Possible Mechanisms by which Stefin B could Regulate Proteostasis and Oxidative Stress


Abstract
:1. Introduction
2. Progressive Myoclonus Epilepsy of Type 1 (EPM1)—Unverricht–Lundborg Disease
3. Disease Development and Progression
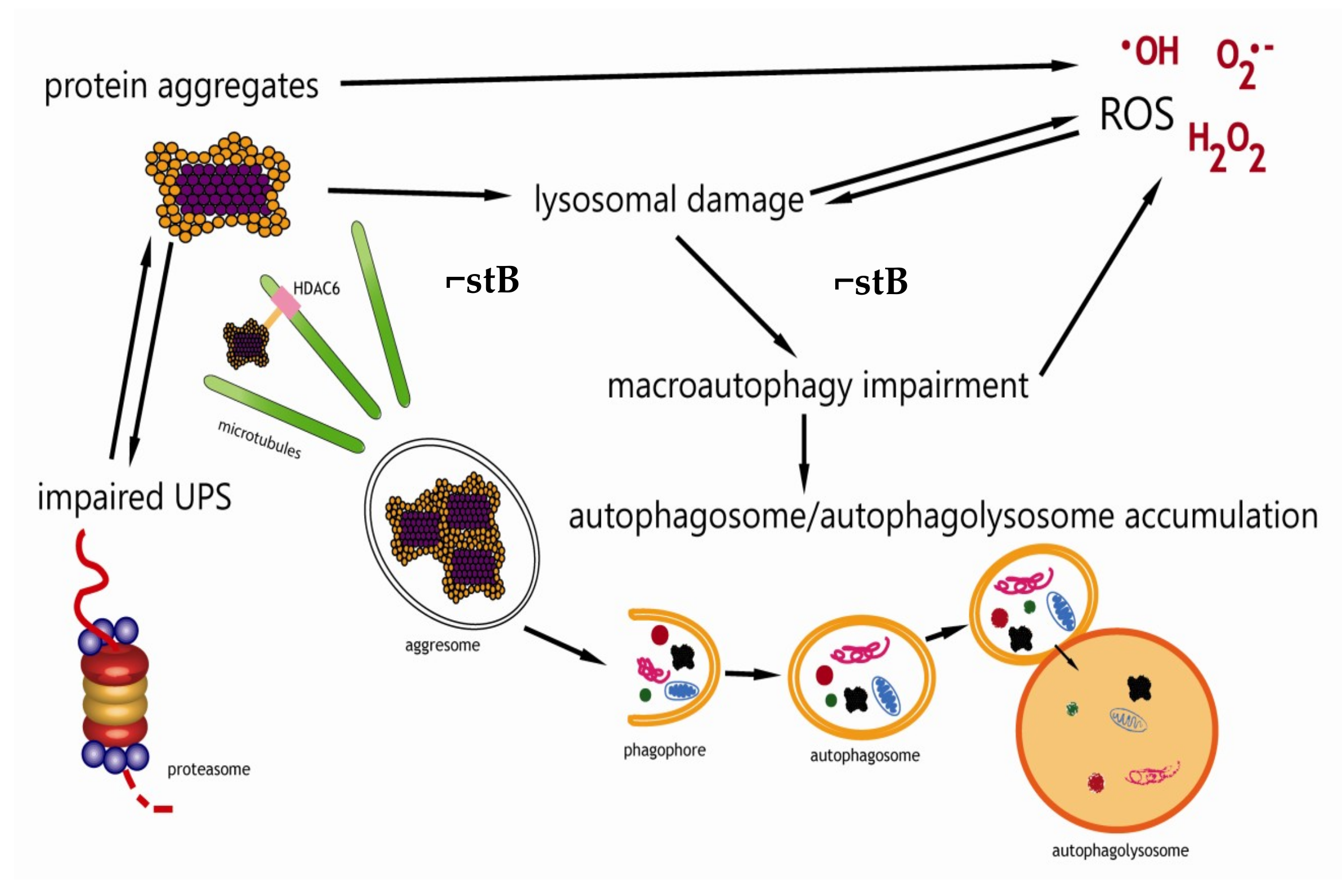
4. Amyloidogenesis of Human Stefin B In Vitro
5. Protein Aggregation in Cells and Oxidative Stress
6. Protein Aggregation and Autophagy
7. Is Protein Aggregation a Modulating Factor in Myoclonal Epilepsies and Broader?
Funding
Conflicts of Interest
References
- Rawlings, N.D. Peptidase inhibitors in the MEROPS database. Biochimie 2010, 96, 1463–1483. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Levy, E. Cystatin C in Alzheimer’s disease. Front. Mol. Neurosci. 2012, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Tizon, B.; Ribe, E.M.; Mi, W.; Troy, C.M.; Levy, E. Cystatin C protects neuronal cells from amyloid-beta-induced toxicity. J. Alzheimers Dis. 2010, 19, 885–894. [Google Scholar] [CrossRef]
- Tizon, B.; Sahoo, S.; Yu, H.; Gauthier, S.; Kumar, A.R.; Mohan, P.; Figliola, M.; Pawlik, M.; Grubb, A.; Uchiyama, Y.; et al. Induction of autophagy by cystatin C: A mechanism that protects murine primary cortical neurons and neuronal cell lines. PLoS ONE 2010, 5, e9819. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Mirotsou, M.; Buresi, C.; Peitsch, M.C.; Rossier, C.; Ouazzani, R.; Baldy-Moulinier, M.; Bottani, A.; Malafosse, A.; Antonarakis, S.E.; et al. Identification of mutations in cystatin B, the gene responsible for the Unverricht-Lundborg type of progressive myoclonus epilepsy (EPM1). Am. J. Hum. Genet. 1997, 60, 342–351. [Google Scholar] [PubMed]
- Pennacchio, L.A.; Lehesjoki, A.E.; Stone, N.E.; Willour, V.L.; Virtaneva, K.; Miao, J.; D’Amato, E.; Ramirez, L.; Faham, M.; Koskiniemi, M.; et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science 1996, 271, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Scott, H.S.; Genton, P.; Grid, D.; Ouazzani, R.; M’Rabet, A.; Ibrahim, S.; Gouider, R.; Dravet, C.; Chkili, T.; et al. The expanded dodecamer repeat in Progressive myoclonus epilepsy (EPM1) is unstable, shows no correlation with age of onset, and results in reduced expression of reporter genes in vitro. Eur. J. Hum. Genet. 1998, 6, 146. [Google Scholar]
- Joensuu, T.; Lehesjoki, A.E.; Kopra, O. Molecular background of EPM1-Unverricht-Lundborg disease. Epilepsia 2008, 49, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Polajnar, M.; Čeru, S.; Jerala, N.K.; Žerovnik, E. Human stefin B normal and patho-physiological role: Molecular and cellular aspects of amyloid-type aggregation of certain EPM1 mutants. Front. Mol. Neurosci. 2012. [Google Scholar] [CrossRef]
- Kenig, M.; Jenko-Kokalj, S.; Tusek-Znidaric, M.; Pompe-Novak, M.; Guncar, G.; Turk, D.; Waltho, J.P.; Staniforth, R.A.; Avbelj, F.; Zerovnik, E. Folding and amyloid-fibril formation for a series of human stefins’ chimeras: Any correlation? Proteins 2006, 62, 918–927. [Google Scholar] [CrossRef]
- Skerget, K.; Vilfan, A.; Pompe-Novak, M.; Turk, V.; Waltho, J.P.; Turk, D.; Zerovnik, E. The mechanism of amyloid-fibril formation by stefin B: Temperature and protein concentration dependence of the rates. Proteins 2009, 74, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Taler-Vercic, A.; Kirsipuu, T.; Friedemann, M.; Noormagi, A.; Polajnar, M.; Smirnova, J.; Znidaric, M.T.; Zganec, M.; Skarabot, M.; Vilfan, A.; et al. The role of initial oligomers in amyloid fibril formation by human stefin B. Int. J. Mol. Sci. 2013, 14, 18362–18384. [Google Scholar] [CrossRef] [PubMed]
- Zerovnik, E. Amyloid-fibril formation. Proposed mechanisms and relevance to conformational disease. Eur. J. Biochem. 2002, 269, 3362–3371. [Google Scholar] [PubMed]
- Zerovnik, E.; Jerala, R.; Kroon-Zitko, L.; Turk, V.; Pain, R.H. Denaturation of stefin B by GuHCl, pH and heat; evidence for molten globule intermediates. Biol. Chem. Hoppe Seyler 1992, 373, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Zerovnik, E.; Jerala, R.; Virden, R.; Kroon, Z.L.; Turk, V.; Waltho, J.P. On the mechanism of human stefin B folding: II. Folding from GuHCl unfolded, TFE denatured, acid denatured, and acid intermediate states. Proteins 1998, 32, 304–313. [Google Scholar] [CrossRef]
- Zerovnik, E.; Virden, R.; Jerala, R.; Kroon-Zitko, L.; Turk, V.; Waltho, J.P. Differences in the effects of TFE on the folding pathways of human stefins A and B. Proteins 1999, 36, 205–216. [Google Scholar] [CrossRef]
- Ceru, S.; Layfield, R.; Zavašnik-Bergant, T.; Repnik, U.; Kopitar-Jerala, N.; Turk, V.; Žerovnik, E. Intracellular aggregation of human stefin B; confocal and electron microscopy study. Biol Cell 2010. [Google Scholar] [CrossRef]
- Polajnar, M.; Zavasnik-Bergant, T.; Kopitar-Jerala, N.; Tusek-Znidaric, M.; Zerovnik, E. Gain in toxic function of stefin B EPM1 mutants aggregates: Correlation between cell death, aggregate number/size and oxidative stress. Biochim. Biophys. Acta 2014, 1843, 2089–2099. [Google Scholar] [CrossRef] [Green Version]
- Lehtinen, M.K.; Tegelberg, S.; Schipper, H.; Su, H.; Zukor, H.; Manninen, O.; Kopra, O.; Joensuu, T.; Hakala, P.; Bonni, A.; et al. Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J. Neurosci. 2009, 29, 5910–5915. [Google Scholar] [CrossRef]
- Polajnar, M.; Zavasnik-Bergant, T.; Skerget, K.; Vizovisek, M.; Vidmar, R.; Fonovic, M.; Kopitar-Jerala, N.; Petrovic, U.; Navarro, S.; Ventura, S.; et al. Human stefin B role in cell’s response to misfolded proteins and autophagy. PLoS ONE 2014, 9, e102500. [Google Scholar] [CrossRef]
- Kaur, G.; Mohan, P.; Pawlik, M.; DeRosa, S.; Fajiculay, J.; Che, S.; Grubb, A.; Ginsberg, S.D.; Nixon, R.A.; Levy, E. Cystatin C rescues degenerating neurons in a cystatin B-knockout mouse model of progressive myoclonus epilepsy. Am. J. Pathol 2010, 177, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, R.; Iivanainen, M.; Stern, R.; Koerber, T.; Wilder, B.J. “Baltic” myoclonus epilepsy: Hereditary disorder of childhood made worse by phenytoin. Lancet 1983, 2, 838–842. [Google Scholar] [CrossRef]
- Kalviainen, R.; Khyuppenen, J.; Koskenkorva, P.; Eriksson, K.; Vanninen, R.; Mervaala, E. Clinical picture of EPM1-Unverricht-Lundborg disease. Epilepsia 2008, 49, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Di Giaimo, R.; Riccio, M.; Santi, S.; Galeotti, C.; Ambrosetti, D.C.; Melli, M. New insights into the molecular basis of progressive myoclonus epilepsy: A multiprotein complex with cystatin B. Hum. Mol. Genet. 2002, 11, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Škerget, K.; Taler-Verčič, A.; Bavdek, A.; Hodnik, V.; Čeru, S.; Tušek-Žnidarič, M.; Kumm, T.; Pitsi, D.; Pompe-Novak, M.; Palumaa, P.; et al. Interaction between oligomers of stefin B and amyloid-beta in vitro and in cells. J. Biol. Chem. 2010, 285, 3201–3210. [Google Scholar] [CrossRef] [PubMed]
- Taler-Vercic, A.; Zerovnik, E. Binding of amyloid peptides to domain-swapped dimers of other amyloid-forming proteins may prevent their neurotoxicity. Bioessays 2010, 32, 1020–1024. [Google Scholar] [CrossRef] [Green Version]
- Anderluh, G.; Gutierrez-Aguirre, I.; Rabzelj, S.; Ceru, S.; Kopitar-Jerala, N.; Macek, P.; Turk, V.; Zerovnik, E. Interaction of human stefin B in the prefibrillar oligomeric form with membranes. Correlation with cellular toxicity. FEBS J. 2005, 272, 3042–3051. [Google Scholar] [CrossRef]
- Pennacchio, L.A.; Bouley, D.M.; Higgins, K.M.; Scott, M.P.; Noebels, J.L.; Myers, R.M. Progressive ataxia, myoclonic epilepsy and cerebellar apoptosis in cystatin B-deficient mice. Nat. Genet. 1998, 20, 251–258. [Google Scholar] [CrossRef]
- Vaarmann, A.; Kaasik, A.; Zharkovsky, A. Altered tryptophan metabolism in the brain of cystatin B-deficient mice: A model system for progressive myoclonus epilepsy. Epilepsia 2006, 47, 1650–1654. [Google Scholar] [CrossRef]
- Korja, M.; Kaasinen, V.; Lamusuo, S.; Parkkola, R.; Nagren, K.; Marttila, R.J. Substantial thalamostriatal dopaminergic defect in Unverricht-Lundborg disease. Epilepsia 2007, 48, 1768–1773. [Google Scholar] [CrossRef]
- Franceschetti, S.; Sancini, G.; Buzzi, A.; Zucchini, S.; Paradiso, B.; Magnaghi, G.; Frassoni, C.; Chikhladze, M.; Avanzini, G.; Simonato, M. A pathogenetic hypothesis of Unverricht-Lundborg disease onset and progression. Neurobiol. Dis. 2007, 25, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Koskenkorva, P.; Khyuppenen, J.; Niskanen, E.; Kononen, M.; Bendel, P.; Mervaala, E.; Lehesjoki, A.E.; Kalviainen, R.; Vanninen, R. Motor cortex and thalamic atrophy in Unverricht-Lundborg disease: Voxel-based morphometric study. Neurology 2009, 73, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Korja, M.; Ferlazzo, E.; Soilu-Hanninen, M.; Magaudda, A.; Marttila, R.; Genton, P.; Parkkola, R. T2-weighted high-intensity signals in the basal ganglia as an interesting image finding in Unverricht-Lundborg disease. Epilepsy Res. 2010, 88, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Ceru, S.; Kokalj, S.J.; Rabzelj, S.; Skarabot, M.; Gutierrez-Aguirre, I.; Kopitar-Jerala, N.; Anderluh, G.; Turk, D.; Turk, V.; Zerovnik, E. Size and morphology of toxic oligomers of amyloidogenic proteins: A case study of human stefin B. Amyloid 2008, 15, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Ceru, S.; Zerovnik, E. Similar toxicity of the oligomeric molten globule state and the prefibrillar oligomers. FEBS Lett. 2008, 582, 203–209. [Google Scholar] [CrossRef]
- Stefani, M. Protein aggregation diseases: Toxicity of soluble prefibrillar aggregates and their clinical significance. Methods Mol. Biol. 2010, 648, 25–41. [Google Scholar]
- Rabzelj, S.; Viero, G.; Gutierrez-Aguirre, I.; Turk, V.; Dalla Serra, M.; Anderluh, G.; Zerovnik, E. Interaction with model membranes and pore formation by human stefin B: Studying the native and prefibrillar states. FEBS J. 2008, 275, 2455–2466. [Google Scholar] [CrossRef]
- Alakurtti, K.; Weber, E.; Rinne, R.; Theil, G.; de Haan, G.J.; Lindhout, D.; Salmikangas, P.; Saukko, P.; Lahtinen, U.; Lehesjoki, A.E. Loss of lysosomal association of cystatin B proteins representing progressive myoclonus epilepsy, EPM1, mutations. Eur. J. Hum. Genet. 2005, 13, 208–215. [Google Scholar] [CrossRef]
- Cipollini, E.; Riccio, M.; Di Giaimo, R.; Dal Piaz, F.; Pulice, G.; Catania, S.; Caldarelli, I.; Dembic, M.; Santi, S.; Melli, M. Cystatin B and its EPM1 mutants are polymeric and aggregate prone in vivo. Biochim. Biophys. Acta 2008, 1783, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Rabzelj, S.; Turk, V.; Zerovnik, E. In vitro study of stability and amyloid-fibril formation of two mutants of human stefin B (cystatin B) occurring in patients with EPM1. Protein Sci. 2005, 14, 2713–2722. [Google Scholar] [CrossRef] [Green Version]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell. Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta 2008, 1782, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Mayes, J.; Tinker-Mill, C.; Kolosov, O.; Zhang, H.; Tabner, B.J.; Allsop, D. β-amyloid fibrils in Alzheimer disease are not inert when bound to copper ions but can degrade hydrogen peroxide and generate reactive oxygen species. J. Biol. Chem. 2014, 289, 12052–12062. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol. 2000, 130, 184–208. [Google Scholar] [CrossRef] [PubMed]
- Zerovnik, E.; Skerget, K.; Tusek-Znidaric, M.; Loeschner, C.; Brazier, M.W.; Brown, D.R. High affinity copper binding by stefin B (cystatin B) and its role in the inhibition of amyloid fibrillation. FEBS J. 2006, 273, 4250–4263. [Google Scholar] [CrossRef] [Green Version]
- Jena, K.K.; Kolapalli, S.P.; Mehto, S.; Nath, P.; Das, B.; Sahoo, P.K.; Ahad, A.; Syed, G.H.; Raghav, S.K.; Senapati, S.; et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Meriin, A.B.; Narayanan, A.; Meng, L.; Alexandrov, I.; Varelas, X.; Cisse, I.I.; Sherman, M.Y. Hsp70-Bag3 complex is a hub for proteotoxicity-induced signaling that controls protein aggregation. Proc. Natl. Acad. Sci. USA 2018, 115, E7043–E7052. [Google Scholar] [CrossRef]
- Zerovnik, E. Putative alternative functions of human stefin B (cystatin B): binding to amyloid-beta, membranes, and copper. J. Mol. Recognit. 2016. [Google Scholar] [CrossRef]
- Polajnar, M.; Zerovnik, E. Impaired autophagy: A link between neurodegenerative diseases and progressive myoclonus epilepsies. Trends Mol. Med. 2011, 17, 293–300. [Google Scholar] [CrossRef]
- Polajnar, M.; Zerovnik, E. Impaired autophagy: A link between neurodegenerative and neuropsychiatric diseases. J. Cell. Mol. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floto, R.A.; Sarkar, S.; Perlstein, E.O.; Kampmann, B.; Schreiber, S.L.; Rubinsztein, D.C. Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington’s disease models and enhance killing of mycobacteria by macrophages. Autophagy 2007, 3, 620–622. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Mutation | Position of Mutation in Gene/Type | Predicted Consequence on Protein |
|---|---|---|
| dodecamer repeat expansion | 5’ UTR/expansion | reduced CSTB expression |
| c.10G>C | exon 1/missense | p.G4R |
| c.67-1G>C | intron 1/splice site | p.delV23_K56 |
| c.149G>A | exon 2/missense | p.G50E |
| c.168>A | exon 2/splice site | aberrant splicing? |
| c.169-2A>G | intron 2/splice site | aberrant splicing? |
| c.168+1_18del | intron 2/deletion | p.delV23_K56 p.V57EfsX28 |
| c.202C>T | exon 3/nonsense | p.R68X |
| c.218_219delTC | exon 3/deletion | p.L73FSX3 |
| c.212A>C | exon 3/missense | p.Q71P |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Žerovnik, E. Possible Mechanisms by which Stefin B could Regulate Proteostasis and Oxidative Stress. Cells 2019, 8, 70. https://doi.org/10.3390/cells8010070
Žerovnik E. Possible Mechanisms by which Stefin B could Regulate Proteostasis and Oxidative Stress. Cells. 2019; 8(1):70. https://doi.org/10.3390/cells8010070
Chicago/Turabian StyleŽerovnik, Eva. 2019. "Possible Mechanisms by which Stefin B could Regulate Proteostasis and Oxidative Stress" Cells 8, no. 1: 70. https://doi.org/10.3390/cells8010070
APA StyleŽerovnik, E. (2019). Possible Mechanisms by which Stefin B could Regulate Proteostasis and Oxidative Stress. Cells, 8(1), 70. https://doi.org/10.3390/cells8010070