome-wide association study for serum urate concentrations and gout among African Americans identifies genomic risk loci and a novel URAT1 loss-of-function allele. Hum. Mol. Genet. 2011, 20, 4056–4068. [Google Scholar] [CrossRef]
- Cai, T.; Ke, Q.; Fang, Y.; Wen, P.; Chen, H.; Yuan, Q.; Luo, J.; Zhang, Y.; Sun, Q.; Lv, Y.; et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. 2020, 11, 390. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapf, A.M.; Woodward, O.M. SGLT2 Inhibitors and Uric Acid Homeostasis. Gout Urate Cryst. Depos. Dis. 2024, 2, 157-172. https://doi.org/10.3390/gucdd2020014
Zapf AM, Woodward OM. SGLT2 Inhibitors and Uric Acid Homeostasis. Gout, Urate, and Crystal Deposition Disease. 2024; 2(2):157-172. https://doi.org/10.3390/gucdd2020014
Chicago/Turabian StyleZapf, Ava M., and Owen M. Woodward. 2024. "SGLT2 Inhibitors and Uric Acid Homeostasis" Gout, Urate, and Crystal Deposition Disease 2, no. 2: 157-172. https://doi.org/10.3390/gucdd2020014