
Universal Paediatric and Newborn Screening for Familial Hypercholesterolaemia—Challenges and Opportunities: An Australian Perspective

 and
and 
Abstract
1. Background
2. Paediatric Screening for FH
2.1. National and International Guideline Recommendations
2.2. Possible Paediatric FH Screening Strategies
3. Large Population Paediatric FH Screening (Non-Newborns)
4. Universal Paediatric FH Screening (Newborns)
4.1. Biochemical Testing
4.1.1. Cord Blood Testing
4.1.2. Heel Prick DBS Testing
4.2. Genetic Testing
4.2.1. Second Tier/Confirmatory Genetic Testing
4.2.2. First Tier/Primary Genetic Testing
5. Opportunities, Challenges, and Ethical Considerations of Newborn Genetic Testing for FH
5.1. Opportunities
5.2. Ethical Considerations and Challenges
6. Local Models of Care
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| apoA1 | Apolipoprotein A1 |
| ASCVD | Atherosclerotic cardiovascular disease |
| ALSPAC | Avon Longitudinal Study of Parents and Children |
| cIMT | carotid intima media thickness |
| DR | Detection rate |
| DBS | Dried blood spots |
| FPR | False positive rate |
| FH | Familial hypercholesterolaemia |
| GP | General Practitioner |
| HDL-C | High density lipoprotein cholesterol |
| LDL | Low-density lipoprotein |
| LDL-C | Low-density lipoprotein cholesterol |
| Lp(a) | Lipoprotein(a) |
| MOM | Multiples of the median |
| NHLBI | National Heart, Lung, and Blood Institute’s Expert Panel |
| NPV | Negative predictive value |
| nonHDL-C | Non-high-density lipoprotein cholesterol |
| PPV | Positive predictive value |
| TC | Total cholesterol |
| TG | Triglycerides |
| UK | United Kingdom |
| VUS | Variants of unknown significance |
| WGS | Whole genome sequencing |
References
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Primers 2017, 3, 17093. [Google Scholar] [CrossRef]
- Austin, M.A.; Hutter, C.M.; Zimmern, R.L.; Humphries, S.E. Genetic Causes of Monogenic Heterozygous Familial Hypercholesterolemia: A HuGE Prevalence Review. Am. J. Epidemiol. 2004, 160, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Di Taranto, M.D.; Fortunato, G. Genetic Heterogeneity of Familial Hypercholesterolemia: Repercussions for Molecular Diagnosis. Int. J. Mol. Sci. 2023, 24, 3224. [Google Scholar] [CrossRef] [PubMed]
- Akioyamen, L.E.; Genest, J.; Shan, S.D.; Reel, R.L.; Albaum, J.M.; Chu, A.; Tu, J.V. Estimating the prevalence of heterozygous familial hypercholesterolaemia: A systematic review and meta-analysis. BMJ Open 2017, 7, e016461. [Google Scholar] [CrossRef] [PubMed]
- Toft-Nielsen, F.; Emanuelsson, F.; Benn, M. Familial Hypercholesterolemia Prevalence Among Ethnicities-Systematic Review and Meta-Analysis. Front. Genet. 2022, 13, 840797. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Raal, F.J.; Hegele, R.A.; Al-Rasadi, K.; Arca, M.; Averna, M.; Bruckert, E.; Freiberger, T.; Gaudet, D.; Harada-Shiba, M.; et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: New treatments and clinical guidance. Eur. Heart J. 2023, 44, 2277–2291. [Google Scholar] [CrossRef]
- Lan, N.S.R.; Martin, A.C.; Brett, T.; Watts, G.F.; Bell, D.A. Improving the detection of familial hypercholesterolaemia. Pathology 2019, 51, 213–221. [Google Scholar] [CrossRef]
- Watts, G.F.; Sullivan, D.R.; Hare, D.L.; Kostner, K.M.; Horton, A.E.; Bell, D.A.; Brett, T.; Trent, R.J.; Poplawski, N.K.; Martin, A.C.; et al. Integrated Guidance for Enhancing the Care of Familial Hypercholesterolaemia in Australia. Heart Lung Circ. 2021, 30, 324–349. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.F.; Gidding, S.S.; Mata, P.; Pang, J.; Sullivan, D.R.; Yamashita, S.; Raal, F.J.; Santos, R.D.; Ray, K.K. Familial hypercholesterolaemia: Evolving knowledge for designing adaptive models of care. Nat. Rev. Cardiol. 2020, 17, 360–377. [Google Scholar] [CrossRef] [PubMed]
- Kusters, D.M.; Wiegman, A.; Kastelein, J.J.P.; Hutten, B.A. Carotid intima-media thickness in children with familial hypercholesterolemia. Circ. Res. 2014, 114, 307–310. [Google Scholar] [CrossRef]
- Ademi, Z.; Norman, R.; Pang, J.; Liew, D.; Zoungas, S.; Sijbrands, E.; Ference, B.A.; Wiegman, A.; Watts, G.F. Health economic evaluation of screening and treating children with familial hypercholesterolemia early in life: Many happy returns on investment? Atherosclerosis 2020, 304, 1–8. [Google Scholar] [CrossRef]
- Meng, R.; Wei, Q.; Zhou, J.; Zhang, B.; Li, C.; Shen, M. A systematic review of cost-effectiveness analysis of different screening strategies for familial hypercholesterolemia. J. Clin. Lipidol. 2024, 18, e21–e32. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, A.; Gidding, S.S.; Watts, G.F.; Chapman, M.J.; Ginsberg, H.N.; Cuchel, M.; Ose, L.; Averna, M.; Boileau, C.; Borén, J.; et al. Familial hypercholesterolaemia in children and adolescents: Gaining decades of life by optimizing detection and treatment. Eur. Heart J. 2015, 36, 2425–2437. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, A.; Hutten, B.A.; de Groot, E.; Rodenburg, J.; Bakker, H.D.; Büller, H.R.; Sijbrands, E.J.G.; Kastelein, J.J.P. Efficacy and Safety of Statin Therapy in Children With Familial Hypercholesterolemia: A Randomized Controlled Trial. JAMA 2004, 292, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Braamskamp, M.J.A.M.; Langslet, G.; McCrindle, B.W.; Cassiman, D.; Francis, G.A.; Gagne, C.; Gaudet, D.; Morrison, K.M.; Wiegman, A.; Turner, T.; et al. Effect of Rosuvastatin on Carotid Intima-Media Thickness in Children With Heterozygous Familial Hypercholesterolemia: The CHARON Study (Hypercholesterolemia in Children and Adolescents Taking Rosuvastatin Open Label). Circulation 2017, 136, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Luirink, I.K.; Wiegman, A.; Kusters, D.M.; Hof, M.H.; Groothoff, J.W.; de Groot, E.; Kastelein, J.J.P.; Hutten, B.A. 20-Year Follow-up of Statins in Children with Familial Hypercholesterolemia. N. Engl. J. Med. 2019, 381, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Anagnostis, P.; Vaitsi, K.; Kleitsioti, P.; Mantsiou, C.; Pavlogiannis, K.; Athyros, V.G.; Mikhailidis, D.P.; Goulis, D.G. Efficacy and safety of statin use in children and adolescents with familial hypercholesterolaemia: A systematic review and meta-analysis of randomized-controlled trials. Endocrine 2020, 69, 249–261. [Google Scholar] [CrossRef]
- Wiegman, A.; Ruzza, A.; Hovingh, G.K.; Santos, R.D.; Mach, F.; Stefanutti, C.; Luirink, I.K.; Bridges, I.; Wang, B.; Bhatia, A.K.; et al. Evolocumab treatment reduces carotid intima-media thickness in paediatric patients with heterozygous familial hypercholesterolaemia. Eur. J. Prev. Cardiol. 2024, zwae369. [Google Scholar] [CrossRef]
- Klevmoen, M.; Bogsrud, M.P.; Retterstøl, K.; Svilaas, T.; Vesterbekkmo, E.K.; Hovland, A.; Berge, C.; Roeters van Lennep, J.; Holven, K.B. Loss of statin treatment years during pregnancy and breastfeeding periods in women with familial hypercholesterolemia. Atherosclerosis 2021, 335, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Dharmayat, K.I.; Stevens, C.A.T.; Sharabiani, M.T.A.; Jones, R.S.; Watts, G.F.; Genest, J.; Ray, K.K.; Vallejo-Vaz, A.J. Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation 2020, 141, 1742–1759. [Google Scholar] [CrossRef]
- Dharmayat, K.I.; Watts, G.F.; Aguilar-Salinas, C.A.; Al-Sayed, N.; Corral, P.; Gaita, D.; Harada-Shiba, M.; Kayikcioglu, M.; Reiner, Ž.; Stoll, M.; et al. Global perspective of familial hypercholesterolaemia: A cross-sectional study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Lancet 2021, 398, 1713–1725. [Google Scholar] [CrossRef]
- Vallejo-Vaz, A.J.; Brandts, J.M.; Lyons, A.R.M.; Groselj, U.; Aguilar-Salinas, C.A.; Almahmeed, W.; Durst, R.; Roeters van Lennep, J.; Lima-Martinez, M.M.; März, W.; et al. Familial hypercholesterolaemia in children and adolescents from 48 countries: A cross-sectional study. Lancet 2024, 403, 55–66. [Google Scholar] [CrossRef]
- Pang, J.; Sullivan, D.R.; Hare, D.L.; Colquhoun, D.M.; Bates, T.R.; Ryan, J.D.M.; Bishop, W.; Burnett, J.R.; Bell, D.A.; Simons, L.A.; et al. Gaps in the Care of Familial Hypercholesterolaemia in Australia: First Report From the National Registry. Heart Lung Circ. 2021, 30, 372–379. [Google Scholar] [CrossRef]
- Wilson, J.M.G.; Jungner, G. Principles and practice of screening for disease. In Public Health Paper No. 34; World Health Organization: Geneva, Switzerland, 1968. [Google Scholar]
- Humphries, S.E.; Ramaswami, U.; Hopper, N. Should Familial Hypercholesterolaemia Be Included in the UK Newborn Whole Genome Sequencing Programme? Curr. Atheroscler. Rep. 2023, 25, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Gidding, S.S.; Ballantyne, C.M.; Cuchel, M.; Ferranti, S.D.E.; Hudgins, L.; Jamison, A.; McGowan, M.P.; Peterson, A.L.; Steiner, R.D.; Uveges, M.K.; et al. It is Time to Screen for Homozygous Familial Hypercholesterolemia in the United States. Glob. Heart 2024, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.K.; Walters, N.; Brangan, A.; Ahmed, C.D.; Wilemon, K.A.; Campbell-Salome, G.; Rahm, A.K.; Gidding, S.S.; Sturm, A.C. Patient experiences align with the familial hypercholesterolemia global call to action. Am. J. Prev. Cardiol. 2022, 10, 100344. [Google Scholar] [CrossRef] [PubMed]
- Gidding, S.S.; Wiegman, A.; Groselj, U.; Freiberger, T.; Peretti, N.; Dharmayat, K.I.; Daccord, M.; Bedlington, N.; Sikonja, J.; Ray, K.K.; et al. Paediatric familial hypercholesterolaemia screening in Europe: Public policy background and recommendations. Eur. J. Prev. Cardiol. 2022, 29, 2301–2311. [Google Scholar] [CrossRef]
- Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents: Summary Report. Pediatrics 2011, 128, S213–S256. [CrossRef] [PubMed]
- Jacobson, T.A.M.D.; Maki, K.C.P.; Orringer, C.E.M.D.; Jones, P.H.M.D.; Kris-Etherton, P.P.; Sikand, G.M.A.; La Forge, R.M.; Daniels, S.R.M.D.P.; Wilson, D.P.M.D.; Morris, P.B.M.D.; et al. National Lipid Association Recommendations for Patient-Centered Management of Dyslipidemia: Part 2. J. Clin. Lipidol. 2015, 9, S1–S122.e121. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.C.; Hopkins, P.N.; Toth, P.P.; Ballantyne, C.M.; Rader, D.J.; Robinson, J.G.; Daniels, S.R.; Gidding, S.S.; De Ferranti, S.D.; Ito, M.K.; et al. Familial hypercholesterolemia: Screening, diagnosis and management of pediatric and adult patients: Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 2011, 5, S133–S138. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2019, 41, 111–188. [Google Scholar] [CrossRef]
- Guirguis-Blake, J.M.; Evans, C.V.; Coppola, E.L.; Redmond, N.; Perdue, L.A. Screening for Lipid Disorders in Children and Adolescents: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2023, 330, 261–274. [Google Scholar] [CrossRef]
- Watts, G.F.; Gidding, S.S.; Hegele, R.A.; Raal, F.J.; Sturm, A.C.; Jones, L.K.; Sarkies, M.N.; Al-Rasadi, K.; Blom, D.J.; Daccord, M.; et al. International Atherosclerosis Society guidance for implementing best practice in the care of familial hypercholesterolaemia. Nat. Rev. Cardiol. 2023, 20, 845–869. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Poulter, E.B.; Bell, D.A.; Bates, T.R.; Jefferson, V.-L.; Hillis, G.S.; Schultz, C.J.; Watts, G.F. Frequency of familial hypercholesterolemia in patients with early-onset coronary artery disease admitted to a coronary care unit. J. Clin. Lipidol. 2015, 9, 703–708. [Google Scholar] [CrossRef] [PubMed]
- De Backer, G.; Besseling, J.; Chapman, J.; Hovingh, G.K.; Kastelein, J.J.P.; Kotseva, K.; Ray, K.; Reiner, Ž.; Wood, D.; De Bacquer, D. Prevalence and management of familial hypercholesterolaemia in coronary patients: An analysis of EUROASPIRE IV, a study of the European Society of Cardiology. Atherosclerosis 2015, 241, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.A.; Bender, R.; Hooper, A.J.; McMahon, J.; Edwards, G.; van Bockxmeer, F.M.; Watts, G.F.; Burnett, J.R. Impact of interpretative commenting on lipid profiles in people at high risk of familial hypercholesterolaemia. Clin. Chim. Acta 2013, 422, 21–25. [Google Scholar] [CrossRef]
- Bender, R.; Edwards, G.; McMahon, J.; Hooper, A.J.; Watts, G.F.; Burnett, J.R.; Bell, D.A. Interpretative comments specifically suggesting specialist referral increase the detection of familial hypercholesterolaemia. Pathology 2016, 48, 463–466. [Google Scholar] [CrossRef]
- Bell, D.A.; Pang, J.; Burrows, S.; Bates, T.R.; van Bockxmeer, F.M.; Hooper, A.J.; O’Leary, P.; Burnett, J.R.; Watts, G.F. Effectiveness of genetic cascade screening for familial hypercholesterolaemia using a centrally co-ordinated clinical service: An Australian experience. Atherosclerosis 2015, 239, 93–100. [Google Scholar] [CrossRef]
- Groselj, U.; Kovac, J.; Sustar, U.; Mlinaric, M.; Fras, Z.; Podkrajsek, K.T.; Battelino, T. Universal screening for familial hypercholesterolemia in children: The Slovenian model and literature review. Atherosclerosis 2018, 277, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Vaz, A.J.; De Marco, M.; Stevens, C.A.T.; Akram, A.; Freiberger, T.; Hovingh, G.K.; Kastelein, J.J.P.; Mata, P.; Raal, F.J.; Santos, R.D.; et al. Overview of the current status of familial hypercholesterolaemia care in over 60 countries—The EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Atherosclerosis 2018, 277, 234–255. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, U.; Futema, M.; Bogsrud, M.P.; Holven, K.B.; Roeters van Lennep, J.; Wiegman, A.; Descamps, O.S.; Vrablik, M.; Freiberger, T.; Dieplinger, H.; et al. Comparison of the characteristics at diagnosis and treatment of children with heterozygous familial hypercholesterolaemia (FH) from eight European countries. Atherosclerosis 2020, 292, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Sustar, U.; Kordonouri, O.; Mlinaric, M.; Kovac, J.; Arens, S.; Sedej, K.; Jenko Bizjan, B.; Trebusak Podkrajsek, K.; Danne, T.; Battelino, T.; et al. Universal screening for familial hypercholesterolemia in 2 populations. Genet. Med. 2022, 24, 2103–2111. [Google Scholar] [CrossRef]
- Wald, D.S.; Bestwick, J.P.; Wald, N.J. Child-parent screening for familial hypercholesterolaemia: Screening strategy based on a meta-analysis. BMJ 2007, 335, 599–603. [Google Scholar] [CrossRef]
- Wald, D.S.; Bestwick, J.P.; Morris, J.K.; Whyte, K.; Jenkins, L.; Wald, N.J. Child–Parent Familial Hypercholesterolemia Screening in Primary Care. N. Engl. J. Med. 2016, 375, 1628–1637. [Google Scholar] [CrossRef]
- Martin, A.C.; Hooper, A.J.; Norman, R.; Nguyen, L.T.; Burnett, J.R.; Bell, D.A.; Brett, T.; Garton-Smith, J.; Pang, J.; Nowak, K.J.; et al. Pilot study of universal screening of children and child-parent cascade testing for familial hypercholesterolaemia in Australia. J. Paediatr. Child Health 2022, 58, 281–287. [Google Scholar] [CrossRef]
- Futema, M.; Cooper, J.A.; Charakida, M.; Boustred, C.; Sattar, N.; Deanfield, J.; Lawlor, D.A.; Timpson, N.J.; Humphries, S.E.; Hingorani, A.D. Screening for familial hypercholesterolaemia in childhood: Avon Longitudinal Study of Parents and Children (ALSPAC). Atherosclerosis 2017, 260, 47–55. [Google Scholar] [CrossRef]
- Ibarretxe, D.; Rodríguez-Borjabad, C.; Feliu, A.; Bilbao, J.Á.; Masana, L.; Plana, N. Detecting familial hypercholesterolemia earlier in life by actively searching for affected children:The DECOPIN project. Atherosclerosis 2018, 278, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Tani, R.; Matsunaga, K.; Inoue, T.; Fu, H.; Ishizawa, M.; Ishikawa, K.; Murakami, K.; Noma, T.; Nishioka, K.; Irie, K.; et al. The effectiveness and barrier of reverse cascade screening for paediatric familial hypercholesterolaemia. Eur. Heart J. 2024, 45, ehae666.2817. [Google Scholar] [CrossRef]
- Besseling, J.; Sjouke, B.; Kastelein, J.J.P. Screening and treatment of familial hypercholesterolemia—Lessons from the past and opportunities for the future (based on the Anitschkow Lecture 2014). Atherosclerosis 2015, 241, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Taageby Nielsen, S.; Mohr Lytsen, R.; Strandkjær, N.; Juul Rasmussen, I.; Sillesen, A.S.; Vøgg, R.O.B.; Axelsson Raja, A.; Nordestgaard, B.G.; Kamstrup, P.R.; Iversen, K.; et al. Significance of lipids, lipoproteins, and apolipoproteins during the first 14-16 months of life. Eur. Heart J. 2023, 44, 4408–4418. [Google Scholar] [CrossRef] [PubMed]
- Higgins, V.; Asgari, S.; Chan, M.K.; Adeli, K. Pediatric reference intervals for calculated LDL cholesterol, non-HDL cholesterol, and remnant cholesterol in the healthy CALIPER cohort. Clin. Chim. Acta 2018, 486, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Balder, J.W.; Lansberg, P.J.; Hof, M.H.; Wiegman, A.; Hutten, B.A.; Kuivenhoven, J.A. Pediatric lipid reference values in the general population: The Dutch lifelines cohort study. J. Clin. Lipidol. 2018, 12, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Steiner, R.; Peterson, A.L. Newborn screening for lipid disorders. Curr. Opin. Lipidol. 2024, 35, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Brody, S.; Carlson, L.A. Plasma lipid concentrations in the newborn with special reference to the distribution of the different lipid fractions. Clin. Chim. Acta 1962, 7, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.; Lee, V.F. Serum lipid levels in infants and mothers at parturition. Clin. Chim. Acta 1965, 12, 258–263. [Google Scholar] [CrossRef]
- Glueck, C.J.; Heckman, F.; Schoenfeld, M.; Steiner, P.; Pearce, W. Neonatal familial type II hyperlipoproteinemia: Cord blood cholesterol in 1800 births. Metabolism 1971, 20, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Tsang, R.C.; Fallat, R.W.; Glueck, C.J. Cholesterol at birth and age 1: Comparison of normal and hypercholesterolemic neonates. Pediatrics 1974, 53, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Mishkel, M.A. Neonatal plasma lipids as measured in cord blood. Can. Med. Assoc. J. 1974, 111, 775–780. [Google Scholar] [PubMed]
- Hardell, L.I. Serum lipids and lipoproteins at birth based on a study of 2815 newborn infants. Acta Paediatrica 1981, 70, 5–10. [Google Scholar] [CrossRef]
- Darmady, J.M.; Fosbrooke, A.S.; Lloyd, J.K. Prospective Study of Serum Cholesterol Levels during First Year of Life. Br. Med. J. 1972, 2, 685–688. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Albers, J.J.; Schrott, H.G.; Hazzard, W.R.; Bierman, E.L.; Motulsky, A.G. Plasma lipid levels and coronary heart disease in adult relatives of newborns with normal and elevated cord blood lipids. Am. J. Hum. Genet. 1974, 26, 727–735. [Google Scholar] [PubMed]
- Boulton, T.J.C.; Craig, I.H.; Hill, G. Screening of cord blood low-density-lipoprotein cholesterol in the diagnosis of familial hypercholesterolaemia: A study of 2000 infants. Acta Paediatr. 1979, 68, 363–370. [Google Scholar] [CrossRef]
- Averna, M.R.; Barbagallo, C.M.; Di Paola, G.; Labisi, M.; Pinna, G.; Marino, G.; Dimita, U.; Notarbartolo, A. Total cholesterol, LDL-cholesterol and apoprotein B in umbilical cord blood: Cross-sectional study. Minerva Pediatr. 1992, 44, 395–399. [Google Scholar] [PubMed]
- Vuorio, A.F.; Turtola, H.; Kontula, K. Neonatal Diagnosis of Familial Hypercholesterolemia in Newborns Born to a Parent With a Molecularly Defined Heterozygous Familial Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3332–3337. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.H.; Liu, Y.J.; Shou, H.C.; Chen, L.J. Cholesterol concentrations in cord blood of newborn infants. Chin. J. Pediatr. 2003, 41, 107–109. [Google Scholar] [PubMed]
- Badiee, Z.; Kelishadi, R. Cord Blood Lipid Profile in a Population of Iranian Term Newborns. Pediatr. Cardiol. 2008, 29, 574–579. [Google Scholar] [CrossRef]
- Pac-Kozuchowska, E.; Rakuś-Kwiatosz, A.; Krawiec, P. Cord blood lipid profile in healthy newborns: A prospective single-center study. Adv. Clin. Exp. Med. 2018, 27, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Pilot Project of Familial Hypercholesterolemia Screening in Newborns in the Czech Republic (CzeCH-IN). Available online: https://clinicaltrials.gov/study/NCT05638022 (accessed on 13 January 2025).
- Asami, T. Screening for hypercholesterolaemia on blood-spotted filterpaper. Lancet 1983, 322, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, W.H.; Forrest, A.R. Screening for hypercholesterolemia by use of blood spotted on filter paper. Clin. Chem. 1985, 31, 648–649. [Google Scholar] [CrossRef]
- Vanbiervliet, J.P.; Vinaimont, N.; Caster, H.; Rosseneu, M.; Belpaire, F. A screening procedure for dyslipoproteinemia in the newborn. Apoprotein quantitation on dried blood spots. Clin. Chim. Acta 1982, 120, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Beeso, J.; Wong, N.; Ayling, R.; Eldridge, P.; Marshall, W.; Sherwood, R.; Peters, T. Screening for hypercholesterolaemia in 10,000 neonates in a multi-ethnic population. Eur. J. Pediatr. 1999, 158, 833–837. [Google Scholar] [CrossRef]
- Wang, X.L.; Dudman, N.P.; Wilcken, D.E. Enzyme-linked immunosorbent assay of apolipoprotein B in blood spotted onto filter paper, suitable for neonatal screening. Clin. Chem. 1989, 35, 1000–1004. [Google Scholar] [CrossRef]
- Ohta, T.; Migita, M.; Yasutake, T.; Matsuda, I. Enzyme-Linked Immunosorbent Assay for Apolipoprotein B on Dried Blood Spot Derived from Newborn Infant: Its Application to Neonatal Mass Screening for Hypercholesterolemia. J. Pediatr. Gastroenterol. Nutr. 1988, 7, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Bangert, S.K.; Eldridge, P.H.; Peters, T.J. Neonatal screening for familial hypercholesterolaemia by immunoturbidimetric assay of apolipoprotein B in dried blood spots. Clin. Chim. Acta 1992, 213, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Dudman, N.P.B.; Blades, B.L.; Wilcken, D.E.L.; Aitken, J.M. Radial immunodiffusion assay of apolipoprotein B in blood dried on filter paper—A potential screening method for familial type II hypercholesterolaemia. Clin. Chim. Acta 1985, 149, 117–127. [Google Scholar] [CrossRef]
- Blades, B.L.; Dudman, N.P.B.; Wilcken, D.E.L. Screening for familial hypercholesterolemia in 5000 neonates: A recall study. Pediatr. Res. 1988, 23, 500–504. [Google Scholar] [CrossRef]
- Wilcken, D.E.L.; Blades, B.L.; Dudman, N.P.B. A neonatal screening approach to the detection of familial hypercholesterolaemia and family-based coronary prevention. J. Inherit. Metab. Dis. 1988, 11, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Blades, B.L.; Dudman, N.P.B.; Wilcken, D.E.L. Variables affecting apolipoprotein B measurements in 3- to 5-day-old babies: A study of 4491 neonates. Pediatr. Res. 1987, 21, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Lakshmy, R.; Gupta, R.; Prabhakaran, D.; Snehi, U.; Reddy, K.S. Utility of Dried Blood Spots for Measurement of Cholesterol and Triglycerides in a Surveillance Study. J. Diabetes Sci. Technol. 2010, 4, 258–262. [Google Scholar] [CrossRef]
- Corso, G.; Papagni, F.; Gelzo, M.; Gallo, M.; Barone, R.; Graf, M.; Scarpato, N.; Dello Russo, A. Development and Validation of an Enzymatic Method for Total Cholesterol Analysis Using Whole Blood Spot. J. Clin. Lab. Anal. 2016, 30, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Eick, G.N.; Kowal, P.; Barrett, T.; Thiele, E.A.; Snodgrass, J.J. Enzyme-Linked Immunoassay-Based Quantitative Measurement of Apolipoprotein B (ApoB) in Dried Blood Spots, a Biomarker of Cardiovascular Disease Risk. Biodemography Soc. Biol. 2017, 63, 116–130. [Google Scholar] [CrossRef]
- Held, P.K.; Campbell, K.; Wiberley-Bradford, A.E.; Lasarev, M.; Horner, V.; Peterson, A. Analytical Validation of Familial Hypercholesterolemia Biomarkers in Dried Blood Spots. Int. J. Neonatal Screen. 2022, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Held, P.K.; Lasarev, M.; Zhang, X.; Wiberley-Bradford, A.E.; Campbell, K.; Horner, V.; Shao, X.; Benoy, M.; Dodge, A.M.; Peterson, A.L. Familial Hypercholesterolemia Biomarker Distribution in Dried Blood Spots. J. Pediatr. 2023, 259, 113469. [Google Scholar] [CrossRef] [PubMed]
- Crimmins, E.M.; Zhang, Y.S.; Kim, J.K.; Frochen, S.; Kang, H.; Shim, H.; Ailshire, J.; Potter, A.; Cofferen, J.; Faul, J. Dried blood spots: Effects of less than optimal collection, shipping time, heat, and humidity. Am. J. Hum. Biol. 2020, 32, e23390. [Google Scholar] [CrossRef] [PubMed]
- Lunke, S.; Bouffler, S.E.; Downie, L.; Caruana, J.; Amor, D.J.; Archibald, A.; Bombard, Y.; Christodoulou, J.; Clausen, M.; De Fazio, P.; et al. Prospective cohort study of genomic newborn screening: BabyScreen+ pilot study protocol. BMJ Open 2024, 14, e081426. [Google Scholar] [CrossRef]
- Stark, Z.; Scott, R.H. Genomic newborn screening for rare diseases. Nat. Rev. Genet. 2023, 24, 755–766. [Google Scholar] [CrossRef]
- Nurchis, M.C.; Altamura, G.; Riccardi, M.T.; Radio, F.C.; Chillemi, G.; Bertini, E.S.; Garlasco, J.; Tartaglia, M.; Dallapiccola, B.; Damiani, G. Whole genome sequencing diagnostic yield for paediatric patients with suspected genetic disorders: Systematic review, meta-analysis, and GRADE assessment. Arch. Public. Health 2023, 81, 93. [Google Scholar] [CrossRef]
- Peterson, A.L.; Held, P.K.; Shao, X.; Lasarev, M.; Zhang, X.; Wiberley-Bradford, A.; Campbell, K.; Benoy, M.; Dodge, A.M.; Horner, V. Abstract 11554: Biochemical and Molecular Newborn Screening for Familial Hypercholesterolemia. Circulation 2022, 146, A11554. [Google Scholar] [CrossRef]
- Peterson, A.L.; Shao, X.; Zhang, X.; Lasarev, M.; Benoy, M.; Held, P.; Horner, V. Abstract 15499: Biochemical and Genetic Newborn Screening for Familial Hypercholesterolemia. Circulation 2023, 148, A15499. [Google Scholar] [CrossRef]
- Shum, B.O.V.; Pretorius, C.J.; Sng, L.M.F.; Henner, I.; Barahona, P.; Basar, E.; McGill, J.; Wilgen, U.; Zournazi, A.; Downie, L.; et al. Feasibility of Targeted Next-Generation DNA Sequencing for Expanding Population Newborn Screening. Clin. Chem. 2023, 69, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Luo, G.; Zhang, A.; Gao, S.; Tang, Y.; Du, Z.; Pan, S. Genetic identification of familial hypercholesterolemia within whole genome sequences in 6820 newborns. Clin. Genet. 2024, 105, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Wald, D.S.; Wald, N.J. Integration of child–parent screening and cascade testing for familial hypercholesterolaemia. J. Med. Screen. 2019, 26, 71–75. [Google Scholar] [CrossRef]
- Wald, D.S.; Bestwick, J.P. Reaching detection targets in familial hypercholesterolaemia: Comparison of identification strategies. Atherosclerosis 2020, 293, 57–61. [Google Scholar] [CrossRef] [PubMed]
- McKay, A.J.; Hogan, H.; Humphries, S.E.; Marks, D.; Ray, K.K.; Miners, A. Universal screening at age 1–2 years as an adjunct to cascade testing for familial hypercholesterolaemia in the UK: A cost-utility analysis. Atherosclerosis 2018, 275, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Jahn, B.; Santamaria, J.; Dieplinger, H.; Binder, C.J.; Ebenbichler, C.; Scholl-Bürgi, S.; Conrads-Frank, A.; Rochau, U.; Kühne, F.; Stojkov, I.; et al. Familial hypercholesterolemia: A systematic review of modeling studies on screening interventions. Atherosclerosis 2022, 355, 15–29. [Google Scholar] [CrossRef]
- Chief Health Officer, Queensland. Newborn Screening. Available online: https://www.choreport.health.qld.gov.au/our-lifestyle/population-screening/newborn-screening (accessed on 19 January 2025).
- McDonagh, E.M.; Cipriani, V.; Ellingford, J.M.; Tucci, A.; Vandrovcova, J.; Chan, G.; Williams, H.J.; Ratnaike, T.; Wei, W.; Stirrups, K.; et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report. N. Engl. J. Med. 2021, 385, 1868–1880. [Google Scholar] [CrossRef]
- Redit, C.; Hura, Z.; Amy, L. UK launches whole-genome sequencing pilot for babies. Nat. Biotechnol. 2023, 41, 4. [Google Scholar] [CrossRef]
- Borry, P.; Evers-Kiebooms, G.; Cornel, M.C.; Clarke, A.; Dierickx, K. Genetic testing in asymptomatic minors Background considerations towards ESHG Recommendations. Eur. J. Hum. Genet. 2009, 17, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Raitakari, O.; Pahkala, K.; Magnussen, C.G. Prevention of atherosclerosis from childhood. Nat. Rev. Cardiol. 2022, 19, 543–554. [Google Scholar] [CrossRef]
- Umans-Eckenhausen, M.A.W.; Oort, F.J.; Ferenschild, K.C.M.P.; Defesche, J.C.; Kastelein, J.J.P.; de Haes, J.C.J.M. Parental attitude towards genetic testing for familial hypercholesterolaemia in children. J. Med. Genet. 2002, 39, e49. [Google Scholar] [CrossRef] [PubMed]
- Tobik, K.; Orland, K.M.; Zhang, X.; Garcia, K.; Peterson, A.L. Parental Attitudes and Ideas Regarding Newborn Screening for Familial Hypercholesterolemia. Matern. Child Health J. 2023, 27, 978–983. [Google Scholar] [CrossRef]
- van Maarle, M.C.; Stouthard, M.E.A.; Marang-van de Mheen, P.J.; Klazinga, M.N.S.; Bonsel, G.J. How Disturbing Is It to Be Approached for a Genetic Cascade Screening Programme for Familial Hypercholesterolaemia? Psychological Impact and Screenees’ Views. Community Genet. 2001, 4, 244–252. [Google Scholar] [CrossRef]
- Marteau, T.; Senior, V.; Humphries, S.E.; Bobrow, M.; Cranston, T.; Crook, M.A.; Day, L.; Fernandez, M.; Horne, R.; Iversen, A.; et al. Psychological impact of genetic testing for familial hypercholesterolemia within a previously aware population: A randomized controlled trial. Am. J. Med. Genet. 2004, 128A, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Meulenkamp, T.M.; Tibben, A.; Mollema, E.D.; van Langen, I.M.; Wiegman, A.; de Wert, G.M.; de Beaufort, I.D.; Wilde, A.A.M.; Smets, E.M.A. Predictive genetic testing for cardiovascular diseases: Impact on carrier children. Am. J. Med. Genet. 2008, 146A, 3136–3146. [Google Scholar] [CrossRef]
- Smets, E.M.A.; Stam, M.M.H.; Meulenkamp, T.M.; van Langen, I.M.; Wilde, A.A.M.; Wiegman, A.; de Wert, G.M.; Tibben, A. Health-related quality of life of children with a positive carrier status for inherited cardiovascular diseases. Am. J. Med. Genet. 2008, 146A, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Brett, T. Universal screening for familial hypercholesterolaemia in newborns: Time for general practice to contribute. Aust. J. Gen. Pract. 2023, 52, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Tiller, J.; Lacaze, P.; Otlowski, M. The Australian moratorium on genetics and life insurance: Evaluating policy compared to Parliamentary recommendations regarding genetic discrimination. Public. Health Res. Pract. 2022, 32, e3242235. [Google Scholar] [CrossRef]
- Ministry of Treasury. Use of Genetic Testing Results in Life Insurance and Underwriting. Available online: https://treasury.gov.au/consultation/c2023-467047 (accessed on 18 November 2024).
- Dowling, G.; Tiller, J.; McInerney-Leo, A.; Belcher, A.; Haining, C.; Barlow-Stewart, K.; Boughtwood, T.; Gleeson, P.; Delatycki, M.B.; Winship, I.; et al. Health professionals’ views and experiences of the Australian moratorium on genetic testing and life insurance: A qualitative study. Eur. J. Hum. Genet. 2022, 30, 1262–1268. [Google Scholar] [CrossRef]
- Tiller, J.; Gleeson, P.; McInerney-Leo, A.M.; Keogh, L.; Nowak, K.; Barlow-Stewart, K.; Boughtwood, T.; Delatycki, M.B.; Winship, I.; Otlowski, M.; et al. Final Stakeholder Report of the Australian Genetics and Life Insurance Moratorium: Monitoring the Effectiveness and Response (A-GLIMMER) Project. Available online: https://bridges.monash.edu/articles/report/_strong_Final_Stakeholder_Report_of_the_strong_em_strong_Australian_Genetics_and_Life_Insurance_Moratorium_Monitoring_the_Effectiveness_and_Response_A-GLIMMER_strong_em_strong_Project_strong_/23564538?file=41361345 (accessed on 18 November 2024).
- Ministry of Treasury. Total Ban on the Use of Adverse Genetic Testing Results in Life Insurance. Available online: https://ministers.treasury.gov.au/ministers/stephen-jones-2022/media-releases/total-ban-use-adverse-genetic-testing-results-life (accessed on 13 November 2024).
- Chora, J.R.; Iacocca, M.A.; Tichý, L.; Wand, H.; Kurtz, C.L.; Zimmermann, H.; Leon, A.; Williams, M.; Humphries, S.E.; Hooper, A.J.; et al. The Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel consensus guidelines for LDLR variant classification. Genet. Med. 2022, 24, 293–306. [Google Scholar] [CrossRef]
- Haralambos, K.; Payne, J.; Datta, D.; McDowell, I.; Cramb, R.; Williams, S.; Cather, M.; Neely, D.; Soran, H.; Miedzybroadzka, Z.; et al. How many patients with a monogenic diagnosis of Familial Hypercholesterolemia are currently known in UK lipid clinics? Atheroscler. Suppl. 2017, 28, e8–e9. [Google Scholar] [CrossRef]
- Guillem, P.-M.; Maria, I.; Giorgio, C.; Raquel, Y.; Elena-Alexandra, T.; Clara, D.M.v.K.; Francjan, J.v.S. Analysis of genomics implementation in newborn screening for inherited metabolic disorders: An IRDiRC initiative. Rare Dis. Orphan Drug J. 2024, 3, 12. [Google Scholar] [CrossRef]
- Thormaehlen, A.S.; Schuberth, C.; Won, H.-H.; Blattmann, P.; Joggerst-Thomalla, B.; Theiss, S.; Asselta, R.; Duga, S.; Merlini, P.A.; Ardissino, D.; et al. Systematic Cell-Based Phenotyping of Missense Alleles Empowers Rare Variant Association Studies: A Case for LDLR and Myocardial Infarction. PLoS Genet. 2015, 11, e1004855. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Larrea-Sebal, A.; Alonso-Estrada, R.; Aguilo-Arce, J.; Ostolaza, H.; Palacios, L.; Martin, C. Mutation type classification and pathogenicity assignment of sixteen missense variants located in the EGF-precursor homology domain of the LDLR. Sci. Rep. 2020, 10, 1727. [Google Scholar] [CrossRef]
- Horton, A.E.; Martin, A.C.; Srinivasan, S.; Justo, R.N.; Poplawski, N.K.; Sullivan, D.; Brett, T.; Chow, C.K.; Nicholls, S.J.; Pang, J.; et al. Integrated guidance to enhance the care of children and adolescents with familial hypercholesterolaemia: Practical advice for the community clinician. J. Paediatr. Child Health 2022, 58, 1297–1312. [Google Scholar] [CrossRef] [PubMed]
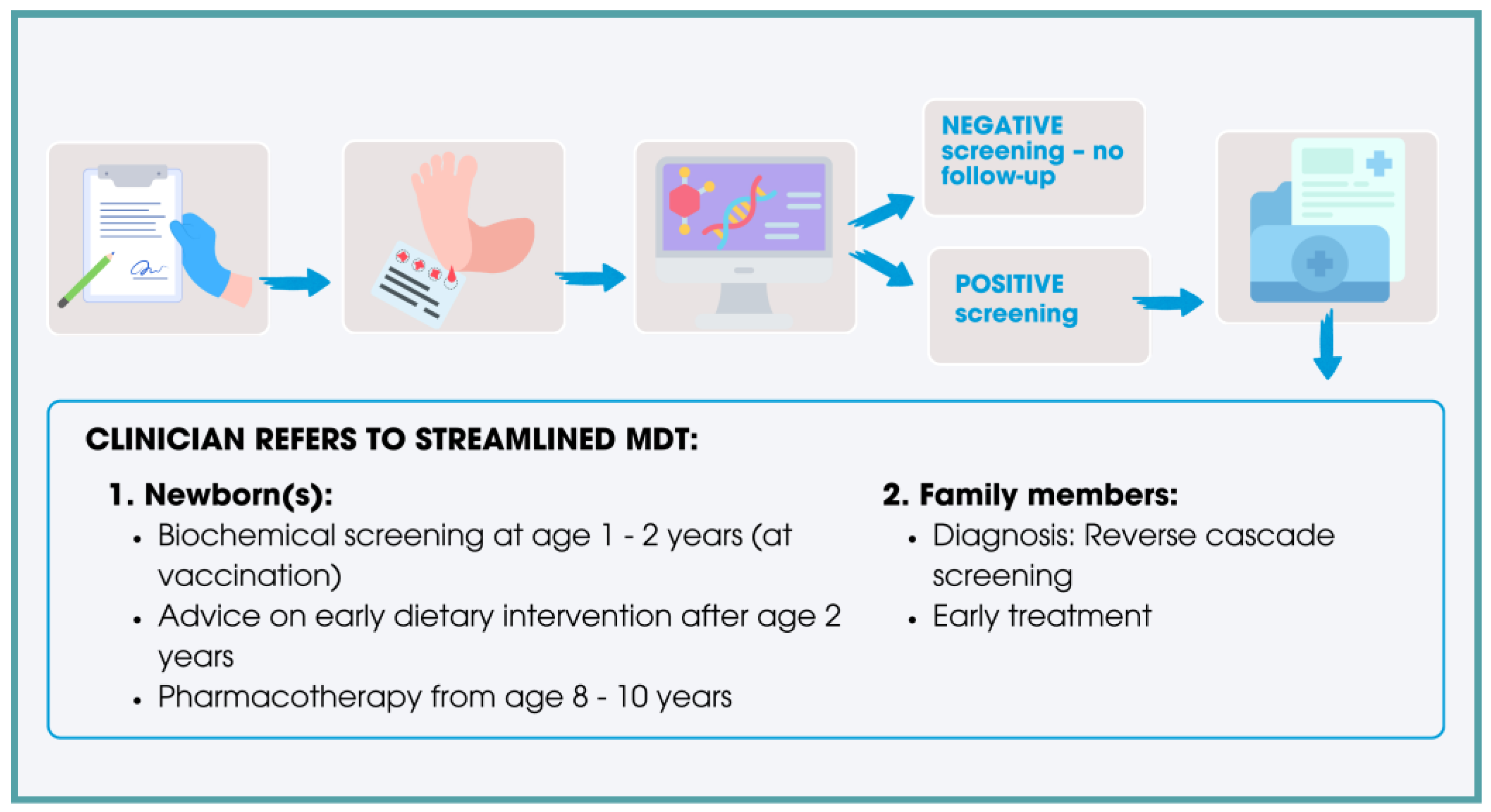

{kind=link}
| Year | Issuing Body | Approach of Screening Age at First Screening | Initial Test | Reference |
|---|---|---|---|---|
| 2011 | National Heart, Lung, and Blood Institute’s Expert Panel (NHLBI) | Selective: age 2–8 Universal: age 9–11 and 17–21 | Lipid profile | [29] |
| 2011 | American Academy of Pediatrics (adopted NHLBI) | Selective: age 2–8 Universal: age 9–11 and 17–21 | Lipid profile | [29] |
| 2011 and 2015 | National Lipid Association | Selective: age 2 Universal: age 9–11, repeat at age 20 or earlier | Lipid profile | [30,31] |
| 2018 | American Heart Association | Selective: age 2 Universal: age 9–11 and 17–21 | Lipid profile | [32] |
| 2015 and 2019 | European Society of Cardiology/European Atherosclerosis Society | Selective: age of 5, or as early as possible if homozygous FH suspected Universal: may be considered | Lipid profile | [13,33] |
| 2021 | FH Australasia Network Consensus Working Group (endorsed by the Australian Atherosclerosis Society) | Selective:
| Lipid profile | [8] |
| 2023 | US Preventative Services Task Force | Insufficient data to screen asymptomatic children aged 20 years or younger | Not applicable | [34] |
| 2023 | International Atherosclerosis Society | Selective:
| Lipid profile | [35] |
| Strategy | Situation | Test | Reference |
|---|---|---|---|
| Selective | |||
| Opportunistic |
| Lipid profile | [36,37] |
| Lipid profile | [35] | |
| Lipid profile | ||
| [38,39] | ||
| |||
| Systematic Cascade testing |
| Lipid profile/genetic testing | [7,40] |
| Universal | |||
| Opportunistic |
| Lipid profile/genetic testing | See Section 3 and Section 4 |
| Systematic Reverse cascade Child parent |
| Lipid profile/genetic testing | See Section 3 and Section 4 |
| Opportunities |
| Reduction in morbidity and mortality from ASCVD due to early treatment |
| Detection of homozygous FH |
| No requirement for additional blood collections |
| Unaffected by prematurity/gestational age, sex, maternal lipid concentrations, illness |
| Possibility of early reverse cascade screening |
| Ability of re-analysis of stored data as knowledge of FH variants expands |
| Equitable access to and high participation rate in newborn screening |
| Challenges |
| Time distance between diagnosis of heterozygous FH and start of treatment |
| Laboratory resource implications for large increase in genomic screening numbers |
| Cost of sequencing FH genes |
| Workforce and infrastructure requirements for follow-up of expected increase in diagnosed FH individuals |
| Informed consent without impact on standard newborn screening |
| Impact on insurance access |
| Data interpretation: variants of unknown significance, variable penetrance and/or expressivity |
| Maintaining genetic privacy and security |
| Concern regarding parental anxiety and impact on parent–child relationship |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bachmeier, C.; Ungerer, J.; Pretorius, C.; Kassianos, A.; Kostner, K.M. Universal Paediatric and Newborn Screening for Familial Hypercholesterolaemia—Challenges and Opportunities: An Australian Perspective. Lipidology 2025, 2, 4. https://doi.org/10.3390/lipidology2010004
Bachmeier C, Ungerer J, Pretorius C, Kassianos A, Kostner KM. Universal Paediatric and Newborn Screening for Familial Hypercholesterolaemia—Challenges and Opportunities: An Australian Perspective. Lipidology. 2025; 2(1):4. https://doi.org/10.3390/lipidology2010004
Chicago/Turabian StyleBachmeier, Caroline, Jacobus Ungerer, Carel Pretorius, Andrew Kassianos, and Karam M. Kostner. 2025. "Universal Paediatric and Newborn Screening for Familial Hypercholesterolaemia—Challenges and Opportunities: An Australian Perspective" Lipidology 2, no. 1: 4. https://doi.org/10.3390/lipidology2010004
APA StyleBachmeier, C., Ungerer, J., Pretorius, C., Kassianos, A., & Kostner, K. M. (2025). Universal Paediatric and Newborn Screening for Familial Hypercholesterolaemia—Challenges and Opportunities: An Australian Perspective. Lipidology, 2(1), 4. https://doi.org/10.3390/lipidology2010004