PML Bodies in Mitosis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. PML Bodies and the Cell Cycle
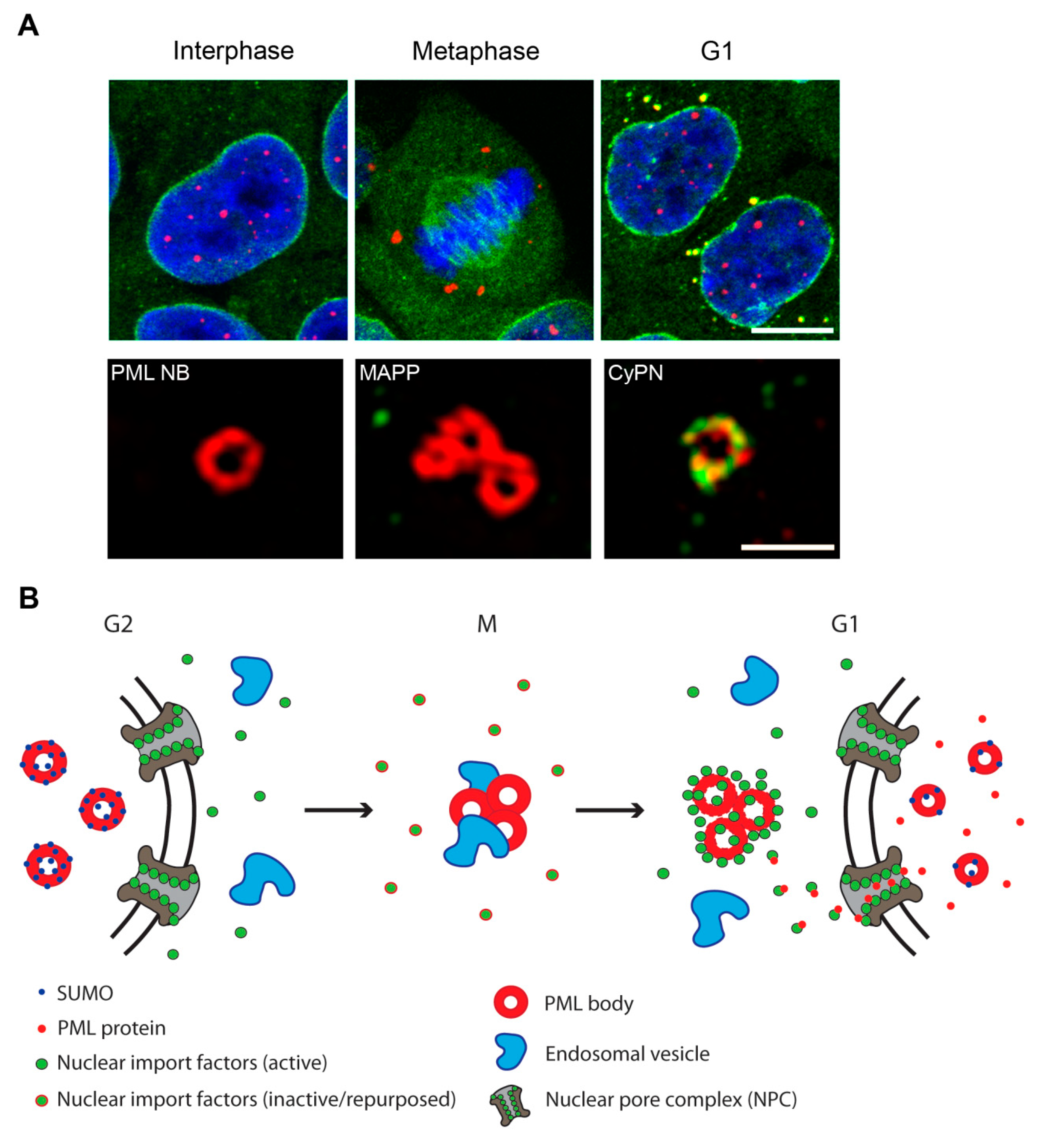
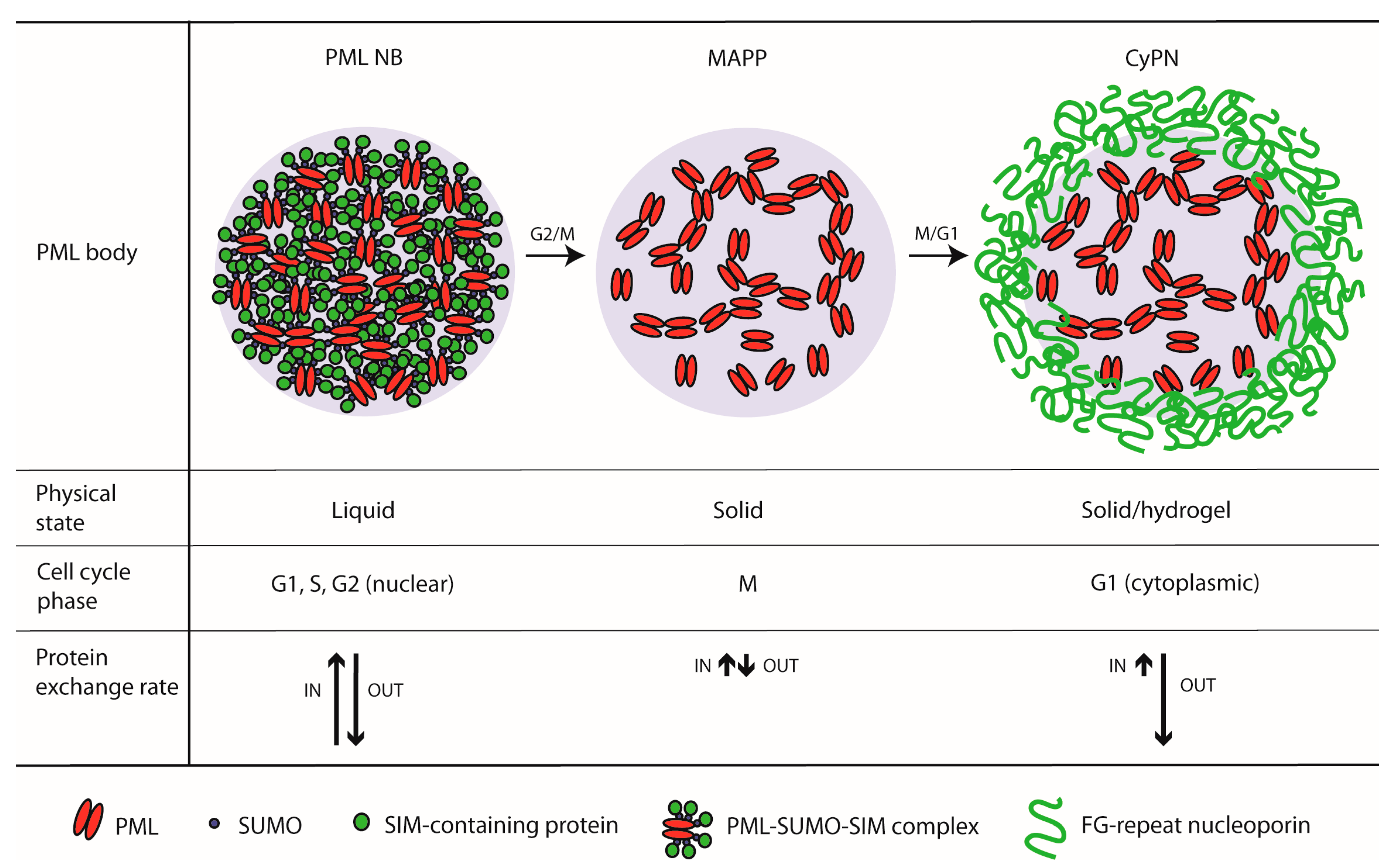
2.1. PML Bodies in Interphase
2.2. PML Bodies at the G2/M-Phase Transition
2.3. PML Bodies at the M/G1-Phase Transition
3. Implications of Mitotic PML Body Trafficking
3.1. Do PML Bodies Undergo a Liquid-to-Solid Transition at Entry into Mitosis?
3.2. The Role of Mitotic PML Bodies in Asymmetric Cell Division and Cell Fate
3.3. The Role of Mitotic PML Bodies in Acute Promyelocytic Leukemia (APL) Therapy
3.4. Cell Compartment Sorting by the Nuclear Localization Signal during Post-Mitotic Nuclear Import
4. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejean, A.; De Th, C.C.H. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor α gene to a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kakizuka, A.; Miller, W.H.; Umesono, K.; Warrell, R.P.; Frankel, S.R.; Murty, V.V.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991, 66, 663–674. [Google Scholar] [CrossRef]
- Longo, L.; Pandolfi, P.P.; Biondi, A.; Rambaldi, A.; Mencarelli, A.; Coco, F.L.; Diverio, D.; Pegoraro, L.; Avanzi, G.; Tabilio, A. Rearrangements and aberrant expression of the retinoic acid receptor alpha gene in acute promyelocytic leukemias. J. Exp. Med. 1990, 172, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Shiels, C.; Freemont, P.S. PML protein isoforms and the RBCC/TRIM motif. Oncogene 2001, 20, 7223–7233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Boil. 2007, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.-S.; Kao, H.-Y. PML: Regulation and multifaceted function beyond tumor suppression. Cell Biosci. 2018, 8, 5. [Google Scholar] [CrossRef]
- Wang, Z.G.; Delva, L.; Gaboli, M.; Rivi, R.; Giorgio, M.; Cordon-Cardo, C.; Grosveld, F.; Pandolfi, P.P. Role of PML in Cell Growth and the Retinoic Acid Pathway. Science 1998, 279, 1547–1551. [Google Scholar] [CrossRef]
- Wang, Z.-G.; Ruggero, D.; Ronchetti, S.; Zhong, S.; Gaboli, M.; Rivi, R.; Pandolfi, P.P. Pml is essential for multiple apoptotic pathways. Nat. Genet. 1998, 20, 266–272. [Google Scholar] [CrossRef]
- Ferbeyre, G.; De Stanchina, E.; Querido, E.; Baptiste, N.; Prives, C.; Lowe, S.W. PML is induced by oncogenic ras and promotes premature senescence. Genome Res. 2000, 14, 2015–2027. [Google Scholar]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Bischof, O.; Kirsh, O.; Pearson, M.; Itahana, K.; Pelicci, P.G.; Dejean, A. Deconstructing PML-induced premature senescence. EMBO J. 2002, 21, 3358–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, N.; Tsujimura, N.; Kumazaki, M.; Shinohara, H.; Taniguchi, K.; Nakagawa, Y.; Naoe, T.; Akao, Y. Colorectal cancer cell-derived microvesicles containing microRNA-1246 promote angiogenesis by activating Smad 1/5/8 signaling elicited by PML down-regulation in endothelial cells. Biochim. et Biophys. Acta (BBA) - Gene Regul. Mech. 2014, 1839, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, Y.; Chu, H.; Kao, H.-Y. Promyelocytic Leukemia Protein (PML) Regulates Endothelial Cell Network Formation and Migration in Response to Tumor Necrosis Factor α (TNFα) and Interferon α (IFNα)*. J. Boil. Chem. 2012, 287, 23356–23367. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.-S.; Zhao, X.; Cheng, X.; Guan, D.; Mahabeleshwar, G.H.; Liu, Y.; Borden, E.; Jain, M.K.; Kao, H.-Y. Dual regulation of Stat1 and Stat3 by the tumor suppressor protein PML contributes to interferon α-mediated inhibition of angiogenesis. J. Boil. Chem. 2017, 292, 10048–10060. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML-PPARδ pathway for fatty acid oxidation regulates haematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef]
- Regad, T.; Bellodi, C.; Nicotera, P.; Salomoni, P. The tumor suppressor Pml regulates cell fate in the developing neocortex. Nat. Neurosci. 2009, 12, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Bøe, S.O.; Haave, M.; Jul-Larsen, Å.; Grudic, A.; Bjerkvig, R.; Lønning, P.E. Promyelocytic leukemia nuclear bodies are predetermined processing sites for damaged DNA. J. Cell Sci. 2006, 119, 3284–3295. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.R.; Munkhjargal, A.; Kim, M.-J.; Park, S.Y.; Jung, E.; Ryu, J.-H.; Yang, Y.; Lim, J.-S.; Kim, Y. The functional roles of PML nuclear bodies in genome maintenance. Mutat. Res. Mol. Mech. Mutagen. 2018, 809, 99–107. [Google Scholar] [CrossRef]
- Boichuk, S.; Hu, L.; Makielski, K.; Pandolfi, P.P.; Gjoerup, O.V. Functional Connection between Rad51 and PML in Homology-Directed Repair. PLoS ONE 2011, 6, e25814. [Google Scholar] [CrossRef] [PubMed]
- Di Masi, A.; Cilli, D.; Berardinelli, F.; Talarico, A.; Pallavicini, I.; Pennisi, R.; Leone, S.; Antoccia, A.; Noguera, N.I.; Lo-Coco, F.; et al. PML nuclear body disruption impairs DNA double-strand break sensing and repair in APL. Cell Death Dis. 2016, 7, e2308. [Google Scholar] [CrossRef] [PubMed]
- Buczek, M.E.; Miles, A.K.; Green, W.; Johnson, C.; Boocock, D.J.; Pockley, A.G.; Rees, R.C.; Hulman, G.; van Schalkwyk, G.; Parkinson, R.; et al. Cytoplasmic PML promotes TGF-beta-associated epithelial-mesenchymal transition and invasion in prostate cancer. Oncogene 2016, 35, 3465–3475. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; De Boer, V.C.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Wang, H.; Zhang, J.; Fu, Y.; Zhu, Z.; Wang, J.; Yin, Y.; Wang, H.; Zhou, Z.; Yang, J.; et al. Telomere Maintenance-Associated PML Is a Potential Specific Therapeutic Target of Human Colorectal Cancer. Transl. Oncol. 2019, 12, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef]
- Ponente, M.; Campanini, L.; Cuttano, R.; Piunti, A.; Delledonne, G.A.; Coltella, N.; Valsecchi, R.; Villa, A.; Cavallaro, U.; Pattini, L.; et al. PML promotes metastasis of triple-negative breast cancer through transcriptional regulation of HIF1A target genes. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Wan, J.; Block, S.; Scribano, C.M.; Thiry, R.; Esbona, K.; Audhya, A.; Weaver, B.A. Mad1 destabilizes p53 by preventing PML from sequestering MDM2. Nat. Commun. 2019, 10, 10. [Google Scholar] [CrossRef]
- Zhang, P.; Chin, W.; Chow, L.T.; Chan, A.S.; Yim, A.P.; Leung, S.-F.; Mok, T.S.; Chang, K.-S.; Johnson, P.J.; Chan, J.Y.; et al. Lack of expression for the suppressor PML in human small cell lung carcinoma. Int. J. Cancer 2000, 85, 599–605. [Google Scholar] [CrossRef]
- Zhou, L.; Shang, Y.; Jin, Z.; Zhang, W.; Lv, C.; Zhao, X.; Liu, Y.; Li, N.; Liang, J. UHRF1 promotes proliferation of gastric cancer via mediating tumor suppressor gene hypermethylation. Cancer Boil. Ther. 2015, 16, 1241–1251. [Google Scholar] [CrossRef] [Green Version]
- Guan, D.; Kao, H.-Y. The function, regulation and therapeutic implications of the tumor suppressor protein, PML. Cell Biosci. 2015, 5, 675. [Google Scholar] [CrossRef]
- Bernstein, R.M.; Neuberger, J.M.; Bunn, C.C.; Callender, G.R.; Hughes, V.; Williams, R. Diversity of autoantibodies in primary biliary cirrhosis and chronic active hepatitis. Clin. Exp. Immunol. 1984, 55, 553–560. [Google Scholar]
- Ascoli, C.A. Identification of a novel nuclear domain. J. Cell Boil. 1991, 112, 785–795. [Google Scholar] [CrossRef]
- Everett, R.D.; Lomonte, P.; Sternsdorf, T.; Van Driel, R.; Orr, A. Cell cycle regulation of PML modification and ND10 composition. J. Cell Sci. 1999, 112, 4581–4588. [Google Scholar]
- Hoischen, C.; Monajembashi, S.; Weisshart, K.; Hemmerich, P. Multimodal Light Microscopy Approaches to Reveal Structural and Functional Properties of Promyelocytic Leukemia Nuclear Bodies. Front. Oncol. 2018, 8, 125. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; De Thé, H. PML Nuclear Bodies. Cold Spring Harb. Perspect. Boil. 2010, 2, a000661. [Google Scholar] [CrossRef]
- Lang, M.; Jegou, T.; Chung, I.; Richter, K.; Münch, S.; Udvarhelyi, A.; Cremer, C.; Hemmerich, P.; Engelhardt, J.; Hell, S.W.; et al. Three-dimensional organization of promyelocytic leukemia nuclear bodies. J. Cell Sci. 2010, 123, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Dellaire, G.; Ching, R.W.; Dehghani, H.; Ren, Y.; Bazett-Jones, D.P. The number of PML nuclear bodies increases in early S phase by a fission mechanism. J. Cell Sci. 2006, 119, 1026–1033. [Google Scholar] [CrossRef] [Green Version]
- Dellaire, G.; Eskiw, C.H.; Dehghani, H.; Ching, R.W.; Bazett-Jones, D.P. Mitotic accumulations of PML protein contribute to the re-establishment of PML nuclear bodies in G1. J. Cell Sci. 2006, 119, 1034–1042. [Google Scholar] [CrossRef] [Green Version]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Everett, R.D.; Weitzman, M.D. The Intrinsic Antiviral Defense to Incoming HSV-1 Genomes Includes Specific DNA Repair Proteins and Is Counteracted by the Viral Protein ICP0. PLoS Pathog. 2011, 7, e1002084. [Google Scholar] [CrossRef]
- Sahin, U.; Ferhi, O.; Jeanne, M.; Benhenda, S.; Berthier, C.; Jollivet, F.; Niwa-Kawakita, M.; Faklaris, O.; Setterblad, N.; De Thé, H.; et al. Oxidative stress–induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J. Cell Boil. 2014, 204, 931–945. [Google Scholar] [CrossRef]
- Sazer, S.; Lynch, M.; Needleman, D. Deciphering the evolutionary history of open and closed mitosis. Curr. Boil. 2014, 24, R1099–R1103. [Google Scholar] [CrossRef]
- Dundr, M.; Misteli, T. Biogenesis of Nuclear Bodies. Cold Spring Harb. Perspect. Boil. 2010, 2, a000711. [Google Scholar] [CrossRef]
- Brown, J.R.; Conn, K.L.; Wasson, P.; Charman, M.; Tong, L.; Grant, K.; McFarlane, S.; Boutell, C. SUMO Ligase Protein Inhibitor of Activated STAT1 (PIAS1) Is a Constituent Promyelocytic Leukemia Nuclear Body Protein That Contributes to the Intrinsic Antiviral Immune Response to Herpes Simplex Virus 1. J. Virol. 2016, 90, 5939–5952. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.-Y.; Huang, Y.-S.; Jeng, J.-C.; Kuo, H.-Y.; Chang, C.-C.; Chao, T.-T.; Ho, C.-C.; Chen, Y.-C.; Lin, T.-P.; Fang, H.-I.; et al. Role of SUMO-Interacting Motif in Daxx SUMO Modification, Subnuclear Localization, and Repression of Sumoylated Transcription Factors. Mol. Cell 2006, 24, 341–354. [Google Scholar] [CrossRef]
- Shen, T.H.; Lin, H.-K.; Scaglioni, P.P.; Yung, T.M.; Pandolfi, P.P. The Mechanisms of PML-Nuclear Body Formation. Mol. Cell 2006, 24, 805. [Google Scholar] [CrossRef]
- Sung, K.S.; Lee, Y.-A.; Kim, E.T.; Lee, S.-R.; Ahn, J.-H.; Choi, C.Y. Role of the SUMO-interacting motif in HIPK2 targeting to the PML nuclear bodies and regulation of p53. Exp. Cell Res. 2011, 317, 1060–1070. [Google Scholar] [CrossRef]
- Banani, S.F.; Rice, A.M.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Cuchet, D.; Sykes, A.; Nicolas, A.; Orr, A.; Murray, J.; Sirma, H.; Heeren, J.; Bartelt, A.; Everett, R.D. PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J. Cell Sci. 2011, 124, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Lang, E.; Grudic, A.; Pankiv, S.; Bruserud, O.; Simonsen, A.; Bjerkvig, R.; Bjoras, M.; Boe, S.O. The arsenic-based cure of acute promyelocytic leukemia promotes cytoplasmic sequestration of PML and PML/RARA through inhibition of PML body recycling. Blood 2012, 120, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Peng, Q.; Wan, X.; Sun, H.; Tang, J. C-terminal motifs in promyelocytic leukemia protein isoforms critically regulate PML nuclear body formation. J. Cell Sci. 2017, 130, 3496–3506. [Google Scholar] [CrossRef]
- Ching, R.W.; Ahmed, K.; Boutros, P.C.; Penn, L.Z.; Bazett-Jones, D.P. Identifying gene locus associations with promyelocytic leukemia nuclear bodies using immuno-TRAP. J. Cell Boil. 2013, 201, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Ching, R.W. PML bodies: A meeting place for genomic loci? J. Cell Sci. 2005, 118, 847–854. [Google Scholar] [CrossRef]
- Kumar, P.P.; Bischof, O.; Purbey, P.K.; Notani, D.; Urlaub, H.; Dejean, A.; Galande, S. Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat. Cell Biol. 2007, 9, 45–56. [Google Scholar]
- Shiels, C.; Islam, S.A.; Vatcheva, R.; Sasieni, P.; Sternberg, M.J.; Freemont, P.S.; Sheer, D. PML bodies associate specifically with the MHC gene cluster in interphase nuclei. J. Cell Sci. 2001, 114, 3705–3716. [Google Scholar]
- Ulbricht, T.; Alzrigat, M.; Horch, A.; Reuter, N.; Von Mikecz, A.; Steimle, V.; Schmitt, E.; Krämer, O.H.; Stamminger, T.; Hemmerich, P. PML promotes MHC class II gene expression by stabilizing the class II transactivator. J. Cell Boil. 2012, 199, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Shiels, C.; Sasieni, P.; Wu, P.J.; Islam, S.A.; Freemont, P.S.; Sheer, D. Promyelocytic leukemia nuclear bodies associate with transcriptionally active genomic regions. J. Cell Boil. 2004, 164, 515–526. [Google Scholar] [CrossRef] [Green Version]
- Dellaire, G.; Bazett-Jones, D.P.; Bazett-Jones, D.P. PML nuclear bodies: Dynamic sensors of DNA damage and cellular stress. BioEssays 2004, 26, 963–977. [Google Scholar] [CrossRef]
- Zhong, S.; Salomoni, P.; Pandolfi, P.P. The transcriptional role of PML and the nuclear body. Nature Cell Bio. 2000, 2, E85–E90. [Google Scholar] [CrossRef]
- Shastrula, P.K.; Sierra, I.; Deng, Z.; Keeney, F.; Hayden, J.E.; Lieberman, P.M.; Janicki, S.M. PML is recruited to heterochromatin during S phase and represses DAXX-mediated histone H3.3 chromatin assembly. J. Cell Sci. 2019, 132, jcs220970. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar]
- Episkopou, H.; Diman, A.; Claude, E.; Viceconte, N.; Decottignies, A. TSPYL5 Depletion Induces Specific Death of ALT Cells through USP7-Dependent Proteasomal Degradation of POT1. Mol. Cell 2019. [Google Scholar] [CrossRef]
- Fu, L.; Gao, Y.-S.; Tousson, A.; Shah, A.; Chen, T.-L.L.; Vertel, B.M.; Sztul, E. Nuclear Aggresomes Form by Fusion of PML-associated Aggregates. Mol. Boil. Cell 2005, 16, 4905–4917. [Google Scholar] [CrossRef]
- Guo, L.; Giasson, B.I.; Glavis-Bloom, A.; Brewer, M.D.; Shorter, J.; Gitler, A.D.; Yang, X. A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell 2014, 55, 15–30. [Google Scholar] [CrossRef]
- Janer, A.; Martin, E.; Muriel, M.-P.; Latouche, M.; Fujigasaki, H.; Ruberg, M.; Brice, A.; Trottier, Y.; Sittler, A. PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins. J. Cell Boil. 2006, 174, 65–76. [Google Scholar] [CrossRef]
- Nakano, Y.; Takahashi-Fujigasaki, J.; Sengoku, R.; Kanemaru, K.; Arai, T.; Kanda, T.; Murayama, S. PML Nuclear Bodies Are Altered in Adult-Onset Neuronal Intranuclear Hyaline Inclusion Disease. J. Neuropathol. Exp. Neurol. 2017, 76, 585–594. [Google Scholar] [CrossRef]
- Qin, Q.; Inatome, R.; Hotta, A.; Kojima, M.; Yamamura, H.; Hirai, H.; Yoshizawa, T.; Tanaka, H.; Fukami, K.; Yanagi, S. A novel GTPase, CRAG, mediates promyelocytic leukemia protein–associated nuclear body formation and degradation of expanded polyglutamine protein. J. Cell Boil. 2006, 172, 497–504. [Google Scholar] [CrossRef]
- Butler, J.T.; Hall, L.L.; Smith, K.P.; Lawrence, J.B. Changing Nuclear Landscape and Unique PML Structures During Early Epigenetic Transitions of Human Embryonic Stem Cells. J. Cell. Biochem. 2009, 107, 609–621. [Google Scholar] [CrossRef]
- Condemine, W.; Takahashi, Y.; Zhu, J.; Puvion-Dutilleul, F.; Guégan, S.; Janin, A.; De Thé, H. Characterization of Endogenous Human Promyelocytic Leukemia Isoforms. Cancer Res. 2006, 66, 6192–6198. [Google Scholar] [CrossRef] [Green Version]
- Jul-Larsen, Å.; Grudic, A.; Bjerkvig, R.; Bøe, S.O. Subcellular distribution of nuclear import-defective isoforms of the promyelocytic leukemia protein. BMC Mol. Boil. 2010, 11, 89. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Kawai, T.; Yoshikawa, Y.; Cheng, J.; Jokitalo, E.; Fujimoto, T. PML isoform II plays a critical role in nuclear lipid droplet formation. J. Cell Boil. 2016, 212, 29–38. [Google Scholar] [CrossRef]
- Berciano, M.T.; Novell, M.; Villagra, N.T.; Casafont, I.; Bengoechea, R.; Val-Bernal, J.F.; Lafarga, M. Cajal body number and nucleolar size correlate with the cell body mass in human sensory ganglia neurons. J. Struct. Boil. 2007, 158, 410–420. [Google Scholar] [CrossRef]
- Sun, J.; Hebert, M.D.; Xu, H.; Subramony, S.H. Interactions between Coilin and PIASy partially link Cajal bodies to PML bodies. J. Cell Sci. 2005, 118, 4995–5003. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, R.; Scaglioni, P.P.; Bergmann, S.; Horn, H.F.; Vousden, K.H.; Pandolfi, P.P. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat. Cell Bio. 2004, 6, 665–672. [Google Scholar] [CrossRef]
- Condemine, W.; Takahashi, Y.; Le Bras, M.; De Thé, H. A nucleolar targeting signal in PML-I addresses PML to nucleolar caps in stressed or senescent cells. J. Cell Sci. 2007, 120, 3219–3227. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, K.; Pokrovskaja, K.; Kiss, C.; Klein, G.; Szekely, L. Proteins associated with the promyelocytic leukemia gene product (PML)-containing nuclear body move to the nucleolus upon inhibition of proteasome-dependent protein degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 1012–1017. [Google Scholar] [CrossRef]
- Foltankova, V.; Matula, P.; Sorokin, D.; Kozubek, S.; Bártová, E. Hybrid Detectors Improved Time-Lapse Confocal Microscopy of PML and 53BP1 Nuclear Body Colocalization in DNA Lesions. Microsc. Microanal. 2013, 19, 360–369. [Google Scholar] [CrossRef]
- Muratani, M.; Gerlich, D.; Janicki, S.M.; Gebhard, M.; Eils, R.; Spector, D.L. Metabolic-energy-dependent movement of PML bodies within the mammalian cell nucleus. Nat. Cell Bio. 2002, 4, 106–110. [Google Scholar] [CrossRef]
- Stixová, L.; Matula, P.; Kozubek, S.; Gombitová, A.; Bártová, E.; Cmarko, D.; Raška, I. Trajectories and nuclear arrangement of PML bodies are influenced by A-type lamin deficiency. Boil. Cell 2012, 104, 418–432. [Google Scholar] [CrossRef]
- Görisch, S.M.; Wachsmuth, M.; Ittrich, C.; Bacher, C.P.; Rippe, K.; Lichter, P. Nuclear body movement is determined by chromatin accessibility and dynamics. Proc. Natl. Acad. Sci. USA 2004, 101, 13221–13226. [Google Scholar] [CrossRef] [Green Version]
- Niwa-Kawakita, M.; Ferhi, O.; Soilihi, H.; Le Bras, M.; Lallemand-Breitenbach, V.; De Thé, H. PML is a ROS sensor activating p53 upon oxidative stress. J. Exp. Med. 2017, 214, 3197–3206. [Google Scholar] [CrossRef] [Green Version]
- Niwa-Kawakita, M.; Wu, H.-C.; De Thé, H.; Lallemand-Breitenbach, V. PML nuclear bodies, membrane-less domains acting as ROS sensors? Semin. Cell Dev. Boil. 2018, 80, 29–34. [Google Scholar] [CrossRef]
- Bongiorno-Borbone, L.; De Cola, A.; Vernole, P.; Finos, L.; Barcaroli, D.; Knight, R.A.; Melino, G.; De Laurenzi, V. FLASH and NPAT positive but not Coilin positive Cajal bodies correlate with cell ploidy. Cell Cycle 2008, 7, 2357–2367. [Google Scholar] [CrossRef] [Green Version]
- Bubulya, P.A.; Prasanth, K.V.; Deerinck, T.J.; Gerlich, D.; Beaudouin, J.; Ellisman, M.H.; Ellenberg, J.; Spector, D.L. Hypophosphorylated SR splicing factors transiently localize around active nucleolar organizing regions in telophase daughter nuclei. J. Cell Boil. 2004, 167, 51–63. [Google Scholar] [CrossRef]
- Dundr, M.; Olson, M.O. Partially Processed pre-rRNA Is Preserved in Association with Processing Components in Nucleolus-derived Foci during Mitosis. Mol. Boil. Cell 1998, 9, 2407–2422. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.A. Differential interaction of splicing snRNPs with coiled bodies and interchromatin granules during mitosis and assembly of daughter cell nuclei. J. Cell Boil. 1994, 126, 11–23. [Google Scholar] [CrossRef]
- Gerace, L.; Blobel, G. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 1980, 19, 277–287. [Google Scholar] [CrossRef]
- Guizzunti, G.; Seemann, J. Mitotic Golgi disassembly is required for bipolar spindle formation and mitotic progression. Proc. Natl. Acad. Sci. USA 2016, 113, E6590–E6599. [Google Scholar] [CrossRef] [Green Version]
- Lucocq, J.M.; Warren, G. Fragmentation and partitioning of the Golgi apparatus during mitosis in HeLa cells. EMBO J. 1987, 6, 3239–3246. [Google Scholar] [CrossRef]
- Axelsson, M.A.B.; Warren, G. Rapid, Endoplasmic Reticulum-independent Diffusion of the Mitotic Golgi Haze. Mol. Boil. Cell 2004, 15, 1843–1852. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Ladinsky, M.S.; Kirchhausen, T. Formation of the postmitotic nuclear envelope from extended ER cisternae precedes nuclear pore assembly. J. Cell Boil. 2011, 194, 425–440. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Ladinsky, M.S.; Kirchhausen, T. Cisternal Organization of the Endoplasmic Reticulum during Mitosis. Mol. Boil. Cell 2009, 20, 3471–3480. [Google Scholar] [CrossRef]
- Chen, Y.-C.M.; Kappel, C.; Beaudouin, J.; Eils, R.; Spector, D.L. Live Cell Dynamics of Promyelocytic Leukemia Nuclear Bodies upon Entry into and Exit from Mitosis. Mol. Boil. Cell 2008, 19, 3147–3162. [Google Scholar] [CrossRef] [Green Version]
- Lång, A.; Eriksson, J.; Schink, K.O.; Lång, E.; Blicher, P.; Połeć, A.; Brech, A.; Dalhus, B.; Bøe, S.O. Visualization of PML nuclear import complexes reveals FG-repeat nucleoporins at cargo retrieval sites. Nucleus 2017, 8, 404–420. [Google Scholar] [CrossRef] [Green Version]
- Palibrk, V.; Lang, E.; Lång, A.; Schink, K.O.; Rowe, A.D.; Bøe, S.O. Promyelocytic leukemia bodies tether to early endosomes during mitosis. Cell Cycle 2014, 13, 1749–1755. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.-K.; Bergmann, S.; Pandolfi, P.P. Cytoplasmic PML function in TGF-β signalling. Nature 2004, 431, 205–211. [Google Scholar] [CrossRef]
- Jul-Larsen, A.; Grudic, A.; Bjerkvig, R.; Bøe, S.O. Cell-cycle regulation and dynamics of cytoplasmic compartments containing the promyelocytic leukemia protein and nucleoporins. J. Cell Sci. 2009, 122, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Lång, E.; Połeć, A.; Lång, A.; Valk, M.; Blicher, P.; Rowe, A.D.; Tønseth, K.A.; Jackson, C.J.; Utheim, T.P.; Janssen, L.M.C.; et al. Coordinated collective migration and asymmetric cell division in confluent human keratinocytes without wounding. Nat. Commun. 2018, 9, 3665. [Google Scholar] [CrossRef]
- Stewart, M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Boil. 2007, 8, 195–208. [Google Scholar] [CrossRef]
- Ditlev, J.A.; Case, L.B.; Rosen, M.K. Who’s In and Who’s Out—Compositional Control of Biomolecular Condensates. J. Mol. Boil. 2018, 430, 4666–4684. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Bosch, L.V.D.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Boil. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Eskiw, C.H.; Dellaire, G.; Bazett-Jones, D.P. Chromatin contributes to structural integrity of promyelocytic leukemia bodies through a SUMO-1-independent mechanism. J. Biol. Chem. 2004, 279, 9577–9585. [Google Scholar] [CrossRef]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Shang, Y.; Araki, Y.; Guo, T.; Huganir, R.L.; Zhang, M. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 2016, 166, 1163–1175.e12. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, J.B.; Gomes, B.F.; Widlund, P.O.; Mahamid, J.; Honigmann, A.; Hyman, A.A. The Centrosome Is a Selective Condensate that Nucleates Microtubules by Concentrating Tubulin. Cell 2017, 169, 1066–1077.e10. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Wang, L.; Ishikawa, R.; Li, Y.; Fiedler, M.; Liu, F.; Calder, G.; Rowan, B.; Weigel, D.; Li, P.; et al. Arabidopsis FLL2 promotes liquid–liquid phase separation of polyadenylation complexes. Nature 2019, 569, 265–269. [Google Scholar] [CrossRef]
- Frey, S.; Richter, R.P.; Görlich, D. FG-Rich Repeats of Nuclear Pore Proteins Form a Three-Dimensional Meshwork with Hydrogel-Like Properties. Science 2006, 314, 815–817. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, F.; Weiss, C.N.; Ito, K. The role of PML in hematopoietic and leukemic stem cell maintenance. Int. J. Hematol. 2014, 100, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Palibrk, V.; Suganthan, R.; Scheffler, K.; Wang, W.; Bjørås, M.; Bøe, S.O. PML regulates neuroprotective innate immunity and neuroblast commitment in a hypoxic–ischemic encephalopathy model. Cell Death Dis. 2016, 7, e2320. [Google Scholar] [CrossRef]
- Li, W.; Ferguson, B.J.; Khaled, W.T.; Tevendale, M.; Stingl, J.; Poli, V.; Rich, T.; Salomoni, P.; Watson, C.J. PML depletion disrupts normal mammary gland development and skews the composition of the mammary luminal cell progenitor pool. Proc. Natl. Acad. Sci. USA 2009, 106, 4725–4730. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Fu, S.; Zhong, W.; Huang, H. PML overexpression inhibits proliferation and promotes the osteogenic differentiation of human mesenchymal stem cells. Oncol. Rep. 2013, 30, 2785–2794. [Google Scholar] [CrossRef]
- Salsman, J.; Rapkin, L.M.; Margam, N.N.; Duncan, R.; Bazett-Jones, D.P.; Dellaire, G. Myogenic differentiation triggers PML nuclear body loss and DAXX relocalization to chromocentres. Cell Death Dis. 2017, 8, e2724. [Google Scholar] [CrossRef]
- Hadjimichael, C.; Chanoumidou, K.; Nikolaou, C.; Klonizakis, A.; Theodosi, G.-I.; Makatounakis, T.; Papamatheakis, J.; Kretsovali, A. Promyelocytic Leukemia Protein Is an Essential Regulator of Stem Cell Pluripotency and Somatic Cell Reprogramming. Stem Cell Rep. 2017, 8, 1366–1378. [Google Scholar] [CrossRef] [Green Version]
- De Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991, 66, 675–684. [Google Scholar] [CrossRef]
- Melnick, A.; Licht, J.D. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 1999, 93, 3167–3215. [Google Scholar]
- Perez, A.; Kastner, P.; Sethi, S.; Lutz, Y.; Reibel, C.; Chambon, P. PMLRAR homodimers: Distinct DNA binding properties and heteromeric interactions with RXR. EMBO J. 1993, 12, 3171–3182. [Google Scholar] [CrossRef]
- Breitman, T.R.; Selonick, S.E.; Collins, S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef]
- Flynn, P.J.; Miller, W.J.; Weisdorf, D.J.; Arthur, D.C.; Brunning, R.; Branda, R.F. Retinoic acid treatment of acute promyelocytic leukemia: In vitro and in vivo observations. Blood 1983, 62, 1211–1217. [Google Scholar]
- Nilsson, B. Probable in vivo induction of differentiation by retinoic acid of promyelocytes in acute promyelocytic leukaemia. Br. J. Haematol. 1984, 57, 365–371. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Chen, Z. Acute promyelocytic leukemia: From highly fatal to highly curable. Blood 2008, 111, 2505–2515. [Google Scholar] [CrossRef]
- Ghavamzadeh, A.; Alimoghaddam, K.; Rostami, S.; Ghaffari, S.H.; Jahani, M.; Iravani, M.; Mousavi, S.A.; Bahar, B.; Jalili, M. Phase II study of single-agent arsenic trioxide for the front-line therapy of acute promyelocytic leukemia. J. Clin. Oncol. 2011, 29, 2753–2757. [Google Scholar] [CrossRef]
- Mathews, V.; George, B.; Chendamarai, E.; Lakshmi, K.M.; Desire, S.; Balasubramanian, P.; Viswabandya, A.; Thirugnanam, R.; Abraham, A.; Shaji, R.V.; et al. Single-Agent Arsenic Trioxide in the Treatment of Newly Diagnosed Acute Promyelocytic Leukemia: Long-Term Follow-Up Data. J. Clin. Oncol. 2010, 28, 3866–3871. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Li, J.; Li, L.; Han, X.; Han, L.; Hu, L.; Wang, S.; Zhao, Y.; Li, X.; et al. Long-term efficacy and safety of arsenic trioxide for first-line treatment of elderly patients with newly diagnosed acute promyelocytic leukemia. Cancer 2013, 119, 115–125. [Google Scholar] [CrossRef]
- Zhang, X.-W.; Yan, X.-J.; Zhou, Z.-R.; Yang, F.-F.; Wu, Z.Y.; Sun, H.-B.; Liang, W.-X.; Song, A.-X.; Lallemand-Breitenbach, V.; Jeanne, M.; et al. Arsenic Trioxide Controls the Fate of the PML-RAR Oncoprotein by Directly Binding PML. Science 2010, 328, 240–243. [Google Scholar] [CrossRef]
- Jeanne, M.; Lallemand-Breitenbach, V.; Ferhi, O.; Koken, M.; Le Bras, M.; Duffort, S.; Peres, L.; Berthier, C.; Soilihi, H.; Raught, B.; et al. PML/RARA Oxidation and Arsenic Binding Initiate the Antileukemia Response of As2O3. Cancer Cell 2010, 18, 88–98. [Google Scholar] [CrossRef]
- Koken, M.; Puvion-Dutilleul, F.; Guillemin, M.; Viron, A.; Linares-Cruz, G.; Stuurman, N.; De Jong, L.; Szostecki, C.; Calvo, F.; Chomienne, C. The t(15;17) translocation alters a nuclear body in a retinoic acid-reversible fashion. EMBO J. 1994, 13, 1073–1083. [Google Scholar] [CrossRef]
- Dyck, J.A.; Maul, G.G.; Miller, W.H.; Chen, J.; Kakizuka, A.; Evans, R.M. A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell 1994, 76, 333–343. [Google Scholar] [CrossRef]
- Isakson, P.; Bjørås, M.; Bøe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; De Thé, H. Arsenic degrades PML or PML–RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Bio. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Tatham, M.H.; Geoffroy, M.-C.; Shen, L.; Plechanovová, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Bio. 2008, 10, 538–546. [Google Scholar] [CrossRef]
- De Thé, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Kitamura, K.; Tanaka, K.; Omura, S.; Miyazaki, T.; Hachiya, T.; Ohno, R.; Naoe, T. Accelerated degradation of PML-retinoic acid receptor alpha (PML-RARA) oncoprotein by all-trans-retinoic acid in acute promyelocytic leukemia: Possible role of the proteasome pathway. Cancer Res. 1996, 56, 2945–2948. [Google Scholar]
- Goto, E.; Tomita, A.; Hayakawa, F.; Atsumi, A.; Kiyoi, H.; Naoe, T. Missense mutations in PML-RARA are critical for the lack of responsiveness to arsenic trioxide treatment. Blood 2011, 118, 1600–1609. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Ito, K.; Lin, H.-K.; Santangelo, C.; Wieckowski, M.R.; Lebiedzinska, M.; Bononi, A.; Bonora, M.; Duszynski, J.; Bernardi, R.; et al. PML Regulates Apoptosis at Endoplasmic Reticulum by Modulating Calcium Release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafà, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef] [Green Version]
- Bellodi, C.; Kindle, K.; Bernassola, F.; Cossarizza, A.; Dinsdale, D.; Melino, G.; Heery, D.; Salomoni, P.; Melino, G. A Cytoplasmic PML Mutant Inhibits p53 Function. Cell Cycle 2006, 5, 2688–2692. [Google Scholar] [CrossRef]
- Slupphaug, G.; Markussen, F.-H.; Olosen, L.C.; Aasland, R.; Aarsæther, N.; Bakke, O.; Krokan, H.E.; Helland, D.E. Nuclear and mitochondrial forms of human uracil-DNA glycosylase are encoded by the same gene. Nucleic Acids Res. 1993, 21, 2579–2584. [Google Scholar] [CrossRef] [Green Version]
- Twyffels, L.; Wauquier, C.; Soin, R.; Decaestecker, C.; Gueydan, C.; Kruys, V. A Masked PY-NLS in Drosophila TIS11 and Its Mammalian Homolog Tristetraprolin. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Truong, K.; Lee, T.D.; Li, B.; Chen, Y. Sumoylation of SAE2 C Terminus Regulates SAE Nuclear Localization*. J. Boil. Chem. 2012, 287, 42611–42619. [Google Scholar] [CrossRef]
- Malek, S. Ikappa Bbeta, but Not Ikappa Balpha, Functions as a Classical Cytoplasmic Inhibitor of NF-kappa B Dimers by Masking Both NF-kappa B Nuclear Localization Sequences in Resting Cells. J. Boil. Chem. 2001, 276, 45225–45235. [Google Scholar] [CrossRef]
- Wagstaff, K.M.; Jans, D.A. Intramolecular masking of nuclear localization signals: Analysis of importin binding using a novel AlphaScreen-based method. Anal. Biochem. 2006, 348, 49–56. [Google Scholar] [CrossRef]
- Takahashi, J.; Fujigasaki, H.; Iwabuchi, K.; Bruni, A.C.; Uchihara, T.; El Hachimi, K.H.; Stevanin, G.; Dürr, A.; Lebre, A.-S.; Trottier, Y.; et al. PML nuclear bodies and neuronal intranuclear inclusion in polyglutamine diseases. Neurobiol. Dis. 2003, 13, 230–237. [Google Scholar] [CrossRef]
- Yasuda, S.; Inoue, K.; Hirabayashi, M.; Higashiyama, H.; Yamamoto, Y.; Fuyuhiro, H.; Komure, O.; Tanaka, F.; Sobue, G.; Tsuchiya, K.; et al. Triggering of neuronal cell death by accumulation of activated SEK1 on nuclear polyglutamine aggregations in PML bodies. Genes Cells 1999, 4, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Chort, A.; Alves, S.; Marinello, M.; Dufresnois, B.; Dornbierer, J.-G.; Tesson, C.; Latouche, M.; Baker, D.P.; Barkats, M.; El Hachimi, K.H.; et al. Interferon beta induces clearance of mutant ataxin 7 and improves locomotion in SCA7 knock-in mice. Brain 2013, 136, 1732–1745. [Google Scholar] [CrossRef] [Green Version]
- Reichelt, M.; Wang, L.; Sommer, M.; Perrino, J.; Nour, A.M.; Sen, N.; Baiker, A.; Zerboni, L.; Arvin, A.M. Entrapment of Viral Capsids in Nuclear PML Cages Is an Intrinsic Antiviral Host Defense against Varicella-Zoster Virus. PLoS Pathog. 2011, 7, e1001266. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lång, A.; Lång, E.; Bøe, S.O. PML Bodies in Mitosis. Cells 2019, 8, 893. https://doi.org/10.3390/cells8080893
Lång A, Lång E, Bøe SO. PML Bodies in Mitosis. Cells. 2019; 8(8):893. https://doi.org/10.3390/cells8080893
Chicago/Turabian StyleLång, Anna, Emma Lång, and Stig Ove Bøe. 2019. "PML Bodies in Mitosis" Cells 8, no. 8: 893. https://doi.org/10.3390/cells8080893
APA StyleLång, A., Lång, E., & Bøe, S. O. (2019). PML Bodies in Mitosis. Cells, 8(8), 893. https://doi.org/10.3390/cells8080893