Mesenchymal Stem/Stromal Cells Derived from Dental Tissues: A Comparative In Vitro Evaluation of Their Immunoregulatory Properties Against T cells

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Obtaining Biological Samples
2.2. Isolation and Culture of BM-MSCs, DP-MSCs, G-MSCs, and PDL-MSCs
2.2.1. Isolation and Culture of BM-MSCs
2.1.2. Isolation and Culture of MSCs from a Dental Tissue Explant Tissue Culture System
2.3. Characterization of Mesenchymal Stem Cells
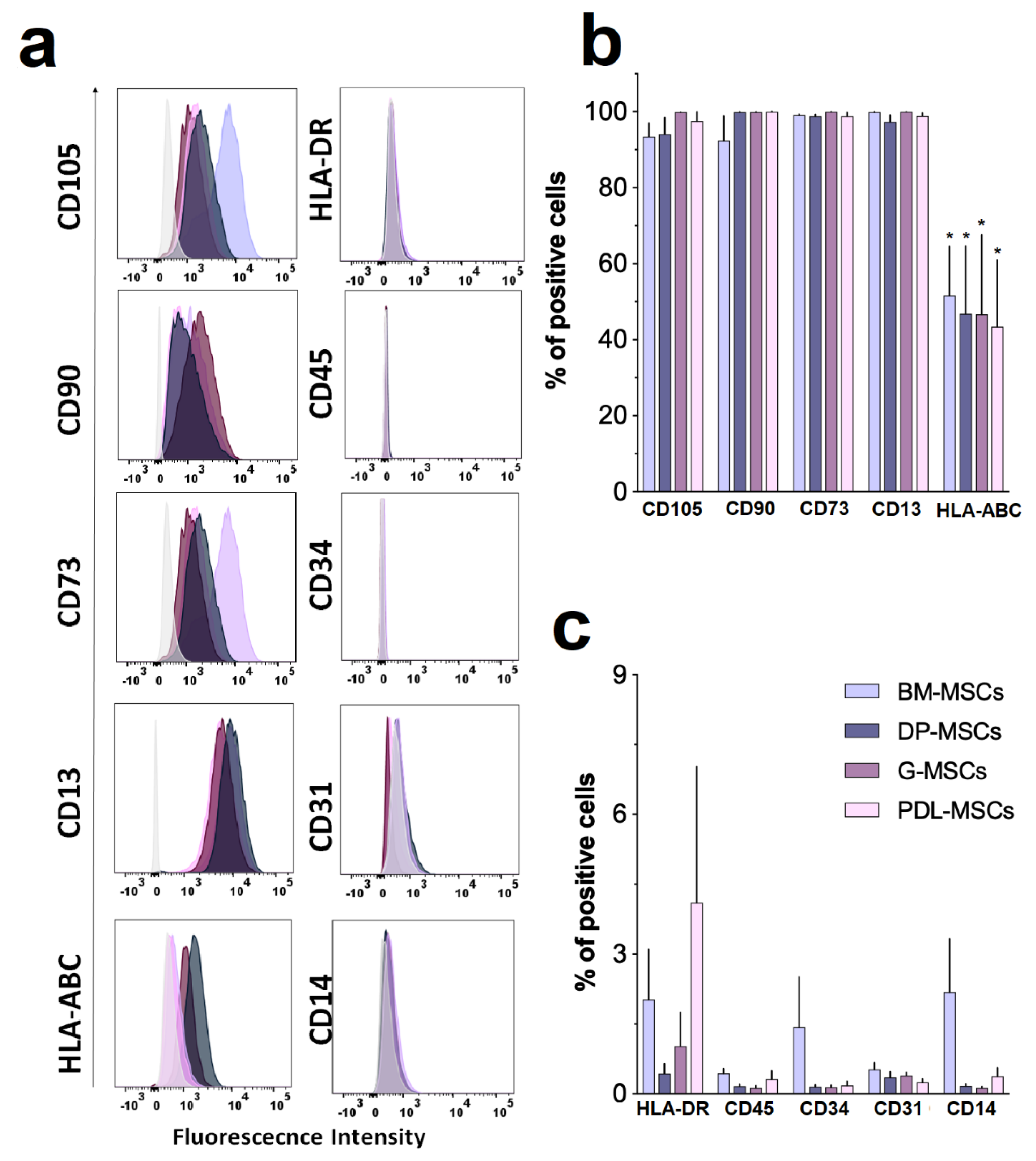
2.3.1. Immunophenotype
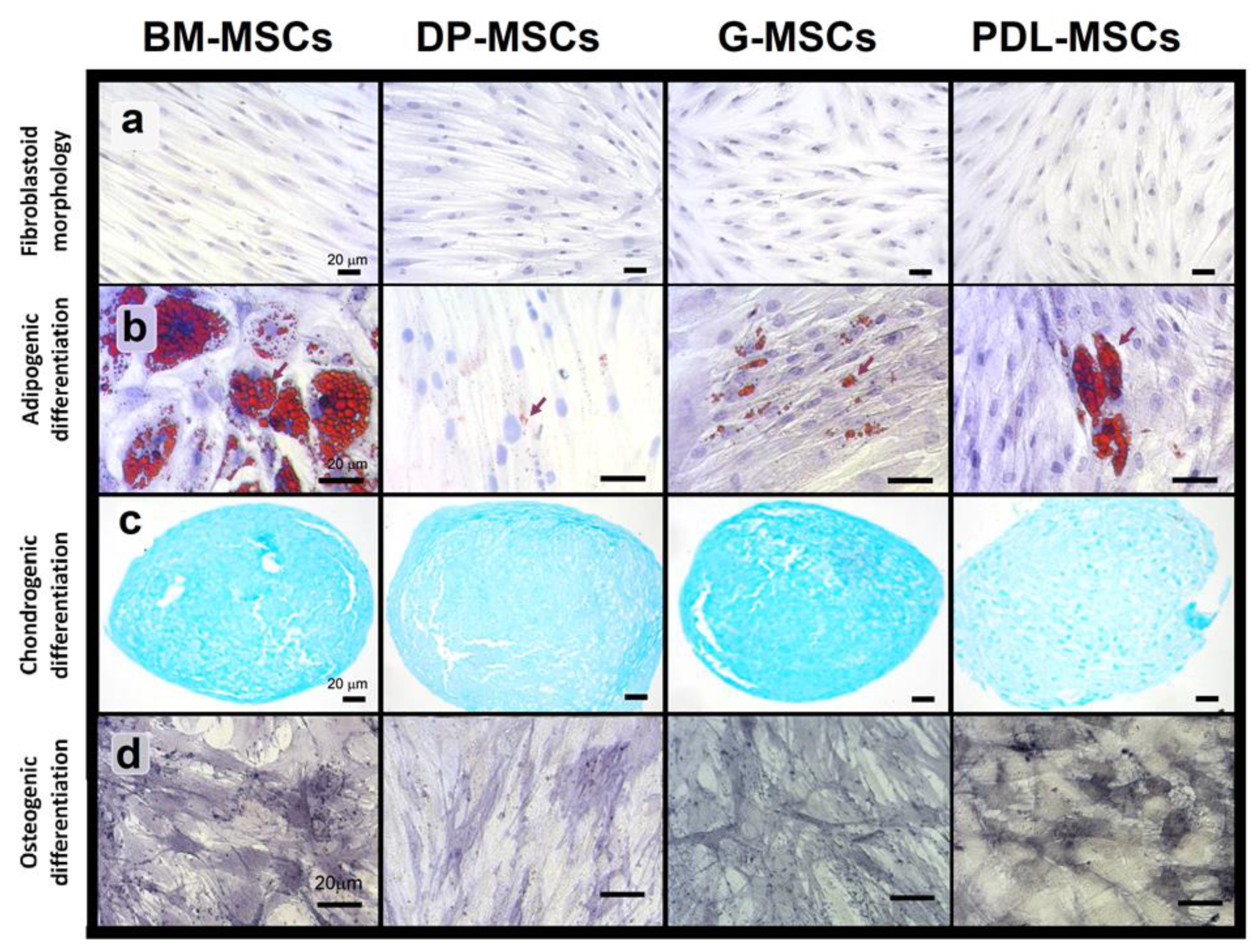
2.3.2. Morphological Analysis
2.3.3. Differentiation Capacity: Adipogenic
2.3.4. Osteogenic
2.3.5. Chondrogenic
2.4. Obtaining CD3+ T-cells
2.5. Activation of CD3+ T-cells
2.6. Co-culture of MSCs/CD3+ T-cells
2.7. Proliferation Assay
2.8. Expression of Immunoregulatory Molecules
2.9. Quantitative Analysis of Soluble Molecules
2.9.1. Cytokines
2.9.2. Prostaglandin E2
2.10. Statistical Analysis
3. Results
3.1. MSCs from Dental Tissues Show Immunophenotypes and Differentiation Capacities Similar to Those from Bone Marrow
3.2. CD4+ and CD8+ T-cell Proliferation Was Similarly Inhibited by Co-culture with MSCs from BM and Dental Tissues
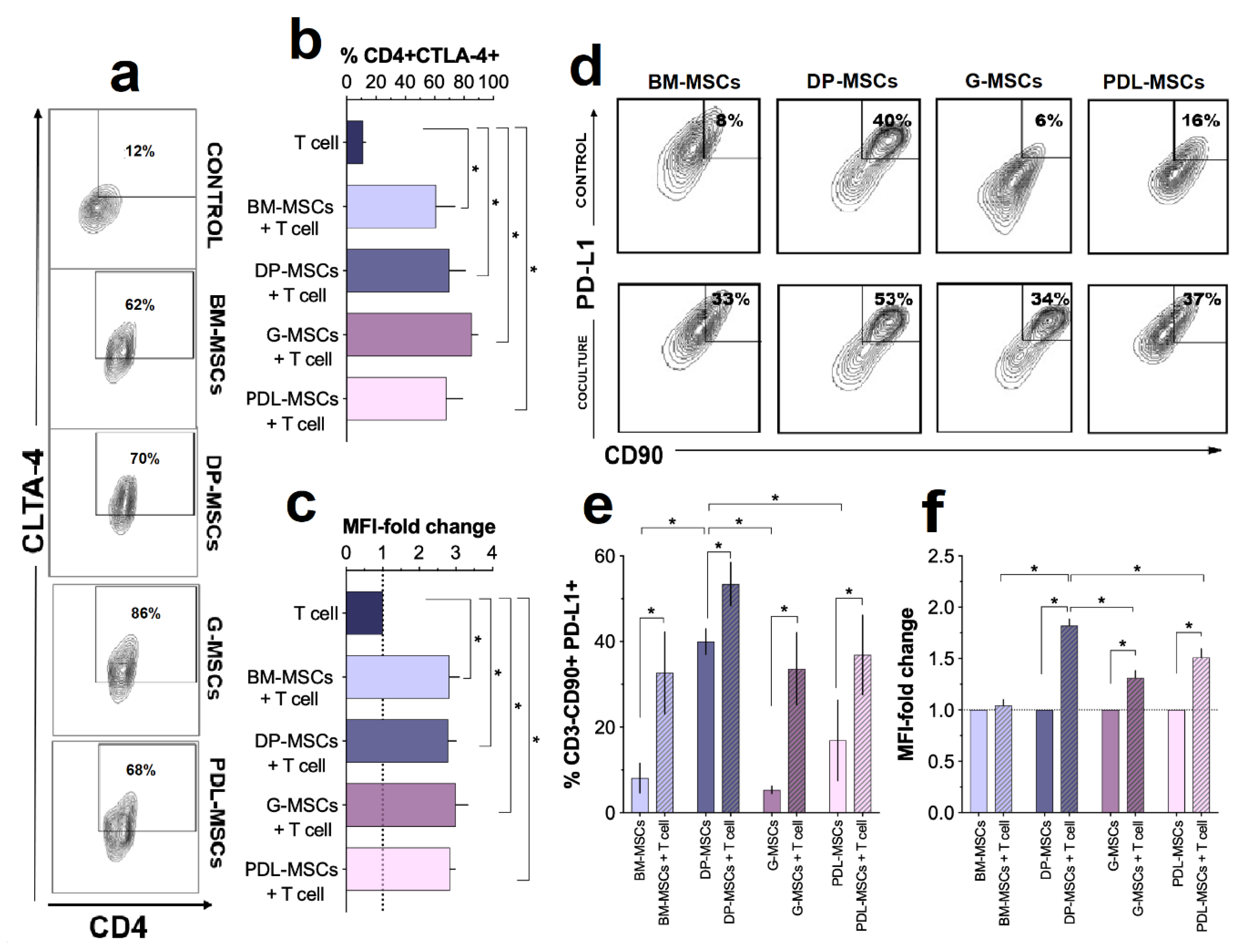
3.3. Increased Expression of the Immunosuppressive Molecules CTLA-4 and PD-L1
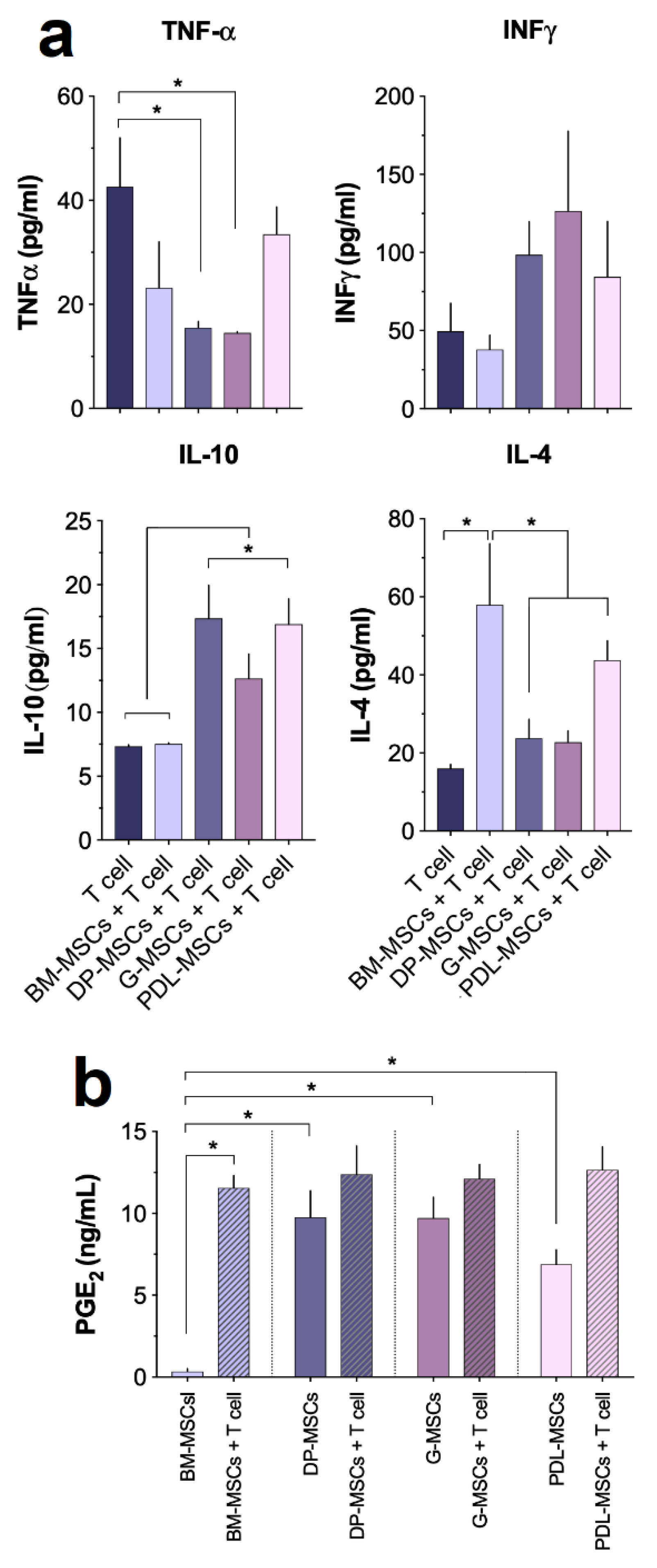
3.4. Cytokine and PGE2 Secretion in MSC Co-cultures
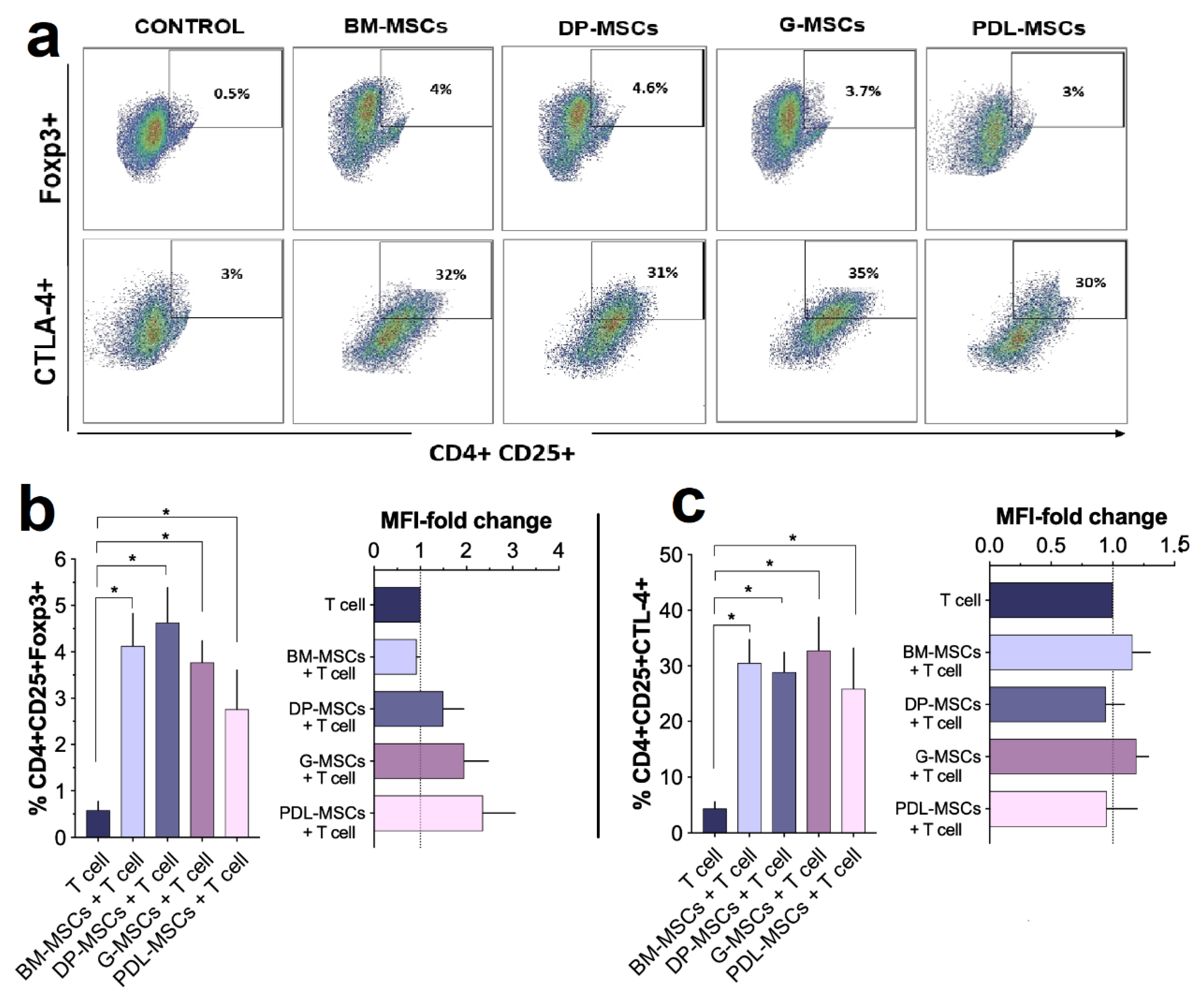
3.5. MSCs from Dental Tissues Induce a Similar Generation of T-cell Subsets Displaying a Regulatory Phenotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Prolif. 1970, 3, 393–403. [Google Scholar] [CrossRef]
- Pittenger, M.F. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Ren, H.; Sang, Y.; Zhang, F.; Liu, Z.; Qi, N.; Chen, Y. Comparative Analysis of Human Mesenchymal Stem Cells from Umbilical Cord, Dental Pulp, and Menstrual Blood as Sources for Cell Therapy. Stem Cells Int. 2016, 2016, 3516574. [Google Scholar] [CrossRef]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef]
- Meisel, R.; Zibert, A.; Laryea, M.; Gobel, U.; Daubener, W.; Dilloo, D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood 2004, 103, 4619–4621. [Google Scholar] [CrossRef]
- Kuzmina, L.A.; Petinati, N.A.; Parovichnikova, E.N.; Lubimova, L.S.; Gribanova, E.O.; Gaponova, T.V.; Shipounova, I.N.; Zhironkina, O.A.; Bigildeev, A.E.; Svinareva, D.A.; et al. Multipotent Mesenchymal Stromal Cells for the Prophylaxis of Acute Graft-versus-Host Disease-A Phase II Study. Stem Cells Int. 2012, 2012, 968213. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Philippe, B.; Luc, S.; Valerie, P.B.; Jerome, R.; Alessandra, B.R.; Louis, C. Culture and Use of Mesenchymal Stromal Cells in Phase I and II Clinical Trials. Stem Cells Int. 2010, 2010, 503593. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Why are MSCs therapeutic? New data: New insight. J. Pathol. 2009, 217, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.E.; Walker, J.T.; Keating, A. Concise Review: Wharton’s Jelly: The Rich, but Enigmatic, Source of Mesenchymal Stromal Cells. Stem Cells Transl. Med. 2017, 6, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Baer, P.C.; Geiger, H. Adipose-derived mesenchymal stromal/stem cells: Tissue localization, characterization, and heterogeneity. Stem Cells Int. 2012, 2012, 812693. [Google Scholar] [CrossRef] [PubMed]
- Riordan, N.H.; Ichim, T.E.; Min, W.P.; Wang, H.; Solano, F.; Lara, F.; Alfaro, M.; Rodriguez, J.P.; Harman, R.J.; Patel, A.N.; et al. Non-expanded adipose stromal vascular fraction cell therapy for multiple sclerosis. J. Transl. Med. 2009, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, J.J.; Flores-Figueroa, E.; Castillo-Medina, S.; Flores-Guzman, P.; Hernandez-Estevez, E.; Fajardo-Orduna, G.; Orozco, S.; Mayani, H. Human mesenchymal stromal cells from adult and neonatal sources: Comparative analysis of their morphology, immunophenotype, differentiation patterns and neural protein expression. Cytotherapy 2009, 11, 163–176. [Google Scholar] [CrossRef]
- Gorski, B. Gingiva as a new and the most accessible source of mesenchymal stem cells from the oral cavity to be used in regenerative therapies. Postepy. Hig. Med. Dosw. 2016, 70, 858–871. [Google Scholar] [CrossRef]
- Zhang, Q.; Shi, S.; Liu, Y.; Uyanne, J.; Shi, Y.; Shi, S.; Le, A.D. Mesenchymal stem cells derived from human gingiva are capable of immunomodulatory functions and ameliorate inflammation-related tissue destruction in experimental colitis. J. Immunol. 2009, 183, 7787–7798. [Google Scholar] [CrossRef]
- Wada, N.; Menicanin, D.; Shi, S.; Bartold, P.M.; Gronthos, S. Immunomodulatory properties of human periodontal ligament stem cells. J. Cell Physiol. 2009, 219, 667–676. [Google Scholar] [CrossRef]
- Yuan, H.; Zhang, H.; Hong, L.; Zhao, H.; Wang, J.; Li, H.; Che, H.; Zhang, Z. MicroRNA let-7c-5p Suppressed Lipopolysaccharide-Induced Dental Pulp Inflammation by Inhibiting Dentin Matrix Protein-1-Mediated Nuclear Factor kappa B (NF-kappaB) Pathway In Vitro and In Vivo. Med. Sci. Monit. 2018, 24, 6656–6665. [Google Scholar] [CrossRef]
- Castro-Manrreza, M.E.; Mayani, H.; Monroy-Garcia, A.; Flores-Figueroa, E.; Chavez-Rueda, K.; Legorreta-Haquet, V.; Santiago-Osorio, E.; Montesinos, J.J. Human mesenchymal stromal cells from adult and neonatal sources: A comparative in vitro analysis of their immunosuppressive properties against T cells. Stem Cells Dev. 2014, 23, 1217–1232. [Google Scholar] [CrossRef]
- Augello, A.; Tasso, R.; Negrini, S.M.; Amateis, A.; Indiveri, F.; Cancedda, R.; Pennesi, G. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur. J. Immunol. 2005, 35, 1482–1490. [Google Scholar] [CrossRef]
- Sheng, H.; Wang, Y.; Jin, Y.; Zhang, Q.; Zhang, Y.; Wang, L.; Shen, B.; Yin, S.; Liu, W.; Cui, L.; et al. A critical role of IFNgamma in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Res. 2008, 18, 846–857. [Google Scholar] [CrossRef]
- Ryan, J.M.; Barry, F.; Murphy, J.M.; Mahon, B.P. Interferon-gamma does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin. Exp. Immunol. 2007, 149, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Selmani, Z.; Naji, A.; Zidi, I.; Favier, B.; Gaiffe, E.; Obert, L.; Borg, C.; Saas, P.; Tiberghien, P.; Rouas-Freiss, N.; et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells 2008, 26, 212–222. [Google Scholar] [CrossRef] [PubMed]
- English, K.; Wood, K.J. Mesenchymal stromal cells in transplantation rejection and tolerance. Cold Spring Harb. Perspect. Med. 2013, 3, 15560. [Google Scholar] [CrossRef]
- Melief, S.M.; Schrama, E.; Brugman, M.H.; Tiemessen, M.M.; Hoogduijn, M.J.; Fibbe, W.E.; Roelofs, H. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells 2013, 31, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.T.; Lin, C.H.; Chiang, B.L.; Jui, H.Y.; Wu, K.K.; Lee, C.M. Prostaglandin E2 potentiates mesenchymal stem cell-induced IL-10+IFN-gamma+CD4+ regulatory T cells to control transplant arteriosclerosis. J. Immunol. 2013, 190, 2372–2380. [Google Scholar] [CrossRef]
- English, K.; Ryan, J.M.; Tobin, L.; Murphy, M.J.; Barry, F.P.; Mahon, B.P. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin. Exp. Immunol. 2009, 156, 149–160. [Google Scholar] [CrossRef]
- Krampera, M.; Cosmi, L.; Angeli, R.; Pasini, A.; Liotta, F.; Andreini, A.; Santarlasci, V.; Mazzinghi, B.; Pizzolo, G.; Vinante, F.; et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 2006, 24, 386–398. [Google Scholar] [CrossRef]
- Montesinos, J.J.; Mora-Garcia Mde, L.; Mayani, H.; Flores-Figueroa, E.; Garcia-Rocha, R.; Fajardo-Orduna, G.R.; Castro-Manrreza, M.E.; Weiss-Steider, B.; Monroy-Garcia, A. In vitro evidence of the presence of mesenchymal stromal cells in cervical cancer and their role in protecting cancer cells from cytotoxic T cell activity. Stem Cells Dev. 2013, 22, 2508–2519. [Google Scholar] [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Interleukin 6 Present in Inflammatory Ascites from Advanced Epithelial Ovarian Cancer Patients Promotes Tumor Necrosis Factor Receptor 2-Expressing Regulatory T Cells. Front Immunol. 2017, 8, 1482. [Google Scholar] [CrossRef]
- Plock, J.A.; Schnider, J.T.; Zhang, W.; Schweizer, R.; Tsuji, W.; Kostereva, N.; Fanzio, P.M.; Ravuri, S.; Solari, M.G.; Cheng, H.Y.; et al. Adipose- and Bone Marrow-Derived Mesenchymal Stem Cells Prolong Graft Survival in Vascularized Composite Allotransplantation. Transplantation 2015, 99, 1765–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawzy El-Sayed, K.M.; Dorfer, C.E. Gingival Mesenchymal Stem/Progenitor Cells: A Unique Tissue Engineering Gem. Stem Cells Int. 2016, 2016, 7154327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, B.P.; Ferre, F.C.; Couty, L.; Lataillade, J.J.; Gourven, M.; Naveau, A.; Coulomb, B.; Lafont, A.; Gogly, B. Multipotent progenitor cells in gingival connective tissue. Tissue Eng. Part A 2010, 16, 2891–2899. [Google Scholar] [CrossRef] [PubMed]
- Gronthos, S.; Mankani, M.; Brahim, J.; Robey, P.G.; Shi, S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 13625–13630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, G.; Niu, J.; Liu, Y. Dental pulp stem cells suppress the proliferation of lymphocytes via transforming growth factor-beta1. Hum. Cell 2015, 28, 81–90. [Google Scholar] [CrossRef]
- Tomic, S.; Djokic, J.; Vasilijic, S.; Vucevic, D.; Todorovic, V.; Supic, G.; Colic, M. Immunomodulatory properties of mesenchymal stem cells derived from dental pulp and dental follicle are susceptible to activation by toll-like receptor agonists. Stem Cells Dev. 2011, 20, 695–708. [Google Scholar] [CrossRef]
- Vasandan, A.B.; Shankar, S.R.; Prasad, P.; Sowmya Jahnavi, V.; Bhonde, R.R.; Jyothi Prasanna, S. Functional differences in mesenchymal stromal cells from human dental pulp and periodontal ligament. J. Cell Mol. Med. 2014, 18, 344–354. [Google Scholar] [CrossRef]
- Sibov, T.T.; Severino, P.; Marti, L.C.; Pavon, L.F.; Oliveira, D.M.; Tobo, P.R.; Campos, A.H.; Paes, A.T.; Amaro, E., Jr.; Gamarra, L.F.; et al. Mesenchymal stem cells from umbilical cord blood: Parameters for isolation, characterization and adipogenic differentiation. Cytotechnology 2012, 64, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Le Blanc, K.; Rasmusson, I.; Gotherstrom, C.; Seidel, C.; Sundberg, B.; Sundin, M.; Rosendahl, K.; Tammik, C.; Ringden, O. Mesenchymal stem cells inhibit the expression of CD25 (interleukin-2 receptor) and CD38 on phytohaemagglutinin-activated lymphocytes. Scand J. Immunol. 2004, 60, 307–315. [Google Scholar] [CrossRef]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Pierdomenico, L.; Bonsi, L.; Calvitti, M.; Rondelli, D.; Arpinati, M.; Chirumbolo, G.; Becchetti, E.; Marchionni, C.; Alviano, F.; Fossati, V.; et al. Multipotent mesenchymal stem cells with immunosuppressive activity can be easily isolated from dental pulp. Transplantation 2005, 80, 836–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, S.; Zibandeh, N.; Genc, D.; Ozcan, E.M.; Goker, K.; Akkoc, T. The Comparison of the Immunologic Properties of Stem Cells Isolated from Human Exfoliated Deciduous Teeth, Dental Pulp, and Dental Follicles. Stem Cells Int. 2016, 2016, 4682875. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, L.; Jin, Y.; Shi, S. Fas ligand regulates the immunomodulatory properties of dental pulp stem cells. J. Dent. Res. 2012, 91, 948–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Shi, S. Transplantation of gingiva-derived mesenchymal stem cells ameliorates collagen-induced arthritis. Arthritis Res. 2016, 18, 262. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zhao, X.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W.; Roberts, A.I.; Le, A.D.; Shi, S.; Shao, C.; et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef]
- Delasos, L.; Desai, A.; Lopetegui Lia, N.; Kethireddy, N.; Ray, C. A Case of Immunotherapy-Induced Colitis Complicated by Perforation and Treated with Infliximab Postoperatively. Case Rep. Oncol. Med. 2019, 2019, 9069354. [Google Scholar] [CrossRef]
- Wing, K.; Yamaguchi, T.; Sakaguchi, S. Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol. 2011, 32, 428–433. [Google Scholar] [CrossRef]
- Gaber, T.; Schonbeck, K.; Hoff, H.; Tran, C.L.; Strehl, C.; Lang, A.; Ohrndorf, S.; Pfeiffenberger, M.; Rohner, E.; Matziolis, G.; et al. CTLA-4 Mediates Inhibitory Function of Mesenchymal Stem/Stromal Cells. Int. J. Mol. Sci. 2018, 19, 2312. [Google Scholar] [CrossRef] [Green Version]
- Pinchuk, I.V.; Powell, D.W. Immunosuppression by Intestinal Stromal Cells. Adv. Exp. Med. Biol. 2018, 1060, 115–129. [Google Scholar]
- Yuan, W.; Wang, X.; Zhang, J.; Zhou, W.; Feng, Y.; Chen, J.; Zhang, P. Programmed death ligand 1 negatively regulates inflammatory response of chronic periodontitis. West China J. Stomatol. 2015, 33, 366–369. [Google Scholar]
- Luz-Crawford, P.; Noel, D.; Fernandez, X.; Khoury, M.; Figueroa, F.; Carrion, F.; Jorgensen, C.; Djouad, F. Mesenchymal stem cells repress Th17 molecular program through the PD-1 pathway. PLoS ONE 2012, 7, 45272. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Cao, K.; Cao, J.; Wang, Y.; Shi, Y. Mesenchymal stem cells and adaptive immune responses. Immunol. Lett. 2015, 168, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Singer, N.G.; Caplan, A.I. Mesenchymal stem cells: Mechanisms of inflammation. Annu. Rev. Pathol. 2011, 6, 457–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tipnis, S.; Viswanathan, C.; Majumdar, A.S. Immunosuppressive properties of human umbilical cord-derived mesenchymal stem cells: Role of B7-H1 and IDO. Immunol. Cell Biol. 2010, 88, 795–806. [Google Scholar] [CrossRef]
- Polchert, D.; Sobinsky, J.; Douglas, G.; Kidd, M.; Moadsiri, A.; Reina, E.; Genrich, K.; Mehrotra, S.; Setty, S.; Smith, B.; et al. IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur. J. Immunol. 2008, 38, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.J.; Yen, M.L.; Chen, Y.C.; Chien, C.C.; Huang, H.I.; Bai, C.H.; Yen, B.L. Placenta-derived multipotent cells exhibit immunosuppressive properties that are enhanced in the presence of interferon-gamma. Stem Cells 2006, 24, 2466–2477. [Google Scholar] [CrossRef]
- Alipour, R.; Masoumi Karimi, M.; Hashemi-Beni, B.; Adib, M.; Sereshki, N.; Sadeghi, F. Indoleamine 2,3-Dioxygenase Is Dispensable for The Immunomodulatory Function of Stem Cells from Human Exfoliated Deciduous Teeth. Cell J. 2017, 18, 597–608. [Google Scholar]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef]
- Luan, X.; Li, G.; Wang, G.; Wang, F.; Lin, Y. Human placenta-derived mesenchymal stem cells suppress T cell proliferation and support the culture expansion of cord blood CD34(+) cells: A comparison with human bone marrow-derived mesenchymal stem cells. Tissue Cell 2013, 45, 32–38. [Google Scholar] [CrossRef]
- Nojehdehi, S.; Soudi, S.; Hesampour, A.; Rasouli, S.; Soleimani, M.; Hashemi, S.M. Immunomodulatory effects of mesenchymal stem cell-derived exosomes on experimental type-1 autoimmune diabetes. J. Cell Biochem. 2018, 119, 9433–9443. [Google Scholar] [CrossRef]
- Li, P.; Zhao, Y.; Ge, L. Therapeutic effects of human gingiva-derived mesenchymal stromal cells on murine contact hypersensitivity via prostaglandin E2-EP3 signaling. Stem Cell Res. 2016, 7, 103. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.W.; Lim, J.H.; Chung, C.J.; Kang, T.J.; Kim, T.Y.; Kim, Y.S.; Roh, T.S.; Lew, D.H. Immune Tolerance of Human Dental Pulp-Derived Mesenchymal Stem Cells Mediated by CD4(+)CD25(+)FoxP3(+) Regulatory T-Cells and Induced by TGF-beta1 and IL-10. Yonsei. Med. J. 2017, 58, 1031–1039. [Google Scholar] [CrossRef]
- Ng, J.; Hynes, K.; White, G.; Sivanathan, K.N.; Vandyke, K.; Bartold, P.M.; Gronthos, S. Immunomodulatory Properties of Induced Pluripotent Stem Cell-Derived Mesenchymal Cells. J. Cell Biochem. 2016, 117, 2844–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, X.; Xia, Y.; Zhou, S.; Peng, H.; Wu, X.; Lu, H.; Wang, H.; Liu, R.; Blazar, B.R.; Gu, J.; et al. Reduction in murine acute GVHD severity by human gingival tissue-derived mesenchymal stem cells via the CD39 pathways. Cell Death Dis. 2019, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naamani, O.; Chaimovitz, C.; Douvdevani, A. Pharmacological preconditioning with adenosine A(1) receptor agonist suppresses cellular immune response by an A(2A) receptor dependent mechanism. Int. Immunopharmacol. 2014, 20, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Sitkovsky, M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol. 2014, 5, 304. [Google Scholar] [CrossRef] [PubMed]
- Fajardo-Orduna, G.R.; Mayani, H.; Montesinos, J.J. Hematopoietic Support Capacity of Mesenchymal Stem Cells: Biology and Clinical Potential. Arch. Med. Res. 2015, 46, 589–596. [Google Scholar] [CrossRef]
- Martin-Manso, G.; Hanley, P.J. Using the quantum cell expansion system for the automated expansion of clinical-grade bone marrow-derived human mesenchymal stromal cells. Methods Mol. Biol. 2015, 1283, 53–63. [Google Scholar]
- Carmelo, J.G.; Fernandes-Platzgummer, A.; Cabral, J.M.; da Silva, C.L. Scalable ex vivo expansion of human mesenchymal stem/stromal cells in microcarrier-based stirred culture systems. Methods Mol.Biol 2015, 1283, 147–159. [Google Scholar]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De la Rosa-Ruiz, M.d.P.; Álvarez-Pérez, M.A.; Cortés-Morales, V.A.; Monroy-García, A.; Mayani, H.; Fragoso-González, G.; Caballero-Chacón, S.; Diaz, D.; Candanedo-González, F.; Montesinos, J.J. Mesenchymal Stem/Stromal Cells Derived from Dental Tissues: A Comparative In Vitro Evaluation of Their Immunoregulatory Properties Against T cells. Cells 2019, 8, 1491. https://doi.org/10.3390/cells8121491
De la Rosa-Ruiz MdP, Álvarez-Pérez MA, Cortés-Morales VA, Monroy-García A, Mayani H, Fragoso-González G, Caballero-Chacón S, Diaz D, Candanedo-González F, Montesinos JJ. Mesenchymal Stem/Stromal Cells Derived from Dental Tissues: A Comparative In Vitro Evaluation of Their Immunoregulatory Properties Against T cells. Cells. 2019; 8(12):1491. https://doi.org/10.3390/cells8121491
Chicago/Turabian StyleDe la Rosa-Ruiz, María del Pilar, Marco Antonio Álvarez-Pérez, Víctor Adrián Cortés-Morales, Alberto Monroy-García, Héctor Mayani, Gladis Fragoso-González, Sara Caballero-Chacón, Daniel Diaz, Fernando Candanedo-González, and Juan José Montesinos. 2019. "Mesenchymal Stem/Stromal Cells Derived from Dental Tissues: A Comparative In Vitro Evaluation of Their Immunoregulatory Properties Against T cells" Cells 8, no. 12: 1491. https://doi.org/10.3390/cells8121491