Functions and Implications of Autophagy in Colon Cancer

Abstract
:1. Introduction
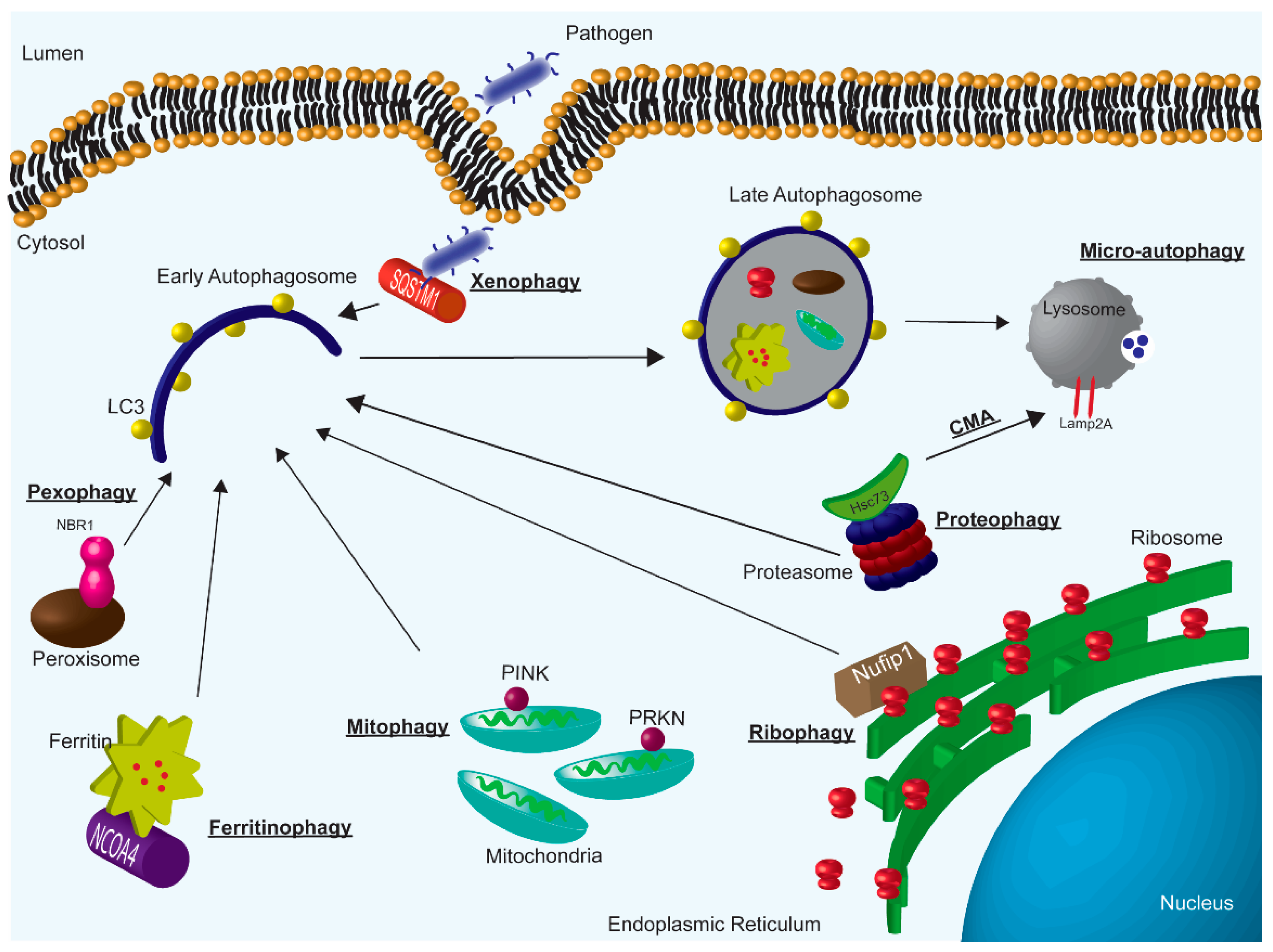
2. Autophagy Subtypes
2.1. Chaperone-Mediated Autophagy
2.2. Micro-Autophagy
2.3. Macro-Autophagy
2.4. Mitophagy
2.4.1. Parkin-Mediated Mitophagy
2.4.2. Parkin-Independent Mitophagy
2.5. Ribophagy
2.6. Proteophagy
2.7. Pexophagy
2.8. Ferritinophagy
2.9. Xenophagy
3. Role of Autophagy in CRC
4. Cellular Cues for Autophagic Activation in Cancer
4.1. Starvation
4.2. Hypoxia
4.3. Microbiota
5. Autophagic Substrates
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) Program Research Data (1973–2014); S.R.P. National Cancer Institute: Rockville, MD, USA, 2017.
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Altekruse, S.F. SEER Cancer Statistics Review, 1975–2013; National Cancer Institute: Rockville, MD, USA, 2016.
- Fearon, E.R. Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Eaden, J.; Abrams, K.; Mayberry, J. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment. Pharm. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef]
- Kameyama, H.; Nagahashi, M.; Shimada, Y.; Tajima, Y.; Ichikawa, H.; Nakano, M.; Sakata, J.; Kobayashi, T.; Narayanan, S.; Takabe, K.; et al. Genomic characterization of colitis-associated colorectal cancer. World J. Surg. Oncol. 2018, 16, 121. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.-O.; Nah, J.; Jung, Y.-K. Molecules and their functions in autophagy. Exp. Mol. Med. 2012, 44, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, J.; Xie, J.; Liu, Z.; Zhou, Y.; Pan, H.; Han, W. The role of autophagy in colitis-associated colorectal cancer. Signal Transduct. Target. Ther. 2018, 3, 31. [Google Scholar] [CrossRef]
- Garbar, C.; Mascaux, C.; Giustiniani, J.; Merrouche, Y.; Bensussan, A. Chemotherapy treatment induces an increase of autophagy in the luminal breast cancer cell MCF7, but not in the triple-negative MDA-MB231. Sci. Rep. 2017, 7, 7201. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, M.; Kang, R.; Wang, Z.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; Lotze, M.T.; et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011, 25, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.-G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Corrigendum: Chaperone-mediated autophagy degrades mutant p53. Genome Res. 2016, 30, 870. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109–117. [Google Scholar] [CrossRef]
- Mejlvang, J.; Olsvik, H.; Svenning, S.; Bruun, J.-A.; Abudu, Y.P.; Larsen, K.B.; Brech, A.; Hansen, T.E.; Brenne, H.; Hansen, T.; et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J. Cell Boil. 2018, 217, 3640–3655. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Boil. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [PubMed]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal Turnover, but not a Cellular Level, of Endogenous LC3 is a Marker for Autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geißler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature 2010, 12, 119–131. [Google Scholar]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; De Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Hollville, É.; Carroll, R.G.; Cullen, S.P.; Martin, S.J. Bcl-2 Family Proteins Participate in Mitochondrial Quality Control by Regulating Parkin/PINK1-Dependent Mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Boil. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Eiyama, A.; Kondo-Okamoto, N.; Okamoto, K. Mitochondrial degradation during starvation is selective and temporally distinct from bulk autophagy in yeast. FEBS Lett. 2013, 587, 1787–1792. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Fung, G.; Deng, H.; Zhang, J.; Fiesel, F.C.; Springer, W.; Li, X.; Luo, H. NBR1 is dispensable for PARK2-mediated mitophagy regardless of the presence or absence of SQSTM1. Cell Death Dis. 2015, 6, e1943. [Google Scholar] [CrossRef]
- Yoo, S.-M.; Jung, Y.-K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [PubMed]
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, Ö.; De Oliveira, T.; Diamanti, M.A.; Müller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity during Tumorigenesis. Cell 2018, 174, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.T.; Wu, M.M.; Wang, C.L.; Su, Z.R.; Cheng, Y.Y.; Zhang, X.J. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol. Immunol. 2019, 105, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, J.; Yang, Y.; Liu, X.; Zhu, Y.; Zou, J.; Peng, S.; Le, T.H.; Chen, Y.; Zhao, S.; et al. Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem. Pharm. 2018, 155, 366–379. [Google Scholar] [CrossRef]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y.; et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef]
- Poulogiannis, G.; McIntyre, R.E.; Dimitriadi, M.; Apps, J.R.; Wilson, C.H.; Ichimura, K.; Luo, F.; Cantley, L.C.; Wyllie, A.H.; Adams, D.J.; et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15145–15150. [Google Scholar] [CrossRef]
- Da Silva-Camargo, C.C.V.; Baldin, R.K.S.; Polli, N.L.C.; Agostinho, A.P.; Olandosk, M.; de Noronha, L.; Sotomaior, V.S. Parkin protein expression and its impact on survival of patients with advanced colorectal cancer. Cancer Biol. Med. 2018, 15, 61–69. [Google Scholar]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Boil. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirane, M.; Nakayama, K.I. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 2003, 5, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Von Stockum, S.; Marchesan, E.; Ziviani, E. Mitochondrial quality control beyond PINK1/Parkin. Oncotarget 2018, 9, 12550–12551. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.A.; Van Wickle, J.; Hill, R.B.; Marchese, A.; Kalyanaraman, B.; Dwinell, M.B. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J. Boil. Chem. 2018, 293, 14891–14904. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, C.; Yan, X.; Lan, B.; Wang, J.; Wei, C.; Cao, X.; Wang, R.; Yao, J.; Zhou, T.; et al. A Novel Bioavailable BH3 Mimetic Efficiently Inhibits Colon Cancer via Cascade Effects of Mitochondria. Clin. Cancer Res. 2016, 22, 1445–1458. [Google Scholar] [CrossRef]
- Yan, C.; Li, T.-S. Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer Res. 2018, 38, 617–621. [Google Scholar] [Green Version]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.-M.; Hu, X.; Ming, Q.-L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef] [Green Version]
- Dany, M.; Gencer, S.; Nganga, R.; Thomas, R.J.; Oleinik, N.; Baron, K.D.; Szulc, Z.M.; Ruvolo, P.; Kornblau, S.; Andreeff, M.; et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 2016, 128, 1944–1958. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Harper, J.W. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 2018, 20, 135–143. [Google Scholar] [CrossRef]
- Wyant, G.A.; Abu-Remaileh, M.; Frenkel, E.M.; Laqtom, N.N.; Dharamdasani, V.; Lewis, C.A.; Chan, S.H.; Heinze, I.; Ori, A.; Sabatini, D.M. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 2018, 360, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, K.A.; Mota-Peynado, A.; Vontz, G.; Roelofs, J. Starvation Induces Proteasome Autophagy with Different Pathways for Core and Regulatory Particles. J. Biol. Chem. 2016, 291, 3239–3253. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Palmer, A.; Rivett, A.J.; Knecht, E. Degradation of Proteasomes by Lysosomes in Rat Liver. JBIC J. Boil. Inorg. Chem. 1995, 227, 792–800. [Google Scholar]
- Dengjel, J.; Høyer-Hansen, M.; Nielsen, M.O.; Eisenberg, T.; Harder, L.M.; Schandorff, S.; Farkas, T.; Kirkegaard, T.; Becker, A.C.; Schroeder, S.; et al. Identification of Autophagosome-associated Proteins and Regulators by Quantitative Proteomic Analysis and Genetic Screens. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zientara-Rytter, K.; Subramani, S.; Katarzyna, Z.-R.; Suresh, S. Autophagic degradation of peroxisomes in mammals. Biochem. Soc. Trans. 2016, 44, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Iwata, J.-I.; Ostrovsky, O.; Berman, B.; Gallagher, J.; Mulloy, B.; Fernig, D.G.; Delehedde, M.; Ron, D. Excess Peroxisomes Are Degraded by Autophagic Machinery in Mammals. J. Boil. Chem. 2006, 281, 4035–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargent, G.; Van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Boil. 2016, 214, 677–690. [Google Scholar] [CrossRef]
- Walter, K.M.; Schönenberger, M.J.; Trötzmüller, M.; Horn, M.; Elsässer, H.-P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif-2α Promotes Degradation of Mammalian Peroxisomes by Selective Autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Sui, S.; Zhang, J.; Xu, S.; Wang, Q.; Wang, P.; Pang, D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 2019, 10, 331. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.-C.; Mazière, J.-C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, R.L.; Davis, F.G.; Sutter, E.; Sobin, L.H.; Kikendall, J.W.; Bowen, P. Body Iron Stores and Risk of Colonic Neoplasia. J. Natl. Cancer Inst. 1994, 86, 455–460. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Conway, K.L.; Kuballa, P.; Song, J.; Patel, K.K.; Castoreno, A.B.; Yilmaz, Ö.H.; Jijon, H.B.; Zhang, M.; Aldrich, L.N.; Villablanca, E.J.; et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 2013, 145, 1347–1357. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, P.; Cheng, S.; Lu, Q.; Nowak, K.; Hopp, A.K.; Li, L.; Shi, X.; Zhou, Z.; Gao, W.; et al. A Bacterial Effector Reveals the V-ATPase-ATG16L1 Axis that Initiates Xenophagy. Cell 2019, 178, 552–566. [Google Scholar] [CrossRef]
- Zheng, Y.T.; Shahnazari, S.; Brech, A.; Lamark, T.; Johansen, T.; Brumell, J.H. The Adaptor Protein p62/SQSTM1 Targets Invading Bacteria to the Autophagy Pathway. J. Immunol. 2009, 183, 5909–5916. [Google Scholar] [CrossRef] [Green Version]
- Al Azzaz, J.; Rieu, A.; Aires, V.; Delmas, D.; Chluba, J.; Winckler, P.; Bringer, M.A.; Lamarche, J.; Vervandier-Fasseur, D.; Dalle, F.; et al. Resveratrol-Induced Xenophagy Promotes Intracellular Bacteria Clearance in Intestinal Epithelial Cells and Macrophages. Front. Immunol. 2018, 9, 3149. [Google Scholar] [CrossRef]
- Lu, C.; Chen, J.; Xu, H.G.; Zhou, X.; He, Q.; Li, Y.L.; Jiang, G.; Shan, Y.; Xue, B.; Zhao, R.X.; et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology 2014, 146, 188–199. [Google Scholar] [CrossRef]
- Cho, D.-H.; Jo, Y.K.; Kim, S.C.; Park, I.J.; Kim, J.C. Down-regulated expression of ATG5 in colorectal cancer. Anticancer Res. 2012, 32, 4091–4096. [Google Scholar] [PubMed]
- Niklaus, M.; Adams, O.; Berezowska, S.; Zlobec, I.; Graber, F.; Slotta-Huspenina, J.; Nitsche, U.; Rosenberg, R.; Tschan, M.P.; Langer, R. Expression analysis of LC3B and p62 indicates intact activated autophagy is associated with an unfavorable prognosis in colon cancer. Oncotarget 2017, 8, 54604–54615. [Google Scholar] [CrossRef] [PubMed]
- Lévy, J.; Cacheux, W.; Bara, M.A.; L’Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; LeMarchand, J.; Durand, A.; Crain, A.-M.; et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nature 2015, 17, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zhu, W.; Huang, W.; Lu, S.-S.; Li, X.-H.; Xiao, Z.-Q.; Yi, H. RACK1 promotes tumorigenicity of colon cancer by inducing cell autophagy. Cell Death Dis. 2018, 9, 1148. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 2018, 44, 555–565. [Google Scholar] [CrossRef]
- Sakitani, K.; Hirata, Y.; Hikiba, Y.; Hayakawa, Y.; Ihara, S.; Suzuki, H.; Suzuki, N.; Serizawa, T.; Kinoshita, H.; Sakamoto, K.; et al. Inhibition of autophagy exerts anti-colon cancer effects via apoptosis induced by p53 activation and ER stress. BMC Cancer 2015, 15, 795. [Google Scholar] [CrossRef]
- You, P.; Wu, H.; Deng, M.; Peng, J.; Li, F.; Yang, Y. Brevilin A induces apoptosis and autophagy of colon adenocarcinoma cell CT26 via mitochondrial pathway and PI3K/AKT/mTOR inactivation. Biomed. Pharm. 2018, 98, 619–625. [Google Scholar] [CrossRef]
- Ahn, C.H.; Jeong, E.G.; Lee, J.W.; Kim, M.S.; Kim, S.H.; Kim, S.S.; Yoo, N.J.; Lee, S.H. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS 2007, 115, 1344–1349. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Weidberg, H.; Gonen, C.; Wilder, S.; Elazar, Z.; Oren, M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 2010, 107, 18511–18516. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Khor, S.; Hackett, S.R.; Rabinowitz, J.D.; Perlman, D.H.; White, E. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol. Cell 2014, 55, 916–930. [Google Scholar] [CrossRef]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Boil. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Alves, S.; Castro, L.; Fernandes, M.S.; Francisco, R.; Castro, P.; Priault, M.; Chaves, S.R.; Moyer, M.P.; Oliveira, C.; Seruca, R.; et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 2015, 6, 30787–30802. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A.; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell Mol. Med. 2010, 14, 2448–2459. [Google Scholar] [CrossRef]
- Yao, C.W.; Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Fernando, P.M.D.J.; Oh, M.C. Reduced Autophagy in 5-Fluorouracil Resistant Colon Cancer Cells. Biomol. Ther. 2017, 25, 315–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-W.; Yang, S.-H.; Huang, G.-D.; Lin, J.-K.; Chen, W.-S.; Jiang, J.-K.; Lan, Y.-T.; Lin, C.-C.; Hwang, W.-L.; Tzeng, C.-H.; et al. Temsirolimus enhances the efficacy of cetuximab in colon cancer through a CIP2A-dependent mechanism. J. Cancer Res. Clin. Oncol. 2014, 140, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Huang, C.; Wu, H.; Huang, J. Estrogen Receptor Beta (ERβ) Mediated-CyclinD1 Degradation via Autophagy Plays an Anti-Proliferation Role in Colon Cells. Int. J. Boil. Sci. 2019, 15, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Kohli, L.; Kaza, N.; Coric, T.; Byer, S.J.; Brossier, N.M.; Klocke, B.J.; Bjornsti, M.-A.; Carroll, S.L.; Roth, K.A. 4-Hydroxytamoxifen induces autophagic death through K-Ras degradation. Cancer Res. 2013, 73, 4395–4405. [Google Scholar] [CrossRef]
- Costa, J.R.; Prak, K.; Aldous, S.; Gewinner, C.A.; Ketteler, R. Autophagy gene expression profiling identifies a defective microtubule-associated protein light chain 3A mutant in cancer. Oncotarget 2016, 7, 41203–41216. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef]
- Koustas, E.; Papavassiliou, A.G.; Karamouzis, M.V. The role of autophagy in the treatment of BRAF mutant colorectal carcinomas differs based on microsatellite instability status. PLoS ONE 2018, 13, e0207227. [Google Scholar] [CrossRef]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Bourhis, L.L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Dai, W.; Xiang, W.; Li, Y.; Feng, Y.; Zhang, L.; Li, Q.; Cai, G. Prognostic and predictive value of an autophagy-related signature for early relapse in stages I-III colon cancer. Carcinogenesis 2019, 40, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Tampakis, A.; Tampaki, E.; Nebiker, C.; Kouraklis, G. Histone deacetylase inhibitors and colorectal cancer: What is new? Anticancer Agents Med. Chem. 2014, 14, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-L.; Shao, B.-Z.; Zhao, S.-B.; Fang, J.; Gu, L.; Miao, C.-Y.; Li, Z.-S.; Bai, Y. Impact of Paneth Cell Autophagy on Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuballa, P.; Nolte, W.M.; Castoreno, A.B.; Xavier, R.J. Autophagy and the immune system. Annu. Rev. Immunol. 2012, 30, 611–646. [Google Scholar] [CrossRef]
- Shao, L.-N.; Xing, C.-G.; Yang, X.-D.; Young, W.; Zhu, B.-S.; Cao, J.-P. Effects of autophagy regulation of tumor-associated macrophages on radiosensitivity of colorectal cancer cells. Mol. Med. Rep. 2016, 13, 2661–2670. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.-M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.-T.; Meng, X.-J.; Li, L.-L.; Liu, Y.; Li, W.-F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 17. [Google Scholar] [CrossRef]
- Mathew, R.; White, E. Eat this, not that! How selective autophagy helps cancer cells survive. Mol. Cell. Oncol. 2015, 2, e975638. [Google Scholar] [CrossRef] [Green Version]
- Nighot, P.K.; Hu, C.-A.A.; Ma, T.Y. Autophagy Enhances Intestinal Epithelial Tight Junction Barrier Function by Targeting Claudin-2 Protein Degradation. J. Boil. Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Kosumi, K.; Masugi, Y.; Yang, J.; Qian, Z.R.; Kim, S.A.; Li, W.; Shi, Y.; Da Silva, A.; Hamada, T.; Liu, L.; et al. Tumor SQSTM1 (p62) expression and T cells in colorectal cancer. OncoImmunology 2017, 6, e1284720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Jones, E.; Inoki, K. Lysosomal Regulation of mTORC1 by Amino Acids in Mammalian Cells. Biomolecules 2017, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genome Res. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, 42. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Boil. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Meijer, A.J.; Lorin, S.; Blommaart, E.F.; Codogno, P. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 2015, 47, 2037–2063. [Google Scholar] [CrossRef]
- Sanchez-Garrido, J.; Sancho-Shimizu, V.; Shenoy, A.R. Regulated proteolysis of p62/SQSTM1 enables differential control of autophagy and nutrient sensing. Sci. Signal. 2018, 11, 6903. [Google Scholar] [CrossRef]
- Shuvayeva, G.; Bobak, Y.; Igumentseva, N.; Titone, R.; Morani, F.; Stasyk, O.; Isidoro, C. Single Amino Acid Arginine Deprivation Triggers Prosurvival Autophagic Response in Ovarian Carcinoma SKOV3. BioMed. Res. Int. 2014, 2014, 505041. [Google Scholar] [CrossRef]
- Schroll, M.M.; Labonia, G.J.; Ludwig, K.R.; Hummon, A.B. Glucose Restriction Combined with Autophagy Inhibition and Chemotherapy in HCT 116 Spheroids Decreases Cell Clonogenicity and Viability Regulated by Tumor Suppressor Genes. J. Proteome Res. 2017, 16, 3009–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroll, M.M.; Liu, X.; Herzog, S.K.; Skube, S.B.; Hummon, A.B. Nutrient Restriction of Glucose or Serum Results in Similar Proteomic Expression Changes in 3D Colon Cancer Cell Cultures. Nutr. Res. 2016, 36, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Choi, S.; Kim, J.O.; Kim, K.K. Autophagy-mediated upregulation of cytoplasmic claudin 1 stimulates the degradation of SQSTM1/p62 under starvation. Biochem. Biophys. Res. Commun. 2018, 496, 159–166. [Google Scholar] [CrossRef]
- Xue, X.; Ramakrishnan, S.K.; Shah, Y.M. Activation of HIF-1alpha does not increase intestinal tumorigenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, 187–195. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Wu, Y.; Zhao, G.-L.; Ye, Z.-Y.; Xing, C.-G.; Yang, X.-D. Inhibition of autophagy by 3-MA promotes hypoxia-induced apoptosis in human colorectal cancer cells. Eur. Rev. Med Pharm. Sci. 2019, 23, 1047–1054. [Google Scholar]
- Che, J.; Wang, W.; Huang, Y.; Zhang, L.; Zhao, J.; Zhang, P.; Yuan, X. miR-20a inhibits hypoxia-induced autophagy by targeting ATG5/FIP200 in colorectal cancer. Mol. Carcinog. 2019, 58, 1234–1247. [Google Scholar] [CrossRef]
- Hu, Y.-L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef]
- Pott, J.; Maloy, K.J. Epithelial autophagy controls chronic colitis by reducing TNF-induced apoptosis. Autophagy 2018, 14, 1460–1461. [Google Scholar] [CrossRef] [PubMed]
- Burger, E.; Araujo, A.; López-Yglesias, A.; Rajala, M.W.; Geng, L.; Levine, B.; Hooper, L.V.; Burstein, E.; Yarovinsky, F. Loss of Paneth Cell Autophagy Causes Acute Susceptibility to Toxoplasma gondii-Mediated Inflammation. Cell Host Microbe 2018, 23, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 9862–9867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Österreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Triner, D.; Devenport, S.N.; Ramakrishnan, S.K.; Ma, X.; Frieler, R.A.; Greenson, J.K.; Inohara, N.; Nunez, G.; Colacino, J.A.; Mortensen, R.M.; et al. Neutrophils Restrict Tumor-Associated Microbiota to Reduce Growth and Invasion of Colon Tumors in Mice. Gastroenterology 2018, 156, 1467–1482. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Cianci, R.; Pagliari, D.; Piccirillo, C.A.; Fritz, J.H.; Gambassi, G. The Microbiota and Immune System Crosstalk in Health and Disease. Mediat. Inflamm. 2018, 2018, 2912539. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; MacPherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.; Zhao, W.; He, C.; Ding, J.; Dai, R.; Xu, K.; Xiao, L.; Luo, L.; Liu, S.; et al. Impaired Autophagy in Intestinal Epithelial Cells Alters Gut Microbiota and Host Immune Responses. Appl. Environ. Microbiol. 2018, 84, e00880-18. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Onodera, J.; Ohsumi, Y. Autophagy Is Required for Maintenance of Amino Acid Levels and Protein Synthesis under Nitrogen Starvation. J. Boil. Chem. 2005, 280, 31582–31586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Davis, T.; Loos, B.; Sishi, B.; Huisamen, B.; Strijdom, H.; Engelbrecht, A.-M. Autophagy is essential for the maintenance of amino acids and ATP levels during acute amino acid starvation in MDAMB231 cells. Cell Biochem. Funct. 2018, 36, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Cao, W.; Bao, L.; Zuo, W.; Xie, G.; Cai, T.; Fu, W.; Zhang, J.; Wu, W.; Zhang, X.; et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nature 2010, 12, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Wiegering, A.; Pfann, C.; Uthe, F.W.; Otto, C.; Rycak, L.; Mäder, U.; Gasser, M.; Waaga-Gasser, A.-M.; Eilers, M.; Germer, C.-T. CIP2A Influences Survival in Colon Cancer and Is Critical for Maintaining Myc Expression. PLoS ONE 2013, 8, e75292. [Google Scholar] [CrossRef] [PubMed]
- Martins, W.K.; Baptista, M.S. Autophagy Modulation for Organelle-Targeting Therapy. In Autophagy in Current Trends in Cellular Physiology and Pathology; IntechOpen: London, UK, 2016. [Google Scholar] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| A | ||||
| Observation | Autophagy | Tumor response | Reference | |
| Activated chaperone-mediated autophagy in tumors | Active | Pro-tumor | [19] | |
| Epithelial mitophagy increases CD8+T-Cells | Active | Anti-tumor | [34] | |
| Loss of PARK2 accelerates tumor development | Inactive | Pro-tumor | [38] | |
| Decreased ATG5 in CRC patients | Inactive | Pro-tumor | [73] | |
| Increased ATG5 yields increased invasion | Active | Pro-tumor | [73] | |
| Active autophagy through LC3B and SQSTM1 | Active | Pro-tumor | [74] | |
| Loss of ATG7 | Inactive | Anti-tumor | [75] | |
| RACK1 induces autophagy | Active | Pro-tumor | [76] | |
| High Beclin-1 in CRC | Active | Pro-tumor | [80] | |
| Increased LC3 with loss of p53 | Active | Anti-tumor | [81] | |
| Autophagy suppresses immune response in KRAS cancer | Active | Pro-tumor | [82] | |
| Autophagy drives glycolysis in RAS cancers | Active | Pro-tumor | [83] | |
| B | ||||
| Treatment | Autophagy | Tumor response | Reference | |
| Mito-CP or Mito-Met10 | Active | Anti-Tumor; Decreased proliferation in KRAS mutant cancers. | [47] | |
| BH3 mimetic and chloroquine | Inactive | Anti-Tumor; Induced apoptosis. | [48] | |
| Bafilomycin A1 or chloroquine | Inactive | Anti-Tumor; Elevated FOXO3a and transcriptional upregulation of pro-apoptotic genes. | [77] | |
| Brevlin A | Active | Anti-Tumor; Promoted expression of LC3-II and induced autophagy. | [79] | |
| KRAS siRNA | Inactive | Anti-Tumor; Inhibiting mutant Kras inhibits autophagy and induces apoptosis. | [84] | |
| Vorinostat with chloroquine | Inactive | Anti-Tumor; Induced apoptosis. | [85] | |
| 5-Fuorouracil and chloroquine | Inactive | Anti-Tumor; 5-FU treatment induced autophagy for resistance. Inhibition of autophagy reduced growth. | [86] | |
| Temsirolimus | Active | Anti-Tumor; Inhibited mTOR to activate autophagy and degrade CIP2A. | [87] | |
| Estrogen Receptor Beta | Active | Anti-Tumor; Autophagy directed CyclinD1 degradation inhibited growth. | [88] | |
| 4-Hydroxytamoxifen | Active | Anti-Tumor; Degradation of KRAS through autophagy induced cel death. | [89] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devenport, S.N.; Shah, Y.M. Functions and Implications of Autophagy in Colon Cancer. Cells 2019, 8, 1349. https://doi.org/10.3390/cells8111349
Devenport SN, Shah YM. Functions and Implications of Autophagy in Colon Cancer. Cells. 2019; 8(11):1349. https://doi.org/10.3390/cells8111349
Chicago/Turabian StyleDevenport, Samantha N, and Yatrik M Shah. 2019. "Functions and Implications of Autophagy in Colon Cancer" Cells 8, no. 11: 1349. https://doi.org/10.3390/cells8111349
APA StyleDevenport, S. N., & Shah, Y. M. (2019). Functions and Implications of Autophagy in Colon Cancer. Cells, 8(11), 1349. https://doi.org/10.3390/cells8111349